
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable surface functionalization of carbonized mesoporous silicon†
Joakim
Riikonen
 abc,
Tuomo
Nissinen
a,
Aino
Alanne
d,
Rinez
Thapa
a,
Philippe
Fioux
bc,
Magali
Bonne
bc,
Séverinne
Rigolet
bc,
Fabrice
Morlet-Savary
bc,
Fabien
Aussenac
e,
Claire
Marichal
bc,
Jacques
Lalevée
bc,
Jouko
Vepsäläinen
d,
Bénédicte
Lebeau
bc and
Vesa-Pekka
Lehto
*a
abc,
Tuomo
Nissinen
a,
Aino
Alanne
d,
Rinez
Thapa
a,
Philippe
Fioux
bc,
Magali
Bonne
bc,
Séverinne
Rigolet
bc,
Fabrice
Morlet-Savary
bc,
Fabien
Aussenac
e,
Claire
Marichal
bc,
Jacques
Lalevée
bc,
Jouko
Vepsäläinen
d,
Bénédicte
Lebeau
bc and
Vesa-Pekka
Lehto
*a
aDepartment of Applied Physics, University of Eastern Finland, Yliopistonranta 1F, 70211 Kuopio, Finland. E-mail: vesa-pekka.lehto@uef.fi
bUniversité de Haute Alsace, CNRS, IS2M UMR 7361, F-68100 Mulhouse, France
cUniversité de Strasbourg, France
dSchool of Pharmacy, University of Eastern Finland, Yliopistonranta 1B, 70211 Kuopio, Finland
eBruker Biospin SA, 34, rue de l'industrie, 67166 Wissembourg Cedex, France
First published on 22nd November 2019
Abstract
Mesoporous silicon (PSi) is an emerging nanomaterial studied in e.g. biomedical, sensor and energy applications. In many applications, a major obstacle in its commercial use is the instability of its surfaces, especially when functionalized with organic molecules. In the present work, we introduce a surface functionalization method for PSi, in which carbonized surface of silicon is functionalized with terminal alkenes. A good surface coverage of 0.3 molecules per nm2 was achieved and the material showed excellent aqueous stability at low and neutral pH. It also withstood a highly basic solution for several days. The developed method was used to graft bisphosphonates on the surface and the material was used for metal adsorption. Because of its excellent stability, the adsorbent material lasted up to 50 adsorption/desorption cycles without a significant deterioration of its performance.
1. Introduction
Mesoporous silicon (PSi) has many potential applications in e.g. drug delivery, sensing, Li-ion batteries and adsorption. It is relatively unstable in its native form because of its high surface area and reactivity of the Si–Si bonds. Therefore, surface of PSi is typically passivated by oxidation, carbonization or hydrosilylation to improve its stability.Many applications of PSi, such as sensing and separation, require grafting of functional molecules on the surface.1,2 The stability of these surfaces has been a focus of several studies.2–6 Among the first functionalization strategies of PSi was grafting of alkenes, or alkynes on PSi by hydrosilylation.7 Although the highly hydrophobic surfaces passivated by hydrosilylation are shown to be stable for several hours even at pH 13,8 the grafting of molecules with functional groups, such as carboxylic acids, leads to significantly less stable materials.6 Good to moderate stability of the hydrosilylated materials in aqueous solutions near neutral pH has been shown only up to few hours3,9 with the exception of a 2-month stability study conducted by Kilian et al.5 They observed dissolution of PSi in phosphate buffered saline visually after one month but also saw significant changes in the structure in the first few days. However, the amount of the grafted molecules on the surface was not studied directly.
Many surface functionalization strategies rely on grafting organic molecules on oxidized PSi via Si–O bonds which are easily hydrolyzed in an aqueous environment and are thus sensitive to degradation.10 A more stable functionalization can be achieved if only stable Si–C and C–C bonds are exposed on the surface. Sciacca et al. and Jalkanen et al. studied hydrophobic PSi that was thermally hydrocarbonized at 500 °C and subsequently functionalized with a carboxylic acid.4,6 Sciacca et al. observed an improved structural stability of the material in phosphate buffered saline for 2 h. Jalkanen et al. studied the stability in harsher conditions and observed complete dissolution of the material in 1 M KOH in 24 h.
Thermal carbonization of PSi at high temperatures (above 800 °C) has been shown to produce highly stable surfaces consisting of silicon carbide, silicon oxycarbide and carbon.11,12 So far, functionalization of thermally carbonized PSi (TCPSi) has been performed via silanization which grafts functional molecules on the surface via Si–O bonds leaving the functional molecules susceptible to hydrolysis.13
In the present study, a new functionalization method for TCPSi is introduced in which terminal alkenes are directly grafted on the carbonized silicon surface leading to functionalization which is highly stable in aqueous solutions for several weeks and resists even high pH solutions for several days.
The developed method is used to functionalize TCPSi with bisphosphonates that are good chelation agents for metals.14 Several mesoporous materials have been proposed for metal adsorption applications15–20 but their repeated use has often not been studied supposedly due to their low stability and diminishing performance. If repeated use is studied, it is reported for less than ten ad/desorption cycles and even the most stable materials lose at least 1% of their capacity per cycle.15–17 The aim of the present study is to provide a material that can be reused several tens of times without significant reduction of its capacity thus essentially reducing the costs in demanding applications.
2. Experimental
2.1. Production of PSi
PSi was produced by electrochemical etching in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HF (38–40%)/EtOH mixture on p+ Si wafers (0.01–0.02 Ω cm) with 40 mA cm−2 current density for 2400 s. HF is highly toxic and a special care should be taken to prevent any contact with the liquid and inhalation of vapor. PSi films were detached from the wafer with a high current pulse and dried at 65 °C for 2 h. The PSi films were milled to microparticles in a planetary ball mill and sieved to 25 μm–75 μm size fraction. PSi was used in different forms, microparticles, freestanding films or films supported on the substrate depending on the demands of measurement in order to obtain high quality data. Films supported on the substrate were produced similarly as above by etching a 5 μm porous layer on the wafer without the high current pulse.
:1 HF (38–40%)/EtOH mixture on p+ Si wafers (0.01–0.02 Ω cm) with 40 mA cm−2 current density for 2400 s. HF is highly toxic and a special care should be taken to prevent any contact with the liquid and inhalation of vapor. PSi films were detached from the wafer with a high current pulse and dried at 65 °C for 2 h. The PSi films were milled to microparticles in a planetary ball mill and sieved to 25 μm–75 μm size fraction. PSi was used in different forms, microparticles, freestanding films or films supported on the substrate depending on the demands of measurement in order to obtain high quality data. Films supported on the substrate were produced similarly as above by etching a 5 μm porous layer on the wafer without the high current pulse.
2.2. Bisphosphonate synthesis
10-Undecenoic acid, oxalyl chloride and P(OSiMe3)3 were purchased from Aldrich. CDCl3 (D 99.96%) used as NMR solvent was purchased from Euriso-Top.The bisphosphonate molecule (Tetrakis(trimethylsilyl) 1-(trimethylsilyloxy)undec-10-ene-1,1-diylbisphosphonate) was synthesized according to the method published by Lecouvey et al. (ESI Fig. S1†).21 10-undecenoic acid (13.56 mmol, 2.50 g) in 20 ml of freshly distilled dichloromethane was placed in dried two necked flask equipped with an argon inlet. Oxalyl chloride (27.10 mmol, 3.44 g) was added dropwise on ice. The reaction mixture was stirred at room temperature for 1 hour, evaporated and the formation of the corresponding acid chloride was observed by NMR. Two equivalents of tris(trimethyl silyl) phosphite (27.10 mmol, 8.09 g) was added dropwise to the acid chloride under argon at 0 °C. Once the addition was complete, the reaction mixture was left under magnetic stirring at room temperature for 2 h. Volatile fractions were then evaporated under reduced pressure. Yellow oily substance with 94% yield was obtained. 1H NMR (500.1 MHz, CDCl3) δ 5.80 (ddt, 1H, 3JHH = 17.2, 10.3 and 6.7 Hz, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –CH2), 4.98 (dd, 1H, 3JHH = 17.2, 2JHH = 2.2, C2CH), 4.92 (d, 1H, 3JHH = 10.3, 2JHH = 2.2, C2CH), 2.07–1.99 (m, 2H), 1.96–1.84 (m, 2H), 1.60–1.51 (m, 2H), 1.41–1.33 (m, 2H), 1.33–1.20 (m, 8H), 0.34–0.25 (m, 45H); 13C NMR (125.8 MHz, CDCl3) δ 139.0, 114.1, 78.2 (t, 1JCP = 166.2 Hz), 35.6, 33.7, 30.3, 29.4, 29.4, 29.0, 28.9, 23.8 (t, 2JCP = 6.5 Hz), 2.8 (3C), 1.4 (6C), 1.3 (6C); 31P NMR (202.5 MHz, CDCl3) δ 1.95 (s). The compound was left in silylated form to achieve good solubility in mesitylene.
–CH2), 4.98 (dd, 1H, 3JHH = 17.2, 2JHH = 2.2, C2CH), 4.92 (d, 1H, 3JHH = 10.3, 2JHH = 2.2, C2CH), 2.07–1.99 (m, 2H), 1.96–1.84 (m, 2H), 1.60–1.51 (m, 2H), 1.41–1.33 (m, 2H), 1.33–1.20 (m, 8H), 0.34–0.25 (m, 45H); 13C NMR (125.8 MHz, CDCl3) δ 139.0, 114.1, 78.2 (t, 1JCP = 166.2 Hz), 35.6, 33.7, 30.3, 29.4, 29.4, 29.0, 28.9, 23.8 (t, 2JCP = 6.5 Hz), 2.8 (3C), 1.4 (6C), 1.3 (6C); 31P NMR (202.5 MHz, CDCl3) δ 1.95 (s). The compound was left in silylated form to achieve good solubility in mesitylene.
