
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyperbranched polymers with step-growth chemistries from transfer-dominated branching radical telomerisation (TBRT) of divinyl monomers†
Savannah R.
Cassin
ab,
Pierre
Chambon
ab and
Steve P.
Rannard
 *ab
*ab
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
bMaterials Innovation Factory, University of Liverpool, Crown Street, L69 7ZD, UK
First published on 20th November 2020
Abstract
The commercial synthesis of polymers is generally limited to two main mechanisms that are typically considered to be mutually exclusive, namely step-growth and chain-growth polymerisation. This also defines the vast number of academic advances in macromolecular synthesis including the increasingly studied reversible deactivation radical polymerisation techniques (chain-growth) and complex polymer architectures such as dendrimers (step-growth). We report here a new synthetic strategy that utilises conventional free radical chain-growth chemistry, under modified telomerisation conditions, to form branched polymers containing chemistries conventionally formed under step-growth conditions. Telomerisation is typically limited to small molecule synthesis and employs addition across the unsaturated bond of a substrate, whilst minimising intermolecular reaction between substrates. Through the careful manipulation of reaction conditions, we have created a ‘transfer dominated branching radical telomerisation’ mechanism that creates branched polymers containing step-growth motifs from multi-vinyl monomers, with molecular weights in excess of 1000 kg mol−1, using industrially relevant free radical chain-growth chemistry. The scope of this approach is considerable, allowing access to entirely new macromolecular structures.
Introduction
Telomerisation has developed over many years as an efficient strategy for the conversion of unsaturated molecules, including renewable feedstocks, to a range of functional raw materials for the chemical industry.1,2 The aims of commercially focussed telomerisation processes are to limit the formation of carbon–carbon, or carbon-heteroatom, bonds to control the synthesis of small molecules whilst minimising the number of products that are formed.3,4 The term telomerisation was first introduced over 70 years ago5,6 and was defined as the reaction of a telogen, designated XY, to an unsaturated taxogen compound, Q, to form a telomer, X(Q)nY. Many common reactions may be technically classified by the term telomerisation such as thiol–ene reactions7 (addition of RS-H telogen across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of a taxogen), the addition of CO2,8 water or hydroxyl functional small molecules to alkenes9 and the Cysteine Lipidation on a Peptide or Amino acid (CLipPA) reactions.10 Within the field of polymerisation, a telomer contains a distribution of products with a low number of Q residues within the final sample; specifically, the telogen fragments X and Y comprise the α and ω chain-ends of each oligomeric chain respectively.11 The formation of short polymer chains (number average degree of polymerisation, DPn, of <100 monomer units; typically <10 monomer units)12 by chain-growth telomerisation reactions was a major focus of research in the 1950–1980 period, and global advances in controlled polymerisation, such as reversible deactivation radical polymerisations, RDRPs, (e.g., atom transfer radical, ATRP,13 and reversible addition–fragmentation chain transfer polymerisations, RAFT14) have undoubtedly benefitted from understanding created during the advancement of telomerisation.
C double bond of a taxogen), the addition of CO2,8 water or hydroxyl functional small molecules to alkenes9 and the Cysteine Lipidation on a Peptide or Amino acid (CLipPA) reactions.10 Within the field of polymerisation, a telomer contains a distribution of products with a low number of Q residues within the final sample; specifically, the telogen fragments X and Y comprise the α and ω chain-ends of each oligomeric chain respectively.11 The formation of short polymer chains (number average degree of polymerisation, DPn, of <100 monomer units; typically <10 monomer units)12 by chain-growth telomerisation reactions was a major focus of research in the 1950–1980 period, and global advances in controlled polymerisation, such as reversible deactivation radical polymerisations, RDRPs, (e.g., atom transfer radical, ATRP,13 and reversible addition–fragmentation chain transfer polymerisations, RAFT14) have undoubtedly benefitted from understanding created during the advancement of telomerisation.
Global polymer manufacture is dominated by chain-growth polymerisation with conventional free-radical polymerisation of monomers containing vinyl double bonds contributing an estimated 40–45% of production.15 The resulting macromolecules generally comprise a carbon–carbon backbone with optional pendant substituent groups which may bear functionality. Alternatively, step-growth polymerisation of An and Bn functional monomers (such as diacids, diamines and diols) is used to form large volumes of polymeric materials bearing linking chemistry (such as esters, amides and carbonates) directly within the backbone itself.16 The fundamental mathematical principles controlling A2 + B2 step-growth polymerisation are simply represented by the elegant Carothers equation that relates DPn to conversion;17 equally elegant modifications of this relationship establish the importance of the average functionality within An + Bn polymerisations, where n > 2, that offer gelation at relatively low conversions, as characterised by the formation of “infinite” molecular weight. Apart from examples such as ring-opening polymerisation18,19 and ‘living’ chain-growth polymerisation using catalyst transfer,20 these two strategies are generally considered as being mutually exclusive; step-growth polymerisations do not utilise vinyl monomers to form high molecular weight polymers, whilst chain-growth mechanisms using vinyl chemistries do not create polymers with backbones containing linking chemistries or heteroatoms.
Within conventional free-radical polymerisation of vinyl monomers the telogen is often referred to as a chain-transfer agent (CTA) and the structure, chemistry and concentration of the chosen CTA has a significant influence on polymerisation outcomes;21 a range of CTA strategies have been reported including addition of compounds such as thiols, coordinative chain transfer,22 the use of cobalt-derived catalytic chain transfer.23 Strictly speaking, the CTA is only considered a telogen if very short chains are formed, comprising chain-ends that are derived solely from the CTA,24 and initiator residues are not found within the oligomeric product.
Telomerisation as a direct route for advanced polymeric materials synthesis has been a relatively overlooked area although preformed telomers have been used to form linear polymers.25 We have studied the potential for new high molecular weight, branched polymer synthesis using telomerisation of divinyl compounds and report here the first example of the formation of novel branched polyesters through the telomerisation of ethylene glycol dimethacrylate (EGDMA) conducted under conventional and industrially-relevant free radical conditions. Telomerisation reactions are dominated by thiol chain-transfer26 and complete conversion of vinyl functionality has been observed within our reactions. This strategy directly forms a branched polymer comprising step-growth chemistries using free radical chain-growth chemistry in a single pot reaction by creating the carbon backbone of the polyacid components of the polyester structure and maintaining a DPn < 2 monomer units, which complies with the modified Carothers equation (see later), thereby avoiding gelation. This is a new concept that contains previously considered mutually incompatible inputs and outputs. Although we refer to these polymers as ‘polyesters’, to clearly indicate the backbone chemistry that is created, it is important to note that a thioether group is generated at each telomerisation step. We present a detailed analysis of the telomerisation reaction, a thorough characterisation of the resulting polymers and a series of model reactions that lead us to propose a chain transfer governed mechanism that avoids termination and loss of radical species; a transfer-dominated branching radical telomerisation (TBRT). Previously, branched vinyl polymers have been generated by the addition of low concentrations (typically <10 mol%) of divinyl compounds to linear polymerisations under conventional CTA-mediated free radical polymerisation conditions;27 TBRT is fundamentally different to these previous chemistries as telomerisation of divinyl monomers, in the absence of monovinyl monomer, results in the formation of branched polymers comprising heteroatoms in the backbone without crosslinking and avoids propagation through vinyl chemistry to a DPn < 2 (see later). This approach has considerable potential for new materials synthesis using commercially available feedstocks, industrially relevant polymerisation processes and facilities.
