
Temperature-mediated molecular ladder self-assembly employing Diels–Alder cycloaddition†
Samuel C.
Leguizamon
 a,
Abdulla F.
Alqubati
a and
Timothy F.
Scott
*bc
a,
Abdulla F.
Alqubati
a and
Timothy F.
Scott
*bc
aDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
bDepartment of Chemical Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: timothy.scott@monash.edu
cDepartment of Materials Science and Engineering, Monash University, Clayton, VIC 3800, Australia
First published on 18th November 2020
Abstract
Dynamic covalent self-assembly processes often exhibit poor capacities for error-correction owing to the relatively low connectivity rearrangement rates of dynamic covalent interactions and the common use of reaction conditions where the equilibrium state remains fixed. Here, we report a dynamic covalent self-assembly technique employing temperature, a conventional, externally-applied stimulus, to mediate the hybridization of peptoid oligomers bearing maleimide- and furan-based pendant groups to afford molecular ladders incorporating Diels–Alder adduct-based rungs. By raising or lowing the reaction temperature, this system enables the equilibrium state to be readily varied without altering reagent concentrations. Both triethylamine and the Lewis acidic scandium triflate were examined as candidate reaction catalysts; however, only scandium triflate increased the rate of single strand conversion. As the Diels–Alder cycloaddition reaction does not liberate a small molecule, a registry-dependent mass change was effected by employing a base-catalyzed thiol-Michael addition reaction between any un-reacted maleimide pendant groups and a low molecular weight thiol to enable the number of Diels–Alder adduct rungs to be readily determined by mass spectrometry. Finally, by employing a slow temperature ramp from high to low temperature, approximating the thermal cycle employed for nucleic acid hybridization, sequence-selective hybridization between model, tetra-functional precursor strands was demonstrated.
Introduction
Molecular self-assembly is commonly associated with relatively weak, intermolecular bonding as the transience of these interactions provides an inherent rearrangement mechanism to enable error correction. Nevertheless, dynamic covalent interactions have emerged as indispensable reactions to mediate nanoarchitecture fabrication, wherein the structural complexity attainable by self-assembly is combined with the strength of covalent bonds.1–3 These dynamic, rearrangeable covalent interactions have been used extensively for the synthesis of structurally diverse, mechanical and chemical robust materials including covalent organic frameworks (COFs),4–6 molecular cages,7–9 and molecular ladders.10–12 In comparison with supramolecular interactions, dynamic covalent reactions suffer from relatively low dissociation rates which impede rearrangement amongst the assembled components and can result in the kinetic trapping of non-equilibrium species.13–16 To overcome this limitation, dynamic covalent assembly processes are generally restricted to those employing constituent precursors bearing few reactive sites, the utilization of harsh reaction conditions,1 or the assistance of secondary intermolecular interactions.17 Conversely, the inherently transient nature of supramolecular interactions permits the use of non-covalent building blocks with high degrees of functionality and mild reaction conditions to generate complex molecular architectures.18–20 Particularly suitable for the formation of multi-dimensional structures, nucleic acids are often employed as sophisticated nanoconstruction media where encoded sequences self-assemble in a predetermined manner upon the gradual cooling of dissociated strands from raised temperatures.21–23 In a recent effort to mimic the hybridization of complementary nucleic acid sequences with precursors bearing dynamic covalent reaction pairs, we reported a step-wise process involving the dissociation of encoded peptoid (i.e., oligo(N-substituted glycine)) strands bearing amine- and aldehyde-pendant species by treatment with large amounts of a Lewis acidic rare-earth metal triflate, Sc(OTf)3, and subsequent extraction of this metal triflate to promote imine formation and error-correction of information-bearing molecular ladders.24 Similarly, base-4-encoded peptoid strands bearing two sets of orthogonal dynamic covalent reaction pairs capable of forming either imine- or boronate ester-rungs, exhibited sequence-selective self-assembly using a mechanism in which acid-dissociated strands were introduced to alkaline conditions to promote rung formation between complementary sequences.25 Whereas these methods moderate the kinetic trapping that prevails in dynamic covalent assembly systems to enable the self-assembly of complementary oligomeric sequences, both processes are limited by an abrupt equilibrium shift from dissociation to association and are dissimilar to the gradual temperature ramp employed for nucleic acid hybridization. The development of a covalent self-assembly system where the reaction equilibrium is progressively shifted is more readily realized with dynamic covalent reactions that are affected by an external stimulus (e.g., temperature, light intensity, etc.) than those influenced by a chemical stimulus (e.g., pH, reagent concentration, etc.). One such thermally-sensitive dynamic covalent interaction, the Diels–Alder cycloaddition between a maleimide and a furan, has been used extensively in self-healing polymeric materials;26,27 however, it is rarely employed as a mechanism to mediate self-assembly processes. Here we demonstrate the temperature-mediated self-assembly of oligomers bearing maleimide- and furan-pendant groups to afford molecular ladders with Diels–Alder adduct-based rungs.Results and discussion
To examine the utility of the Diels–Alder cycloaddition reaction as a dynamic covalent interaction capable of mediating molecular self-assembly, molecular ladders were employed as target structures owing to their demonstrated capacity for sequence-selective assembly from information-bearing precursors with large numbers of reactive sites, and their facile characterization.24,28,29 By employing the sub-monomer solid phase synthetic method, sequence-specific peptoid precursor strands bearing furan and maleimide pendant groups were synthesized from readily available primary amines.30To preclude potential hydrolysis or Michael addition side reactions, or premature Diels–Alder cycloaddition during the synthesis and purification of maleimide-bearing oligomers, a furan-protected maleimide (3) was synthesized as outlined in Fig. 1a. The Diels–Alder reaction commonly generates a mixture of endo and exo diastereomers, where the cyclo-reversion of the endo adduct proceeds at a lower temperature (∼60 °C) than the exo adduct (∼110 °C).31,32 As the peptoid synthesis is performed at 75 °C, the endo form is ill-suited as a maleimide-protecting group here owing to its potential for undergoing premature deprotection. To preclude the deleterious influence of endo adducts, we employed an approach to selectively yield the exo-Diels–Alder cycloaddition adduct (1) from furan and maleic anhydride by toluene precipitation, described elsewhere.33N-Boc-ethylenediamine was added to compound 1via a ring-opening mechanism and subsequently cyclized in ethanol under reflux to afford compound 2 which was then treated with trifluoroacetic acid (TFA) in dichloromethane to yield the final, furan-protected maleimide monomer (3). Furfuryl amine, 2-methoxyethylamine, and compound 3, were employed as furan-bearing, inert spacer, and protected maleimide-bearing primary amine monomers, respectively, in the microwave-assisted synthesis of a small library of peptoids (Fig. S4†). The deprotonated form of compound 3 was generated in situ by the addition of N,N-diisopropylethylamine to the reaction mixture, ensuring reactivity of the monomer in the nucleophilic displacement of the terminal bromide.
