
Highly efficient oxidation of various thioethers catalyzed by organic ligand-modified polyoxomolybdates†
Haiyan
An
 *,
Yujiao
Hou
,
Shenzhen
Chang
,
Jie
Zhang
and
Qingshan
Zhu
*,
Yujiao
Hou
,
Shenzhen
Chang
,
Jie
Zhang
and
Qingshan
Zhu
College of Chemistry, Dalian University of Technology, Dalian 116023, P. R. China. E-mail: anhy@dlut.edu.cn; Fax: +86-411-84657675; Tel: +86-411-84657675
First published on 17th October 2019
Abstract
The selective oxidation of thioethers is among the most straightforward and common methods to obtain the synthetic intermediates sulfoxides for application in chemical industry, medicinal chemistry and biology. In this study, we prepared four hybrid compounds, i.e. Cs4[M(H2O)4][PMo6O21(PABA)3]2·nH2O 1–4 (M = Co, Mn, Ni, Zn; PABA = p-aminobenzoic acid), based on carboxylic acid-modified polyoxomolybdates and metal cations with wonderful catalytic performance for the selective oxidation of thioethers. The compounds 1–4 comprise PABA ligand-modified polyoxomolybdates [PMo6O21(PABA)3]3− as building units and the Co2+/Mn2+/Ni2+/Zn2+ cations as linkers to generate novel dimeric architectures. Using compound 1 as a pre-catalyst, methyl phenyl sulfides were nearly entirely converted to sulfoxides with the selectivity of 98% at room temperature within 20 minutes. Satisfactory catalytic effects were also observed in the selective oxidation of various phenyl sulfides with substituent groups; in particular, another typical thioether, i.e. the chemical warfare agent simulant 2-chloroethyl ethyl sulfide (CEES), could be completely degraded to nontoxic 2-chloroethyl ethyl sulfoxide (CEESO) with the selectivity of 99% within 12 minutes at room temperature. These pre-catalysts could be recycled at least 10 times by simple filtration with a negligible decrease in conversion and selectivity.
1 Introduction
Oxidative transformations of organic compounds using green oxidants are of both industrial and academic interest.1 In this area, the oxidation of sulfides to their corresponding sulfoxides and sulfones is a research field that is attracting extensive attention from researchers. On the one hand, this transformation can produce important intermediates for the construction of various medically and chemically active molecules. On the other hand, this oxidation process affords a sustainable way to a green environment via the oxidative decontamination of toxic chemicals, such as the sulfur mustard simulant 2-chloroethyl ethyl sulfide (CEES), and the desulfurization of oil.2 To date, various materials, such as inorganic oxides, metal–organic frameworks (MOFs) and modified zeolites, have been synthesized and demonstrated for catalyzing the selective oxidation of sulfides. However, the current reported materials always display limited selectivity and take a long time to finish the catalytic reaction. Thus, the search for environmentally benign and more efficient materials that can highly selectively and rapidly oxidize sulfides is a challenging issue.Polyoxometalates (POMs), as well-defined inorganic metal oxide clusters, possess significant applications in catalysis, optics, medicine, etc.3–18 Among these applications, oxidation catalysis has been one of the most attractive aspects in the last few years owing to the resistance of POMs to oxidation. As an important branch, the selective oxidation of various sulfides catalyzed by POMs has been explored by Hill, Hu, Song, Niu and other groups.19–35 In the early years, some homogeneous materials, such as [(n-C4H9)4N]4(α-Mo8O26), [{CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)6Si}xOySiWwOz]4−, and TBA4H2[BW11Mn(H2O)O39]·H2O, based on POMs have been developed for the selective oxidation of a wide range of thioethers.21–23 Subsequently, to solve the problem of recyclability, supported heterogeneous materials based on POMs and some solid materials, such as zeolites, polymers, or silica, have been extensively studied and have gained ever-increasing attention. In 2016, Zhao et al. have reported that the POM anion {SiV2W10} can be combined with a linear positively charged polymer, poly(diallyldimethylammonium chloride) (PDDA), to obtain the composite material PDDA-SiV2W10 through electrostatic interaction, which can efficiently catalyze the oxidation of various thioethers.24 The same year, Song and coworkers prepared a supported POM pre-catalyst by employing the facile condensation reaction between polytungstate {P2W15} and γ-Al2O3 spheres.25 This supported material P2W15-γ-Al2O3 is active for the oxygenation of thioethers, and displays high stability, excellent reactivity and selectivity. Recently, Hu's group successively obtained several composite materials based on POMs, [CnH2n+1N(CH3)3]7HNb6O19, Zn2Cr-LDH-PW11M and Mg3Al-LDH-Nb6, which exhibited good catalytic activities for the oxidative degradation of CEES.26–28 However, most of these supported materials always suffer from a low loading efficiency, ill-defined structure and inhomogeneous pre-catalyst binding sites, which result in differences in both activity and selectivity. In addition, previous investigations mostly centered on the design and synthesis of heterogeneous POM materials, which can selectively catalyze various thioethers into their corresponding sulfones. Although sulfone compounds are of great importance, high selectivity to obtain sulfoxides in the oxidation reaction is also expected. Current catalytic systems based on POMs displayed low selectivity for the oxidation of sulfides to sulfoxides, such as PW12@Al-MCFen, {As4W40O140(Ru2(Ac)2)} and VV17VIV12(C6H8O4)8.19,29,30 Therefore, it is still highly desirable to exploit alternative catalytic materials based on POMs with high reactivity and selectivity to obtain sulfoxide compounds.
CH(CH2)6Si}xOySiWwOz]4−, and TBA4H2[BW11Mn(H2O)O39]·H2O, based on POMs have been developed for the selective oxidation of a wide range of thioethers.21–23 Subsequently, to solve the problem of recyclability, supported heterogeneous materials based on POMs and some solid materials, such as zeolites, polymers, or silica, have been extensively studied and have gained ever-increasing attention. In 2016, Zhao et al. have reported that the POM anion {SiV2W10} can be combined with a linear positively charged polymer, poly(diallyldimethylammonium chloride) (PDDA), to obtain the composite material PDDA-SiV2W10 through electrostatic interaction, which can efficiently catalyze the oxidation of various thioethers.24 The same year, Song and coworkers prepared a supported POM pre-catalyst by employing the facile condensation reaction between polytungstate {P2W15} and γ-Al2O3 spheres.25 This supported material P2W15-γ-Al2O3 is active for the oxygenation of thioethers, and displays high stability, excellent reactivity and selectivity. Recently, Hu's group successively obtained several composite materials based on POMs, [CnH2n+1N(CH3)3]7HNb6O19, Zn2Cr-LDH-PW11M and Mg3Al-LDH-Nb6, which exhibited good catalytic activities for the oxidative degradation of CEES.26–28 However, most of these supported materials always suffer from a low loading efficiency, ill-defined structure and inhomogeneous pre-catalyst binding sites, which result in differences in both activity and selectivity. In addition, previous investigations mostly centered on the design and synthesis of heterogeneous POM materials, which can selectively catalyze various thioethers into their corresponding sulfones. Although sulfone compounds are of great importance, high selectivity to obtain sulfoxides in the oxidation reaction is also expected. Current catalytic systems based on POMs displayed low selectivity for the oxidation of sulfides to sulfoxides, such as PW12@Al-MCFen, {As4W40O140(Ru2(Ac)2)} and VV17VIV12(C6H8O4)8.19,29,30 Therefore, it is still highly desirable to exploit alternative catalytic materials based on POMs with high reactivity and selectivity to obtain sulfoxide compounds.
As a special subclass of hybrid POMs that can be rationally designed and synthesized, and increase the stability and activity of the precursors, organic ligand covalently modified POMs have recently attracted great attention in catalysis. Owing to their intrinsic nature and flexible tunability, a few hybrid POMs, such as a hybrid polymer based on polyoxovanadates and 1,3,5-benzenetricarboxamide and mixed carboxylic acid-modified polyoxomolydates, have been evidenced to be privileged pre-catalysts that enable the selective oxidative degradation of CEES.31,32 Inspired by this idea, we predicted that the obtained carboxylic acid-modified POMs with the exposed metal cation linkers Cs4[M(H2O)4][PMo6O21(PABA)3]2·nH2O 1–4 (M = Co, Mn, Ni, Zn) would also be good candidates for catalyzing the selective oxidation of thioethers. As expected, these compounds displayed excellent ability to selectively oxidize various sulfide substrates. Within 20 minutes, various phenyl sulfides could be rapidly converted to the corresponding sulfoxide with high selectivity using these compounds as pre-catalysts. More importantly, the sulfur mustard simulant CEES can also be highly selectively and rapidly degraded to nontoxic CEESO within 12 minutes.