2.3. Surface modifications
Three independent surface modifications were performed for each sample type. First, hydrogen terminated PSi (HTPSi) was prepared by briefly immersing PSi in 1:1HF/EtOH mixture and drying at 65 °C for 2 h.
The carbonization was performed to the HTPSi in a quartz tube under continuous N2 flow of 1 L min−1 throughout the process. Samples were first flushed with nitrogen for 30 min then acetylene flow of 1 L min−1 was added for 15 min. The sample was then placed into a preheated oven at 500 °C. After 15 min at 500 °C, the acetylene flow was terminated and the tube was removed from the oven and sample were let to cool down to room temperature (in nitrogen flow). At room temperature, acetylene flow (1 L min−1) was applied again for 9 min 40 s and the sample was inserted into the oven at 820 °C 20 s after the acetylene flow was terminated. Sample was heated in the oven for 10 min and then cooled down to room temperature (in nitrogen flow).
For surface functionalization of TCPSi, the neat alkene (undecylenic acid or decene) was added on the TCPSi in the quartz tube and the tube was heated at 120 °C for 16 h. Bisphosphonate functionalization was performed with a mesitylene solution22 of BP (9 wt%, 1:1 mass ratio of BP and PSi). The undecylenic acid grafted TCPSi is labeled UnTCPSi, the decene grafted TCPSi DeTCPSi and the bisphosphonate grafted TCPSi BPTCPSi. The structures of all the PSi materials used in the study are presented in ESI Fig. S2.† The UnTCPSi samples were rinsed thrice with 25 ml chloroform to remove excess undecylenic acid and then six times with 25 ml of a 1:1 mixture of EtOH and 1 M NaOH in order to remove physisorbed or dimerized undecylenic acid. The particles were then washed with water and 1 M HCl to convert grafted undecylenic acid from Na salt to the acid form and finally rinsed with water to remove HCl and dried at 65 °C. DeTCPSi was washed eight times with 25 ml of chloroform and dried at 65 °C for 1 h. BPTCPSi was washed eight times with 25 ml of chloroform, then with methanol and dried at 65 °C for 1 h.
To prepare undecylenic acid grafted HTPSi (UnHTPSi), HTPSi was immersed in neat undecylenic acid and heated at 120 °C for 16 h. The material was washed eight times with 25 ml of chloroform and dried at 65 °C for 1 h.
2.4. Liquid NMR
1H, 31P and 13C NMR spectra were measured on a Bruker Avance 500 DRX spectrometer operating at 500.1, 202.5 and 125.8 MHz, respectively. The NMR spectra measured in CDCl3 were calibrated on the solvent residual signals at 7.26 ppm for 1H and 77.1 ppm for 13C and 85% H3PO4 was used as an external standard in the 31P measurements.2.5. Nitrogen sorption
Prior to the measurements, the samples were degassed in vacuum at 65 °C for 2 h. The nitrogen adsorption and desorption isotherms were measured with Micromeritics Tristar II 3020. Surface area was calculated using BET (Brunauer, Emmett and Teller) theory, pore size distributions were calculated with BJH (Barrett, Joyner and Halenda) theory and pore volume was calculated from a single point at relative pressure of 0.95.2.6. Electron paramagnetic resonance (EPR)
TCPSi microparticles which were not exposed to air were transferred under nitrogen atmosphere into a glove box with Ar atmosphere. In the glove box, the sample was transferred into 6 mm cylindrical quartz tubes and closed. Electron paramagnetic resonance experiments were performed using an X-Band spectrometer (Bruker EMX-plus Biospin). The radicals were observed under Ar atmosphere at room temperature. The experimental procedure has previously been described in detail.23 The double integral of the EPR spectrum is proportional to the concentration of paramagnetic centers. A calibration was first done using TEMPO to ensure that this procedure was usable in solid state. TEMPO was also used as standard for calibration of the g-factor (g = 2.0061). To measure change in paramagnetic species caused by addition of undecylenic acid on the TCPSi particles that had not been exposed to air, undecylenic acid was injected into the quartz tube and the EPR spectrum was measured immediately. In addition to the above measurements TCPSi that had been in contact with air was measured in air atmosphere.2.7. Solid state NMR characterization
1H–29Si CPMAS NMR experiments were performed at room temperature, with a Bruker Avance II 300 spectrometer operating at B0 = 7.1 T (Larmor frequency: ω0(29Si) = 59.61 MHz and ω0(1H) = 300.08 MHz) with a Bruker double channel 7 mm probe at a spinning frequency of 4 kHz, with a proton π/2 pulse duration of 4.1 μs, a contact time of 4 ms and a recycle delay of 5 s according to T1 measurements. 29Si Chemical shifts were referenced to tetramethylsilane (TMS).31P solid-state MAS NMR experiments were performed at room temperature on Bruker Avance II 400 spectrometer operating at B0 = 9.4 T equipped with a Bruker double channel 4 mm probe at a Larmor frequency of 161.99 MHz. The spectrum was recorded with a π/2 pulse duration of 3 μs and a recycling delay of 5 s at a spinning frequency of 12 kHz. 31P spin lattice relaxation times (T1) were measured with the saturation-recovery pulse sequence. 31P spectrum was referenced to H3PO4 (85% in water).
2.8. DNP NMR characterization
Solid State DNP experiments were performed with a Bruker 263 GHz Solid-State NMR DNP spectrometer with a gyrotron microwave source,24,25 Ascend DNP 400 WB NMR magnet, Avance III NMR console and a low-temperature 3.2 mm triple resonance 1H/X/Y DNP MAS probe. The NMR 400 MHz Ascend DNP magnet is equipped with a sweep coil for optimization of the frequency match between the NMR and the electron frequencies. The sample temperature reported was 108 K with microwave irradiation. The microwave power was optimized to achieve the highest DNP signal enhancement. 1H–13C cross polarization (CP) MAS experiments were performed with Spinal64261H decoupling during acquisition time. During the CP contact time, both 1H and 13C R.F. field were set to 60 kHz, the 1H decoupling R.F. field strength was set to 95 kHz during the acquisition time. The contact time during the CP step was set to 1 ms. The sample spinning frequency was set to 12 kHz. 8192 scans were acquired with a recycle delay of 6 s which corresponds to a total acquisition time of about 13.5 h.DNP polarization buildup time (TDNP) measurements were performed using saturation-recovery experiments and results were fit in the T1/T2 relaxation module of Topspin software.
Approximately 10 mg of TCPSi microparticles were loaded in a 3.2 mm sapphire rotor. The rotor was sealed with a Teflon top insert and a Zirconia cap. Chemical shifts were referenced to tetramethylsilane (TMS).
2.9. XPS analysis
X-ray photoelectron spectroscopy (XPS) spectra were recorded with a VG SCIENTA SES-2002 spectrometer equipped with a concentric hemispherical analyzer. The incident radiation used was generated by a monochromatic Al Kα X-ray source (1486.6 eV) operating at 420 W (14 kV; 30 mA). Photo-emitted electrons were collected at a take-off angle of 90° from the surface substrate, with electron detection in the constant analyser energy mode (FAT). Widescan spectrum signal was recorded with a pass energy of 500 eV and for high resolution spectra (Si 2p, C 1s and Cu 2p) pass energy was set to 100 eV. Approximately 24 mm2 area of TCPSi film on Si substrate was analysed. The base pressure in the analysis chamber during experimentation was ca. 10−9 mbar. The spectrometer energy scale was calibrated using the Ag 3d5/2, Au 4f7/2 and Cu 2p3/2 core level peaks, set respectively at binding energies of 368.2, 84.0 and 932.6 eV. Spectra were subjected to a Shirley baseline and peak fitting was made with mixed Gaussian–Lorentzian components with equal full-width-at-half-maximum (FWHM) using CASAXPS version 2.3.17 software. The surface composition expressed in atom% was determined using integrated peak areas of each component and take into account transmission factor of the spectrometer, mean free path and Scofield sensitivity factors of each atom. All the binding energies (BE) are referenced to the C 1s peak at 284.5 eV and given with a precision of 0.1 eV. The measured samples were porous films on silicon substrate except for the BPTCPSi samples which were measured as microparticles.2.10. FTIR analysis
FTIR measurements were done in transmission mode with freestanding PSi films with Thermo Scientific Nicolet 8700. The spectral range was 500–4000 cm−1 and resolution 4 cm−1.2.11. Thermogravimetry
Microparticles were measured by TA TGA Q50 under nitrogen atmosphere. The samples were equilibrated at 80 °C for 30 min and then heated up to 700 °C with 20 °C min−1 heating rate.2.12. Scanning electron microscopy
Zeiss HD|VP HR-SEM was utilized for investigation of the morphologies of the particles. The particles were attached to standard aluminum SEM sample holders with conductive carbon adhesive. The imaging was performed with acceleration voltage of 5 kV and InLens detector.2.13. Stability study
One hundred mg of UnHTPSi and UnTCPSi microparticles were first wetted with EtOH and then immersed in 45 ml of 1 M HCl, deionized water or 1 M NaOH. The samples were kept at 30 °C. Part of the samples were removed at predefined time points and washed on a filter paper 3× with 50 ml of deionized water, 3× with 50 ml of 1:1 mixture of deionized water and ethanol and 4× with 50 ml of EtOH. The particles were then dried at 65 °C. At each time point the liquid in which the particles were immersed was replaced with a fresh one.