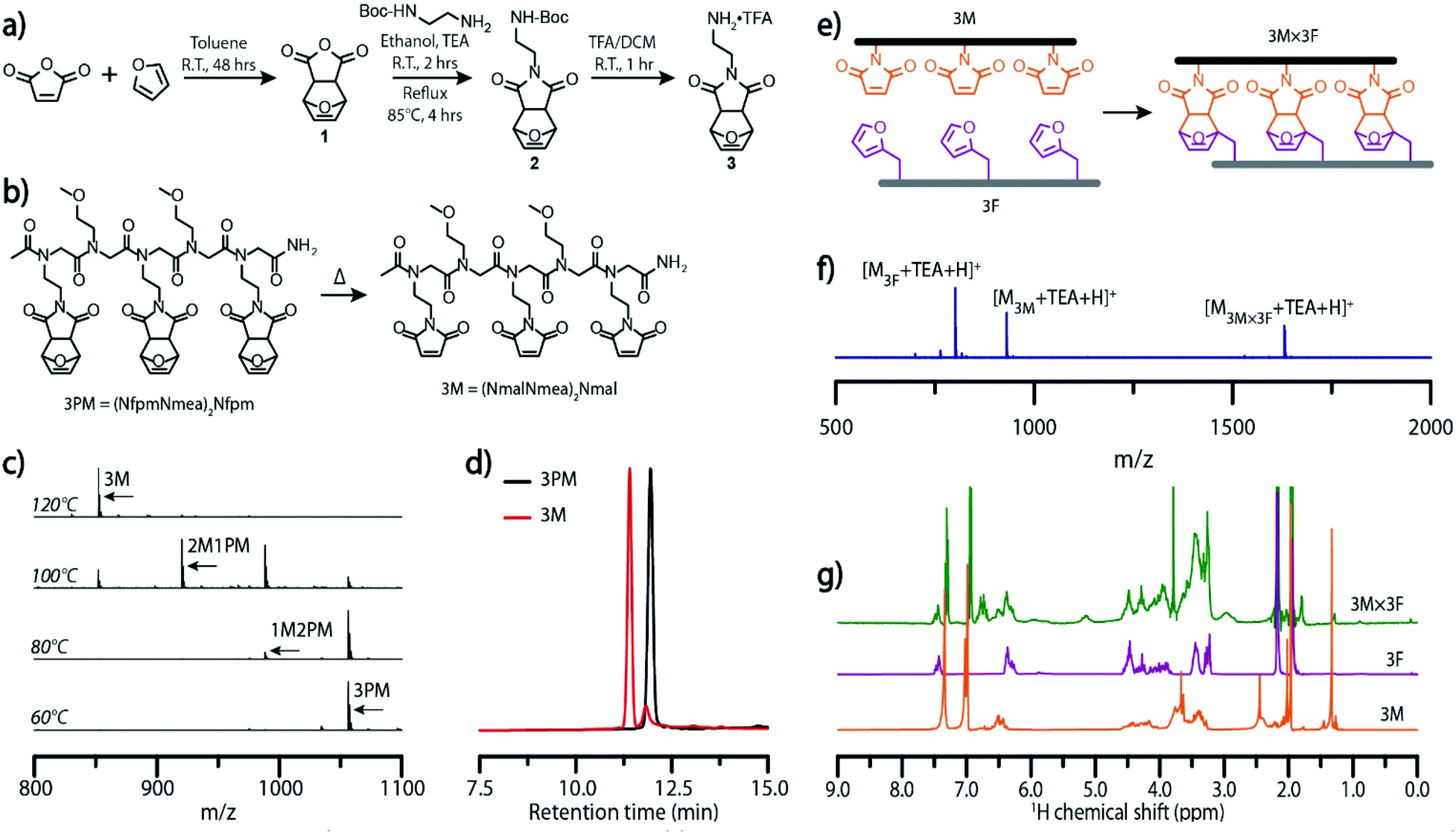 | ||
| Fig. 1 Dynamic covalent assembly of Diels–Alder-based molecular ladders. (a) Synthetic scheme for the furan-protected, maleimide-bearing peptoid monomer, 3 and (b) chemical structures depicting the Diels–Alder adduct-bearing peptoid 3PM (synthesized from 3), and the deprotection of 3PM to afford the maleimide-bearing oligomer 3M. (c) ESI mass spectra generated after exposing solutions of 3PM strands to 60 °C, 80 °C, 100 °C, or 120 °C for one hour. Here, 3M is the fully deprotected oligomer, while 2M1PM and 1M2PM represent partially deprotected oligomers bearing one and two furan-protected maleimide groups, respectively. (d) HPLC traces of a solution of 3PM single strands prior to (black) and after deprotection at 140 °C for 30 minutes (red). (e) Idealized schematic diagram of the hybridization of complementary, trifunctional maleimide- and furan-bearing oligomers (3M and 3F, respectively) to afford a peptoid-based molecular ladder bearing three Diels–Alder adduct rungs (3M × 3F). (f) ESI mass spectrum of a 3PM and 3F hybridization reaction mixture after heating at 140 °C for 30 minutes, then at 60 °C for five days. Expected exact masses: [M3F + TEA + H]+ = 802.4; [M3M + TEA + H]+ = 931.4; [M3M×3F + TEA + H]+ = 1631.7. Exact masses found: [M3F + TEA + H]+ = 802.4; [M3M + TEA + H]+ = 931.5; [M3M×3F + TEA + H]+ = 1631.8. (g) 1H-NMR spectra of the maleimide-bearing peptoid 3M (bottom, orange), the furan-bearing peptoid 3F (middle, purple), and a 3M × 3F molecular ladder reaction mixture (top, green). 1H-NMR solvent signals: anisole = 7.29 ppm, 6.93 ppm, 3.75 ppm; water = 2.13 ppm; acetonitrile = 1.94 ppm. | ||
A peptoid oligomer bearing three furan-protected maleimide pendant groups, 3PM, was used to evaluate the extent of deprotection at elevated temperatures as incomplete deprotection would impede the subsequent self-assembly process (Fig. 1b). Characterization of 3PM sequences deprotected for one hour at various elevated temperatures in anhydrous anisole, used as a high boiling-point solvent, was performed by electrospray ionization mass spectrometry (ESI-MS), configured with low cone voltage to eliminate in-source fragmentation. Here, the deprotection of a maleimide pendant group via the retro-Diels–Alder reaction yielded a mass change of −68 owing to the loss of furan. Mass spectra indicated an onset of the retro-Diels–Alder reaction at 80 °C with complete deprotection at temperatures of 120 °C (Fig. 1c) and above, consistent with similar studies on exo-Diels–Alder isomers.32 Notably, the maleimide pendant groups remained deprotected after cooling of the reaction mixture, attributable to the high volatility and consequent evaporation of furan at raised temperature. Reverse-phase HPLC of 3PM oligomers and the corresponding deprotected oligomer, 3M, confirmed the formation of a new species with a decreased retention time (Fig. 1d). The chromatogram of the post-thermal cycle 3M reaction mixture did show a small peak with a retention time similar to that of the 3PM sequence; however, this peak could not be identified by ESI-MS. Further characterization by 1H-NMR demonstrated the quantitative deprotection of 3PM strands, as shown by the absence of peaks around 5.14 and 2.86 corresponding to the (–CHCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH–), and (OCCH) protons, respectively, of the Diels–Alder adduct (Fig. S5†).