2 Experimental
Syntheses of Cs4X[M(H2O)4][PIIIMo6O21(PABA)3]2·nH2O (M = Co 1, Mn 2, Ni 3, Zn 4; n = 25 1, 27 2; X = Na21)
In a typical synthesis procedure for 1–4, Na2MoO4 (0.145 g, 0.6 mmol), Na2HPO3·5H2O (0.0216 g, 0.1 mmol), and PABA (0.0411 g, 0.3 mmol) were dissolved in 20 mL water. The pH value of the solution was adjusted from the original pH value of 4.5 to 4.0 with HCl solution under stirring. The solution was stirred for one hour at room temperature. Then, CoCl2·6H2O (0.0714 g, 0.3 mmol)/MnCl2·2H2O (0.0486 g, 0.3 mmol)/NiCl2·6H2O (0.0678 g, 0.3 mmol)/ZnSO4·7H2O (0.0861 g, 0.3 mmol) and CsCl (0.034 g, 0.2 mmol) were successively added into the reaction system under stirring. Finally, the solution was heated and stirred for one hour at 80 °C. The filtrate was kept undisturbed for two weeks under ambient conditions, and then crystals of 1–4 were isolated with about 45% yield (based on Mo). Elemental analyses: calcd for 1: Mo, 29.79; P, 1.60; Co, 1.52; Cs, 13.75; C, 13.05; N, 2.18; H, 2.58 (%). Found: Mo, 30.03; P, 1.82; Co, 1.66; Cs, 14.15; C, 13.53; N, 2.47; H, 2.54 (%). FTIR data (cm−1) for 1: 3357 (s), 1605 (m), 1544 (s), 1413 (s), 1275 (w), 1179 (m), 928 (w), 894 (w), 780 (m), 672 (s), 567 (w), 539 (w), 444 (m). Calcd for 2: Mo, 29.89; P, 1.61; Mn, 1.43; Cs, 13.80; C, 13.10; N, 2.18; H, 2.90 (%). Found: Mo, 29.76; P, 1.28; Mn, 1.55; Cs, 13.43; C, 12.58; N, 2.34; H, 3.29 (%). FTIR data (cm−1) for 2: 3447 (s), 1606 (m), 1545 (m), 1415 (s), 1274 (w), 1178 (m), 927 (w), 894 (m), 782 (w), 671 (s), 536 (w), 440 (m). FTIR data (cm−1) for 3: FTIR data (cm−1): 3444 (s), 1606 (w), 1547 (m), 1417 (s), 1272 (w), 1178 (m), 929 (w), 892 (w), 782 (m), 678 (s), 565 (w), 535 (w), 438 (m). FTIR data (cm−1) for 4: FTIR data (cm−1): 3434 (s), 1607 (w), 1549 (m), 1417 (s), 1271 (w), 1175 (m), 929 (w), 890 (w), 784 (m), 679 (s), 564 (w), 537 (w), 439 (m).General methods for catalyzing the selective oxidation of methyl phenyl sulfide and CEES
Methyl phenyl sulfide oxidation: Compound 1 (2.5 μmol), H2O2 (0.3 mmol), methyl phenyl sulfide (0.25 mmol), and ethanol (0.5 mL) were put in a glass bottle. The catalytic reaction was carried out at room temperature for 20 minutes. CEES oxidation: Compound 1 (2.5 μmol), H2O2 (0.3 mmol), CEES (0.25 mmol), and ethanol (0.5 mL) were put in a glass bottle. The catalytic reaction was carried out at room temperature for 12 minutes. After the catalytic reaction was finished, GC-FID, GC-MS and 1H NMR were used to analyze the resulting mixture.3 Results and discussion
Synthesis and characterization of these materials
In this paper, four hybrid compounds were assembled in a simple one-pot system of Na2MoO4, Na2HPO3·5H2O, PABA and metal cation TM2+ at a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3:3 under reflux at 80 °C. These four hybrid POMs displayed similar dimeric structures. We did not obtain the single crystal data of compounds 3 and 4 due to the poor crystal quality, but IR and XRD results of the four compounds confirmed the isostructural architectures. Rare earth metal cations, such as La3+, Ce3+, and Nd3+, were also attempted as linkages to synthesize the similar compounds, but only the poor-quality crystal Cs3[PMo6O21(PABA)3]·nH2O (compound 5) with Cs+ as the counter cation was produced. When we introduced Cu2+ into the reaction system, a coordination complex constructed from PABA and Cu2+ was obtained. More importantly, other counter cations, such as K+ and Rb+, were also introduced into the process in the presence of transition metal cations, but no crystal was obtained without Cs+. Thus, it can be seen that both metal cations and counter cations play an essential part in constructing these hybrid compounds. In addition, if PABA was replaced by other ligands, such as 3-aminobenzoic acid, 4-hydroxybenzoic acid, 3-hydroxybenzoic acid and salicylic acid, no similar dimeric structures were isolated. The parallel experiments described above demonstrated that the metal cations Co2+/Mn2+/Ni2+/Zn2+, counter cation Cs+ and the organic ligands play important roles for the construction of the dimeric structures with carboxylic acid ligand-modified POM units.
:1:3:3 under reflux at 80 °C. These four hybrid POMs displayed similar dimeric structures. We did not obtain the single crystal data of compounds 3 and 4 due to the poor crystal quality, but IR and XRD results of the four compounds confirmed the isostructural architectures. Rare earth metal cations, such as La3+, Ce3+, and Nd3+, were also attempted as linkages to synthesize the similar compounds, but only the poor-quality crystal Cs3[PMo6O21(PABA)3]·nH2O (compound 5) with Cs+ as the counter cation was produced. When we introduced Cu2+ into the reaction system, a coordination complex constructed from PABA and Cu2+ was obtained. More importantly, other counter cations, such as K+ and Rb+, were also introduced into the process in the presence of transition metal cations, but no crystal was obtained without Cs+. Thus, it can be seen that both metal cations and counter cations play an essential part in constructing these hybrid compounds. In addition, if PABA was replaced by other ligands, such as 3-aminobenzoic acid, 4-hydroxybenzoic acid, 3-hydroxybenzoic acid and salicylic acid, no similar dimeric structures were isolated. The parallel experiments described above demonstrated that the metal cations Co2+/Mn2+/Ni2+/Zn2+, counter cation Cs+ and the organic ligands play important roles for the construction of the dimeric structures with carboxylic acid ligand-modified POM units.
Single crystal X-ray diffraction analyses demonstrated that 1 and 2 were isostructural and crystallized in the same space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) constructed from the same polyoxoanion unit {PMo6O21}. The asymmetric unit of 1 contained one crystallographically independent [PMo6O21(PABA)3]3− anion, half of one Co2+ cation, one Na+ cation and two Cs+ cations (Fig. S1†). The unique Co(1) ion displayed a distorted octahedral geometry, defined by two terminal O atoms derived from two polyoxoanions [Co–O 2.136(4)–2.151(5) Å] and four water molecules [Co–OW 1.962(3)–2.144(4) Å]. First, the {PMo6O21} units were covalently coordinated to the carboxylic acid ligand PABA to produce the single-side modified POMs [PMo6O21(PABA)3]3− (Fig. 1a). Then, the two modified POMs were jointed together to form a dimer via the Co–O–Mo bond. Such dimers were next linked together to generate a 1D supramolecular chain through the hydrogen bonds (N1–H⋯O25 = 2.954 Å in 1) between the N atoms of PABA and the O atoms of the {PMo6O21} units (Fig. 1c). Finally, these 1D chains were further joined together to obtain a 2D supramolecular layer via the hydrogen bonds (N2–H⋯O20 = 2.744 Å, N2–H⋯O13 = 2.938 Å in 1) existing between the N atoms of PABA and the O atoms of the {PMo6O21} units (Fig. 1d).