2.14. Metal adsorption
Metal adsorption capacity towards copper was investigated using ∼200 mg BPTCPSi particles primed in 20 ml of 5 M HCl for 1 h at RT. After washing thrice with 20 ml water, the particles were immersed in 20 ml of 200 ppm Cu2+ solution (from CuCl2) at pH = 4.02 with constant mixing. The contact time was 24 h at RT. Cu concentration of the solution was measured before and after adsorption using total reflection X-ray fluorescence spectroscopy, tXRF (S2 Picofox, Bruker). The adsorbed amount was calculated from the concentration difference of the solutions. The reusability of the material was studied by repeating 50 adsorption/desorption cycles. 50 mg of BP-TCPSi particles were placed inside a syringe filter and the following solutions were passed through the particles by gravitation in each cycle: 5 ml of 10 ppm Cu2+, 5 ml of deionized H2O, 5 ml of 0.1 M HCL and 5 ml of deionized H2O. Cu concentrations of the solution before and after each cycle was measured with PerkinElmer 5100 flame atomic absorption spectrometer (AAS) with a wavelength of 327.4 nm to calculate the adsorbed/desorbed amounts of Cu.3. Results and discussion
Thermal carbonization of PSi was performed under nitrogen atmosphere with acetylene gas in a two-step process at 500 and 820 °C.11 After carbonization, the material is cooled to room temperature in nitrogen gas. According to a previous observation, the material will react strongly with a sudden exposure to air and emit bright light (see video in ESI†). A TCPSi sample, that had not been exposed to air, was measured with EPR and observed a high number of carbon radicals in the material (3.4 × 1018 g−1). We hypothesized that these radical groups could be utilized to functionalize the material by free radical addition. Therefore, undecylenic acid or 1-decene was added on the TCPSi immediately after carbonization before exposing the material to air. The mixture was then heated at 120 °C for 16 h to further enhance the grafting. The undecylenic acid and 1-decene modified TCPSi samples are referred to as UnTCPSi and DeTCPSi, respectively. Bisphosphonate (BP) grafted TCPSi (BPTCPSi) was also prepared in a similar way for metal adsorption experiments (see Materials and methods for details). A reference sample was prepared by grafting undecylenic acid on hydrogen terminated PSi (UnHTPSi) using conventional hydrosilylation by immersing hydrogen terminated PSi (HTPSi) in undecylenic acid at 120 °C for 16 h. All the surface functionalizations were performed in triplicates to assess repeatability of the method.The amount of grafted molecules on the samples, measured by thermogravimetry (TG), was 2.3 ± 0.3 wt% in UnTCPSi, 4.6 ± 0.3 wt% in DeTCPSi and 2.7 ± 0.8 wt% in UnHTPSi corresponding to grafting densities of 0.3, 0.9 and 0.4 molecules per nm2, respectively (Table 1). Analysis of UnHTPSi was difficult because mass increase was taking place during the measurement presumably because of oxidation of the material by residual oxygen in the instrument. Very little mass loss was observed in UnTCPSi under 350 °C proving that undecylenic acid was covalently grafted on the surface whereas in DeTCPSi mass loss begun already at 200 °C indicating that some physisorbed molecules were also present (Fig. 1). A control sample was also prepared by exposing a TCPSi sample to air after the carbonization and subsequently immersing it in undecylenic acid at 120 °C for 16 h. In this sample, a significant amount of undecylenic acid was grafted on the TCPSi surface but the mass of the organic layer was 46% smaller than in the sample not exposed to air before functionalization.
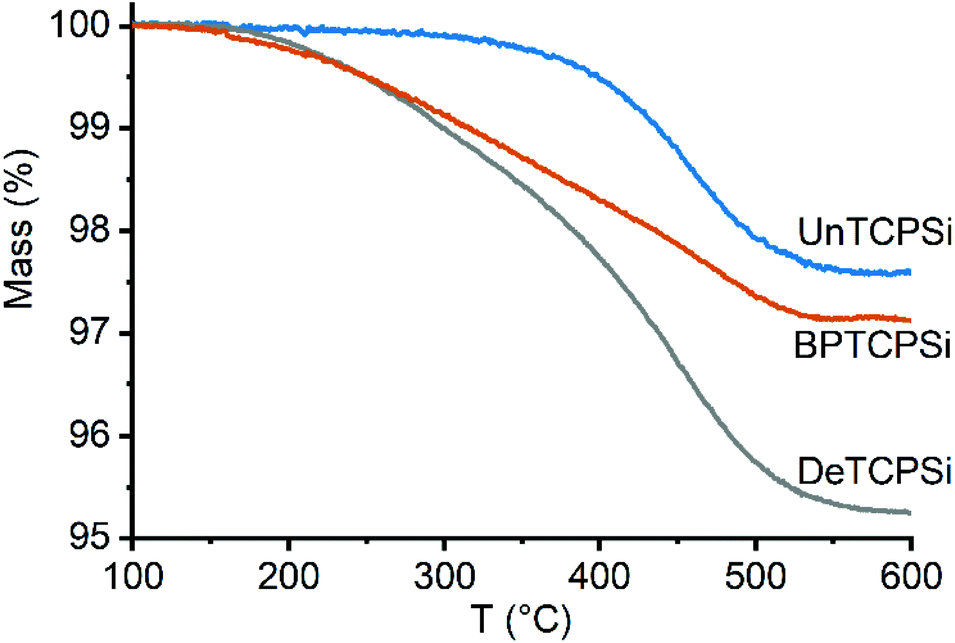 | ||
| Fig. 1 TG curves of functionalized TCPSi samples showing mass loss due to decomposition of the functional layer. | ||
| A (m2 g−1) |
C
BETb |
V (cm3 g−1) | D (nm) | m (%) | Coveragef (nm−2) | |
|---|---|---|---|---|---|---|
| a Specific surface area from nitrogen adsorption calculated by BET theory. b BET constant. c Specific pore volume area calculated from a single point (p/p0 = 0.95) on nitrogen adsorption isotherm. d Average pore diameter calculated by BJH theory from nitrogen desorption isotherms. e Mass of functional molecules divided by the total mass of the sample as measured by TG. f Number of functional molecules per surface area. | ||||||
| HTPSi | 242 ± 1 | 28.8 ± 0.3 | 1.33 ± 0.01 | 17.0 ± 0.1 | — | — |
| UnHTPSi | 211 ± 7 | 35.0 ± 0.4 | 0.94 ± 0.08 | 15.2 ± 0.2 | 2.7 ± 0.8 | 0.4 ± 0.2 |
| TCPSi | 240 ± 5 | 73.9 ± 0.6 | 0.98 ± 0.02 | 15.8 ± 0.1 | — | — |
| UnTCPSi | 223 ± 4 | 57 ± 6 | 0.91 ± 0.01 | 15.6 ± 0.2 | 2.3 ± 0.3 | 0.34 ± 0.04 |
| DeTCPSi | 220 ± 20 | 35 ± 3 | 0.9 ± 0.1 | 15.2 ± 0.5 | 4.7 ± 0.3 | 0.90 ± 0.08 |
| BPTCPSi | 224 ± 2 | 50 ± 1 | 0.87 ± 0.01 | 14.5 ± 0.6 | 2.9 ± 0.5 | 0.23 ± 0.04 |
The effect of surface functionalization on the pore structure was measured by nitrogen sorption. The surface area, BET constant, pore volume and average pore diameter for HTPSi and TCPSi samples before and after functionalization are shown in Table 1 and the isotherms are shown in ESI S3.† Functionalization caused a decrease in surface area, pore volume and average pore diameter. These values decreased between 10 and 30% after functionalization of HTPSi and between 1 and 12% after functionalization of TCPSi showing that the new functionalization method did not affect the pore structure substantially. Functionalization also affected the BET constant (CBET) of the materials. HTPSi had a small CBET value of 28.8 that increased to 34.4 after the functionalization with undecylenic acid. The opposite is observed with TCPSi that had a relatively high CBET of 73.9. After the functionalizations with undecylenic acid or decene, the CBET decreased to 57 and 35, respectively, indicating that the functionalized surface had weaker interactions with the N2 molecule than the non-functionalized surface.