CHCH–), and (OCCH) protons, respectively, of the Diels–Alder adduct (Fig. S5†).
Initial ladder formation was established by heating reaction mixtures of complementary trifunctional furan-protected maleimide- and furan-bearing oligomers, 3PM and 3F, respectively, to 140 °C to simultaneously deprotect the maleimide-bearing peptoid and dissociating the two sequences, then subsequently annealing the reaction mixture at 60 °C for five days (Fig. 1e). Characterization of the resulting hybridization mixtures by ESI-MS confirmed the generation of the target ladder species (3M × 3F). Although the 3M × 3F molecular ladder and residual 3M oligomer both exhibited low intensity signals (Fig. S6†), addition of triethylamine (TEA), an ionizing agent, to a reaction mixture which was subjected to the aforementioned thermal dissociation/annealing process increased the peak intensities (Fig. 1f). 1H-NMR and diffusion ordered spectroscopy (DOSY) NMR further verified the occurrence of a binding event with the appearance of adduct signals around 5.14 (CHCHCH) and 2.95 (OCCH) and a decrease in the diffusion coefficient upon complexation (Fig. 1g and Fig. S7†).
Signal intensity of ESI-MS analytes is dependent on a variety of factors including sample concentration, ease of analyte ionization, and matrix or solute interference, preventing the quantitative analysis of dissimilar species (e.g., single strand and dimeric ladder species).34 Consequently, gel permeation chromatography (GPC), was employed to examine the formation of dimeric and high-order ladder species. Interestingly, the protected maleimide-bearing peptoids, mimicking the Diels–Alder-rung molecular ladder species, were undetectable by GPC analysis using a UV-Vis detector at 254 or 313 nm, while the furan-bearing peptoid presented only a minor peak (Fig. S8†). Nevertheless, peptoids bearing deprotected-maleimide species were found to absorb strongly at 313 nm, enabling the unreacted maleimide concentration in single strand or hybrid species to be monitored over time by spiking reaction mixtures with inert, low dispersity polystyrene at a known concentration as an internal standard. Oligomer sequences with four reactive species per strand were employed for this study to augment the absorption signal (Fig. 2a). As the Diels–Alder reaction proceeds slowly at room temperature, the period each aliquot experienced in the GPC system was assumed to have negligible effects on the overall conversion. This assumption was verified by analyzing aliquots of a hybridization reaction mixtures reacted at room temperature with and without a catalyst (Fig. S9†), where negligible formation of dimeric or multimeric ladder species was observed even after 72 hours.