Results and discussion
Initial exploratory experiments and theoretical considerations
Telomerisation of methacrylate monomers may be controlled to yield singly modified monomers in a distribution of dimers, trimers and tetramers,28 or allowed to form longer functional chains if required;29 in some reports, thiols have been used to create hydroxyl-functional macromonomer precursors.25 For a successful telomerisation, the selection of telogen is important, as are reaction conditions and the free radical source; however, the basic principles of addition of the telogen across either one double bond, or coupled/reacted carbon–carbon structure, are typically upheld and residues of the radical source are not found in the final product (Fig. 1a).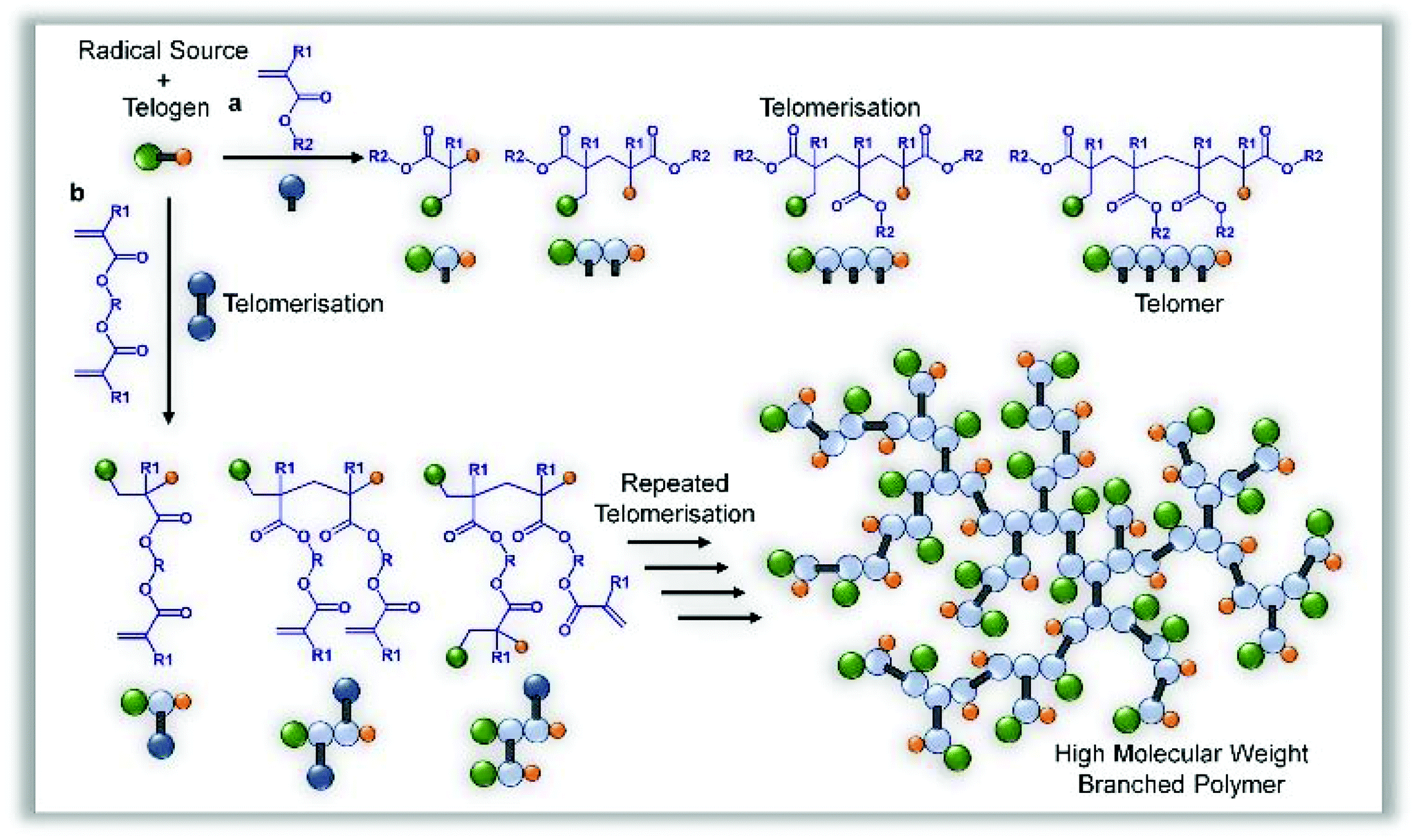 | ||
| Fig. 1 Comparison of the formation of telomers using free radical polymerisation of vinyl monomers and the TBRT of divinyl monomers to form high molecular weight branched polymer structures. (a), Simple propagation and rapid chain transfer leads to the formation of low molecular weight products containing both segments of a telogen at the initiation and termination sites. (b), Identical telomerisation conditions applied to divinyl monomers leads to telomers with unreacted functional groups that further react together to form high molecular weight branched polymers with multiple telogen residues. | ||
Our hypothesis relies upon the formation of an extended macromolecular architecture through repeated telomerisations using a divinyl methacrylate monomer. By only allowing telomers to form during very limited propagation, we expected to establish a branched macromolecular architecture by carbon–carbon bond formation, forming structurally important units of restricted size within an extended branched structure, connected by the ester links between the divinyl functionality (Fig. 1b). In a conventional free radical polymerisation targeting high molecular weight polymers, a low telogen (or CTA) concentration is used and the long linear chains would be expected to be initiated by a mixture of initiator radicals and radicals derived from the telogen; this has also been reported for RAFT polymerisations which utilise conventional free radical sources to initiate polymerisation.30 Under these conventional conditions, the homopolymerisation of a divinyl monomer would reach a gel point at very low vinyl group conversions as predicted by Flory-Stockmayer theory.31,32 The synthesis of branched polymers via divinyl monomer homopolymerisation has been studied using several strategies33 including RDRP methods to control the kinetic chain length of the component propagations within a controlled radical polymerisation using deactivation-enhanced ATRP (DE-ATRP)34,35 or RAFT (at a 35 wt% solids content);36 in both cases, the polymerisations led to gelation if allowed to reach conversions of approximately 70% and termination prior to the gel point produced soluble polymers with considerable numbers of pendant unreacted vinyl functionality. DE-ATRP utilised a 39 wt% solids content, high initiator and catalyst concentrations and careful handling of the final vinyl-functional polymers was required to prevent further reaction and sample crosslinking.37
Our chosen conventional free-radical strategy utilises the well-studied 2,2′-azobis(2-methylpropionitrile) (AIBN) as a radical source at a typical concentration of 1.5 mol% based on vinyl functional groups, EGDMA as the taxogen, dodecanethiol (DDT) as the telogen and toluene as solvent. Our first reactions, with relatively low telogen concentrations (but higher than typical linear polymerisation values), did indeed lead to gelation as predicted (Table 1; ESI Table 1†); however, increases in telogen concentrations to levels more typically used in successful linear telomerisations, led to the formation of fully soluble polymer products from reactions conducted at 50 wt% solids with full vinyl conversion, as determined by 1H and 13C nuclear magnetic resonance spectroscopy (NMR; see ESI Fig. 1–5†). By increasing the telogen concentration, the formation of transfer-dominated branching radical telomerisation conditions could be exploited to prevent gelation, enable full conversion, and yield useful branched polymer products. As mentioned above, the concentration of telogen required under these conditions is typically >1 molar equivalent with respect to divinyl compound as would be expected when considering the telomerisation conditions that need to be established (Table 1).
| 1H NMR (CDCl3) | TD-SECd (THF/TEA) | ||||
|---|---|---|---|---|---|
| [EGDMA]0/[DDT]0a | Conv.b (%) | [EGDMA]Final/[DDT]Finalc | M w (g mol−1) | M n (g mol−1) | Đ |
| a Determined by 1H NMR of polymerisation mixture at t0. b Determined by 1H NMR of crude sample after 24 h. c Determined by 1H NMR of purified and dried material. d Determined by triple-detection size exclusion chromatography. | |||||
| 0.50 | >99 | 0.93 | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 485 |
11747 |
2.51 |
| 0.75 | >99 | 1.04 | 206784 |
6540 | 31.6 |
| 0.80 | >99 | 1.02 | 209265 |
5924 | 35.3 |
| 0.85 | >99 | 1.02 | 1822000 |
23635 |
77.1 |
| 1.00 | Gel | Gel | Gel | Gel | Gel |
The homopolymerisation depicted in Fig. 1b will theoretically lead to full consumption of vinyl functionality, without gelation, if the rate of propagating radical capping, after telogen addition to the vinyl bond, is rapid enough to maintain low DPn values and prevent extensive vinyl group propagation (Fig. 2a). Although carbon–carbon bond formation occurs during this polymer synthesis, the longest chain structures within the branched polymer architecture are aliphatic polyesters, derived from joining the diester units present within the starting divinyl monomer, (Fig. 2a, highlighted chain). In principle, the telomerisation reaction can be considered as having formed the carbon–carbon backbones of a distribution of multifunctional acid monomer residues. Hypothetically, the same polymer structure would result from a polyesterifcation reaction containing ethylene glycol (A2) and a complex mixture of B, B2, B3, B4,…Bn acid-functional monomers (Fig. 2b); experimentally, and industrially, this would be extremely difficult to achieve.
