
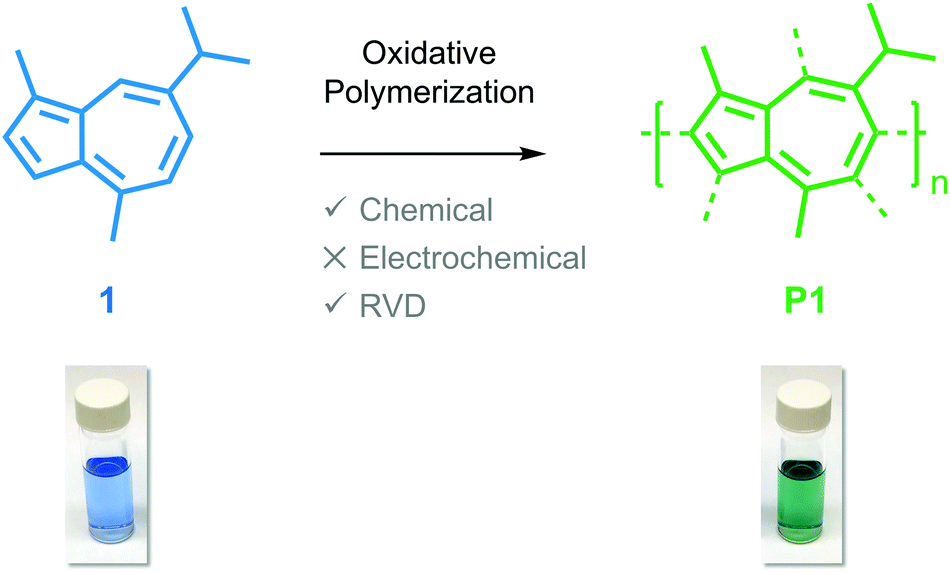
Guaiazulene revisited: a new material for green-processed optoelectronics†
David
Bilger
a,
Kwang-Won
Park
 a,
Ali
Abdel-Maksoud
b and
Trisha L.
Andrew
*ac
a,
Ali
Abdel-Maksoud
b and
Trisha L.
Andrew
*ac
aDepartment of Chemistry University of Massachusetts Amherst, Amherst, MA 01003, USA. E-mail: tandrew@umass.edu
bDepartment of Electrical Engineering, University of Massachusetts Amherst, Amherst, MA 01003, USA
cDepartment of Chemical Engineering, University of Massachusetts Amherst, Amherst, MA 01003, USA
First published on 27th October 2020
Abstract
Green-processed conjugated materials can reduce the cost of optoelectronic devices and simultaneously minimize their ecological footprint. Here, we use both solution and vapor phase chemistry to oxidatively polymerize the natural hydrocarbon dye, guaiazulene, yielding the more functional material poly(guaiazulene). We chemically characterize oligomers of poly(guaiazulene) using nuclear magnetic resonance spectroscopy, gel-permeation chromatography, laser-desorption ionization mass spectroscopy, and ultraviolet-visible absorption spectroscopy. The optical properties of poly(guaiazulene) oligomers are studied via electronic structure calculations and are contrasted to those of standard poly(azulene). We show that poly(guaiazulene) films synthesized from the vapor phase exhibit enhanced optical properties compared to counterparts synthesized in solution. Collectively, this work outlines a green reaction process that consists of a single step and uses earth-abundant reagents to yield a hitherto unreported polymer for optoelectronic applications.