constructed from the same polyoxoanion unit {PMo6O21}. The asymmetric unit of 1 contained one crystallographically independent [PMo6O21(PABA)3]3− anion, half of one Co2+ cation, one Na+ cation and two Cs+ cations (Fig. S1†). The unique Co(1) ion displayed a distorted octahedral geometry, defined by two terminal O atoms derived from two polyoxoanions [Co–O 2.136(4)–2.151(5) Å] and four water molecules [Co–OW 1.962(3)–2.144(4) Å]. First, the {PMo6O21} units were covalently coordinated to the carboxylic acid ligand PABA to produce the single-side modified POMs [PMo6O21(PABA)3]3− (Fig. 1a). Then, the two modified POMs were jointed together to form a dimer via the Co–O–Mo bond. Such dimers were next linked together to generate a 1D supramolecular chain through the hydrogen bonds (N1–H⋯O25 = 2.954 Å in 1) between the N atoms of PABA and the O atoms of the {PMo6O21} units (Fig. 1c). Finally, these 1D chains were further joined together to obtain a 2D supramolecular layer via the hydrogen bonds (N2–H⋯O20 = 2.744 Å, N2–H⋯O13 = 2.938 Å in 1) existing between the N atoms of PABA and the O atoms of the {PMo6O21} units (Fig. 1d).
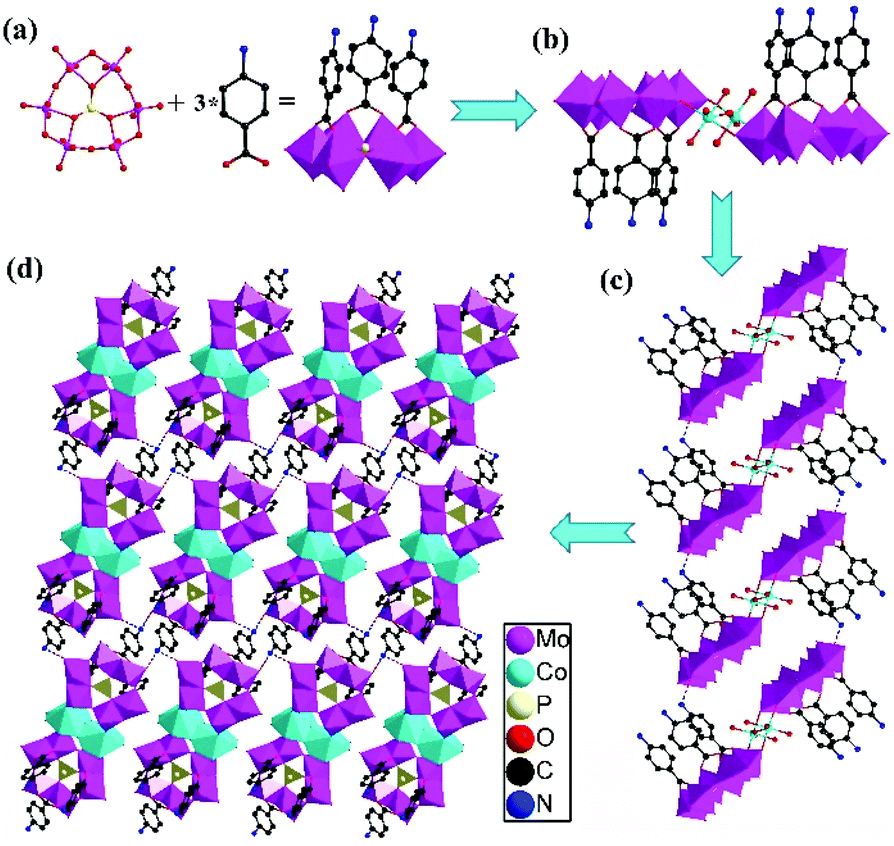 | ||
| Fig. 1 (a) The direct self-assembly protocol for the synthesis of carboxylic acid ligand-modified POMs; (b) ball-stick and polyhedral representation of the dimeric polyanion for 1; (c) schematic of the 1D supramolecular chain showing the hydrogen bonds; and (d) schematic of the 2D supramolecular sheet showing the hydrogen bonds (color code: P: light yellow, Mo: purple, O: red, Co: light blue, N: blue, and C: black). | ||
Bond valence sum (BVS) calculations indicated that the oxidation states of Mo, and P were +6, and +3, respectively, and those of Co/Mn were +2.
Fig. S2† displays the IR spectra of compounds 1–4, indicating the isostructural characteristic of these compounds. The terminal Mo–O bonds and the bridging M–O–Mo units (M = P and Mo) were distributed at 850–950 cm−1 and 580–690 cm−1, respectively. The peaks from 1000 and 1610 cm−1 are ascribed to the carboxylic acid ligands. The absorption maxima at 1605, 1541, and 1415 cm−1 for 1 are assigned to the vas(COO) and vs(COO) stretching frequencies, which indicates the bidentate coordination manner of the carboxylate group of PABA. The diffuse reflectance spectra of 1–4 are shown in Fig. S5† to spot the absorption bands distributed in the ultraviolet and visible regions. Taking compound 1 as an example, the UV region (200–400 nm) exhibits two intense absorption peaks, 234 and 315 nm, which are attributed to the O → Mo charge transfer for the {PMo6O21} polyoxoanions. In addition, one absorption band located at 525 nm in the visible region is assigned to 4T1g → 4T2g and 4A2g → 4T1g for the low-spin Co2+ cations.
Fig. S6† displays the TGA of 1 and 2, which demonstrated multistep weight loss processes. The total weight loss of 34.9% for 1 and 35.2% for 2 are in agreement with the calculated weight losses of 35.4% and 35.6% from 50 to 850 °C. The PXRD patterns of these samples match the calculated peaks, which manifested the phase purities of 1 and 2. In addition, the similar diffraction patterns of 1–4 further indicate the isostructural characteristic (Fig. S7 and S8†).
Catalytic oxidation study for methyl phenyl sulfide
Given that sulfoxides are very important as intermediates for functional materials, obtaining sulfoxides is desirable through the selective oxidation of sulfides. Herein, the selective oxidation of various sulfides to sulfoxides using H2O2 as the oxidant and 1–4 as the pre-catalysts were tested in detail.The selective oxidation of methyl phenyl sulfide was chosen as a model catalytic reaction to evaluate the performance of compounds 1–4. To implement the catalytic oxidation reaction, methyl phenyl sulfide (0.25 mmol), the pre-catalyst (2.5 μmol), H2O2 (oxidant, 0.3 mmol), and naphthalene (internal standard, 0.25 mmol) were performed in ethanol (0.5 ml) at room temperature, and the results of this catalytic system were monitored by gas chromatography (GC). As expected, methyl phenyl sulfide was efficiently catalyzed to the corresponding sulfoxide within 20 min, and compound 1 displayed a relatively high catalytic effect (Fig. 2b). Indeed, compound 1 displayed more notable activity than most reported POM-based materials, and could oxidize 98.7% of methyl phenyl sulfide into sulfoxide with 98% selectivity within 20 min. The performances of these compounds were compared with the heterogeneous catalytic materials based on POMs in recent years, such as the organic–inorganic hybrid POM materials {Cu3(ptz)4(Co2Mo10)}, {[Cu(mIM)4]V2O6}, VV17VIV12(C6H8O4)8, {As4W40O140(Ru2(Ac)2)} and the supported materials PDDA-SiV2W10, P2W15-Al2O3, PW12@Al-MCFen, and SBA-15/K6P2W18O62. As summarized in Table S1,† the compound 1 displayed significantly higher activity than the first three hybrid POM compounds and the first composite material (obtained using TBHP or UHP as the oxidant or long reaction time); this catalytic system also showed a little higher activity than other pre-catalysts that were obtained using a little longer reaction time and displayed relatively low selectivity. Fig. 2c shows the recorded progress of the oxidation of methyl phenyl sulfide in the presence of 1 through GC signals, which illustrated that methyl phenyl sulfide was almost entirely oxidized to the corresponding sulfoxide. To further verify the accuracy of the catalytic process, 1H NMR spectroscopy was employed to monitor the conversion and selectivity (Fig. 3a). The catalytic reaction at 4, 8, 16 and 20 minutes was investigated via1H NMR spectroscopy in CDCl3. In addition, the pure methyl phenyl sulfide, methyl phenyl sulfoxide, and methyl phenyl sulfone were also tested to further illuminate the reaction. The comparison of the 1H NMR spectra of these actual processes and the pure substrate or products demonstrates that methyl phenyl sulfide can be almost converted to methyl phenyl sulfoxide within 20 minutes.