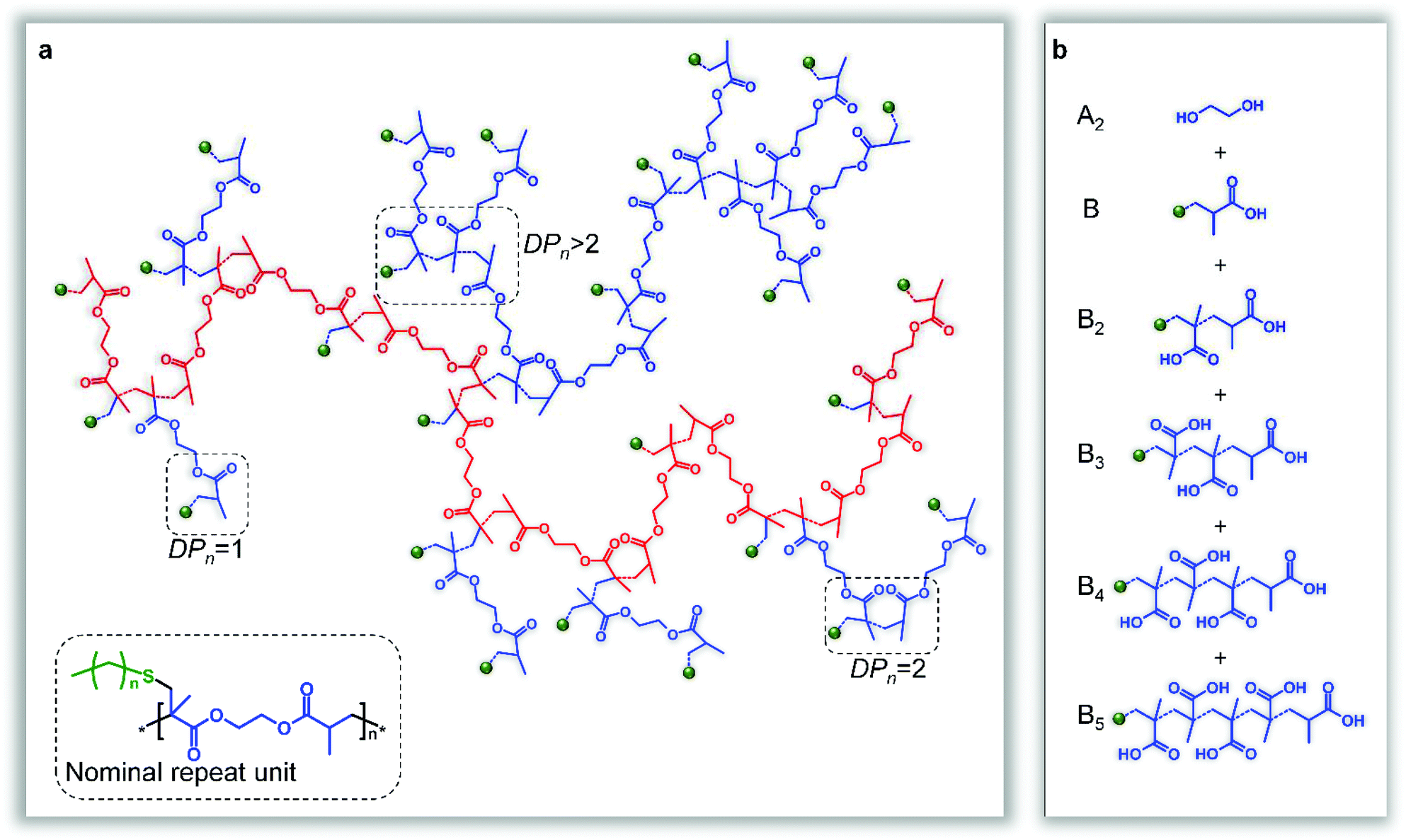 | ||
| Fig. 2 Product structure from the TBRT of ethylene glycol dimethacrylate. (a), The structure formed by the TBRT of a dimethacrylate is best represented as a branched polyester (shown in red), with a nominal repeat unit derived from the addition of one telogen to the dimethacrylate, and structural subunits created by differing degrees of propagation before chain transfer. (b), The branched polyester may theoretically be synthesised by the step-growth polymerisation of a mixture of mono- and polyacids with ethylene glycol. Only the initiation segment of the telogen is shown (in green) for clarity; dotted bonds show new bond formation during telomerisation. | ||
Exploring branched EGDMA/DDT polyester synthesis using TBRT
The potential to form branched polyesters under conventional free radical telomerisation conditions is of considerable fundamental and industrial interest, therefore, the reaction was explored in detail to establish the principles and scope available. As stated above, our early reactions in toluene with a relatively low concentration of telogen, led to gel formation, but systematic increases of DDT relative to EGDMA (50 wt% solids) led to fully soluble polymer at telogen/taxogen molar ratios >1:1 (Table 1; ESI Table 1†). 1H NMR studies of the crude reaction mixture showed no detectable residual vinyl functionality whilst the purified polymer was shown to contain an approximate 1:1 molar ratio of telogen to taxogen in the final product (see ESI Fig. 4†); the 1:1 DDT/EGDMA adduct is therefore the average repeat unit of the resultant polymer (Fig. 2a insert). Additionally, triple detection size exclusion chromatography (TD-SEC) confirmed the high molecular weight of the soluble branched polyesters and the ability to vary weight average molecular weight (Mw) values through manipulation of the telogen/taxogen ratio at a constant solid content (see ESI Fig. 6 and Table 1†). Branched polyesters containing no residual vinyl functionality with Mw ranging from 1822–29 kg mol−1 were readily synthesised with dispersity (Đ) values ranging from 77.10–2.51 and Mark–Houwink–Sakurada (MHS) α values of from 0.340 0.283; low MHS α values of this order are highly indicative of the densely branched nature of the polymer products38 It is worth noting that number average molecular weight (Mn) values remained relatively low (<25 kg mol−1) but the 1:1 ratio of taxogen and telogen in the final purified products was maintained throughout all samples. The significant Mw variation suggests that increasing the taxogen content in the reaction enhances intermolecular propagation, resulting in an extended macromolecular structure within the limits of a soluble polymeric material.
Toluene residues, from possible chain transfer to solvent, were not seen in the polymer products, therefore, repetition of these syntheses at varying telogen/taxogen ratios and 50 wt% solids in ethyl acetate, as a more environmentally acceptable solvent, was conducted. These reactions provided very similar outcomes to those carried out in toluene, with complete conversion of vinyl functionality, a 1:1 ratio of taxogen and telogen in the purified polymers, similar Mn and MHS α values, but lower Mw values (ranging from 1578–12 kg mol−1) at each telogen/taxogen ratio used within the reaction mixture (see ESI Table 3†). One clear advantage of TBRT in the synthesis of branched polyesters is, therefore, the use of ester solvents at relatively low temperature; a synthesis condition not typically available to step-growth polyester production where considerable transesterification would be expected.
A conventional linear free radical polymerisation is well known to be able to create polymer chains of significant DPn values (>1000 monomer units) within seconds or milliseconds of initiation dependent on reaction conditions and monomer chemistry.39 An EGDMA/DDT telomerisation (1.5 mol% AIBN) in ethyl acetate was therefore monitored by removal of samples at regular intervals with subsequent TD-SEC and 1H NMR analysis of the crude samples (see ESI Fig. 15–17 and Table 5†) to determine molecular weight evolution and conversion. The reaction was essentially complete within 2.5 hours, reaching >99% conversion as determined by measurement of the vinyl bond resonances; indeed >80% conversion was seen within the first 60 minutes of the reaction. A semilogarithmic plot showed a slight upward curve that is indicative of an increasing number of radical species within the polymerisation (see ESI Fig. 16†) and would be consistent with a steadily thermally decomposing initiator in the absence of termination reactions.
Within linear free radical polymerisations, chain transfer is known to lead to termination of propagating chains with conservation of radical reactive centres;25 the high concentration of telogen here also provides an identical outcome. However, and in contrast to reactions utilising monovinyl monomer, under TBRT conditions the presence of pendent unreacted vinyl functionality allows the structures formed in the early stages of polymerisation to act as macromonomers for later growth of branched macromolecules; TD-SEC analysis showed low molecular weight polymers being formed in the early stages of the reaction, with molecular weight increasing with time and conversion followed by a rapid growth after approximately 80% monomer conversion (see ESI Fig. 31†). These data collectively suggest an intermolecular reaction of vinyl functional telomers is the route to the formation of the final high molecular weight branched polyesters under TBRT conditions.
It is important at this stage to contrast structures formed by TBRT with other reported and related chain-growth routes to branched polymers. Interestingly, previous RDRP reports of EGDMA homopolymerisation were forced to terminate the reaction at approximately 70% conversion to avoid gelation and yielded vinyl-functional polymers which require post-functionalisation to avoid further reaction;24,25 this is likely due to the difficulty of truly controlling RDRP polymerisations at very low DPn values. TBRT, however, allows the continued growth of branched polymer through to full vinyl consumption via a homo-telomerisation of divinyl monomer. Lightly branched polymers, using conventional40 and controlled radical processes,41 have also been described previously using the incorporation of a low concentration of multi-vinyl monomers into a linear polymerisation, resulting in theoretically less than one brancher per primary structural chain to avoid gelation; this has been referred to as the “Strathclyde” or “modified Strathclyde” strategy. Despite the analogous molecular weight development with respect to conversion, the macromolecular structures formed during a TBRT reaction are fundamentally different. For example, the repeat unit structure is formed during the reaction, there is an absence of extended C–C backbones, a very high number of chain-ends is formed and, importantly, the number average vinyl propagation is limited to <2 monomer units, Fig. 3.