The materials were characterized by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FTIR), 29Si cross polarization magic angle spinning nuclear magnetic resonance spectroscopy (CP-MAS NMR) and 13C CP-MAS dynamic nuclear polarization (DNP) NMR in order to characterize the chemical groups present on the materials. According to XPS, the samples consisted of Si, C, O and trace level impurities of Na and F (Table 2). As reported earlier, TCPSi consisted mainly of silicon, graphitic carbon, SiC and silicon oxycarbides.12 No silicon dioxide was detected. An increased amount of COOH was detected for undecylenic acid functionalized samples, UnHTPSi containing more COOH compared with UnTCPSi. It is worth noting that three measurements were performed on each sample revealing a good reproducibility for the TCPSi samples whereas it is not the case for the HTPSi samples indicating higher instability and/or heterogeneity. Relatively high carbon content from 21 to 65 atomic % was due to the surface sensitive nature of the measurements and impurities adsorbed from the air and does not reflect accurately the overall carbon content.
| HTPSi (%) | UnHTPSi (%) | TCPSi (%) | UnTCPSi (%) | DeTCPSi (%) | BPTCPSi (%) | BPTCPSi + Cu (%) | |
|---|---|---|---|---|---|---|---|
| Total C | 21 | 52 | 65 | 63 | 63 | 36 | 36 |
| SiC | 0 | 0 | 20 | 15 | 14 | 19 | 17 |
| C–C | 90 | 83 | 72 | 70 | 76 | 57 | 50 |
| C–OR | 7 | 6 | 5 | 9 | 6 | 11 | 20 |
| CO |
1 | 1 | 1 | 3 | 2 | 4 | 7 |
| COOH | 2 | 10 | 2 | 3 | 1 | 2 | 2 |
| Total Si | 74 | 26 | 23 | 21 | 24 | 38 | 38 |
| C3SiO | 3 | 7 | 0 | 0 | 0 | 0 | 0 |
| CSiO3 | 0 | 10 | 7 | 10 | 5 | 0 | 0 |
| SiO4 | 0 | 21 | 0 | 0 | 0 | 14 | 23 |
| C2SiO2 | 0 | 0 | 18 | 17 | 17 | 16 | 19 |
| SiC | 0 | 0 | 53 | 57 | 56 | 19 | 17 |
| Si–Si | 97 | 62 | 22 | 16 | 22 | 51 | 41 |
| Total O | 4 | 21 | 11 | 15 | 13 | 23 | 24 |
| Total F | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| Total Na | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total P | 0 | 0 | 0 | 0 | 0 | Traces | Traces |
| Total Cu | 0 | 0 | 0 | 0 | 0 | 0 | 0.3 |
According to the FTIR spectra (Fig. 2), amount of CHx species increased after grafting. Similar increase in CO species was observed in undecylenic acid functionalized samples (UnTCPSi and UnHTPSi). Increase in CH species present in free undecylenic acid and decene molecules were not observed.
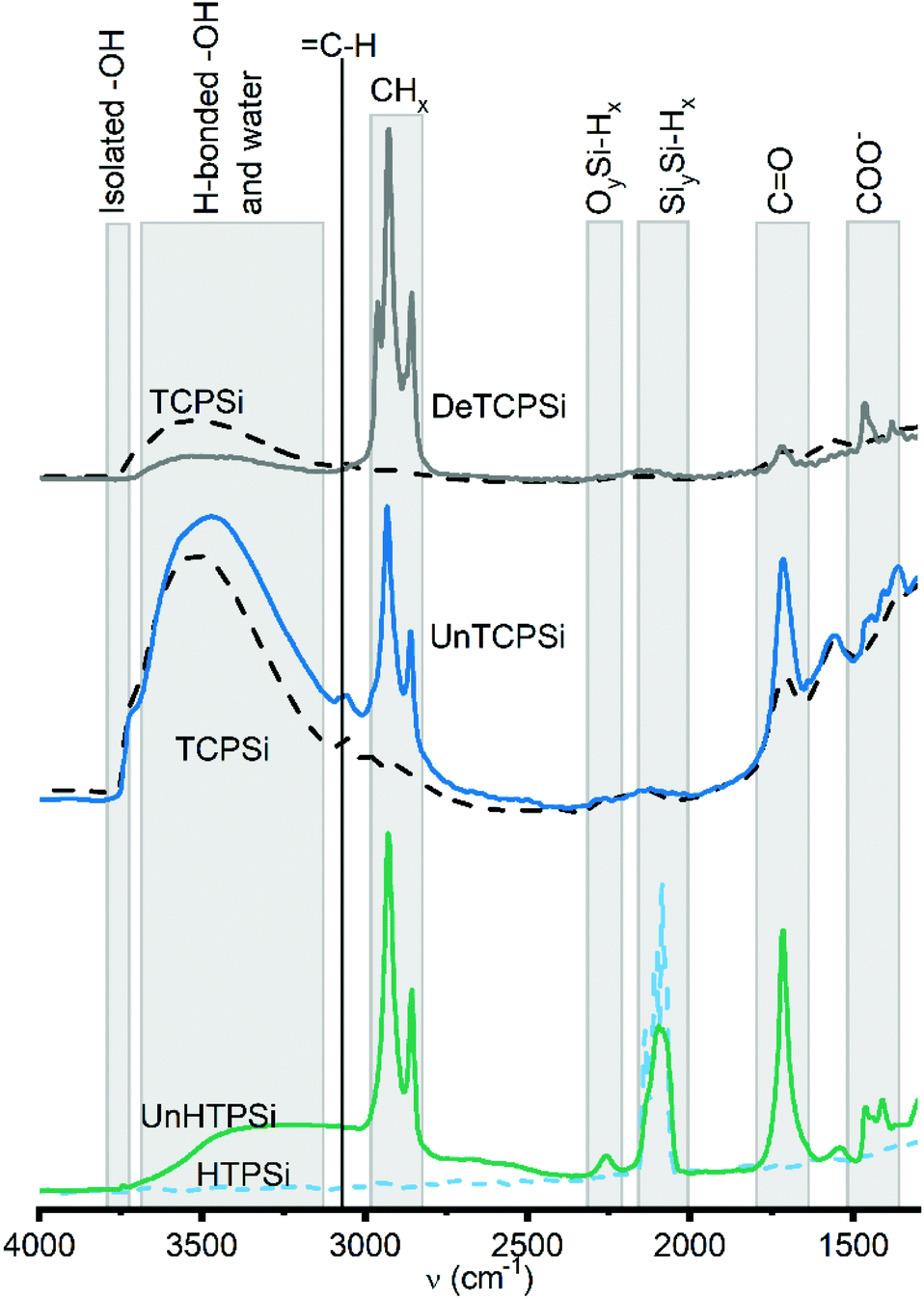 | ||
| Fig. 2 FTIR spectra of functionalized (solid lines) and unfunctionalized (dashed lines) TCPSi and HTPSi samples. | ||
29Si CP-MAS NMR spectrum of TCPSi (Fig. 3) shows three prominent features. The broad resonance between −45 and +25 ppm could correspond to several species such as: (i) Mm species [Mm = Si(R,R′,R′′)(OH)1−m(OSi)m, (0 ≤ m ≤ 1) where R, R′ and R′′ correspond to C in this particular case] that are expected between 0 and 15 ppm, (ii) Dn species [Dn = (R,R′)-Si(OH)2−n(OSi)n, 0 ≤ n ≤ 2] expected in the −25 to −5 ppm range27 and/or (iii) SiC (from −27 to −10 ppm (ref. 28)). The broad resonance may also be due to oxidized silicon species such as [Si(Si)n(OH)4−n, n = 2–3] expected at −14 and −12 ppm or Si(Si)2(OH)(H) or Si(Si)(OSi)(OH)(H) expected at −22 and −24 ppm, respectively.29 These findings are supported by the XPS results in which Si–Si groups, SiC species and C2SiO2 (D) species are the major components (see Table 2).
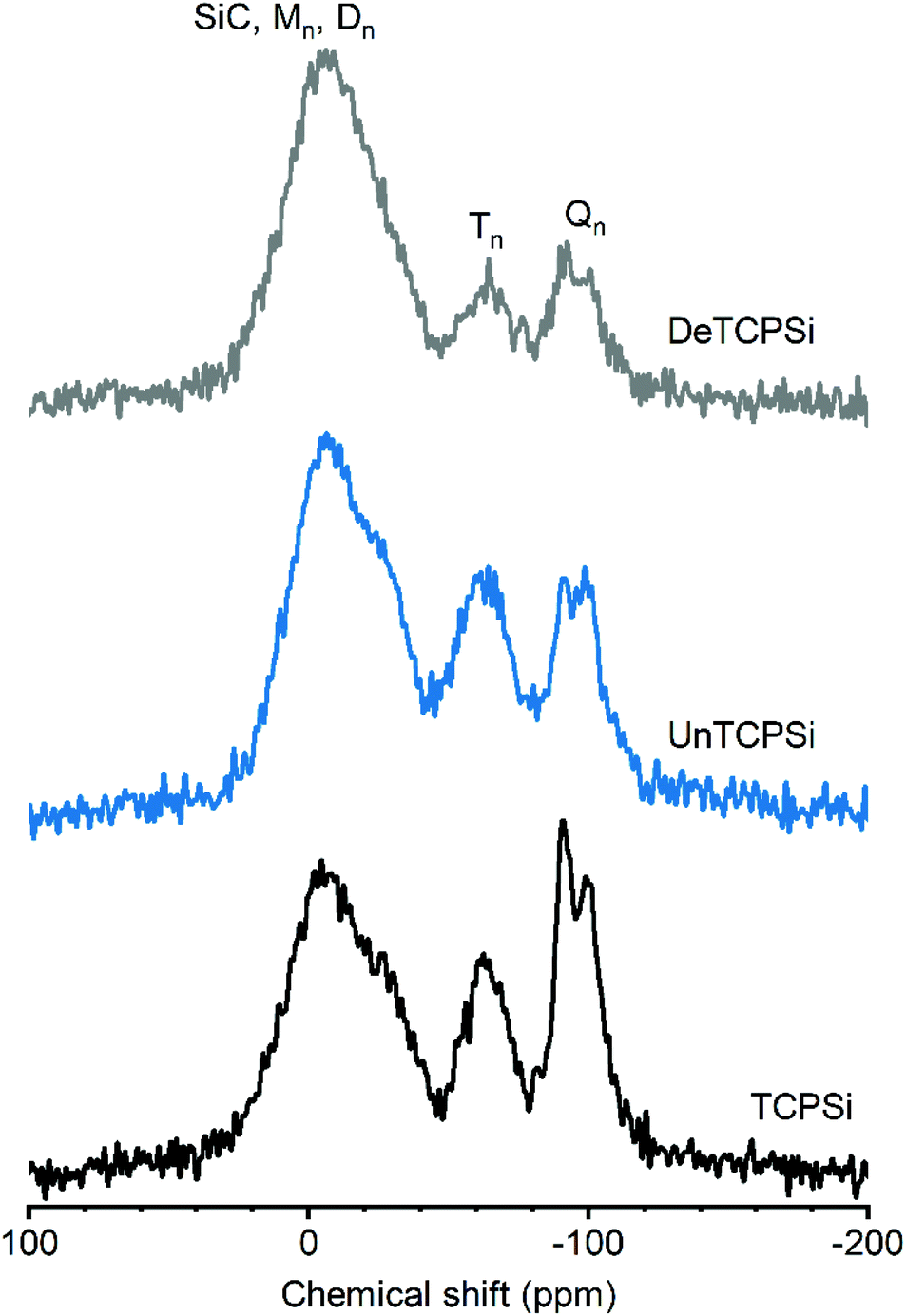 | ||
| Fig. 3 29Si CP-MAS NMR spectra of functionalized and unfunctionalized TCPSi. The intensity of the spectra is normalized according to the height of the highest peak (Mm = Si(R,R′,R′′)(OH)1−m(OSi)m, (0 ≤ m ≤ 1); Dn = (R,R′)-Si(OH)2−n(OSi)n, 0 ≤ n ≤ 2; Tn = R-Si(OH)3−n(OSi)n, 0 ≤ n ≤ 3; Qn = Si(OSi)n(OH)4−n, 0 ≤ n ≤ 4). | ||
The broad resonance between −50 and −80 ppm is assigned to Tn groups [Tn = R-Si(OH)3−n(OSi)n, 0 ≤ n ≤ 3] such as CSiO3 species that were also detected by XPS.