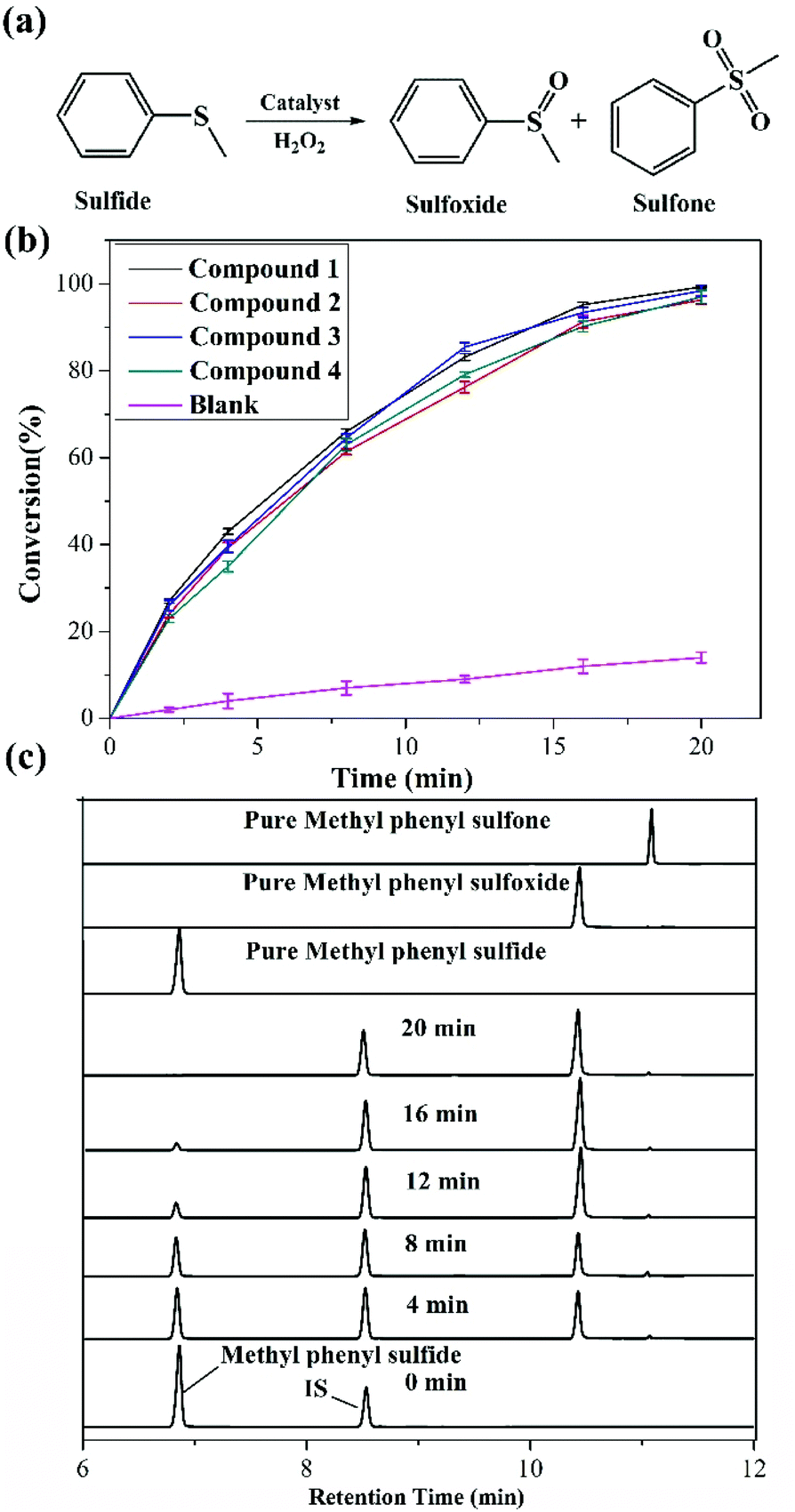 | ||
| Fig. 2 (a) Catalytic oxidation pathway of methyl phenyl sulfide to methyl phenyl sulfoxide and methyl phenyl sulfone; (b) the conversion of methyl phenyl sulfide via oxidation using the compounds 1–4 and blank. Reaction conditions: 0.25 mmol sulfide, 2.5 μmol pre-catalyst, 0.25 mmol naphthalene (internal standard), 0.3 mmol H2O2 and 0.5 mL ethanol at room temperature for 20 min; (c) GC-FID signals indicating the progress of the oxidation of methyl phenyl sulfide (6.89 min) to methyl phenyl sulfoxide (10.38 min) and methyl phenyl sulfone (11.02 min), and the internal standard (8.495 min, naphthalene) in the presence of 1. | ||
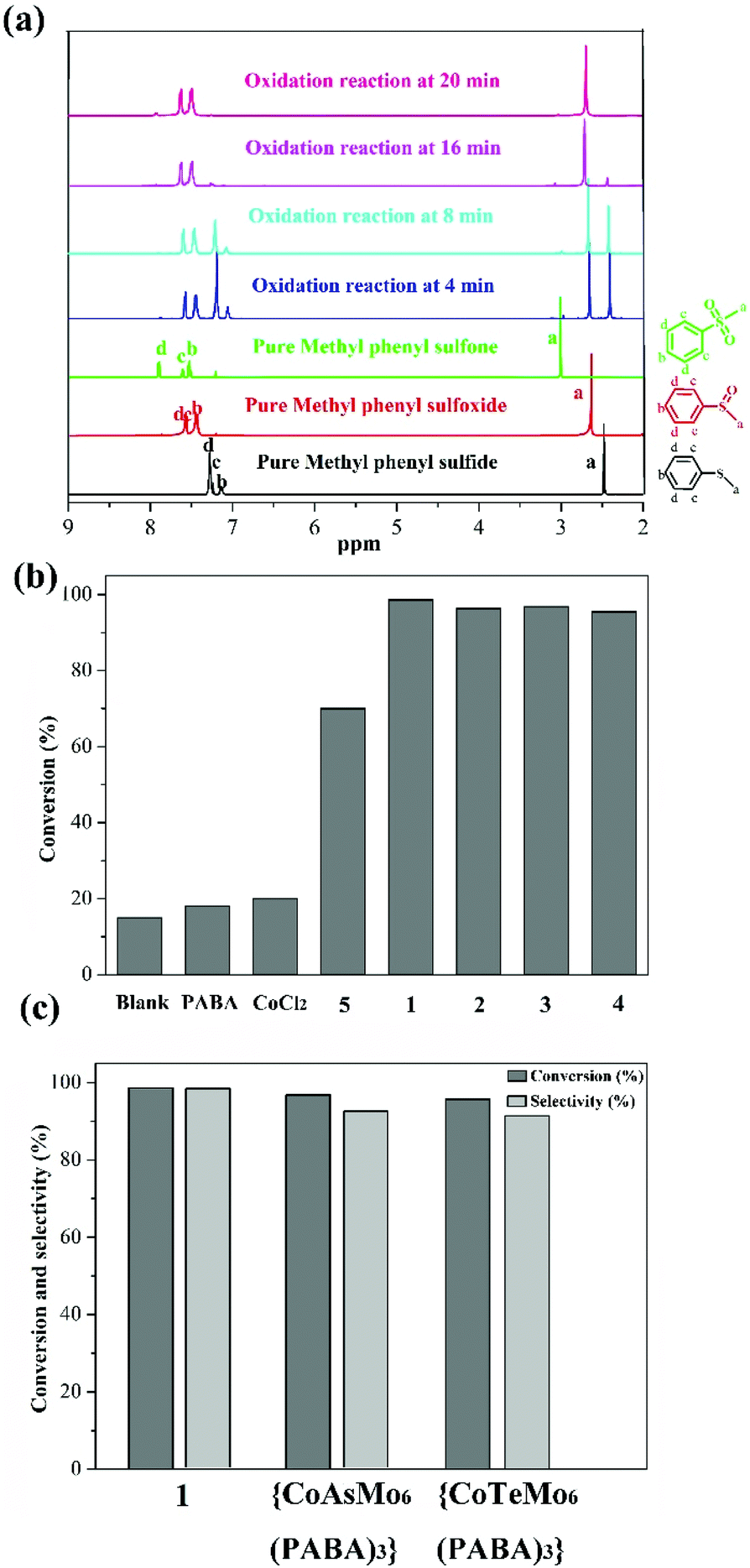 | ||
| Fig. 3 (a) Selected regions of the 1H NMR spectra of methyl phenyl sulfide, methyl phenyl sulfoxide, methyl phenyl sulfone, oxidation reaction (without internal standard) at 4 min, 8 min 16 min and 20 min in CDCl3. The peaks used for calculation are labeled with chemical shift; (b) the oxidation of sulfide by different pre-catalysts; (c) the conversion and selectivity of methyl phenyl sulfide oxidation for different pre-catalysts. | ||
Then, to understand the active center of compound 1 for the selective oxidation of methyl phenyl sulfide, we further investigated the catalytic performance of PABA, CoCl2 salt, and compound 5 (Cs3[PMo6O21(PABA)3]·nH2O). Compound 5 was confirmed by IR spectroscopy and EDS data because of the poor crystal quality (Fig. S3†). Fig. 3b reveals that both PABA and CoCl2 have converted very little methyl phenyl sulfide, and the compound 5 originating from the PABA-modified POMs can catalyze 72% of methyl phenyl sulfide with a selectivity of 89%, which is far behind that in the case of 1. Thus, it can be seen that integrating the Co site and PABA-modified POMs together can improve the catalytic oxidation reaction with rapid and excellent selectivity for the oxidation of methyl phenyl sulfide. Furthermore, a comparison of the catalytic oxidation of compound 1 with those of {CoAsMo6(PABA)3} and {CoTeMo6(PABA)3}, which possessed different central atoms but similar architectures to the compound 1, was conducted to explore how the {ZMo6} unit impacted the catalytic activity. {CoAsMo6(PABA)3} and {CoTeMo6(PABA)3} converted 96.8% and 95.5% of methyl phenyl sulfide with a selectivity of 96.1% and 96.3% under similar conditions, respectively, which indicates that the central heteroatoms played a certain role for the selective oxidation of methyl phenyl sulfide and the corresponding order is PMo > AsMo > TeMo (Fig. 3c). A similar phenomenon has been investigated by the Mizuno and Su's group.36,37