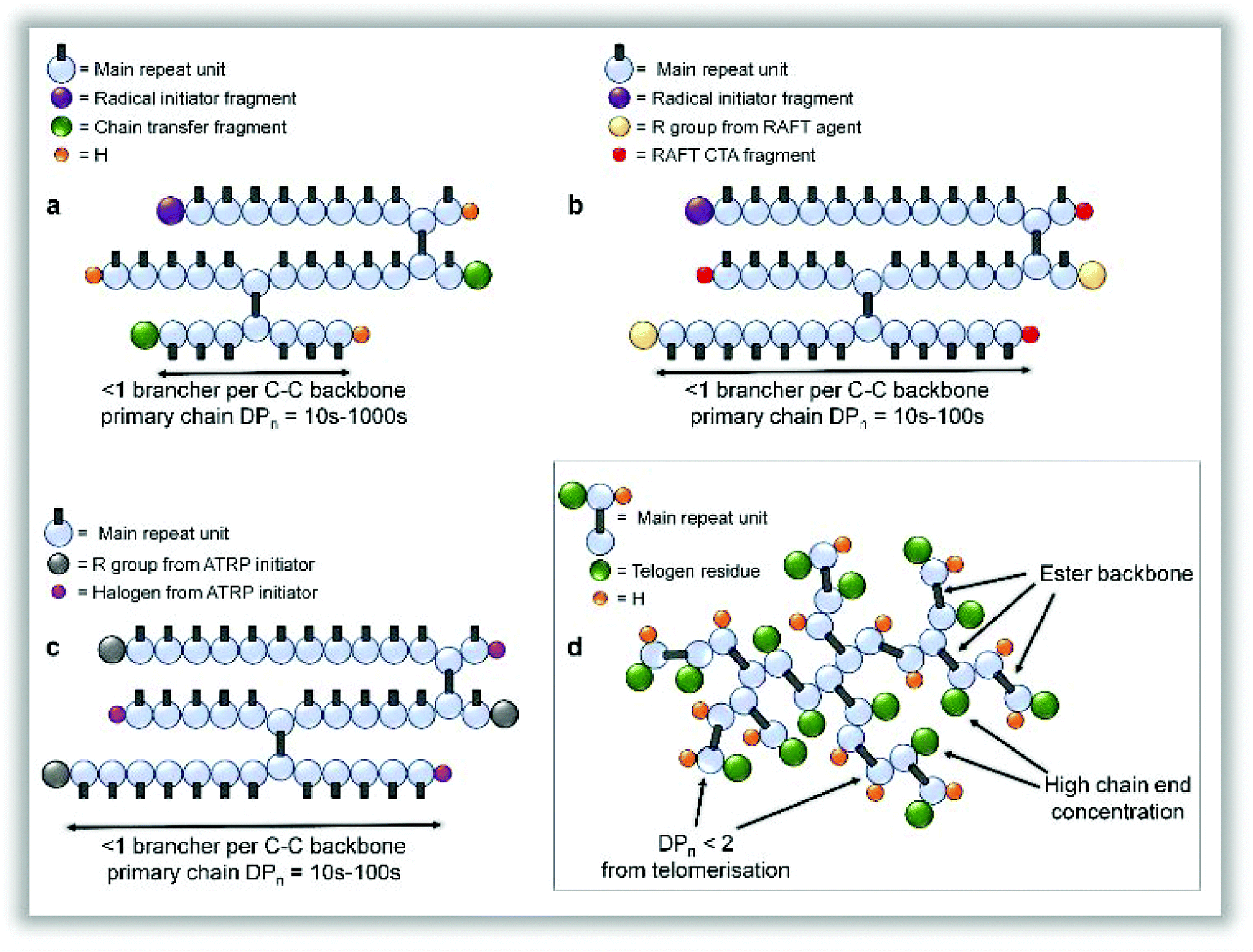 | ||
| Fig. 3 Comparison of branched polymer structures formed by free radical (including RDRP) approaches. (a), “Strathclyde” strategy incorporating conventional free radical approaches and <1 brancher per (C–C) primary chain. (b) “Modified Strathclyde” strategy using RAFT polymerisations techniques to provide control of primary chain uniformity. (c) “Modified Strathclyde” strategy using ATRP polymerisations techniques. (d) TBRT structure created by homo-telomerisation of divinyl monomers and generating a polyester backbone chemistry. Structures a–c have extended C–C backbones and a conventional repeat unit derived from a vinyl monomer; structure d has a repeat unit that is created during polymerisation and an extended backbone derived from chemistry linking the vinyl groups of the divinyl monomer. | ||
Telomerisations using radical sources and thiol telogens are known to have an absence of apparent termination due to the domination of chain transfer reactions;25 this provides an opportunity to investigate the TBRT of DDT/EGDMA using reduced concentrations of AIBN. In this way, a slower polymerisation would result that may be more readily monitored across a longer time-period. A ten-fold decrease to 0.15 mol% AIBN, relative to monomer vinyl functionality, led to the observation that polymerisation not only decreased in rate, with >80% conversion now requiring 6 hours, but achievement of >99% conversion was still possible when the reaction was left to stir (heated) overnight. The semilogarithmic plot for this reaction showed a linear relationship over the first 6 hours (see ESI Fig. 29–31 and Table 8†), suggesting a steady concentration of propagating centres, and a growth of Mw that was initially linear with respect to conversion before rapidly increasing after 75% monomer consumption (approximately 4 hours reaction time). The initial apparent linear relationship is presumably due to the limits of TD-SEC at low conversion; however, the rapid growth of Mw again indicates intermolecular telomerisation-derived coupling of low molecular weight vinyl-functional species that are formed at relatively low conversion. It is apparent, therefore, that the formation of the large numbers of chain-ends and the low DPn values within the telomerisation does not require high concentrations of initiator as reported for DE-ATRP.33,34
Clearly, high molecular weight branched macromolecule formation occurs predominantly when TBRT polymerisations reach high conversions. In the absence of termination during a transfer dominated telomerisation, further reductions of AIBN concentration should be possible as continued reaction would not be reliant on the steady introduction of radicals from the thermally decomposing initiator. Under these conditions, a steady state concentration of radicals would be preserved by the ongoing and rapid transfer reactions between radical centres and telogen molecules; however, highly extended reaction times would be expected. AIBN was therefore reduced to 0.05 mol% AIBN, relative to EGDMA vinyl functionality, and conversion was observed to only reach <65% after 6 hours. Residual vinyl resonances were observed in the 1H NMR spectrum of the crude samples taken after 24 hours, with continued reaction to >99% vinyl conversion after 48 hours (see ESI Fig. 32†) without addition of further AIBN (AIBN 10-hour half-life temperature is 65 °C in toluene); the semilogarithmic plot showed a linear relationship over the first 24 hours (see ESI Fig. 33†). TD-SEC analysis of the resulting purified polymers showed near-identical Mw values at each AIBN concentration (identical telogen/taxogen ratio) and >99% conversion (Mw (1.5 mol% AIBN) = 141000 g mol−1; Mw (0.15 mol% AIBN) = 144000 g mol−1; Mw (0.05 mol% AIBN) = 139000 g mol−1), (see ESI Table 10†), but the MHS α values were remarkably consistent with variation from 0.305–0.312 (see ESI Table 10†). Elemental microanalysis and Fourier-transform infrared (FT-IR) spectroscopy were conducted on a series of polymer samples and no nitrogen residues or nitrile groups were detected respectively, as may be expected from the inclusion of AIBN fragments (see ESI Fig. 7, 14 & 36 and Tables 2, 4 & 11†).
NMR spectra of the branched polymers are complicated by the numerous chemical and architectural environments within the final structures (see ESI Fig. 11 and 12†). Conventional 1H and 13C NMR spectroscopy was combined with distortionless enhancement by polarization using a 135° decoupler pulse (DEPT-135) experiment and heteronuclear single quantum correlation (HSQC) analysis to simplify assignment of the numerous resonances (Fig. 4).