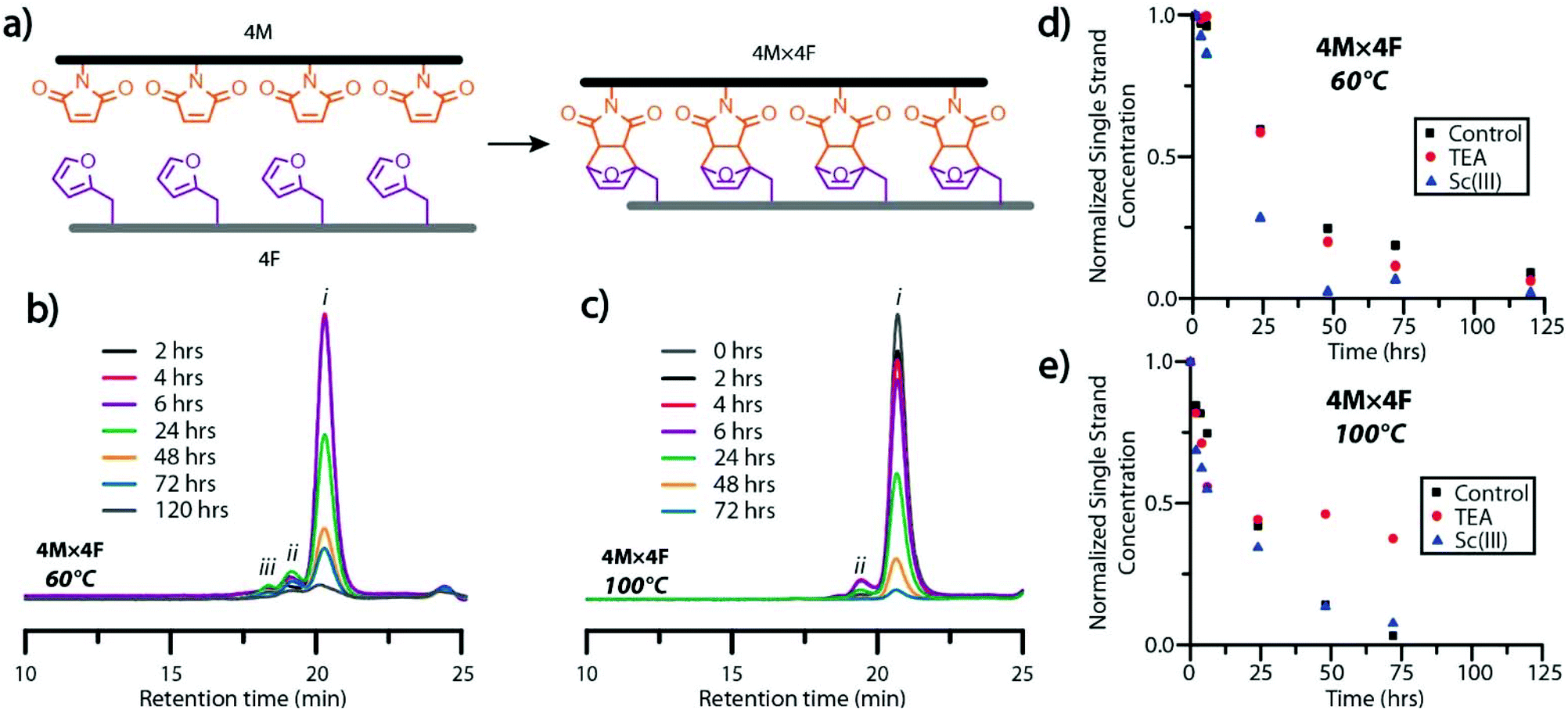 | ||
| Fig. 2 Time-resolved characterization of molecular ladder formation. (a) Idealized schematic diagram of the self-assembly of tetramaleimide (4M) and tetrafuran (4F) precursor strands to afford a molecular ladder bearing four Diels–Alder adduct-based rungs (4M × 4F). GPC traces, normalized to a polystyrene internal standard, of aliquots periodically sampled from 4M × 4F hybridization reaction mixtures reacted at (b) 60 °C and (c) 100 °C, where i, ii, and iii indicate single stranded, dimeric, and multimeric species, respectively, bearing residual maleimide groups. Normalized concentrations of single strand species in hybridization mixtures with and without catalysts and reacted at (d) 60 °C and (e) 100 °C, determined from deconvolution of overlapping GPC trace peaks by Gaussian curve fitting. | ||
GPC analysis of aliquots from hybridization reaction mixtures at 60 °C and 100 °C showed the generation of dimeric ladder species within 2 hours, and high-order, multimeric species at 4 hours (Fig. 2b and c). Multimeric molecular ladder species were consistently observed in aliquots of the 60 °C reaction mixture (Fig. 2b), indicating the persistence of out-of-registry ladders throughout the reaction. In contrast, negligible amounts of dimeric and multimeric species were observed in aliquots of the 100 °C reaction mixture from 48 hours (Fig. 2c), indicating the complete consumption of residual maleimide groups associated with out-of-registry structures and thus an increased capacity for strand rearrangement to form in-registry-ladders at this temperature. This increased capacity for strand rearrangement is reflected in the larger Diels–Alder adduct dissociation rate constant for the reaction between a maleimide and furan at 100 °C than 60 °C (1.44 × 10−3 s−1 and 4.42 × 10−5 s−1, respectively26). Furthermore, the equilibrium constants at 100 °C and 60 °C (1.15 M−1 and 5.43 M−1 respectively26) both favor rung formation; nevertheless, the significant difference between the rates of Diels–Alder adduct formation and dissociation at 60 °C suggests sluggish annealing of out-of-registry species at this temperature, evidenced by the slow disappearance of peaks attributable to multimeric species in GPC at extended periods. To examine the influence of the ionizing agent or a catalyst on the rate of molecular ladder formation, either TEA or scandium triflate35 was added to 4M × 4F reaction mixtures immediately following maleimide-deprotection at 140 °C, and then cooled to either 60 °C or 100 °C. Aliquots of hybridization solutions spiked with polystyrene were collected and the single strand maleimide conversions were determined by GPC (Fig. 2d and e). The addition of TEA did not significantly affect the rate of ladder formation at 60 °C, suggesting that the signal enhancement in mass spectra of molecular ladder species in 3M × 3F reaction mixtures observed above was a result of increased analyte ionization in the presence of TEA. This was confirmed by ESI-MS analysis of 3M × 3F reactions immediately before and after addition of a catalytic amount of TEA to the solutions, yielding mass spectra with increased peak intensities attributable to molecular ladder species (Fig. S10†). Interestingly, evidenced by a discoloration of the reaction solution and a conversion plateau around 24 hours (Fig. 2e), the 4M × 4F reaction solutions at 100 °C incorporating TEA as a catalyst experienced a deleterious side reaction, prompting the exclusion of TEA as a hybridization reaction catalyst. Nevertheless, TEA was added at room temperature prior to characterization by mass spectrometry to improve analyte ionization. At both 60 °C and 100 °C, the addition of scandium triflate increased the rate of single strand conversion, particularly within the first six hours.