In view of previous research studies and the abovementioned results, we speculate that the peroxo-metal species originating from 1 and H2O2 play a crucial part in the catalytic oxidation process, which have been widely investigated based on POMs.26–28,38 In addition, to exclude the free radical mechanism, radical scavengers (such as diphenylamine and 1,4-benzoquinone) were added into the catalytic reactions, and no change of the conversion was observed. Hence, we proposed a possible mechanism of methyl phenyl sulfide selective oxidation in Fig. 4a. To verify the interaction between 1 and H2O2, UV-Vis spectra of the mixed solution were measured before and after the addition of H2O2 into solution 1. Indeed, two red shifts in the ultraviolet region (276 → 284 nm) and in the visual region (540 → 544 nm), and a new band at 760 nm confirm the interaction between compound 1 and H2O2 (Fig. 4b and c). As shown in Fig. 4a, the active peroxomolybdenum and peroxocobalt species derived from compound 1 and H2O2 worked on the S atom of the sulfides to yield the corresponding products.
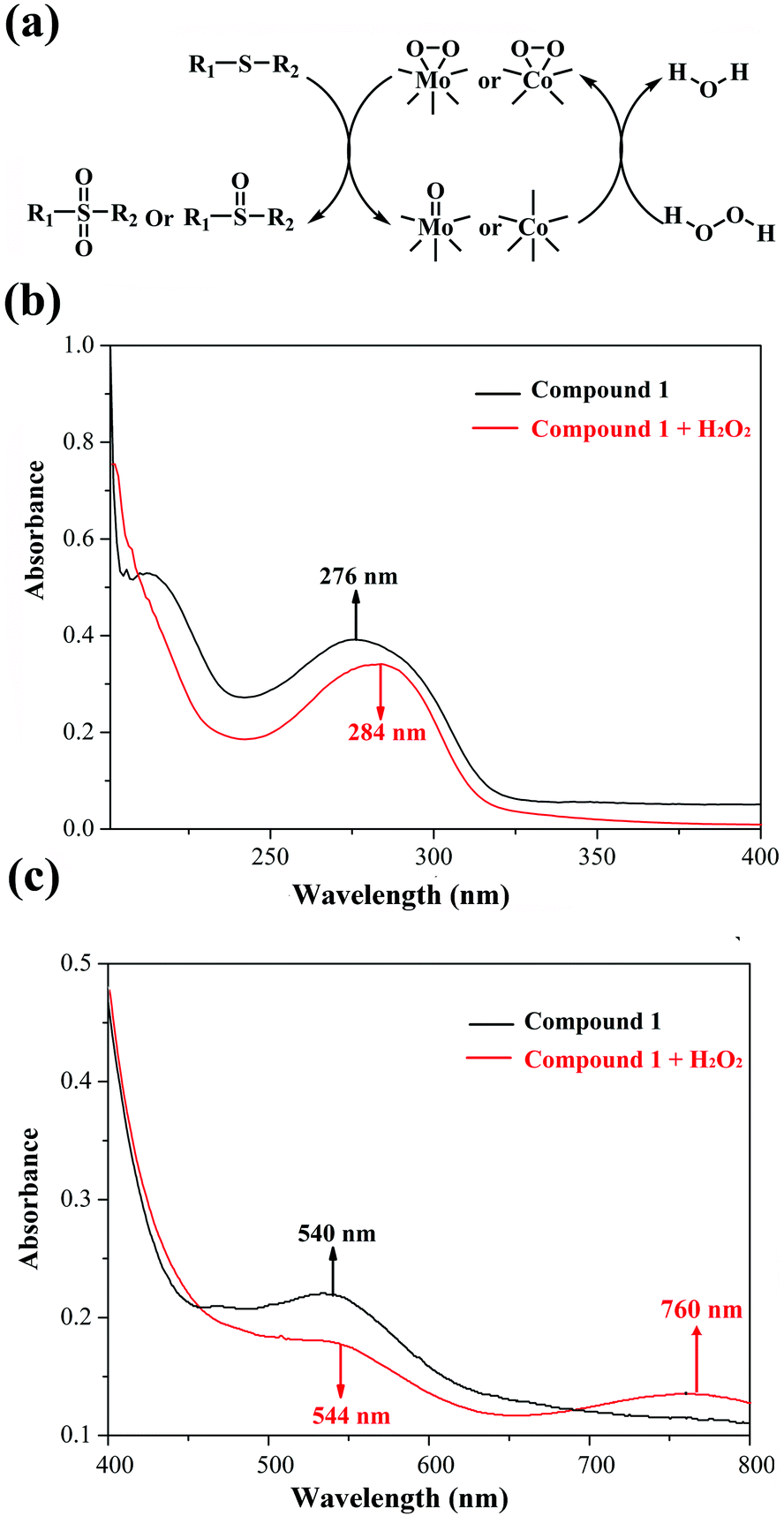 | ||
| Fig. 4 (a) Proposed mechanism of the catalytic oxidation of sulfides; (b) UV-Vis spectra of the compound 1 in dimethyl sulfoxide with one-drop portions of H2O2 in the UV region (200–400 nm); (c) UV-Vis spectra of the compound 1 in dimethyl sulfoxide with one-drop portions of H2O2 in the visible region (400–800 nm). | ||
Subsequently, we also investigated the recyclability and stability of this pre-catalyst. Just as in Fig. 5a, the conversion of methyl phenyl sulfide remained almost steady in the following 10 cycles, and only a negligible decrease occurred using the recovered pre-catalyst by simple filtration. No catalytic activity was observed when the pre-catalyst was filtered. Furthermore, no distinct change could be detected from the IR spectra and PXRD patterns before and after the catalytic reaction (Fig. 5b and c). From the abovementioned results, we can conclude that these compounds are excellent heterogeneous oxidation pre-catalysts.