The third broad resonance between −110 and −80 ppm could correspond to Qn species [Qn = Si(OSi)n(OH)4−n, 0 ≤ n ≤ 4] and/or hydrogenated silicon species such as O3SiH (expected at −84 ppm), Si3SiH (−91 ppm) and Si2SiH2 (−102 ppm).30 Presence of Qn species in NMR spectra despite their absence in XPS spectra can be explained by the fact that the signal corresponding to Si atoms spatially close to hydrogen nuclei such as SiOH species are enhanced in the 1H–29Si CP-MAS spectrum.
Undecylenic acid and decene functionalized TCPSi show enhanced intensity of the M, D and T species relative to Q/hydrogenated silicon species. This indicates that the surface functionalization occurs on silicon carbides/oxycarbides species because grafting of a decyl carbon chain close to Si atoms will enhance the magnetization transfer from 1H to 29Si. The intensity increase is more significant for the DeTCPSi sample than for the UnTCPSi, in agreement with the higher grafting yield for DeTCPSi. The more pronounced enhancement of the resonance between −20 and +25 ppm with respect to the others suggests grafting of several carbon atoms on one Si atom is favored.
For the UnHTPSi sample, the reaction of SiH with undecyl acid creates a Si–C bond. Indeed, resonances corresponding to T or D species are seen on the 29Si CP-MAS NMR spectrum of UnHTPSi as expected (ESI S4†).
Measuring 13C CP-MAS NMR spectra for these samples was challenging due to relatively low amount of carbon in the samples and inherently low sensitivity of the method. As reported earlier, we were able to use dynamic nuclear polarization with the endogenous radicals in the samples to obtain significantly higher quality spectra.12 Spectra of TCPSi and UnTCPSi are shown in Fig. 4. The resonance corresponding to sp2 carbon from graphitic carbon is observed in both samples between 150 and 110 ppm (ref. 31) as well as the resonance corresponding to sp3 carbon in SiC and silicon oxycarbide between −10 and 40 ppm.32,33 As expected, UnTCPSi showed an additional resonance centered at 29 ppm from CH2 groups and at 178 ppm characteristic of COOH groups.
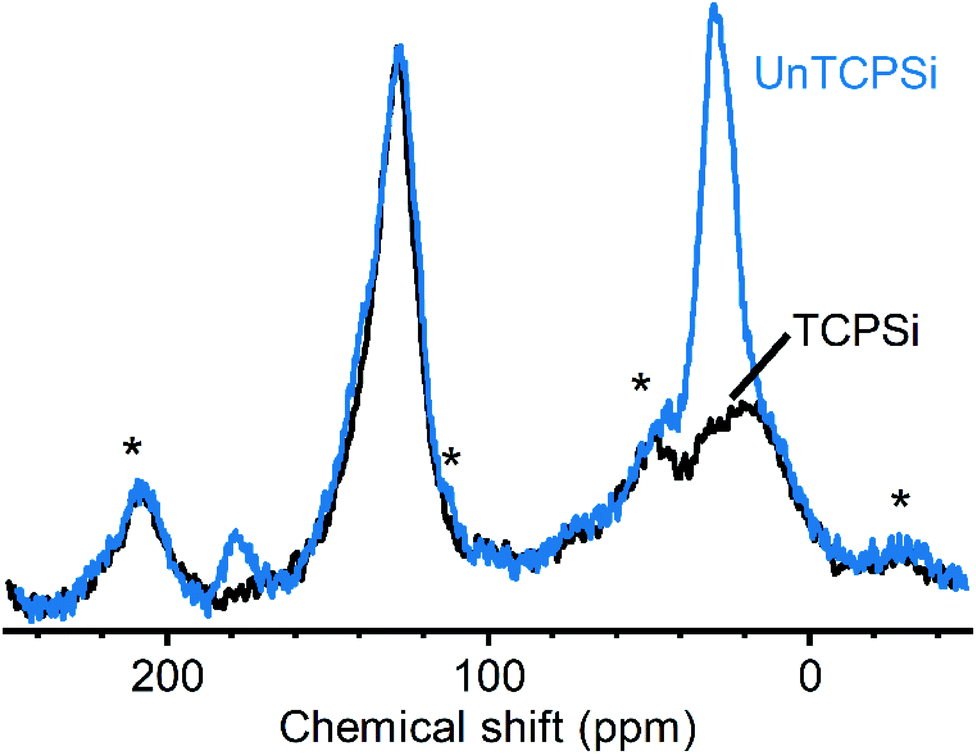 | ||
| Fig. 4 13C CP-MAS DNP spectra of TCPSi and UnTCPSi. Spinning sidebands are marked with an asterisk. | ||
In order to gain insight into the grafting mechanism, EPR measurements were performed for the TCPSi samples stored under inert atmosphere before and immediately after addition of undecylenic acid. The profile fitting revealed that the spectra consisted of two Lorentzian lines with g-factors of 2.0034 ± 0.0002 and 2.0043 ± 0.0002 (ESI S5†). There was no statistically significant increase in the total intensity of the resonance with addition of undecylenic acid. This is not surprising since there is no obvious termination reaction for the radical addition. Interestingly, the proportional intensity of the two lines reversed after addition of undecylenic acid on the material. Before the addition of undecylenic acid, the ratio between the peak at lower and higher g-values was 0.09 ± 0.02 and after the addition 1.2 ± 0.4. This suggests that there is a transfer of radicals from one species to another. It is challenging to identify the species definitively because of the broad resonances and disordered nature of the material. It has been shown that in Six−1Cx the spins form a strongly interacting system with g-factors decreasing from 2.0055 to 2.0037 as x increases from 0 to 1.34–36 The low g-factor component corresponds likely to radicals in the parts of the material with a high carbon content and the high g-factor component corresponds to Si rich parts. Therefore, increase in the intensity of the lower g-value peak indicates increasing C content in Si rich parts because of the functionalization. Alternatively, the shift towards lower g-values may be due to transfer of radicals from Si rich parts to the organic molecule typically having g-factor of approximately 2.0023.37 Similar reversal in the intensities of the relative intensities of the two Lorentzian lines was not observed when undecylenic acid was added on the TCPSi which was not kept under inert conditions.
The 29Si CP-MAS NMR and EPR results suggest formation of chemical bonds between the surface Si atoms and undecylenic acid molecules. However, neither of these results rule out formation of bonds between surface carbon atoms and undecylenic acid molecules. In fact, reaction with the surface carbon atoms is highly probable because carbonization of PSi results in a carbon rich surface12 and the radical groups in the SiC composites are more likely to be found on carbon than silicon atoms.34,36 Furthermore, undecylenic acid molecules can react with other undecylenic molecules which have already reacted with the surface forming molecular chains. This does not seem to take place excessively because the pore size is not significantly reduced after grafting.
The main aim of the new functionalization method is to provide functionalized PSi that is stable in aqueous solutions in a wide pH range. To determine the stability, the UnHTPSi and UnTCPSi microparticles were immersed into 1 M HCl, deionized water and 1 M NaOH at 30 °C. After 1, 3, 6 and 20 days, part of the powder was collected from the liquid, washed to remove any physisorbed molecules and dried. The liquid was changed to a fresh one at each time point. Microparticles were measured with TG to determine the amount of the remaining grafted species in the particles. The results of the stability studies (Fig. 5) show that 68 to 79% of the functional molecules in UnTCPSi remain even after 20 days in 1 M HCl and water, respectively, whereas in UnHTPSi only 24 and 39% of the molecules remains after 6 days in 1 M HCl and water, respectively. In 1 M NaOH solution, UnHTPSi dissolved in just few minutes and no stability data was gathered. However, the functionalization in UnTCPSi remained relatively stable for 6 days. At 20 d time point, a clear increase in the amount of functionalization was observed. This counterintuitive result is due to dissolution of silicon underneath the carbonized and functionalized layer, which decreased the total mass of the sample. Because the organic layer itself was relatively stable, its mass compared to the total mass of the sample increased leading to erroneous values. This conclusion is supported by nitrogen sorption results. The surface area of UnTCPSi sample more than doubled after immersion in 1 M NaOH for 20 days indicating that dissolution of Si below the carbonized layer revealed new surface. The UnTCPSi samples immersed in HCl and water for 20 d showed less than 4% change in specific surface area, pore volume or average pore diameter. Pore structure of UnHTPSi sample experienced significantly larger changes. For example, the specific surface area increased 20 and 40% during 20 d immersion in HCl and water, respectively. SEM images were taken of the UnTCPSi particles before and after 20 d incubation in NaOH (Fig. 6). The typical fir tree pore structure can still be recognized in the particles even after the 20 days incubation (Fig. 6D), but also some pits were formed in the particles (Fig. 6C and D). It can be concluded, that a highly stable surface functionalization was achieved. However, during a long immersion at very basic conditions the silicon structure under the functionalized surface begun to degrade.