Introduction
Conjugated molecules exhibit intriguing optical properties and can be processed into large area thin films using solution or vapor based techniques. Through meticulous molecular design and judicious selection of processing conditions, researchers have incorporated conjugated materials into a myriad of functional optoelectronic devices, such as bulk-heterojunction and dye-sensitized solar cells, to name a few.1 However, these advances have been accompanied by multiple detriments: (1) functional conjugated materials are often synthesized via multi-step pathways that have a linear relationship between cost and the number of steps required for production.2 (2) The prevailing solvents used in the synthesis and solution processing of conjugated materials are halogen-based, such as chloroform, chlorobenzene and dichlorobenzene, which are detrimental to human health and the environment.3 (3) Reliable C–C bond formation between (hetero)aryl halide groups is frequently achieved using organometallic reactions, primarily Stille and Suzuki cross-coupling reactions, that use catalysts derived from palladium, a precious Earth metal.4 These detriments collectively make clear the need for new, sustainable conjugated materials produced from simplified synthetic protocols that use earth-abundant reagents and non-hazardous solvents.Over the past decade, a paradigm shift towards the green synthesis and/or processing of conjugated materials has taken hold within the chemical community.5,6 Extensive efforts have been made to reduce the use of organic, halogenated processing solvents in exchange for non-halogenated alternatives that are comparatively less toxic.7 Other investigations have focused on effecting cross coupling using direct arylation polymerization and heterogeneous catalysis.8,9 Additional approaches, such as using vapor phase chemistry, go so far as to afford solvent-free reaction and deposition conditions with near negligible waste following work-up.10–13 In addition, researchers have used natural dyes in optoelectronic devices, thereby minimizing synthetic cost and environmental waste.8,14
Guaiazulene (1) is a red-absorbing, non-alternate hydrocarbon dye that is naturally derived from various sources, such as the Australian cypress-pine Callitris intratropica.15 The unsaturated architecture of 1, which consists of a five membered ring fused to a seven membered ring, induces asymmetry within its HOMO and LUMO energy levels. This orbital asymmetry is responsible for the remarkably dark blue color of 1,16 and has encouraged its use as an FDA-approved color additive in cosmetics.17 Guaiazulene could also serve as a sustainable, naturally-derived photoactive component in optoelectronic devices given its low cost (MilliporeSigma: $5.75 per g) and unique optical properties; however, it is a low melting point (mp. 28 °C), oily solid on account of its peripheral alkyl groups, which complicates film forming kinetics and results in poor control over the morphology of bulk heterojunctions. As a result, the focus of the chemical community has veered towards investigating the optoelectronic properties of azulene (mp. 99 °C), the parent compound of guaiazulene, despite its arduous synthesis and corresponding high cost (MilliporeSigma: $479.00 per g).18,19
Here, we report on the oxidative polymerization of 1 both in solution and in the vapor phase. To the best of our knowledge, there are no reports indicating that 1 has been polymerized previously via oxidative methods or otherwise. We characterize oligomers of poly(guaiazulene) (P1) using nuclear magnetic resonance spectroscopy (NMR), gel-permeation chromatography (GPC), laser-desorption ionization mass spectroscopy (LDI-MS), and ultraviolet-visible (UV-Vis) spectroscopy. We find that P1 forms uniform films from both solution and the vapor phase. The optical properties of poly(azulene) and P1 oligomers are simulated using electronic structure calculations to rationalize their similarities and differences. We find that thin films fabricated from the vapor phase exhibit higher absorption cross-sections than those fabricated from solution. Collectively, our findings outline a green reaction process that consists of a single step and uses earth-abundant reagents to yield a hitherto unreported polymer for optoelectronic applications.
Experimental
Materials
All solvents and reagents were purchased from MilliporeSigma and used as received without further purification. Guaiazulene (99%) was purchased from MilliporeSigma and its measured ultraviolet-visible (UV-Vis) absorption spectrum was consistent with previous reports.Solution synthesis of poly(guaiazulene) (P1)
Iron(III) chloride (FeCl3) (6.30 mmol) was added to a solution of chloroform (CHCl3) containing 1 (6.50 mmol) and sonicated for 15 minutes at room temperature. After another 5 minutes, the solution was quenched with methanol, washed with potassium carbonate, brine and EDTA, dried over magnesium sulfate and concentrated under vacuum to yield an oily green/blue residue that was purified via flash column chromatography using 5% ethyl acetate in hexanes as the eluent. Insoluble residues remained on the column. Oligomers of P1 were isolated as a vibrant green solid (40% yield) after drying under vacuum.Reactive vapor deposition (RVD) of P1
Glass substrates used for the RVD of P1 were cleaned according to the following procedure: (1) place the substrates in a detergent solution and sonicate for 5 min; (2) rinse the substrates with DI water and sonicate them for 5 min; (3) immerse the substrates (2×) in boiling isopropanol for 5 min; (4) treat the substrates with UV-ozone for 30 min.The reactive vapor deposition of P1 was carried out according to an oxidative chemical vapor deposition procedure.101 was placed in a glass ampule articulated with a Swagelok needle valve via a glass-to-metal seal. The needle valve was then, in turn, articulated with a glass inlet on the RVD reactor. An excess of FeCl3 was loaded into a ceramic crucible and positioned inside the RVD reactor. The distance between the crucible and reactor inlet was kept constant throughout the deposition. The reactor was then sealed and pumped down using an Edwards direct-drive vacuum pump, and a nitrogen gas inlet was used to maintain the reactor pressure between 80–200 mTorr. 1, FeCl3, and the glass substrates were heated using resistive heating tapes and maintained at temperatures of 140 °C, 190 °C, and 160 °C, respectively. As vapors of FeCl3 crossed over the monomer inlet, vapors of 1 were released into the chamber to maximize the intersectional area of both reagents, effecting the polymerization. The as-deposited films were washed with methanol for 10 min to remove any residual metal salts.