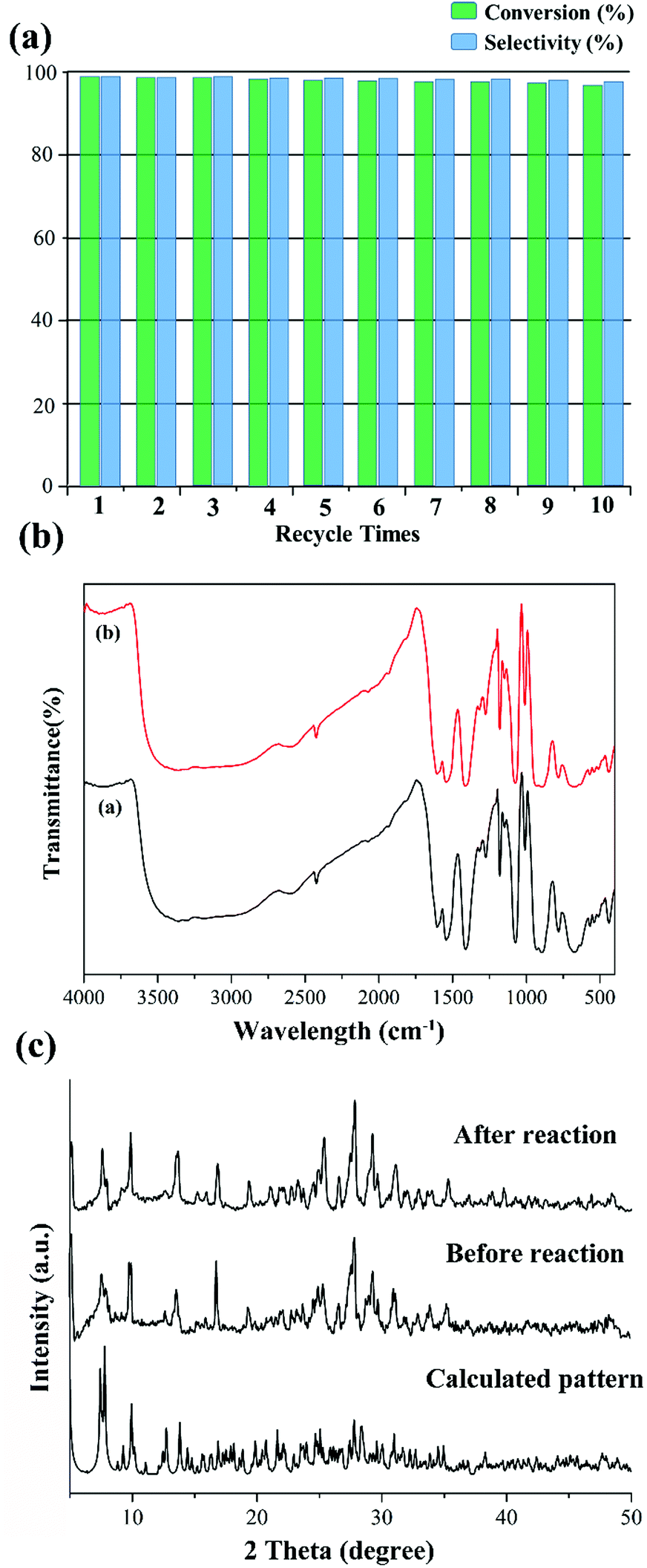 | ||
| Fig. 5 (a) Recycling of the catalytic system for the oxidation of methyl phenyl sulfide using compound 1; (b) IR spectra for the (a) as-synthesized compound 1 and (b) recovered pre-catalyst after catalytic reaction; (c) powder X-ray diffraction (PXRD) patterns of 1: calculated pattern from crystal data, experimental pattern before reaction and experimental pattern of the recovered pre-catalyst after the oxidation of methyl phenyl sulfide. | ||
Given that compounds 1–4 exhibited excellent catalytic activities in the oxidation of methyl phenyl sulfide, and compound 1 outperformed other compounds with respect to the catalytic activity, various organic sulfides with different electronic and steric effects were continuously investigated using compound 1 (Table 1). The conversion of these substrates did not obviously decrease despite the introduction of the electron donor or acceptor units with less steric hindrance into monophenyl sulfides. We were also delighted to find that even the sulfides with large steric hindrance, such as dibenzyl sulfide and diphenyl sulfide, did not greatly influence the catalytic activities. The above results demonstrate that these compounds possessed superior catalytic capability in the oxidation of sulfides. In addition, to further clarify the function of the central heteroatoms of the {ZMo6} units, the selective oxidation of these organic sulfides were also performed in the presence of {CoAsMo6(PABA)3} and {CoTeMo6(PABA)3} (Tables S2–S7†), which indicated that the heteroatoms indeed made a difference in the catalytic activity. We also investigated the catalytic oxidation reaction kinetics of methyl phenyl sulfide. The time profiles for the selective oxidation reaction process of methyl phenyl sulfide are shown in Fig. 2. A plot of the reaction time against ln(Ct/C0) displays a linear correlation, which shows that the oxidation of methyl phenyl sulfide is in accordance with the first order reaction. The first-order kinetics constants k1–k4 for 1–4 are 0.23842 min−1, 0.1624 min−1, 0.20015 min−1, and 0.17483 min−1, respectively (Fig. S9†).
| Entry | Substrates | Products | Con. (%) | Sel.b (%) |
|---|---|---|---|---|
| a Reaction conditions: Sulfide (0.25 mmol, 1 equiv.), pre-catalyst (1 mol%), H2O2 (1.2 equiv.), ethanol (0.5 mL), for 20 min, at room temperature. b Selectivity to sulfoxides, the byproduct was sulfone. | ||||
| 1 |
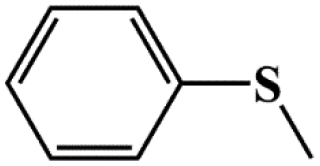
|
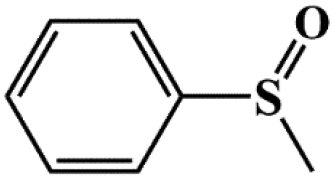
|
99.5 | 98 |
| 2 |
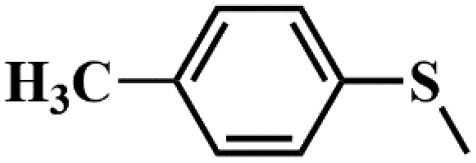
|
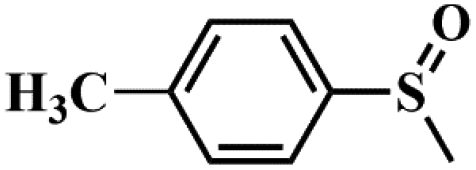
|
98.4 | 97.5 |
| 3 |
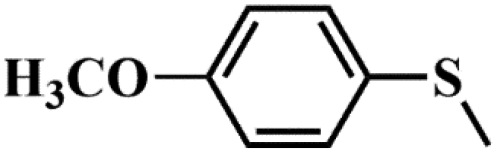
|
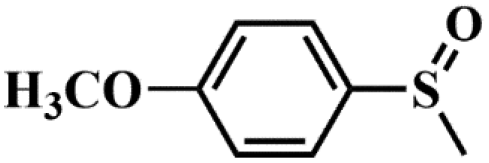
|
98 | 96.5 |
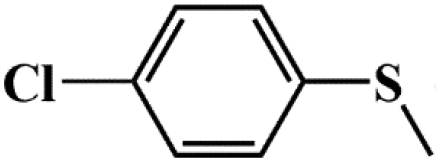
| 4 |
|
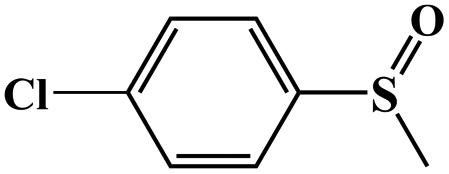
|
97.8 | 95.7 |
| 5 |
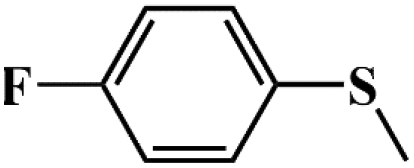
|
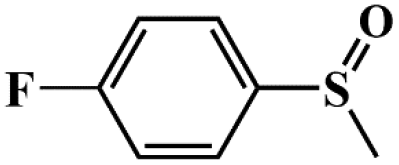
|
98.6 | 96.5 |
| 6 |
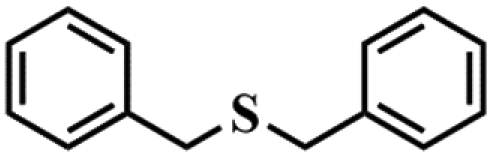
|
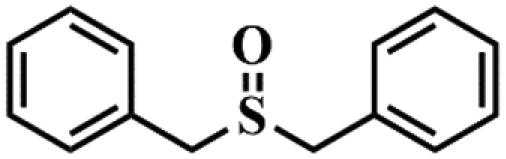
|
98.1 | 94.3 |
| 7 |
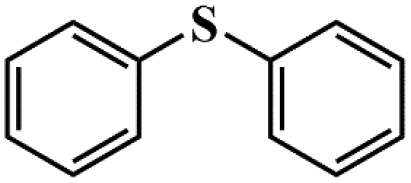
|
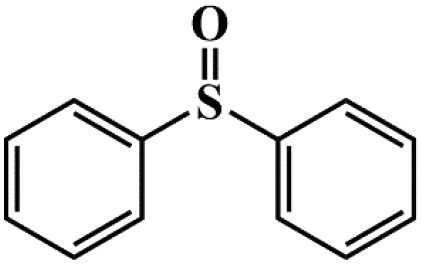
|
95.1 | 92.9 |
Catalytic oxidation study for CEES