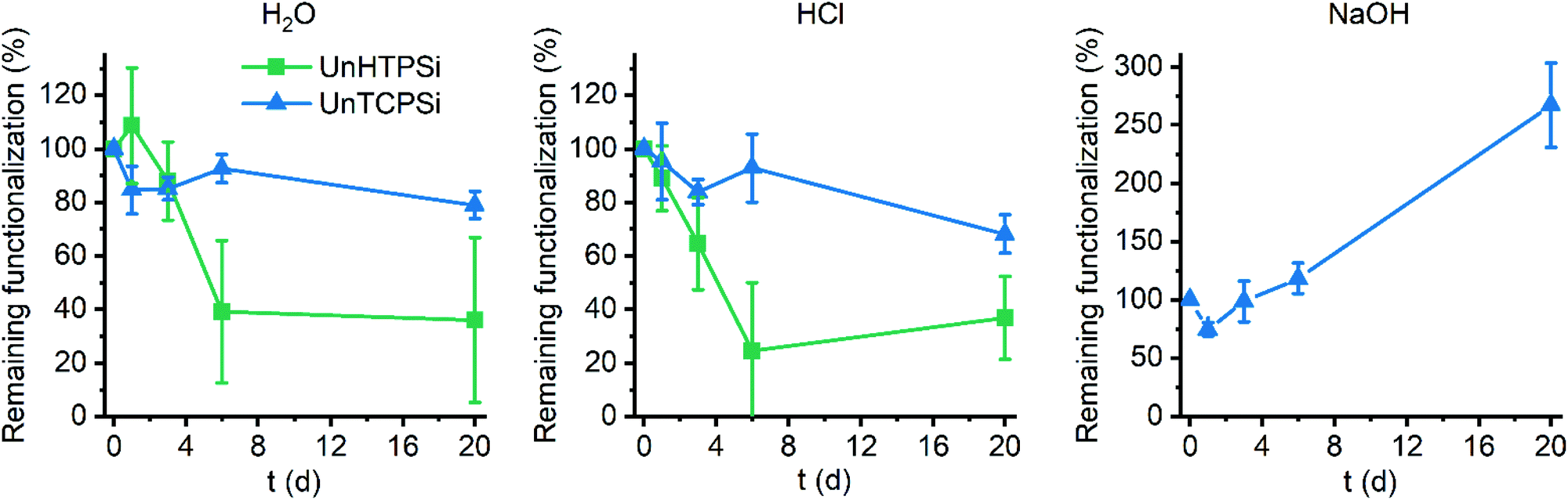 | ||
| Fig. 5 Results of stability study in deionized H2O (left), 1 M HCl (middle) and 1 M NaOH (right). | ||
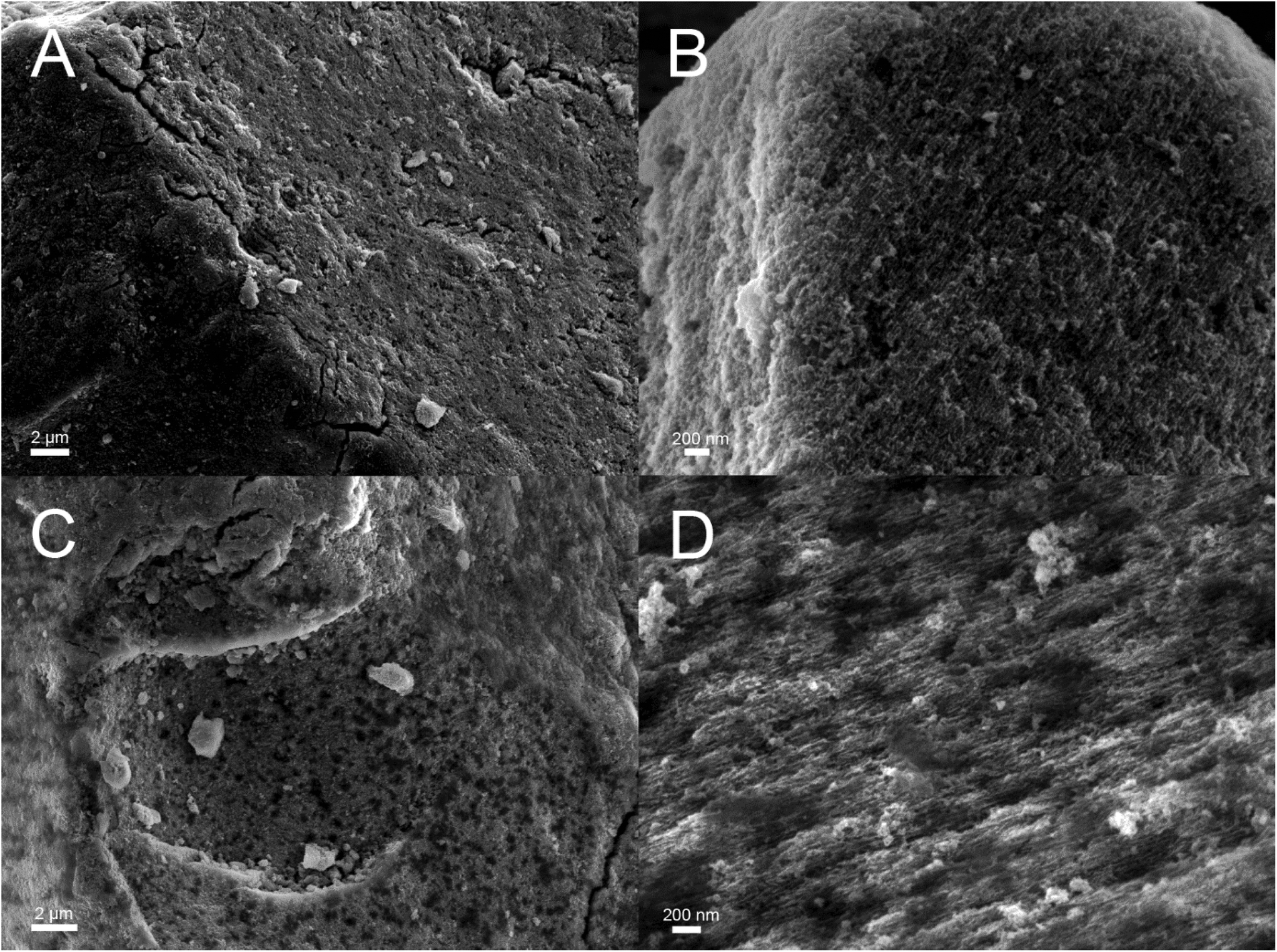 | ||
| Fig. 6 Scanning electron microscopy images of UnTCPSi before (A and B) and after (C and D) 20 days incubation in 1 M NaOH solution. | ||
The developed functionalization method was used to graft bisphosphonates (Fig. 7) on the surface of TCPSi (ESI Fig. S2†). The BPTCPSi samples contained 2.9 ± 0.5 wt% of BP molecules (Fig. 1) corresponding to coverage of 0.23 molecules per nm2. Results of the nitrogen sorption and XPS measurements are shown in Tables 1 and 2. 29Si CP-MAS NMR and Fourier transform infrared spectra of the material are shown in ESI Fig. S6 and S7,† respectively, and the nitrogen sorption isotherm in ESI S3.† The BPTCPSi sample was characterized by 31P MAS NMR (Fig. 8). The spectrum shows an intense resonance at 20 ppm from the phosphorus atoms of the bisphosphonate. In addition to the main resonance, decomposition of the signal suggests at least three other components at −11, 1 and 11 ppm indicating different protonation states, conformations or possible side products of the grafting reaction.38 The sharp resonance at 1 ppm corresponds to the silylated bisphosphonate based on liquid state NMR indicating that some silyl groups that were used as protective groups during the functionalization remained in the material.