Chemical characterization
Absorption spectra were collected on an Agilent 8453 spectrophotometer. Samples were prepared in a suitable solvent for dissolution and diluted to a concentration with a maximum absorption of less than or equal to 1. Mass spectra were acquired using an ultrafleXtreme MALDI-TOF mass spectrometer (Bruker Instruments, Billerica MA) equipped with a Smartbeam™ laser at 355 nm. All samples were run without a matrix in reflectron mode. GPC measurements were conducted using an Agilent 1260 series gel permeation chromatograph with a refractive index (RI) detector. THF was used as an eluent with a flow rate of 1.0 mL min−1 and incubated at 40 °C with toluene serving as a flow marker. Monodisperse polystyrene was used as a calibration standard. 1H NMR spectra were collected on a Bruker Avance 400 MHz nuclear magnetic resonance spectrometer and all samples were prepared in deuterated chloroform.The surface morphology of the thin films prepared by RVD were probed using an Asylum Jupiter XR atomic force microscope (AFM). Images were acquired under tapping mode with a PPP-NCHR cantilever (force constant = 42 N m−1, Nanosensors, Switzerland). The RVD and drop-cast samples used to measure absorption cross-sections had film thicknesses of 350 nm and 1000 nm, respectively.
Electronic structure calculations
Electronic structure calculations were performed with the Gaussian 09’ software suite. All molecular structures were optimized to their global minimum prior to performing any calculations, unless otherwise stated. Density functional theory (DFT) calculations of molecular orbitals were conducted using the B3LYP functional and a 6-311+G (2d, p) basis set. Time-dependent density functional theory (TD-DFT) calculations were run using the CAM-B3LYP functional and a 6-311+G (2d, p) basis set.Results and discussion
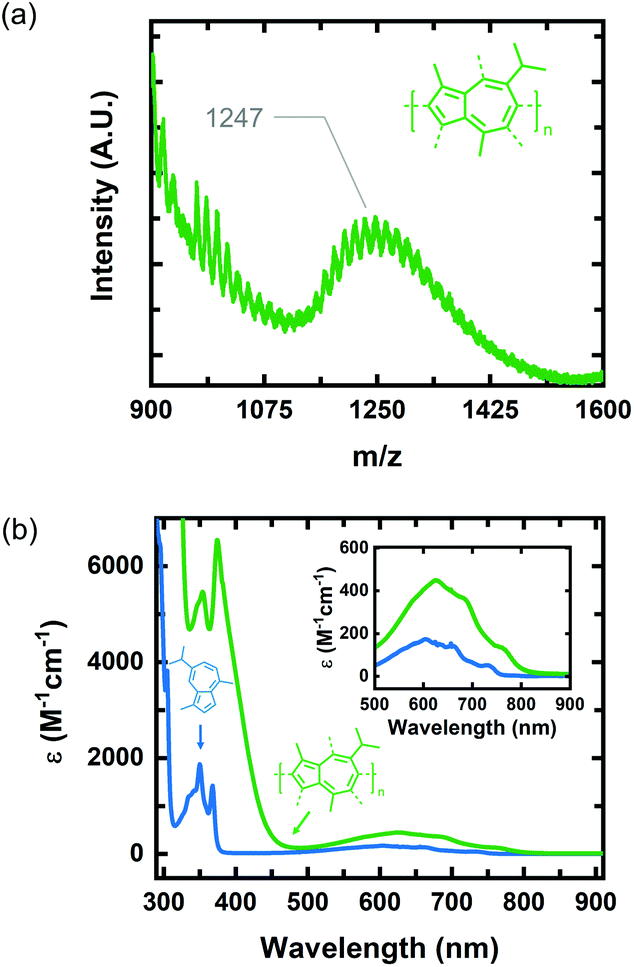
We identified 1 as a promising natural dye for optoelectronics given its low-cost and broad absorption at high wavelengths (>600 nm). Prior attempts to polymerize 1via electrochemical means have been unsuccessful,20 and efforts using a chemical oxidant are, to the best of our knowledge, unreported. In order to increase the processing potential of 1, we attempted to oxidatively polymerize it using iron(III) chloride (FeCl3) as an oxidant. We chose FeCl3 as it is an earth-abundant reagent that has been used extensively in both solution and vapor phase oxidative polymerizations.21We oxidatively polymerized 1 in chloroform using FeCl3, thereby yielding oligomers of P1 (Scheme 1). P1 oligomers exhibit a vibrant green appearance that contrast well with the characteristic blue color of their monomer. We confirmed that 1 polymerized in the presence of FeCl3 using both GPC and LDI-MS (Fig. 1a). The number average molecular weights (Mn) were 1186 g mol−1 and 1247 g mol−1 as determined from GPC and LDI-MS, respectively. Results obtained from 1H NMR spectroscopic studies confirmed the repeat unit structure of P1 oligomers are consistent with that of their monomer (Fig. S1†). Signals corresponding to the aliphatic protons of P1 oligomers match well with those of the methyl and isopropyl substituents of 1.22 The signals in the aromatic region of the 1H NMR spectrum of P1 oligomers suggest that this material is regiorandom, with a statistical mixture of linking points, as is expected from an oxidative polymerization reaction.
