
In situ formation of porous LiCuVO4/LiVO3/C nanotubes as a high-capacity anode material for lithium ion batteries†
 *ab
*ab
Abstract
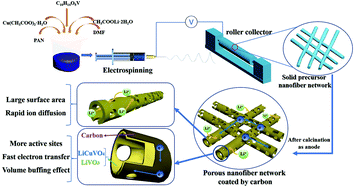
LiCuVO4 has attracted increasing attention as an anode material for lithium ion batteries due to its good structural stability. However, the rate capability is limited by the inferior electronic conductivity and low lithium ion diffusion efficiency. Herein, we report the synthesis of LiCuVO4/LiVO3/C porous nanotubes by an electrospinning method with subsequent calcination in air. The unique morphology, carbonaceous material in the composite and the existence of LiVO3 enable the as-prepared composite to have high capacity, superior cycling stability and rate capability. The LiCuVO4/LiVO3/C electrode possesses a capacity of 636 mA h g−1 which is higher than the theoretical capacity of LiCuVO4 (576 mA h g−1). Moreover, it can retain 77% of the capacity at 0.5 A g−1 after 300 cycles. These results demonstrate that LiCuVO4/LiVO3/C nanotubes are promising anode materials for lithium ion batteries.
- This article is part of the themed collection: 2019 Inorganic Chemistry Frontiers HOT articles
 Please wait while we load your content...
Please wait while we load your content...