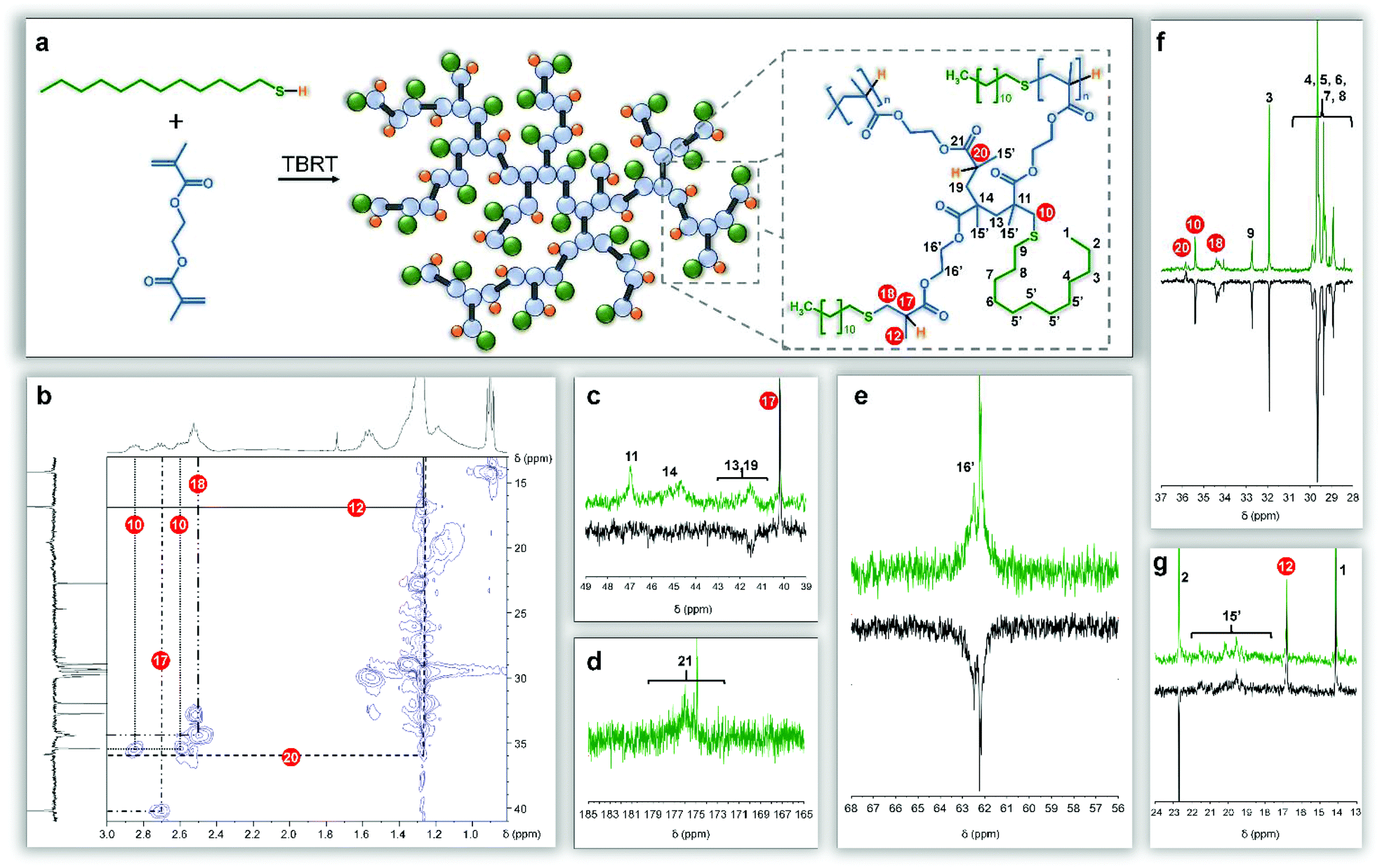 | ||
| Fig. 4 Detailed NMR structural analysis of the product of TBRT of ethylene glycol dimethacrylate and dodecanethiol. (a), Schematic representation of the structural components within the branched polyester product. (b), HSQC correlation aiding assignment of resonances within the complex spectra. (c–g), 13C NMR spectra (with DEPT-135 comparison) showing key NMR resonances. | ||
Resonances at 0.8 and 4.0–4.5 ppm within the 1H NMR spectra were readily assigned to the methyl protons of DDT (Fig. 4, H1) and the oxymethylene protons of EGDMA (Fig. 4, H16′) respectively. Secondary carbon resonances resulting from addition of the thiolate telogen radical to the unsubstituted carbon of the vinyl group of EGDMA were observed with distinctly different shifts and were assigned to structural features resulting from either a single addition forming DPn = 1 substructures (34.4 ppm; Fig. 2a, 4a, b & f C18), or DPn ≥ 2 (35.4 ppm; Fig. 2a, 4a, b & f C10) if propagation occurred. Similarly, telogen addition to the double bond leads to either a tertiary carbon at the substituted side of the vinyl bond if a DPn = 1 substructure is formed (40.7 ppm; Fig. 4a–c C17), or a quaternary carbon (46.9 ppm; Fig. 4a & c C11) if propagation to DPn ≥ 2 occurs. Additional tertiary carbons are formed at the end of each definitive telomer X(Q)nY structure, after hydrogen abstraction from thiol, and show distinctly different resonances to the DPn = 1 substructure, due to the varying proximity of the thioether (35.9 ppm; Fig. 4a, b & f C20). The formation of single telomerisation species (DPn = 1) is critical to prevent gelation and this is analogous to introducing a monofunctional acid into the hypothetical polyesterifcation detailed above (Fig. 2b). Pendant methyl groups of the EGDMA residues could also be separated into those generated due to propagation (19.2–21.7 ppm; Fig. 4a & g C15′) and those resulting from DPn = 1 substructures (16.8 ppm; Fig. 4a, b & g C12).
Mechanistic understanding of TBRT polymerisation
Two comparative experiments were conducted in toluene to investigate the telomerisation reaction in greater detail: firstly, methyl methacrylate (MMA; Fig. 5a) was used as a monofunctional EGDMA analogue, and secondly, an acid-sensitive bifunctional monomer, 1,4-butanediol di(methacryloyloxy)-ethyl ether (BDME; Fig. 5b),42 was substituted for EGDMA. SEC (THF) analysis of the crude MMA telomerisation, conducted using oligomer columns, showed a distribution of several defined species (see ESI Fig. 37†), with deconvolution and molecular weight extrapolation clearly indicating that the sample has very little material above a molecular weight of approximately 2500 g mol−1, and approximately 80 wt% of the sample comprising structures of <600 g mol−1 (DPn ∼ 5). The MMA telomerisation reaction was also studied using matrix-assisted laser desorption ionisation–time of flight (MALDI-TOF) mass spectrometry (Fig. 5c) and revealed a distribution of species, varying by 100 mass units, that strongly reflects the oligomer SEC analysis, with no detectable species greater than m/z [Na+] = 2422 g mol−1 (DPn = 22) and a predominance of species at m/z [Na+] values < 1000 g mol−1 (as detected under these conditions).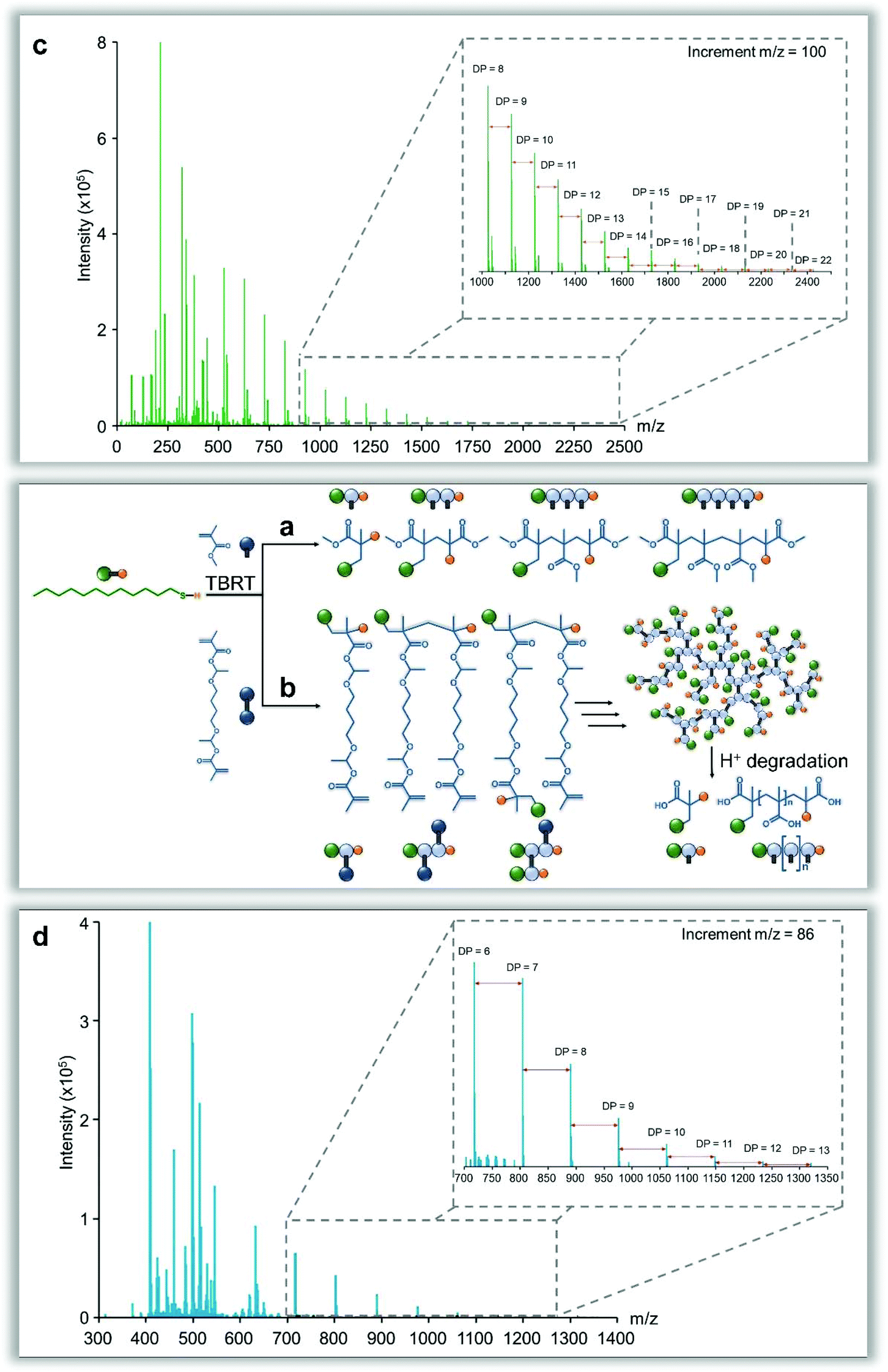 | ||
| Fig. 5 Matrix-assisted laser desorption ionisation–time of flight mass spectrometry analysis of model telomerisation reactions. Schematic representations of (a), the telomerisation of methyl methacrylate and (b), TBRT of an acid sensitive dimethacrylate monomer with subsequent degradation to form the constituent telomer structures. (c), Mass spectrum (positive ion mode) of methyl methacrylate telomers showing a repeating methyl methacrylate distribution. (d), Mass spectrum (negative ion mode) of TBRT polymer product after degradation showing similar distribution of products varying by a mass equivalent to methacrylic acid. | ||
After BDME was polymerised under identical TBRT conditions the resulting soluble, high molecular weight polymer was treated with trifluoroacetic acid and subjected to MALDI-TOF analysis, acquired in negative ion mode in order to detect the poly(methacrylic acid) oligomers present in the final sample (Fig. 5d). The major peak series exhibited a peak-to-peak mass increment of 86 mass units, as would be expected for the methacrylic acid repeat unit; species corresponding to DPn = 2–13 were readily identified. The MALDI-TOF analysis clearly suggests that the propagation of the bifunctional BDME occurred in a near-identical manner to the monofunctional MMA monomer at the individual vinyl group level, yielding telomeric structures derived from carbon–carbon bond formation. The high molecular weight of the final p(BDME) polymer sample is, therefore, a consequence of the combination of telomers into larger branched polymer architectures by the chemistry linking the vinyl groups within the bifunctional monomer, whilst avoiding gelation and allowing full vinyl conversion.