 | ||
| Scheme 1 The oxidative polymerization of guaiazulene (1) to poly(guaiazulene) (P1). | ||
 | ||
| Fig. 1 (a) LDI-MS of P1 oligomers synthesized via oxidative polymerization in CHCl3. (b) UV-vis spectra of monomer 1 (blue curve) and P1 oligomers (green curve) in hexane. | ||
The optical properties of P1 oligomers and the monomer 1 were compared in solution using UV-Vis absorption spectroscopy (Fig. 1b). Two sharp peaks are observed in the high-energy S0–S2 band of P1 oligomers (green) and 1 (blue). These transitions, in the spectrum of 1, have wavelength maxima at 350 nm and 368 nm. In the spectrum of P1 oligomers, these transitions display a slight bathochromic shift, residing at 354 nm and 374 nm. Indeed, the wavelength maximum of the S0–S1 transition of P1 oligomers is also bathochromically shifted compared to 1, with maxima at 624 nm and 604 nm for P1 oligomers and 1, respectively. The absorption coefficient of P1 oligomers is also much higher than that of 1, with the absorption coefficient of the S0–S1 transition maximum increasing by nearly threefold. Collectively, the optical properties of P1 oligomers in solution suggest that chemical species with extended conjugation lengths, relative to 1, are present in the sample.
Previous reports on the synthesis and characterization of poly(azulene) record that, while its absorption spectrum exhibits a very strong S0–S2 transition, its high-wavelength S0–S1 transition is lost, meaning that poly(azulene) lacks the vibrant blue color of its corresponding monomer.23,24 However, we do not observe similar behavior in poly(guaiazulene) oligomers, which is surprising given the similar chemical structures of 1 and azulene (Fig. S2†). To gain a better understanding of why P1 maintains its green color while poly(azulene) is mostly colorless, we performed time-dependent density functional theory (TD-DFT) calculations on guaiazulene and azulene trimers using a CAM-B3LYP functional and 6-311+G (2d, p) basis set. The CAM-B3LYP functional has been used successfully for a range of applications beyond time-dependent excitations, including the polarization of long chains and computation of excited state charge transfer species.25 To reduce computational costs, we simulated the UV-vis spectrum of azulene trimers and simplified (methyl-substituted) guaiazulene trimers linked at either the 1,3- or 1,7-positions and containing varying torsion angles between repeat units. The calculated UV-vis absorption profiles for a series of five trimers with different linking positions are depicted in Fig. S3.† All the trimers were predicted to display similar high-energy S0–S2 transitions, regardless of whether they were linked at the 1,3 or 1,7 positions. The results of these simulations indicate that the absorption profiles of poly(azulene) and P1 are not significantly dependent on where their repeat units are linked. However, while the calculated intensities of the low-energy S0–S1 transitions for the azulene trimers were weak, those of the guaiazulene trimers were comparatively strong, consistent with the experimental absorption profile of P1 oligomers.