In light of the excellent catalytic activity and selectivity of compounds 1–4 in the oxidation of methyl phenyl sulfide, it is of great interest to explore their catalytic performance in the selective oxidation of sulfur mustard (one of the most toxic chemical warfare agents). Owing to the high toxicity of sulfur mustard, the simulant molecule 2-chloroethyl ethyl sulphide (CEES) with a similar architecture is always employed. The catalytic reaction condition was similar to methyl phenyl sulfide: CEES (0.25 mmol), pre-catalyst (2.5 μmol), H2O2 (oxidant 0.3 mmol), naphthalene (internal standard 0.25 mmol) and the solvent ethanol (0.5 ml) at room temperature. As expected, these compounds exhibited superb catalytic activity for the selective oxidation of CEES. The results demonstrate that CEES can be almost entirely degraded within 12 min (Fig. 6b). As the best pre-catalyst among these compounds, compound 1 can oxidize 98.8% of CEES into the nontoxic 2-chloroethyl ethyl sulfoxide (CEESO) with the selectivity of 99.0%. In addition, the process of the selective oxidation of CEES in the presence of 1 was also recorded by GC-FID signals, and it indicated that CEES was almost completely catalyzed within 12 min under mild conditions (Fig. 6c). Similarly, 1H NMR spectroscopy monitored the oxidation of CEES (Fig. 6d). Also, for assessing the recyclability and stability of this hybrid compound, the pre-catalyst (recovered by simple filtration) can be reused in selective oxidation reactions at least ten times with no significant loss of activity (Fig. 6e).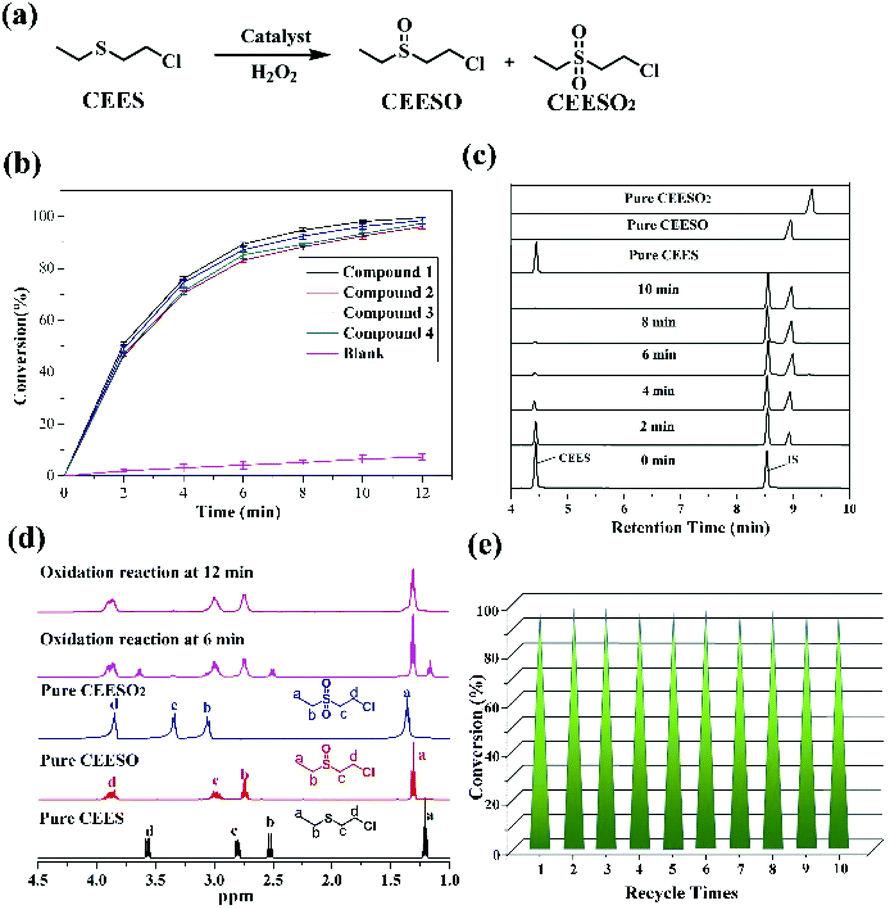 | ||
| Fig. 6 (a) Catalytic oxidation pathway of CEES to CEESO and CEESO2; (b) the conversion of CEES via oxidation using the compounds 1–4 and blank. Reaction conditions: 0.25 mmol CEES, 2.5 μmol pre-catalyst, 0.25 mmol naphthalene (internal standard), 0.3 mmol H2O2 and 0.5 mL ethanol at room temperature for 12 min; (c) GC-FID signals indicating the progress of the oxidation of CEES (4.56 min) to CEESO (8.92 min), CEESO2 (9.23) and internal standard naphthalene (8.495 min) in the presence of 1; (d) selected regions of the 1H NMR spectra of CEES, CEESO, CEESO2 oxidation reaction (without internal standard) at 6 min and oxidation reaction (without internal standard) at 12 min with two compounds in CDCl3. The peaks used for calculation are labeled with the chemical shifts; (e) recycling of the catalytic system for the oxidation of CEES using the compound 1. | ||
4. Conclusions
In conclusion, four hybrid compounds were prepared through a simple one-pot reaction and used as the oxidative pre-catalyst for selectively catalyzing various phenyl sulfides and the sulfur mustard simulant CEES. As a superb pre-catalyst, these compounds can rapidly and selectively convert methyl phenyl sulfides to their corresponding sulfoxide, as well as catalyze the degradation of CEES to the nontoxic CEESO. Such remarkable catalytic activities are possibly ascribed to the synergistic effect between the carboxylic acid modified POMs and metal cations. In addition, the catalytic performance of heteropolymolybdates can be also influenced by altering the heteroatom of {ZMo6} in the order: {PIIIMo} > {AsIIIMo} > {TeIVMo}. More importantly, the pre-catalyst can be recycled at least ten times with no obvious loss of activity. These results encourage us to design and prepare the highly active and novel heterogeneous hybrid materials and investigate the catalytic oxidation performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21371027, 20901013), Natural Science Foundation of Liaoning Province (2015020232) and Fundamental Research Funds for the Central Universities (DUT15LN18, DUT19LK01).References
- (a) J. E. Backvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, Germany, 2010 CrossRef; (b) M. Hudlucky, Oxidations in Organic Chemistry, ACS Monograph Series, American Chemical Society, Washington, DC, 1990 Search PubMed.