Recent advances in diffusion-ordered spectroscopy (DOSY) offers an interpretation of NMR spectra relative to varying diffusion coefficients, and subsequently molecular size, to give insight into molecular weight and functional group assignment within mixed samples; this includes the presence of unreacted monomer and the formation of functional dimers, trimers and higher oligomers.43,44 DOSY reports the translational diffusion coefficients for individual resonances relating to the hydrodynamic radius of a given molecular species and is, therefore, complimentary to SEC methods. As such, DOSY was utilised to characterise the telomerisation mechanism of MMA further by studying the reaction at t = 0 and 4 hours (95% vinyl conversion), with several parameters, such as diffusion times, requiring optimisation for each timepoint in order to obtain valid and consistent results. Through diffusion coefficient-molecular weight analysis, based on the Stokes–Einstein Gierer-Wirtz estimation method,43,45,46 formula weights of oligomeric species could be estimated; the molecular weight of unreacted MMA monomer vinyl residues at the different timepoints were determined as 100 ± 33 g mol−1 (see ESI Fig. 45†). As indicated by MALDI-TOF analysis, the telomerisation of MMA generated oligomers with varying degrees of polymerisation which have indiscernible differences in their 1H NMR spectra due to overlapping resonances and chemical similarity; DOSY was able to readily separate the individual species based on their diffusion coefficients, including DPn = 1 and 2 telomerisation products (see ESI Fig. 46†), correlating well with MALDI-TOF analysis (see ESI Table 15†). Under these conditions, therefore, a high concentration of low molecular weight species, of varying chain lengths, appears to be formed due to the high telogen concentration and the very rapid end-capping of propagating chains.
The ready observation of various species during MMA telomerisation suggested the potential to directly correlate vinyl functionality with the growing species within the TBRT of EGDMA (Fig. 6a); DOSY analysis of crude samples taken during the reaction should allow identification of species present during the early stages of polymerisation and the manner in which free monomer is consumed.
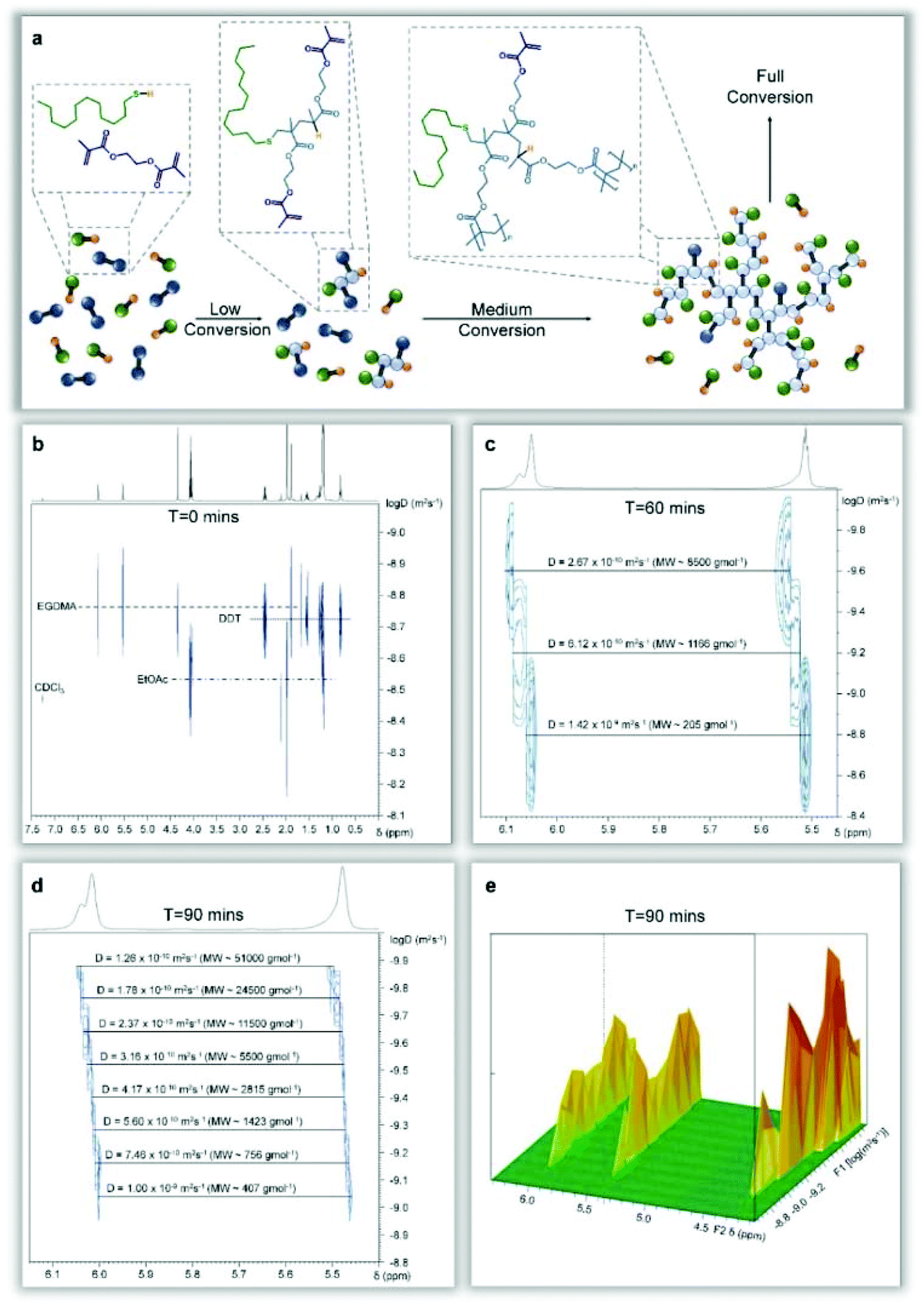 | ||
| Fig. 6 Diffusion-ordered spectroscopy studies of the TBRT of ethylene glycol dimethacrylate and dodecanethiol. (a), Schematic representation of the TBRT reaction showing intermediate vinyl functional structures generated at various levels of conversion. (b), DOSY-NMR spectrum of the starting mixture of reactants prior to initiator addition showing clear separation of species and the ready identification of vinyl functionality within the monomer, without overlap with other reaction components. (c), Expansion of the vinyl region of the spectrum at 60 min reaction time showing several vinyl functional species with differing diffusion characteristics and estimated molecular weights. (d), Vinyl resonances at 90 min reaction time clearly demonstrating the formation of high molecular weight material with unreacted double bonds. (e), 3-Dimensional representation of the DOSY-NMR spectrum at 90 min reaction time showing the variation in resonances from δ = 4–6 ppm, showing variation of diffusion coefficients correlated to structural sub-units. | ||
In contrast, 1H NMR conversion studies of final reaction mixtures allow confirmation of the disappearance of vinyl functionality but provides no indication of the structural or molecular origin of any detected unsaturated functionality. DOSY analysis of the DDT/EGDMA TBRT reaction in ethyl acetate (AIBN = 1.5%) at t = 0 was able to identify all components of the starting reaction mixture at different diffusion coefficients (Fig. 6b); specifically, the vinyl 1H resonances of EGDMA were identified at 5.5–6.0 ppm. Crude reaction samples at t = 60 min showed multiple diffusion coefficients for the vinyl resonances (Fig. 6c); fully unreacted EGDMA could readily be seen at conversions >70% and identification of species containing pendant vinyl groups with molecular weights of approximately 8500 g mol−1 was achieved 75% monomer conversion.