We hypothesized that differences in the torsion angle between repeat units was primarily responsible for the absence or presence of visible color in poly(azulene) versus poly(guaiazulene), respectively. As illustrated by the simulated absorption spectra in Fig. 2, the geometry-optimized trimeric structure of azulene, shown in blue, has a torsion angle of 50°. On the other hand, the geometry-optimized trimeric structure of guaiazulene, shown in green, has a torsion angle of 80°. The simulated UV-vis absorption profile of the geometry-optimized azulene trimer with a torsion angle of 50° does not display a strong S0–S1 transition, whereas that of the guaiazulene trimer (torsion angle 80°) displays a prominent S0–S1 transition. If we deliberately freeze the torsion angle of the azulene trimer at an artificial angle of 80° (shown in red), we find the simulated absorption spectrum evolves into a profile identical to that of the guaiazulene trimer. Therefore, we concluded that the advantageous, red-absorbing characteristic of P1 oligomers results from substantial torsion between its repeat units, which allows the internal charge transfer interaction inherent to the azulene chemical structure (cyclopentadienyl anion–tropelium cation) to retain a high oscillator strength.26
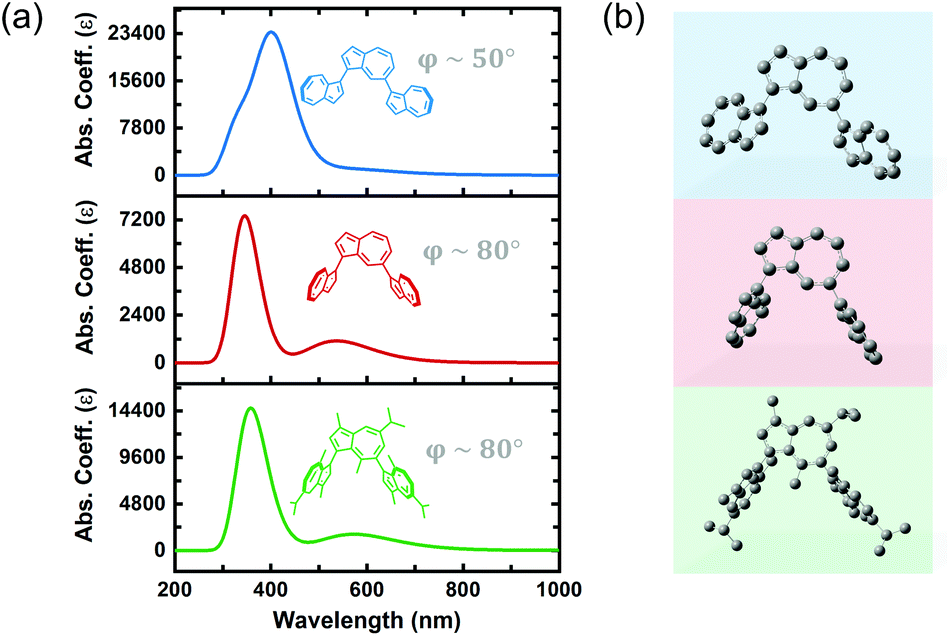 | ||
| Fig. 2 (a) Simulated UV-vis absorption spectra (TD-DFT, CAM-B3LYP/6-311G+) of guaiazulene and azulene trimers with varying torsion angles between repeat units. (b) 3D ball and stick models of the calculated structures with hydrogen atoms omitted for visual clarity. | ||
We adapted the oxidative polymerization reaction of 1 to the vapor phase using a previously reported, custom-built hot-wall reactor. The parameters used for the reaction are shown in Fig. 3a and have been outlined extensively in previous reports on reactive vapor deposition (RVD) protocols.27 RVD is a solvent-free process where a vapor phase chemical reaction and deposition are performed simultaneously, thereby eliminating the need for hazardous solvents, such as chloroform, in this case. We simultaneously heated 1 (140 °C) and FeCl3 (190 °C) under reduced pressure (80–200 mTorr) so their vapors would intersect over glass substrates placed down the long axis of our reactor. The as-deposited RVD films exhibited a black appearance, as shown by the image in Fig. S4,† and were insoluble in a number of organic solvents, including chloroform and dichloromethane, suggesting the samples are composed of higher molecular weight species. Upon washing the RVD films with methanol to remove residual metal salts (following previously explored procedures),10,27 the films adopted a green/blue color (Fig. 3b), consistent with the appearance of solution polymerized P1 oligomers dissolved in hexane. A topographical image of a washed RVD film was acquired using atomic force microscopy and is shown in Fig. 3b. The RVD film exhibits a smooth and uniform texture uncharacteristic of conjugated thin films fabricated using other methods.28 Such uniform films could prove useful in enhancing adhesion between different materials in optoelectronic devices, such as dye and semiconductor components in DSSCs, without the need for anchoring functional groups.29,30