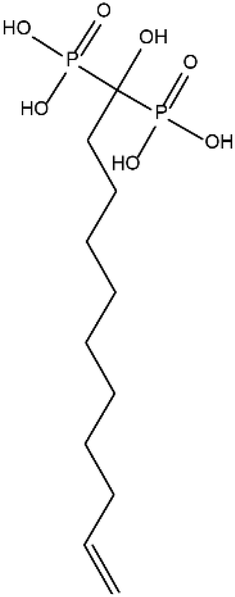 | ||
| Fig. 7 Bisphosphonate molecule grafted on BPTCPSi. To prevent a reaction between the bisphosphonate group and the surface of TCPSi, the –OH groups were protected by replacing the hydrogen atoms with trimethylsilyl groups. The OH groups were deprotected by washing the BPTCPSi particles with methanol after grafting. | ||
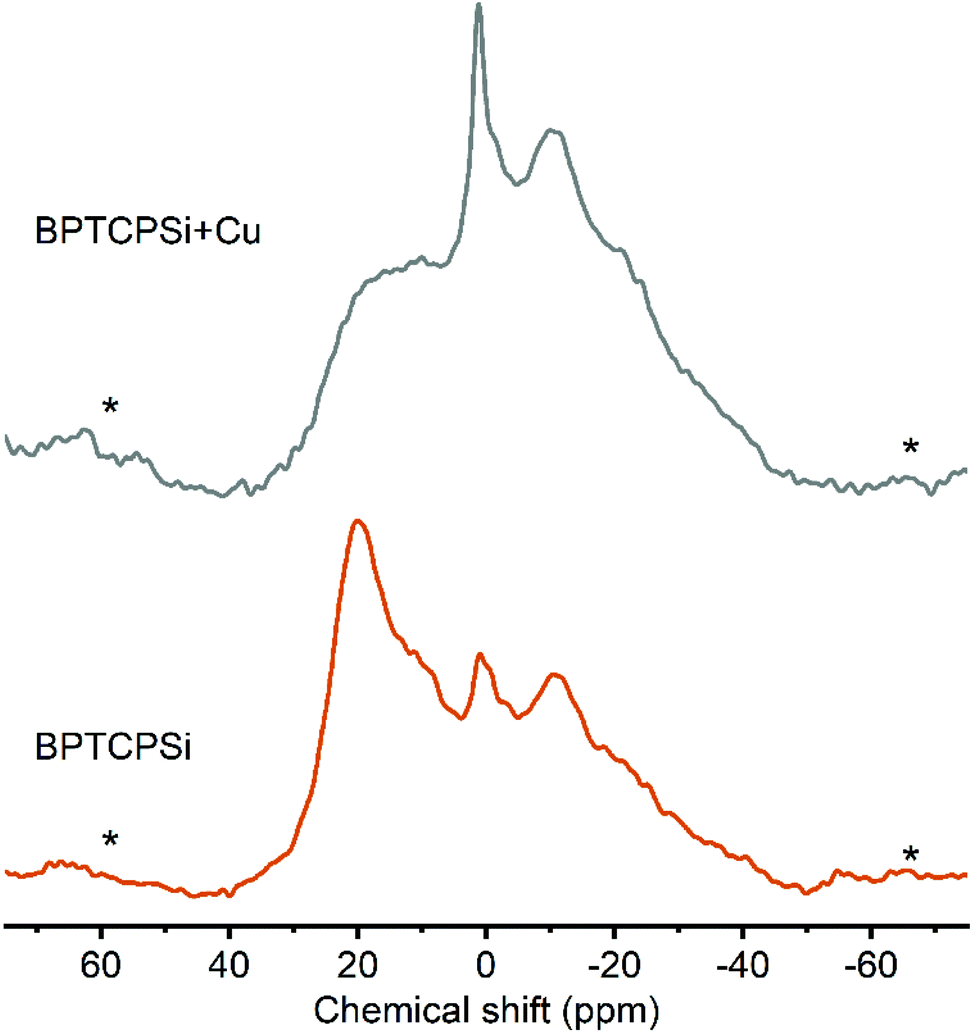 | ||
| Fig. 8 31P MAS spectra of BPTCPSi before (bottom) and after (top) adsorption of Cu2+. The line at 20 ppm corresponds to 31P nuclei in bisphosphonates. Spinning sidebands are marked with an asterisk. | ||
The performance of the material in metal adsorption applications was tested with CuCl2 solution. The maximum capacity of the material towards chelation of Cu2+ was 40 ± 4 μmol g−1 corresponding to 0.46 Cu2+ ions per bisphosphonate molecule. This is a slightly higher ratio than was reported for solid bisphosphonates (0.35).14 After the Cu2+ adsorption the intensity of the resonance at 20 ppm in the 31P MAS NMR spectrum decreases significantly (Fig. 7). This suggests interaction between copper and the grafted bisphosphonates as the presence of paramagnetic species such as Cu2+ is known to broaden and shift the resonances.39
The ability of the material to repeatedly adsorb copper ions from aqueous solution and subsequently to release them into 0.1 M HCl solution was tested by exposing the material to 50 consecutive adsorption–desorption cycles. Between 60 to 80% of the copper ions were adsorbed by BPTCPSi (Fig. 9). Importantly, the results show only small decrease in the performance of the material within the 50 cycles. Similar experiment was also done with unfunctionalized TCPSi. Approximately 90% of Cu2+ ions were adsorbed and 30% desorbed (relative to amount of Cu2+ ions initially in the solution) in the first cycle (Fig. 9). However, subsequent cycles showed adsorption or desorption values smaller than 20%. The results suggest that there is some irreversible adsorption of Cu2+ ions on TCPSi surface as well as small reversible adsorption. However, BP molecules are mainly responsible for the reversible adsorption of Cu2+ in BPTCPSi.
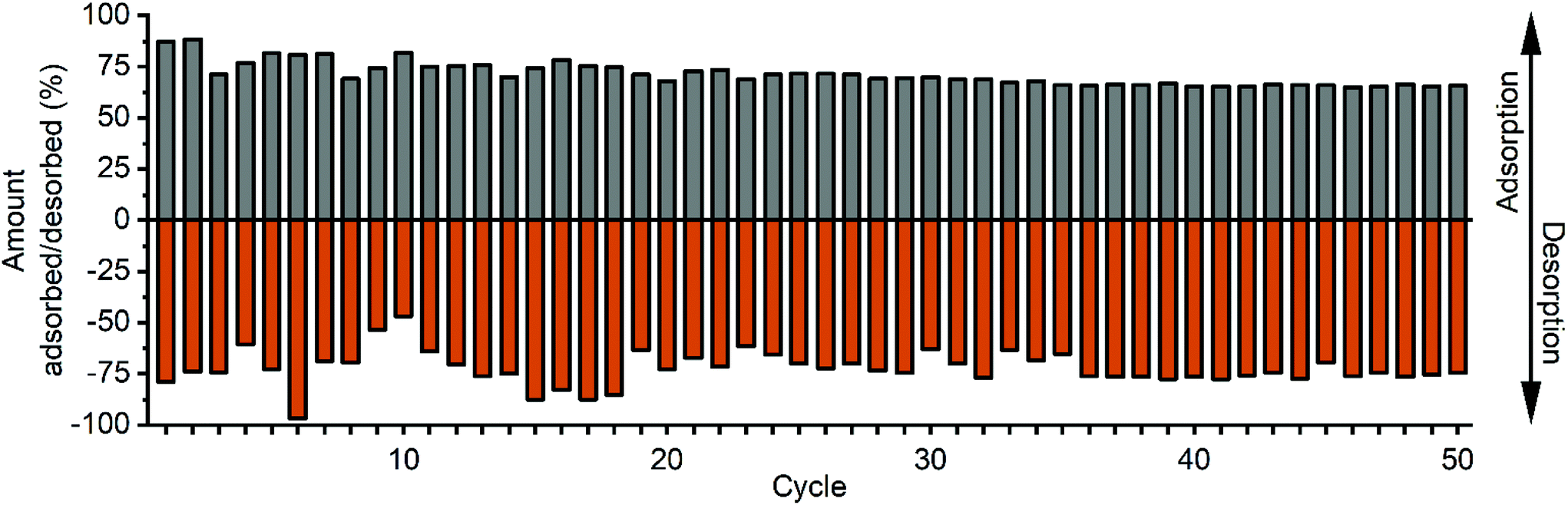 | ||
| Fig. 9 Fifty adsorption–desorption cycles of Cu2+ ions on TCPSi and BPTCPSi. The amount adsorbed or desorbed is presented relative to the amount of Cu2+ available for adsorption initially. The positive values represent adsorption and negative values desorption. | ||
4. Conclusions
Thermally carbonized porous silicon was functionalized by grafting terminal alkenes directly on the surface. The functionalization was performed with three kinds of molecules; a linear alkane, a carboxylic acid and a bisphosphonate. The resulting material has an unprecedented stability. It can withstand immersion in aqueous solutions at low and neutral pH up to 20 days with loss of less than a third of the functionalized layer. It can also withstand highly basic solutions up to 6 days. This surface functionalization is highly beneficial in applications such as sensing and adsorption that require extended stability of the material in aqueous solutions. Applicability of the bisphosphonate functionalized material was demonstrated in metal adsorption. The material was able to adsorb and desorb copper ions from solution repeatedly for 50 cycles without significant loss of performance.Conflicts of interest
J. R. T. N. and V.-P. L. are shareholders of 3AWater Oy which uses and has patent applications regarding the kinds of materials reported in the manuscript.Acknowledgements
MSc. Mejor Lama's contribution to the metal adsorption experiments and MSc. Tomi Miettinen's contribution to sample preparation is acknowledged.This work was supported by the Saastamoinen Foundation, Finnish Cultural foundation, Academy of Finland [project numbers 292601, 288531, 314552] and Tekes [HybREC project].
References
- L. Velleman, C. J. Shearer, A. V. Ellis, D. Losic, N. H. Voelcker and J. G. Shapter, Fabrication of self-supporting porous silicon membranes and tuning transport properties by surface functionalization, Nanoscale, 2010, 2, 1756–1761 RSC.
- K. A. Kilian, T. Böcking and J. J. Gooding, The importance of surface chemistry in mesoporous materials: Lessons from porous silicon biosensors, Chem. Commun., 2009, 2009, 630–640 RSC.
- T. Böcking, K. A. Kilian, K. Gaus and J. J. Gooding, Modifying porous silicon with self-assembled monolayers for biomedical applications: The influence of surface coverage on stability and biomolecule coupling, Adv. Funct. Mater., 2008, 18, 3827–3833 CrossRef.