Although the diffusion coefficient of the ethyl acetate reaction solvent was constant across the samples studied, the measured value (2.37 × 10−9 m2 s−1) was slightly above predicted values (1.45 × 10−9 m2 s−1) which may indicate the presence of sample convection as previously reported for DOSY studies conducted in CDCl3.40 DOSY is not conventionally used to study highly disperse polymerisation reactions, therefore a variation of gradient range was required to create ideal decay curves (see ESI Fig. 20, 24 & 27†) and allow study of the higher molecular weight, slowly diffusing species formed in the later stages of the telomerisation reaction; this results in an artificial exclusion of smaller molecules from the DOSY analysis but allows a more detailed probing of larger molecular reaction products. To be clear, quantification of the various branched polyester species within the telomerisation reaction has not been attempted, but confirmatory evidence was sought that shows vinyl-functional branched structures are present throughout the TBRT reaction; through this approach, vinyl functional branched polyester structures could be identified with molecular weights spanning 400–51000 g mol−1 (Fig. 6d & e) within crude reaction samples after 1.5 hours (>90% conversion). This, again, supports a mechanism involving significant intermolecular combination of vinyl-functional species of varying molecular weights, creating a sudden acceleration of Mw development (high conversion) as very large macromolecules are formed, and unreacted EGDMA monomer is consumed.
TBRT from the perspective of step-growth and chain growth polymerisation
An ideal step-growth polymerisation of bifunctional monomers, such as a diacid and diol, forms a linear polymer; functional group stoichiometry and monomer purity are critical to achieving relatively long polymer chains and eqn (1) and (2) describe the relationship of DPn to conversion, p, and stoichiometry, q assuming pure monomers.47,48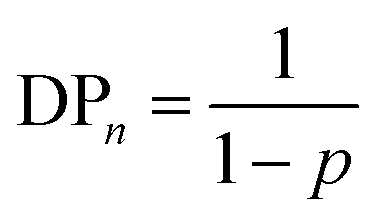 | (1) |
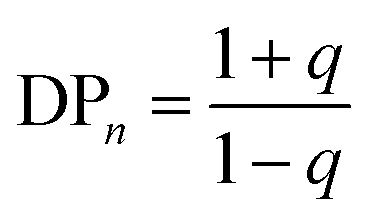 | (2) |
Branching, and ultimately crosslinking, of a step-growth polymerisation may be achieved by adding multifunctional (n > 2) monomers, such as triols or triacids, to the polymerisation and a relatively simple modification of eqn (1) shows the relationship to the average functionality, f, of the polymerisation mixture, eqn (3). Clearly, an average functionality f >2 will lead to infinite molecular weight and gelation at relatively low values of p.
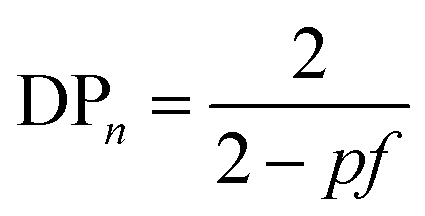 | (3) |
Within the idealised polymer structure shown in Fig. 2a, the hypothetical polyesterification would require perfect stoichiometry of 50 acid groups and 50 hydroxyl groups to be distributed across 51 individual molecules (13 monoacids, 5 diacids, 6 triacids, 1 tetra-acid, 1 penta-acid and 25 ethylene glycol molecules) leading to a value of f = 1.96. If the perfect mixture of this combination of mono and multifunctional acids could be balanced experimentally with ethylene glycol in a step-growth polymerisation, and all functional groups react equally, a soluble, high molecular weight branched polymer would theoretically be formed at 100% conversion (p = 1) of functional groups to esters. The monoacids are critical to this hypothetical polymerisation as these molecules would act as capping agents and remove chain-end functionality whilst the final polymer would contain no residual acid or hydroxyl functionality, unlike commercial step-growth polymers.
From the perspective of chain-growth polymerisation, the simplest product that can be formed from the idealised telomerisation reaction envisaged in Fig. 1, with complete reaction of vinyl functionality, is a single EGDMA residue with a single DDT added to each vinyl bond and no resulting propagation forming extended branched macromolecules; in this case, the telogen/taxogen ratio in the final product would be 2/1. Propagation to include just one additional EGDMA would lead to the requirement of just one additional DDT under telomerisation conditions, therefore the telogen/taxogen ratio in the final dimeric product would decrease to 3/2. This leads to a simple (n + 1)/n arithmetic progression with increasing taxogen (EGDMA), where n is the number of taxogen molecules in the product structure and assuming no intramolecular loop formation. Such a progression will tend to a ratio of 1/1 at high values of n (see ESI Fig. 54†), and this ratio is indeed observed via the 1H NMR spectroscopy conducted here (see ESI Fig. 4 & 11†), and maintained under different conditions of reaction solvent, initiator concentration and initial telogen/taxogen ratios. Interestingly, during an ideal telomerisation, a theoretical kinetic chain length,49 resulting from propagation through the vinyl functionality and carbon–carbon bond formation, may also be calculated; as each of the n + 1 telogen residues is a direct indication of the number of taxogen residues in the macromolecule, n, and there are two vinyl bonds per taxogen, the number average kinetic chain length, ![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) , may be simply calculated as the number of vinyl group residues, 2n, divided by the number of telogens, or 2n/(n + 1). This progression suggests an ideal structure, assuming no intramolecular loop formation, containing a number average kinetic chain length that tends to <2 monomer units at high values of n (see ESI Fig. 54†). The chain-growth consideration of , or DPn, <2 monomer units is in direct accordance with the calculations presented above when considering the analogous, if theoretical, step-growth combination of polyacids that would avoid gelation when reacted with ethylene glycol; an observed DPn value of <2 within the telomer sub structures would correspond directly to f = < 2.
, may be simply calculated as the number of vinyl group residues, 2n, divided by the number of telogens, or 2n/(n + 1). This progression suggests an ideal structure, assuming no intramolecular loop formation, containing a number average kinetic chain length that tends to <2 monomer units at high values of n (see ESI Fig. 54†). The chain-growth consideration of , or DPn, <2 monomer units is in direct accordance with the calculations presented above when considering the analogous, if theoretical, step-growth combination of polyacids that would avoid gelation when reacted with ethylene glycol; an observed DPn value of <2 within the telomer sub structures would correspond directly to f = < 2.
A proposed TBRT mechanism
The model reactions, chromatographic and spectroscopic analysis and theoretical considerations strongly support the model proposed in Fig. 1; telomerisation, in this case the addition of the thiol telogen to the divinyl taxogen functionality, appears to progress via a radical process that limits the carbon–carbon bond formation derived from conventional free radical propagation to a DPn < 2 units. The formation of a low number of relatively long chains (<15 monomer units) does appear to be possible; however, a large number of DPn = 1 groups are created, and this controls and prevents the formation of gelled/crosslinked products. Clearly, intramolecular loop formation may also occur, aiding the avoidance of gelation; however, intramolecular loops have not been identified and the 1:1 telogen/taxogen ratio in the final product suggest that this may be a relatively minor occurrence under these conditions. Vinyl functional branched polyesters are formed during the polymerisation and the overall population of high molecular weight branched polymers is mainly generated towards the end of the reaction via the coupling of vinyl functional groups pendant to the larger macromolecules. The telomerisation of pendant groups, in the absence of propagation, is likely to occur throughout the reaction.
`The kinetic analyses suggest a mechanism that is strongly dominated by the relatively high concentration of telogen (chain transfer agent), and the reaction of the telogen and taxogen (DDT and EGDMA) is conducted under conditions that minimise termination and allows the radical concentration to be governed by the transfer reaction between either thiol and thiolate or thiol and carbon-centred radicals from the carbon–carbon bond forming reactions (Fig. 7).