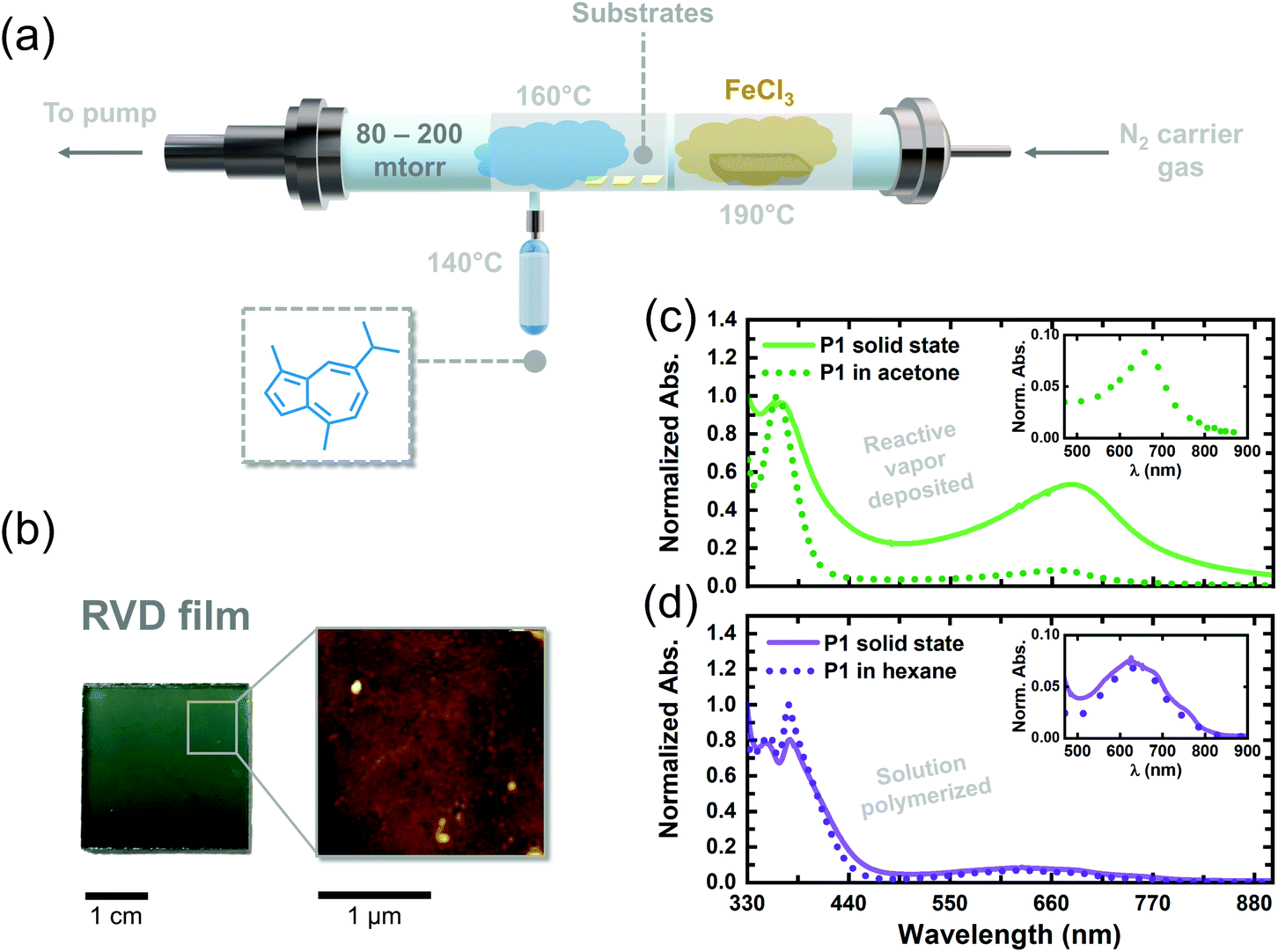 | ||
| Fig. 3 (a) Schematic of RVD reactor and conditions used for the vapor-phase polymerization of P1. (b) Optical image of a rinsed RVD film (left) and corresponding AFM image (right) showing the topography of the film. (c) UV-vis spectra of RVD polymerized P1 measured in solution (dotted green line) and in the solid state (solid green line). (d) UV-vis spectra of solution polymerized P1 measured in solution (dotted purple line) and in the solid state (solid purple line). | ||
We attempted to dissolve the RVD film from the walls of our reactor to perform conventional chemical analyses. However, a substantial amount of insoluble material remained in the reactor, thereby inhibiting us from preparing a sufficiently concentrated solution to acquire an 1H NMR spectrum. Mass spectroscopy of the soluble portions revealed a Mn of only 780 g mol−1 (Fig. S5†), confirming that we were unable to solubilize the higher molecular weight portions that were formed during RVD. The UV-vis absorption spectrum of the soluble portions obtained from the RVD films in acetone exhibited a S0–S1 transition at 665 nm trailed by a strong S0–S2 transition at 362 nm, consistent with solution polymerized P1 oligomers. Given the similarities between the solution spectra of P1 oligomers and the RVD films, we tentatively concluded that the guaiazulene repeat unit remains intact in the RVD films following polymerization in the vapor phase. However, we posit the presence of tri- and tetra-substituted guaiazulene repeat units in the RVD films (depicted by dotted bonds in Scheme 1), on account of the higher temperatures and superstoichiometric equivalents of the iron oxidant used during RVD.
We compared the absorption profiles of the soluble RVD fractions to those of the insoluble RVD thin films (Fig. 3c, solid green line). The solid-state RVD films exhibit much broader S0–S1 and S0–S2 transitions as compared to the soluble, low molecular-weight fractions dissolved in acetone. Indeed, we expect to see broader absorption in the RVD thin films since they likely contain higher molecular weight species. In addition, the S0–S1 transition of the solid state RVD films is stronger compared to the low molecular-weight fractions. The thin film absorption profile of P1 oligomers that were chemically synthesized in solution was also compared to the films created using RVD. Notably, the average absorption cross-section of the RVD thin films is an order of magnitude higher (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−3) than that of dropcast films of P1 oligomers synthesized in solution (2000 cm−3). On the basis of our TD-DFT calculations, we propose that the larger absorption cross-section of the RVD films stems from enhanced torsion strain between repeat units in the sample. Though a complete understanding of this phenomenon requires further investigation, the larger oscillator strength of these films qualifies RVD as a superior synthesis and processing technique for elaborating red-absorbing optoelectronic devices containing poly(guaiazulene) (Table 1).