In imine- and boronate ester-based, self-assembled molecular ladder systems examined previously, the number of rungs (i.e., inter-strand bonds) formed between complementary oligomeric sequences was readily determined by mass spectrometry, where each imine or boronate ester bond formed yielded a mass change of −18 or −36, respectively, owing to the loss of water generated upon amine/aldehyde or boronic acid/diol condensation.28,36 As the Diels–Alder cycloaddition reaction does not liberate a small molecule, in- and out-of-registry constructs are indistinguishable by conventional mass spectrometry. To afford a registry-dependent mass change, we employed a base-catalyzed thiol-Michael addition reaction between any un-reacted maleimide pendant groups and methyl 3-mercaptopropionate (S) as a reactive, low-molecular weight thiol (Fig. 3a). Yielding a mass change of +120 (i.e., the exact mass of S) for each available maleimide pendant group that arises from out-of-registry ladder species, this approach enables the number of Diels–Alder adduct rungs to be readily determined by mass spectrometry. The efficiency of the thiol addition to strands bearing multiple reactive sites was initially examined by adding various amounts of S and a catalytic amount of TEA to solutions of 3M in anhydrous anisole. Mass spectra of the solutions demonstrated that, whereas incompletely reacted oligomers could be detected by ESI MS when the thiol was the limiting reagent, the efficient and near-complete maleimide consumption was observed when three equivalents of S to maleimide groups was employed to yield the fully-substituted strand 3M + 3·S (Fig. 3b). This fully-substituted strand was subjected to evaporation under reduced pressure to remove the solvent, excess S, and TEA, then redissolved in deuterated acetonitrile and analyzed by 1H-NMR (Fig. 3c) and HPLC (Fig. S11†). These revealed a 93% maleimide conversion, evident by a decrease in the 1H-NMR maleimide peak around 6.5 ppm and increase in peaks attributable to the conjugated thiol between 2.6 and 3.2 ppm. Whereas, the deprotected peptoid 3M afforded a peak with a retention time of 11.4 minutes in reverse phase HPLC, the progressive addition of thiol diminished the prevalence of the 3M peak and yielded an emergent peak at an increased retention time of 14.4 minutes, attributable to the generated 3M + 3·S.
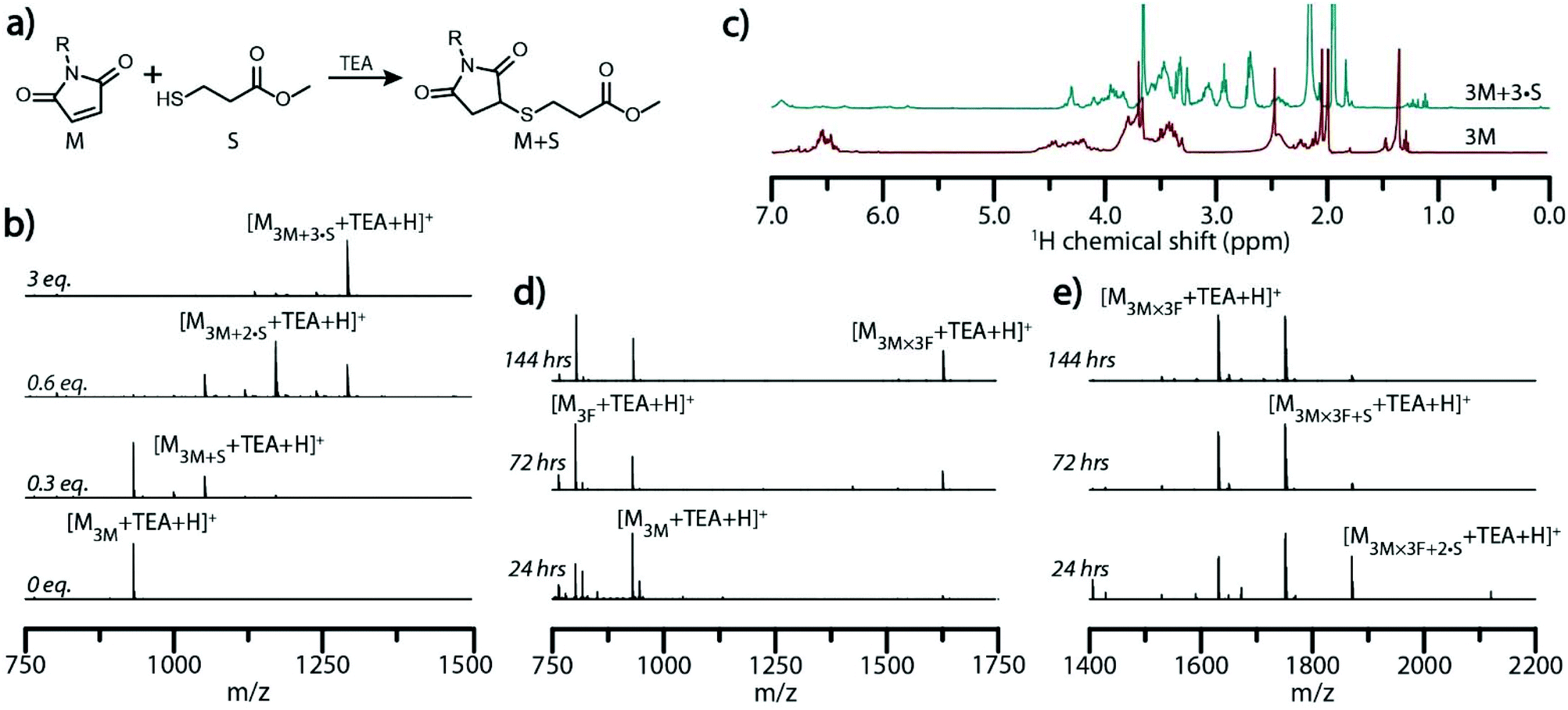 | ||
| Fig. 3 Molecular ladder registry. (a) Reaction scheme for the base catalyzed thiol-Michael addition between a maleimide and methyl 3-mercaptopropionate (S). (b) ESI mass spectra of thiol-Michael addition reaction mixtures between the peptoid 3M, bearing three maleimide pendant groups, and reacted for 30 minutes with various equivalents of S to maleimide groups. (c) 1H-NMR spectra of a solution of 3M strands before (bottom, red) and after (top, blue) reaction with 3 equivalents of S to maleimide groups. ESI mass spectra of 3M × 3F hybridization mixtures (d) after reaction at 60 °C for various periods as shown, and (e) after reaction at 60 °C for various periods as shown, then subjected to a thiol-Michael addition with a threefold excess of S. Expected exact masses: [M3F + TEA + H]+ = 802.4; [M3M + TEA + H]+ = 931.4; [M3M×3F + TEA + H]+ = 1631.7; [M3M+S + TEA + H]+ = 1051.4; [M3M+2·S + TEA + H]+ = 1171.3; [M3M+3·S + TEA + H]+ = 1291.3; [M3M×3F+S + TEA + H]+ = 1751.7; [M3M×3F+2·S + TEA + H]+ = 1871.7. Exact masses found: [M3F + TEA + H]+ = 802.4; [M3M + TEA + H]+ = 931.4; [M3M×3F + TEA + H]+ = 1631.7; [M3M+S + TEA + H]+ = 1051.5; [M3M+2·S + TEA + H]+ = 1171.5; [M3M+3·S + TEA + H]+ = 1291.5; [M3M×3F+S + TEA + H]+ = 1751.8; [M3M×3F+2·S + TEA + H]+ = 1871.8. | ||