- B. Sciacca, S. D. Alvarez, F. Geobaldo and M. J. Sailor, Bioconjugate functionalization of thermally carbonized porous silicon using a radical coupling reaction, Dalton Trans., 2010, 39, 10847–10853 RSC.
- K. A. Kilian, T. Böcking, K. Gaus, M. Gal and J. J. Gooding, Si–C linked oligo(ethylene glycol) layers in silicon-based photonic crystals: Optimization for implantable optical materials, Biomaterials, 2007, 28, 3055–3062 CrossRef CAS PubMed.
- T. Jalkanen, E. Mäkilä, T. Sakka, J. Salonen and Y. H. Ogata, Thermally promoted addition of undecylenic acid on thermally hydrocarbonized porous silicon optical reflectors, Nanoscale Res. Lett., 2012, 7, 1–7 CrossRef PubMed.
- J. M. Buriak and M. J. Allen, Lewis acid mediated functionalization of porous silicon with substituted alkenes and alkynes, J. Am. Chem. Soc., 1998, 120, 1339–1340 CrossRef CAS.
- R. Boukherroub, S. Morin, D. D. M. Wayner, F. Bensebaa, G. I. Sproule, J. M. Baribeau and D. J. Lockwood, Ideal passivation of luminescent porous silicon by thermal, noncatalytic reaction with alkenes and aldehydes, Chem. Mater., 2001, 13, 2002–2011 CrossRef CAS.
- M. J. Sweetman, F. J. Harding, S. D. Graney and N. H. Voelcker, Effect of oligoethylene glycol moieties in porous silicon surface functionalisation on protein adsorption and cell attachment, Appl. Surf. Sci., 2011, 257, 6768–6775 CrossRef CAS.
- H. Ouyang, C. C. Striemer and P. M. Fauchet, Quantitative analysis of the sensitivity of porous silicon optical biosensors, Appl. Phys. Lett., 2006, 88, 163108 CrossRef.
- J. Salonen, E. Laine and L. Niinistö, Thermal carbonization of porous silicon surface by acetylene, J. Appl. Phys., 2002, 91, 456–461 CrossRef CAS.
- J. Riikonen, S. Rigolet, C. Marichal, F. Aussenac, J. Lalevée, F. Morlet-Savary, P. Fioux, C. Dietlin, M. Bonne, B. Lebeau and V. P. Lehto, Endogenous Stable Radicals for Characterization of Thermally Carbonized Porous Silicon by Solid-State Dynamic Nuclear Polarization 13C NMR, J. Phys. Chem. C, 2015, 119, 19272–19278 CrossRef CAS.
- W. Xu, J. Riikonen, T. Nissinen, M. Suvanto, K. Rilla, B. Li, Q. Wang, F. Deng and V. P. Lehto, Amine Surface Modifications and Fluorescent Labeling of Thermally Stabilized Mesoporous Silicon Nanoparticles, J. Phys. Chem. C, 2012, 116, 22307–22314 CrossRef CAS.
- P. A. Turhanen, J. J. Vepsäläinen and S. Peräniemi, Advanced material and approach for metal ions removal from aqueous solutions, Sci. Rep., 2015, 5, 8992 CrossRef CAS PubMed.
- M. Mureseanu, A. Reiss, I. Stefanescu, E. David, V. Parvulescu, G. Renard and V. Hulea, Modified SBA-15 mesoporous silica for heavy metal ions remediation, Chemosphere, 2008, 73, 1499–1504 CrossRef CAS PubMed.
- Y. Wang, D. Liu, J. Lu and J. Huang, Enhanced adsorption of hexavalent chromium from aqueous solutions on facilely synthesized mesoporous iron–zirconium bimetal oxide, Colloids Surf., A, 2015, 481, 133–142 CrossRef CAS.
- P. Rekha, V. Sharma and P. Mohanty, Synthesis of cyclophosphazene bridged mesoporous organosilicas for CO2 capture and Cr(VI) removal, Microporous Mesoporous Mater., 2016, 219, 93–102 CrossRef CAS.
- A. Muoz Garca, A. J. Hunt, V. L. Budarin, H. L. Parker, P. S. Shuttleworth, G. J. Ellis and J. H. Clark, Starch-derived carbonaceous mesoporous materials (Starbon) for the selective adsorption and recovery of critical metals, Green Chem., 2015, 17, 2146–2149 RSC.
- Z. Wang, L. Chen, X. Du, G. Zou and X. Wang, A pillared process to construct graphitic carbon nitride based functionalized mesoporous materials, RSC Adv., 2016, 6, 15605–15609 RSC.
- L. Wang, C. Cheng, S. Tapas, J. Lei, M. Matsuoka, J. Zhang and F. Zhang, Carbon dots modified mesoporous organosilica as an adsorbent for the removal of 2,4-dichlorophenol and heavy metal ions, J. Mater. Chem. A, 2015, 3, 13357–13364 RSC.
- M. Lecouvey, I. Mallard, T. Bailly, R. Burgada and Y. Leroux, A mild and efficient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphonic acids. Preparation of new tripod ligands, Tetrahedron Lett., 2001, 42, 8475–8478 CrossRef CAS.
- A. B. Sieval, V. Vleeming, H. Zuilhof and E. J. R. Sudhölter, Improved method for the preparation of organic monolayers of 1-alkenes on hydrogen-terminated silicon surfaces, Langmuir, 1999, 15, 8288–8291 CrossRef CAS.
- J. Lalevée, F. Dumur, C. R. Mayer, D. Gigmes, G. Nasr, M. A. Tehfe, S. Telitel, F. Morlet-Savary, B. Graff and J. P. Fouassier, Photopolymerization of n -vinylcarbazole using visible-light harvesting iridium complexes as photoinitiators, Macromolecules, 2012, 45, 4134–4141 CrossRef.
- M. Rosay, L. Tometich, S. Pawsey, R. Bader, R. Schauwecker, M. Blank, P. M. Borchard, S. R. Cauffman, K. L. Felch, R. T. Weber, R. J. Temkin, R. G. Griffin and W. E. Maas, Solid-state dynamic nuclear polarization at 263 GHz: Spectrometer design and experimental results, Phys. Chem. Chem. Phys., 2010, 12, 5850–5860 RSC.
- M. Blank, P. Borchard, S. Cauffman, K. Felch, M. Rosay and L. Tometich, High-frequency CW gyrotrons for NMR/DNP applications, IEEE Int. Vac. Electron. Conf., 2012, 327–328 Search PubMed.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- B. Alonso and C. Marichal, Solid-state NMR studies of micelle-templated mesoporous solids, Chem. Soc. Rev., 2013, 42, 3808–3820 RSC.
- L. Gu, D. Ma, S. Yao, X. Liu, X. Han, W. Shen and X. Bao, Template-synthesized porous silicon carbide as an effective host for zeolite catalysts, Chem. – Eur. J., 2009, 15, 13449–13455 CrossRef CAS PubMed.
- R. A. Faulkner, J. A. DiVerdi, Y. Yang, T. Kobayashi and G. E. Maciel, The surface of nanoparticle silicon as studied by solid-state NMR, Materials, 2013, 6, 18–46 CrossRef CAS PubMed.
- W. K. Chang, M. Y. Liao and K. K. Gleason, Characterization of porous silicon by solid-state nuclear magnetic resonance, J. Phys. Chem., 1996, 100, 19653–19658 CrossRef CAS.
- C. Hontoria-Lucas, A. J. López-Peinado, J. D. D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Study of oxygen-containing groups in a series of graphite oxides: Physical and chemical characterization, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- C. T. Brigden, I. Farnan and P. R. Hania, Multi-nuclear NMR study of polytype and defect distribution in neutron irradiated silicon carbide, J. Nucl. Mater., 2014, 444, 92–100 CrossRef CAS.
- S. J. Widgeon, S. Sen, G. Mera, E. Ionescu, R. Riedel and A. Navrotsky, 29Si and 13C Solid-state NMR spectroscopic study of nanometer-scale structure and mass fractal characteristics of amorphous polymer derived silicon oxycarbide ceramics, Chem. Mater., 2010, 22, 6221–6228 CrossRef CAS.
- P. J. Gaczi and D. C. Booth, ESR in CVD silicon and silicon-carbon alloys, Sol. Energy Mater., 1981, 4, 279–289 CrossRef CAS.
- T. Shimizu, M. Kumeda and Y. Kiriyama, ESR studies on sputtered amorphous SiC, SiGe and GeC films, Solid State Commun., 1981, 37, 699–703 CrossRef CAS.
- N. Ishii, M. Kumeda and T. Shimizu, A simple molecular orbital calculation of ESR g-values for amorphous Si1-xCx, Si1-xGex and Ge1-xCx, Solid State Commun., 1982, 41, 143–146 CrossRef CAS.
- C. F. Young, E. H. Poindexter and G. J. Gerardi, Electron paramagnetic resonance of porous silicon: Observation and identification of conduction-band electrons, J. Appl. Phys., 1997, 81, 7468–7470 CrossRef CAS.
- M. S. Ironside, M. J. Duer, D. G. Reid and S. Byard, Bisphosphonate protonation states, conformations, and dynamics on bone mineral probed by solid-state NMR without isotope enrichment, Eur. J. Pharm. Biopharm., 2010, 76, 120–126 CrossRef CAS PubMed.
- M. Tang, K. Mao, S. Li, J. Zhuang and K. Diallo, Paramagnetic effects on the NMR spectra of isotropic bicelles with headgroup modified chelator lipids and metal ions, Phys. Chem. Chem. Phys., 2016, 18, 15524–15527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi01140d |
| This journal is © the Partner Organisations 2020 |