000 cm−3) than that of dropcast films of P1 oligomers synthesized in solution (2000 cm−3). On the basis of our TD-DFT calculations, we propose that the larger absorption cross-section of the RVD films stems from enhanced torsion strain between repeat units in the sample. Though a complete understanding of this phenomenon requires further investigation, the larger oscillator strength of these films qualifies RVD as a superior synthesis and processing technique for elaborating red-absorbing optoelectronic devices containing poly(guaiazulene) (Table 1).
| Compound | Solvent | λ max (nm) | Absorption coefficient (M−1 cm−1) | Absorption cross-section (cm−3) |
|---|---|---|---|---|
| 1 | Hexane | 606 | 170 | — |
| P1 (solution polymerized | Hexane | 624 | 450 | — |
| P1 (solution polymerized) | — | 629 | — | 2000 |
| P1 (RVD soluble fractions) | Acetone | 665 | 160 | — |
| P1 (RVD) | — | 678 | — | 11000 |
Conclusions
Oxidative polymerization of the naturally-occurring hydrocarbon dye guaiazulene with iron(III) chloride creates the advantageously red-absorbing polymer poly(guaiazulene). To the best of our knowledge, there are no reports indicating that guaiazulene has been polymerized previously via oxidative methods or otherwise. We characterized the reaction product, poly(guaiazulene), using nuclear magnetic resonance spectroscopy (NMR), gel-permeation chromatography (GPC), laser-desorption ionization mass spectroscopy (LDI-MS), and ultraviolet-visible (UV-Vis) absorption spectroscopy. Electronic structure calculations reveal that a large torsion angle between repeat units is responsible for preserving the vibrant green color in poly(guaiazulene) films, whereas structurally-similar poly(azulene) films are reported as colorless (i.e., primarily UV-absorbing). Further, we find that poly(guaiazulene) films fabricated from the vapor phase exhibit higher absorption cross-sections in the visible region, as compared to thin films created using conventional solution processing protocols. Collectively, our findings outline a green reaction process that consists of a single step and uses earth-abundant reagents to yield a hitherto unreported polymer for optoelectronic applications.Since conjugated materials with strong absorption features above 650 nm are sparse, and because many known examples of red-absorbing conjugated materials are donor–acceptor polymers with multi-step syntheses,30 we expect that the simple, naturally-derived polymer reported here will be useful as a photoactive layer for next-generation solar energy harvesting devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation under CHEM MSN 1807743. Mass spectroscopy data were obtained at the University of Massachusetts Mass Spectrometry Center.Notes and references
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS.