- (a) I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef; (b) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS.
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239–272 CrossRef.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- A. M. Khenkin, I. J. Efremenko, M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2013, 135, 19304–19310 CrossRef CAS.
- K. Kamata, Y. Yonehara, K. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–966 CrossRef CAS.
- D. L. Long, E. Burkholder and L. Cronin, Polyoxometalate Clusters, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- W. J. Luo, J. Hu, H. L. Diao, B. Schwarz, C. Streb and Y. F. Song, Angew. Chem., Int. Ed., 2017, 56, 4941–4944 CrossRef CAS.
- Y. J. Tang, M. R. Gao, C. H. Liu, S. L. Li, H. L. Jiang, Y. Q. Lan, M. Han and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928–12932 CrossRef CAS.
- H. Yu, S. Ru, G. Y. Dai, Y. Y. Zhai, H. L. Lin, S. Han and Y. G. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867–3871 CrossRef CAS.
- J. Zhang, J. Hao, Y. G. Wei, F. P. Xiao, P. C. Yin and L. S. Wang, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS.
- (a) M. Zhao, X. W. Zhang and C. D. Wu, ACS Catal., 2017, 7, 6573–6580 CrossRef CAS; (b) Y. J. Wang, W. L. Chen, L. Chen, X. T. Zheng, S. S. Xu and E. B. Wang, Inorg. Chem. Front., 2017, 4, 559–565 RSC; (c) T. Zhang, L. K. Yan, S. Cong, W. Guan and Z. M. Su, Inorg. Chem. Front., 2014, 1, 65–70 RSC; (d) T. Zhang, W. Guan, T. Y. Ma, Y. Lin, L. K. Yan and Z. M. Su, Inorg. Chem. Front., 2015, 2, 544–549 RSC.
- (a) Y. Zhang, L. L. Wu, X. Y. Zhao, Y. N. Zhao, H. Q. Tan, X. Zhao, Y. Y. Ma, Z. Zhao, S. Y. Song, Y. H. Wang and Y. G. Li, Adv. Energy Mater., 2018, 1801139 CrossRef; (b) X. L. Hao, Y. Y. Ma, H. Y. Zang, Y. H. Wang, Y. G. Li and E. B. Wang, Chem. – Eur. J., 2015, 21, 3778–3784 CrossRef CAS; (c) J. W. Zhang, Y. C. Huang, J. Hao and Y. G. Wei, Inorg. Chem. Front., 2017, 4, 1215–1218 RSC.
- M. Zheng, Y. Ding, X. H. Cao, T. Tian and J. Q. Lin, Appl. Catal., B, 2018, 237, 1091–1100 CrossRef CAS.
- (a) Y. Wang, B. Li, H. J. Qian and L. X. Wu, Inorg. Chem., 2016, 55, 4271–4277 CrossRef CAS; (b) G. P. Yang, S. X. Shang, B. Yu and C. W. Hu, Inorg. Chem. Front., 2018, 5, 2472–2477 RSC; (c) J. X. Wang, Y. D. Wang, M. J. Wei, H. Q. Tan, Y. H. Wang, H. Y. Zang and Y. G. Li, Inorg. Chem. Front., 2018, 5, 1213–1217 RSC.
- P. T. Ma, F. Hu, J. P. Wang and J. Y. Niu, Coord. Chem. Rev., 2019, 378, 281–309 CrossRef CAS.
- X. B. Han, Y. G. Li, Z. M. Zhang, H. Q. Tan, Y. Lu and E. B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS.
- R. Fazaeli, H. Aliyan, M. A. Ahmadi and S. Hashemian, Catal. Commun., 2012, 29, 48–52 CrossRef CAS.
- Z. Karimi and A. R. Mahjoub, Catal. Commun., 2011, 12, 984–988 CrossRef CAS.
- C. B. Yang, Q. Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. G. Deng, Green Chem., 2009, 11, 1401–1405 RSC.
- M. Carraro, G. L. Fiorani, L. Mognon, F. Caneva, M. Gardan, C. Maccato and M. Bonchio, Chem. – Eur. J., 2012, 18, 13195–13202 CrossRef CAS.
- T. A. G. Duarte, S. M. G. Pires, I. C. M. S. Santos, M. M. Q. Simões, M. G. P. M. S. Neves, A. M. V. Cavaleirob and J. A. S. Cavaleiroa, Catal. Sci. Technol., 2016, 6, 3271–3278 RSC.
- W. Zhao, C. X. Yang, Z. G. Cheng and Z. H. Zhang, Green Chem., 2016, 18, 995–998 RSC.
- (a) L. L. Hong, P. Win, X. Zhang, W. Chen, H. N. Miras and Y. F. Song, Chem. – Eur. J., 2016, 22, 11232–11238 CrossRef CAS; (b) Z. X. Yao, H. N. Miras and Y. F. Song, Inorg. Chem. Front., 2016, 3, 1007–1013 RSC.
- X. Q. Lia, J. Liu, H. F. Dong, X. R. Sun, Y. N. Chi and C. W. Hu, J. Hazard. Mater., 2018, 344, 994–999 CrossRef.
- X. R. Sun, J. Dong, Z. Li, H. F. Liu, X. T. Jing, Y. N. Chi and C. W. Hu, Dalton Trans., 2019, 48, 5285–5291 RSC.
- J. Dong, H. J. Lv, X. R. Sun, Y. Wang, Y. M. Ni, B. Zou, N. Zhang, A. X. Yin, Y. N. Chi and C. W. Hu, Chem. – Eur. J., 2018, 24, 19208–19215 CrossRef CAS.
- (a) M. D. Han, Y. J. Niu, R. Wan, Q. F. Xu, J. K. Lu, P. T. Ma, C. Zhang, J. Y. Niu and J. P. Wang, Chem. – Eur. J., 2018, 24, 11059–11066 CrossRef CAS; (b) M. L. Mei, X. X. Xu, Y. Wang, X. J. Wang and Y. Q. Huo, Inorg. Chem. Front., 2018, 5, 819–826 RSC.
- K. Wang, Y. J. Niu, D. Y. Zhao, Y. X. Zhao, P. T. Ma, D. D. Zhang, J. P. Wang and J. Y. Niu, Inorg. Chem., 2017, 56, 14053–14059 CrossRef CAS.
- K. P. Sullivan, W. A. Neiwert, H. D. Zeng, A. K. Mehta, Q. S. Yin, D. A. Hillesheim, S. Vivek, P. C. Yin, D. L. Collins-Wildman, E. R. Weeks, T. B. Liu and C. L. Hill, Chem. Commun., 2017, 53, 11480–11483 RSC.
- Y. J. Hou, H. Y. An, Y. M. Zhang, T. Hu, W. Yang and S. Z. Chang, ACS Catal., 2018, 8, 6062–6069 CrossRef CAS.
- H. Y. An, Y. J. Hou, L. Wang, Y. M. Zhang, W. Yang and S. Z. Chang, Inorg. Chem., 2017, 56, 11619–11632 CrossRef CAS.
- J. K. Li, X. Q. Huang, S. Yang, Y. Q. Xu and C. W. Hu, Cryst. Growth Des., 2015, 15, 1907–1914 CrossRef CAS.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS.
- K. Kamata, T. Hirano and N. Mizuno, Chem. Commun., 2009, 26, 3958–3960 RSC.
- B. Zhu, L. K. Yan, W. Guan and Z. M. Su, Dalton Trans., 2015, 44, 9063–9070 RSC.
- (a) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 8251–8254 CrossRef CAS; (b) N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358–2370 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP drawing of 1; IR spectra, UV–Vis diffuse reflective spectra, TG plots, PXRD figures for 1–4; kinetic analysis of methyl phenyl sulfide; comparison of methyl phenyl sulfide oxidation in recent years; oxidation of various sulfides to the corresponding sulfoxides and sulfones; crystal data and structure refinement for 1 and 2; selected distances and angles for 1 and 2. CCDC 1949665 and 1949666. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01098j |
| This journal is © the Partner Organisations 2020 |