Hybridization solutions containing 3M and 3F sequences at 60 °C and 100 °C were quenched at various times by rapid cooling to room temperature and subsequently reacted with three equivalents of the thiol S to maleimide groups in the presence of TEA to ensure quantitative conjugation with residual maleimides. Aliquots of the mixture before and after addition of S were characterized by ESI-MS to assess the ladder formation and determine the extent of strand alignment (Fig. 3d, e and Fig. S12†). Mass spectrum peaks attributable to dimeric molecular ladder species progressively emerged with increased reaction times at both temperatures examined, signifying further conversion over time and supporting the previous GPC analysis. Upon addition of S, ESI-MS revealed that hybridization of 3M and 3F at both 60 °C and 100 °C does not yield complete registry of molecular ladders within 24 hours, but rather a combination of ladder species bearing 1, 2, or 3 rungs (Fig. 3e and Fig. S12†). At extended reaction times, the peak attributable to the in-registry, 3-rung ladder does increases relative to the other ladder species for both reaction conditions, indicating rearrangement of the constituent precursor strands towards the target, in-registry ladder product, albeit at an enhanced rate at 100 °C.
Having demonstrated a capacity for effective error correction and rearrangement of the inter-strand Diels–Alder adduct-based rungs at raised temperature, we examined a temperature-mediated approach, analogous to the thermal cycling employed to effect sequence-selective nucleic acid hybridization, to achieve the hybridization selectivity of maleimide- and furan-bearing precursor strands to yield multiple target molecular ladders in a single pot. Here, several unique, sequence-defined oligomers in a single pot reaction mixture were deprotected and dissociated at high temperature, then allowed to react and anneal under lower temperature conditions. One of the sequences employed was a peptoid bearing two furan-protected maleimide and two furan pendant groups (2PM2F), mass-labeled with additional 2-ethoxyethoxyethylamine and 2-methoxyethylamine residues at its N- and C-terminal ends, respectively; upon in situ deprotection, this oligomeric sequence should undergo self-hybridization to afford the molecular ladder 2M2F × 2M2F. Concurrent hybridization selectivity for complementary peptoid pairs was examined by allowing the tetrafunctional, mass-labeled sequences 2PM2F, 4PM, and 4F, in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio, to react simultaneously in a single pot reaction mixture to yield two distinct target molecular ladder species (Fig. 4a). Initially, reaction mixtures were heated at 140 °C for 30 minutes, then cooled and maintained at either 60 °C or 100 °C while aliquots were collected, thermally quenched, and characterized by ESI-MS (Fig. 4b and c, respectively). At a reaction temperature of 60 °C, all possible dimeric combinations between and complementary and non-complementary sequences (i.e., 4M × 4F, 4F × 2M2F, 4M × 2M2F, and 2M2F × 2M2F) were observed (Fig. 4b), even after prolonged reaction times (Fig. S13†), indicating that oligomer hybridization was unselective for sequence under these reaction conditions. At a reaction temperature of 100 °C, ESI-MS showed the generation of only the 4M × 4F species after 24 hours; however, all other possible combinations were present after 48 hours (Fig. 4c), again showing unselective oligomer hybridization. Importantly, as these reaction conditions involve near-step temperature changes that provide insufficient time for connectivity rearrangement, misconfigured ladder species between non-complementary precursor strands remain present in the reaction mixtures. To better approximate the slow cooling step of a thermal cycle employed for nucleic acid hybridization, a modified temperature ramp profile was employed where the reaction mixture temperature was initially reduced from 140 °C to 100 °C and held at that temperature for 24 hours, then slowly reduced further to 60 °C and held there for up to 48 hours. As shown in Fig. 4d, ESI-MS revealed that this temperature profile exclusively yielded the two target molecular ladder assemblies (i.e., 4M × 4F and 2M2F × 2M2F) after 24 hours at 60 °C, and that this distribution of reaction products was maintained after a further 24 hours at 60 °C, confirming the capacity of this Diels–Alder adduct-based system for temperature-mediated, sequence-selective hybridization.