- T. Osedach, T. L. Andrew and V. Bulovic, Energy Environ. Sci., 2013, 6, 711–718 RSC.
- S. Zhang, L. Ye, H. Zhang and J. Hou, Mater. Today, 2016, 19, 533–543 CrossRef CAS.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS.
- L. Giraud, S. Grelier, E. Grau, G. Hadziioannou, C. Brochon, H. Cramail and E. Cloutet, J. Mater. Chem. C, 2020, 8, 9792–9810 RSC.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053–2066 RSC.
- C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS.
- D. Bilger, S. Z. Homayounfar and T. L. Andrew, J. Mater. Chem. C, 2019, 7, 7159–7174 RSC.
- D. C. Borrelli, M. C. Barr, V. Bulovic and K. K. Gleason, Sol. Energy Mater. Sol. Cells, 2012, 99, 190–196 CrossRef CAS.
- Y. Xu, X. Wang, J. Zhou, B. Song, Z. Jiang, E. M. Y. Lee, S. Huberman, K. K. Gleason and G. Chen, Sci. Adv., 2018, 4, eaar3031 CrossRef.
- M. H. Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1801564 CrossRef.
- G. Richhariya, A. Kumar, P. Tekasakul and B. Gupta, Renewable Sustainable Energy Rev., 2017, 69, 705–718 CrossRef CAS.
- L. Doimo, J. Essent. Oil Res., 2001, 13, 25–29 CrossRef CAS.
- R. P. Steer, J. Photochem. Photobiol., C, 2019, 40, 68–80 CrossRef CAS.
- United States Food and Drug Administration . Code of Federal Regulations, Title 21.
- H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945–956 CrossRef CAS.
- D. M. Lermal and G. D. Goldman, J. Chem. Educ., 1988, 65, 923–925 CrossRef.
- J. Bargon, S. Mohmand and R. J. Waltman, Mol. Cryst. Liq. Cryst., 1983, 93, 279–291 CrossRef CAS.
- A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC.
- S. Carret, A. Blanc, Y. Coquerel, M. Berthod, A. E. Greene and J.-P. Deprés, Angew. Chem., Int. Ed., 2005, 44, 5130–5133 CrossRef CAS.
- F. Wang, Y.-H. Lai, N. M. Kocherginsky and Y. Y. Kosteski, Org. Lett., 2003, 5, 995–998 CrossRef CAS.
- M. Suominen, S. Lehtimäki, R. Yewale, P. Damlin, S. Tuukkanen and C. Kvarnström, J. Power Sources, 2017, 356, 181–190 CrossRef CAS.
- R. Maity, D. Mandal and A. Misra, Mol. Phys., 2019, 117, 1781–1789 CrossRef CAS.
- W. Walker, B. Veldman, R. Chiechi, S. Patil, M. Bendikov and F. Wudl, Macromolecules, 2008, 41, 7278–7280 CrossRef CAS.
- N. Cheng, L. Zhang, J. J. Kim and T. L. Andrew, J. Mater. Chem. C, 2017, 5, 5787–5796 RSC.
- L. Allison, S. Hoxie and T. L. Andrew, Chem. Commun., 2017, 53, 7182–7193 RSC.
- D. H. Kim, M. D. Losego, K. Hanson, L. Alibabaei, K. Lee, T. J. Meyer and G. N. Parsons, Phys. Chem. Chem. Phys., 2014, 16, 8615–8622 RSC.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743–768 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data, molecular orbitals, pictorial process descriptions. See DOI: 10.1039/d0py01355b |
| This journal is © The Royal Society of Chemistry 2020 |