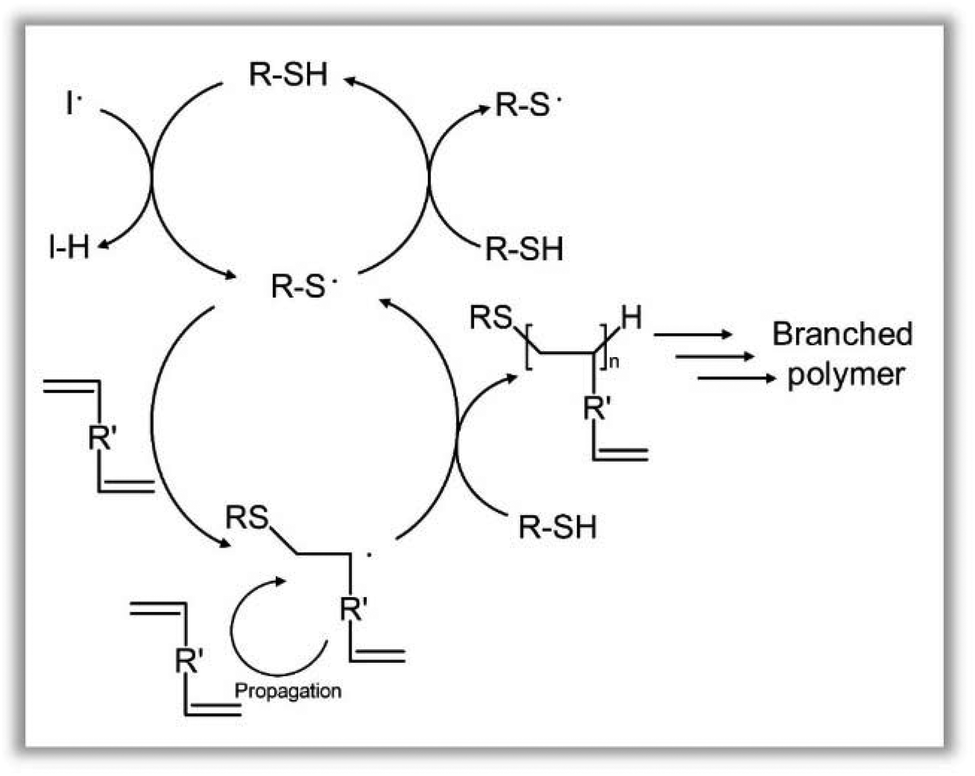 | ||
| Fig. 7 Proposed mechanism of TBRT. An excess of thiol controls the longevity of the radical concentration through thiol-to-thiol chain transfer, whilst mediating telomerisation to a number average degree of polymerisation of 2 monomer units within the branched polymer structure. Pendant vinyl groups allow intermolecular branching to occur and high molecular weight polymers to form. | ||
Conclusions
TBRT of multi-vinyl monomers utilises conventional free radical C–C bond formation to create small sub-structures of large and complex macromolecular architectures. The products do not, however, resemble vinyl polymers but share their dominant backbone chemistry with step-growth polymers that would normally be practically and experimentally inaccessible. The synthesis obeys chain-growth considerations whilst the product polymer structures can be disconnected to constituent mono and multifunctional monomer residue mixtures that also obey step-growth stoichiometry rules. As such, TBRT opens opportunities not available to either step-growth or conventional chain-growth polymerisation strategies. Telomerisation reactions have been optimised for the formation of small molecules through limiting the number of taxogen compounds combined within the final products. By utilising the clear benefits of free radical chemistry and multi–vinyl monomers (speed, lack of catalyst usage, functional group tolerance and structural diversity of commercial monomers) telomerisation approaches have been extended to the formation of branched polymers which strongly resemble the products of step-growth strategies.To address the issue of high thiol use within TBRT reactions, and concerns of toxicity, DDT has a reported oral median lethal dose within rodents, LD50(oral, murine), of 4225 mg kg−1 which compares to values for common monomers such as MMA (5300 mg kg−1) and EGDMA (2000 mg kg−1) and solvents such as toluene (2600–7500 mg kg−1);50 many thiols are regularly used within industry and consumer products, such as agriculture, food, electronics, health, beauty and hygiene, therefore the thiol telogens would not be prohibitive for scale-up and commercialisation. The presence of unreacted telogen within the reaction mixture has not proved problematic during polymer purification; although removal has not been optimised at this stage, the products do not appear to maintain a residual odour possibly due to the very low level after purification and the low volatility of DDT in these examples.
For the first time, we have introduced the potential of telomerisation reactions to form high molecular weight branched polymers with complete vinyl monomer conversion under industrially applicable conditions. Given the adoption of branched polymers such as the BoltornTM, HybraneTM and Lupasol® ranges sold by Perstorp, DSM and BASF respectively, the TBRT reaction is expected to provide the opportunity to create numerous new materials that may open opportunities for future applications across many academic and commercial fields, including the formation of degradable polymers from sustainable feedstocks.
Conflicts of interest
The authors are co-inventors on patents that protect the TBRT chemistry; these patents have been licensed to Scott Bader and form the basis of Polymer Mimetics Ltd (Company number 12598928).Acknowledgements
This manuscript is based on work supported by the Engineering & Physical Sciences Research Council (EPSRC) through grant EP/R010544/1 and the coauthors are grateful for the funding award. SRC would also like to thank EPSRC for a vacation burasary. The authors would like to thank Konstantin Luzyanin for his expertise and aid during NMR spectroscopy experiments and the Materials Innovation Factory (University of Liverpool) for access to, and training in, MALDI-TOF analysis.Notes and references
- A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598–3614 CrossRef CAS.
- T. A. Faßbach, A. J. Vorholt and W. Leitner, ChemCatChem, 2019, 11, 1153–1166 CrossRef.
- A. Gordillo, L. Durán Pachón, E. de Jesus and G. Rothenberg, Adv. Synth. Catal., 2009, 351, 325–330 CrossRef CAS.
- D. Vogelsang, T. A. Faßbach, P. P. Kossmann and A. J. Vorholt, Adv. Synth. Catal., 2018, 360, 1984–1991 CrossRef CAS.
- M. D. Peterson and A. G. Weber, United States Patent and Trademark Office, US2395292, 19 February 1946 Search PubMed.
- W. E. Handford, United States Patent and Trademark Office, US2396786, 19 March 1946 Search PubMed.
- S. Norouzbahari and R. Gharibi, Ind. Eng. Chem. Res., 2020, 59, 6078–6089 CrossRef CAS.
- L. Chen, Y. Li, S. Yue, J. Ling, X. Ni and Z. Shen, Macromolecules, 2017, 50, 9598–9606 CrossRef CAS.
- R. Palkovits, A. N. Parvulescu, P. J. C. Hausoul, C. A. Kruithof, R. J. M. Klein Gebbink and B. M. Weckhuysen, Green Chem., 2009, 11, 1155–1160 RSC.
- S.-H. Yang, P. W. R. Harris, G. M. Williams and M. A. Brimble, Eur. J. Org. Chem., 2016, 2016, 2608–2616 CrossRef CAS.
- B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3235–3243 CrossRef CAS.
-
IUPAC. Compendium of Chemical Terminology, in Gold Book, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, Online version (2019) created by S. J. Chalk, ISBN 0-9678550-9-8, DOI:10.1351/goldbook.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- S.-H. Liu, J. Hazard. Mater., 2020, 384, 121427 CrossRef CAS.
- A. P. Dove and M. A. R. Meier, Macromol. Chem. Phys., 2014, 215, 2135–2137 CrossRef CAS.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456–4462 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS.
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3660 CrossRef CAS.
- I. Goettker-Schnetmann, P. Kenyon and S. Mecking, Angew. Chem., Int. Ed., 2019, 58, 17777–17781 CrossRef CAS.
- Z. Guan, J. Am. Chem. Soc., 2002, 124, 5616–5617 CrossRef CAS.
- M. D. Costioli, D. Berdat, R. Freitag, X. Andre and A. H. E. Mueller, Macromolecules, 2005, 38, 3630–3637 CrossRef CAS.
- H. Mutlu, A. N. Parvulescu, P. C. A. Bruijnincx, B. M. Weckhuysen and M. A. R. Meier, Macromolecules, 2012, 45, 1866–1878 CrossRef CAS.
- C. Boyer, G. Boutevin, J. J. Robin and B. Boutevin, Polymer, 2004, 45, 7863–7876 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- J. Liu, F. Zhan, Q. Fu, X. Zhu and W. Shi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7543–7555 CrossRef CAS.
- C. Zhang, L. Li, H. Cong and S. Zheng, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 952–962 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- W. H. Stockmayer and H. Jacobson, J. Chem. Phys., 1943, 11, 393–393 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- Y. Gao, D. Zhou, J. Lyu, A. Sigen, Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS.
- W. Wang, Y. Zheng, E. Roberts, C. J. Duxbury, L. Ding, D. J. Irvine and S. M. Howdle, Macromolecules, 2007, 40, 7184–7194 CrossRef CAS.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef.
- M. L. Koh, D. Konkolewicz and S. Perrier, Macromolecules, 2011, 44, 2715–2724 CrossRef CAS.
- Y. Zheng, H. Cao, B. Newland, Y. Dong, A. Pandit and W. Wang, J. Am. Chem. Soc., 2011, 133, 13130–13137 CrossRef CAS.
- G. Saunders, P. A. G. Cormack, S. Graham and D. C. Sherrington, Macromolecules, 2005, 38, 6418–6422 CrossRef CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CrossRef CAS.
- S. Graham, S. P. Rannard, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2007, 17, 545–552 RSC.
- T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495–1501 CrossRef CAS.
- E. Ruckenstein and H. Zhang, Macromolecules, 1999, 32, 3979–3983 CrossRef CAS.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
- A. Einstein, Ann. Phys., 1905, 17, 549–560 CrossRef CAS.
- A. Spernol and K. Wirtz, Naturforscher, 1953, 8, 522–532 Search PubMed.
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC.
- P. J. Flory, Chem. Rev., 1946, 39, 137–197 CrossRef CAS.
- K. A. Berchtold, T. M. Lovestead and C. N. Bowman, Macromolecules, 2002, 35, 7968–7975 CrossRef CAS.
- Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; [2004] – [cited 2020 May 01]. Available from: https://pubchem.ncbi.nlm.nih.gov/.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and characterisation. See DOI: 10.1039/d0py01309a |
| This journal is © The Royal Society of Chemistry 2020 |