:1:1 molar ratio, to react simultaneously in a single pot reaction mixture to yield two distinct target molecular ladder species (Fig. 4a). Initially, reaction mixtures were heated at 140 °C for 30 minutes, then cooled and maintained at either 60 °C or 100 °C while aliquots were collected, thermally quenched, and characterized by ESI-MS (Fig. 4b and c, respectively). At a reaction temperature of 60 °C, all possible dimeric combinations between and complementary and non-complementary sequences (i.e., 4M × 4F, 4F × 2M2F, 4M × 2M2F, and 2M2F × 2M2F) were observed (Fig. 4b), even after prolonged reaction times (Fig. S13†), indicating that oligomer hybridization was unselective for sequence under these reaction conditions. At a reaction temperature of 100 °C, ESI-MS showed the generation of only the 4M × 4F species after 24 hours; however, all other possible combinations were present after 48 hours (Fig. 4c), again showing unselective oligomer hybridization. Importantly, as these reaction conditions involve near-step temperature changes that provide insufficient time for connectivity rearrangement, misconfigured ladder species between non-complementary precursor strands remain present in the reaction mixtures. To better approximate the slow cooling step of a thermal cycle employed for nucleic acid hybridization, a modified temperature ramp profile was employed where the reaction mixture temperature was initially reduced from 140 °C to 100 °C and held at that temperature for 24 hours, then slowly reduced further to 60 °C and held there for up to 48 hours. As shown in Fig. 4d, ESI-MS revealed that this temperature profile exclusively yielded the two target molecular ladder assemblies (i.e., 4M × 4F and 2M2F × 2M2F) after 24 hours at 60 °C, and that this distribution of reaction products was maintained after a further 24 hours at 60 °C, confirming the capacity of this Diels–Alder adduct-based system for temperature-mediated, sequence-selective hybridization.
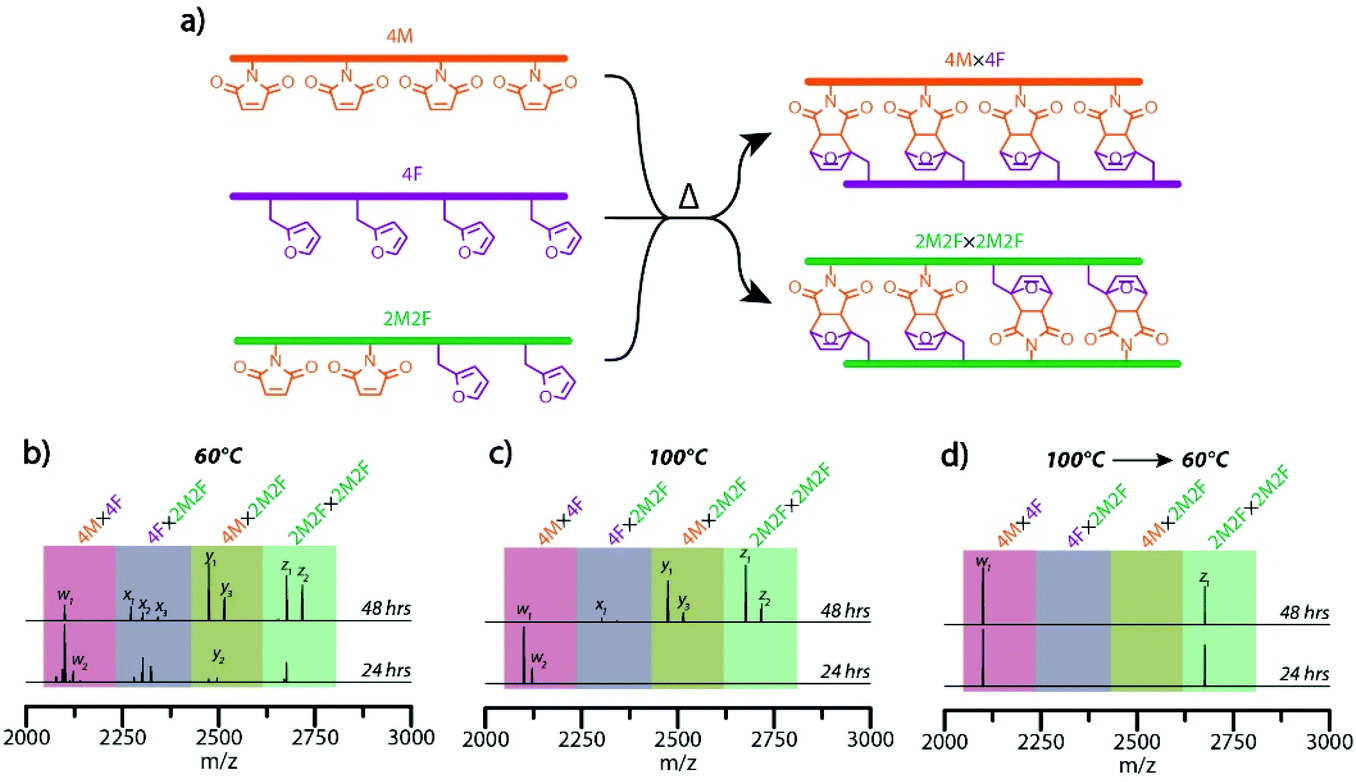 | ||
| Fig. 4 Dynamic covalent assembly of sequence-defined molecular ladders (a) idealized schematic diagram of the concurrent self-assembly of 4PM, 4F, and 2PM2F precursor strands to afford the four-rung molecular ladders 4M × 4F and 2M2F × 2M2F. ESI mass spectra of molecular ladder reaction mixture aliquots removed from hybridization solutions heated at 140 °C for 30 minutes then cooled and maintained at (b) 60 °C, (c) 100 °C, or (d) 100 °C for 24 hours, allowed to slowly cool, then maintained at 60 °C. Expected exact masses: w1 = [M4M×4F + Na]+ = 2099.8; w2 = [M4M×4F + 2Na − H]+ = 2121.8; x1 = [M4F×2M2F + H]+ = 2280.0; x2 = [M4F×2M2F + Na]+ = 2303.0; x3 = [M4F×2M2F + 2Na − H]+ = 2324.0; y1 = [M4M×2M2F + Na]+ = 2474.0; y2 = [M4M×2M2F + 2Na − H]+ = 2496.0; y3 = [M4M×2M2F + MeCN + Na]+ = 2515.1; z1 = [M2M2F×2M2F + Na]+ = 2676.2; z2 = [M2M2F×2M2F + MeCN + Na]+ = 2717.2. Exact masses found: w1 = [M4M×4F + Na]+ = 2099.8; w2 = [M4M×4F + 2Na − H]+ = 2121.8; x1 = [M4F×2M2F + H]+ = 2280.0; x2 = [M4F×2M2F + Na]+ = 2303.0; x3 = [M4F×2M2F + 2Na − H]+ = 2324.0; y1 = [M4M×2M2F + Na]+ = 2474.0; y2 = [M4M×2M2F + 2Na − H]+ = 2496.0; y3 = [M4M×2M2F + MeCN + Na]+ = 2515.0; z1 = [M2M2F×2M2F + Na]+ = 2676.2; z2 = [M2M2F×2M2F + MeCN + Na]+ = 2717.1. | ||
Conclusions
We have described the self-assembly of molecular ladder species from oligomeric precursor strands employing the thermally-reversible Diels–Alder cycloaddition reaction. Sequence-defined peptoids were synthesized bearing maleimide- and furan-based pendant groups by employing furan as a thermally-labile maleimide protecting group to preclude side reactions or premature hybridization. In conjunction with ESI mass spectrometry, GPC was used to monitor the progress of the hybridization reaction between complementary oligomers. To enable the number of Diels–Alder adduct rungs to be determined by mass spectrometry, a thiol-Michael addition reaction between un-reacted maleimide pendant groups and a low-molecular weight thiol was employed to provide a registry-dependent mass change. This approach revealed that, whereas hybridization of trifunctional maleimide- and furan-bearing oligomers did not exclusively yield the completely formed, in-registry molecular ladder within 24 hours, rearrangement towards the target, in-registry ladder product proceeded over extended reaction times. By employing a slow temperature ramp from high to low temperature, sequence-selective hybridization in a model mixture of sequence-specific peptoids was obtained. We anticipate that the sequence-selective oligomer hybridization using a thermally-reversible Diels–Alder cycloaddition reaction demonstrated here will advance the utilization of dynamic covalent interactions mediated by an external stimulus for the fabrication of robust, complex nanostructures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award # DE-SC0012479. S.C.L. would like to acknowledge the National Science Foundation Graduate Research Fellowship Program and the Monash Analytical Platform. A.F.A acknowledges support from Abu Dhabi National Oil Company (ADNOC).References
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- J. C. Furgal, M. Dunn, T. Wei and T. F. Scott, in Dynamic Covalent Chemistry: Principles, Reactions, and Applications, 2017, pp. 389–434 Search PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dyn. Covalent Chem., 2002, 41, 898–952 Search PubMed.
- N. W. Ockwig, A. P. Co, M. O. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1171 CrossRef.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- X. Ma and T. F. Scott, Commun. Chem., 2018, 1, 1–15 CrossRef.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS.
- A. Granzhan, C. Schouwey, T. Riis-Johannessen, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2011, 133, 7106–7115 CrossRef CAS.
- C. S. Hartley, E. L. Elliott and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 4512–4513 CrossRef CAS.
- T. Wei, J. H. Jung and T. F. Scott, J. Am. Chem. Soc., 2015, 137, 16196–16202 CrossRef CAS.
- H. Iida, K. Ohmura, R. Noda, S. Iwahana, H. Katagiri, N. Ousaka, T. Hayashi, Y. Hijikata, S. Irle and E. Yashima, Chem. – Asian J., 2017, 12, 927–935 CrossRef CAS.
- E. L. Elliott, C. S. Hartley and J. S. Moore, Chem. Commun., 2011, 47, 5028–5030 RSC.
- G. Zhu, Y. Liu, L. Flores, Z. R. Lee, C. W. Jones, D. A. Dixon, D. S. Sholl and R. P. Lively, Chem. Mater., 2018, 30, 262–272 CrossRef CAS.
- J. Baram, H. Weissman and B. Rybtchinski, J. Phys. Chem. B, 2014, 118, 12068–12073 CrossRef CAS.
- Q. Wang, C. Yu, C. Zhang, H. Long, S. Azarnoush, Y. Jin and W. Zhang, Chem. Sci., 2016, 7, 3370–3376 RSC.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS.
- B. Gong, Acc. Chem. Res., 2012, 45, 2077–2087 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- S. I. Stupp, V. Lebonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384–389 CrossRef CAS.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- S. C. Leguizamon and T. F. Scott, Nat. Commun., 2020, 11, 784 CrossRef CAS.
- S. C. Leguizamon, M. Dunn and T. F. Scott, Chem. Commun., 2020, 56, 7817–7820 RSC.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- T. Wei, J. C. Furgal, J. H. Jung and T. F. Scott, Polym. Chem., 2017, 8, 520–527 RSC.
- J. F. Reuther, J. L. Dees, I. V. Kolesnichenko, E. T. Hernandez, D. V. Ukraintsev, R. Guduru, M. Whiteley and E. V. Anslyn, Nat. Chem., 2018, 10, 45–50 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- V. Froidevaux, M. Borne, E. Laborbe, R. Auvergne, A. Gandini and B. Boutevin, RSC Adv., 2015, 5, 37742–37754 RSC.
- E. H. Discekici, A. H. St Amant, S. N. Nguyen, I. H. Lee, C. J. Hawker and J. Read De Alaniz, J. Am. Chem. Soc., 2018, 140, 5009–5013 CrossRef CAS.
- S. R. Mane, V. Rao N, K. Chatterjee, H. Dinda, S. Nag, A. Kishore, J. Das Sarma and R. Shunmugam, J. Mater. Chem., 2012, 22, 19639–19642 RSC.
- C. Ho, C. Lam, M. Chan, R. Cheung, L. Law, L. Lit, K. Ng, M. Suen and H. Tai, Clin. Biochem. Rev., 2003, 24, 3–12 CAS.
- S. Fukuzumi, J. Yuasa, T. Miyagawa and T. Suenobu, J. Phys. Chem. A, 2005, 109, 3174–3181 CrossRef CAS.
- M. F. Dunn, T. Wei, R. N. Zuckermann and T. F. Scott, Polym. Chem., 2019, 10, 2337–2343 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01296c |
| This journal is © The Royal Society of Chemistry 2020 |