
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational molecular design of anion exchange membranes functionalized with alicyclic quaternary ammonium cations
Thanh Huong
Pham
 ,
Andrit
Allushi
,
Joel S.
Olsson
and
Patric
Jannasch
*
,
Andrit
Allushi
,
Joel S.
Olsson
and
Patric
Jannasch
*
Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
First published on 19th October 2020
Abstract
High alkaline stability is critical for polymeric anion exchange membranes (AEMs) and ionomers for use in alkaline electrochemical energy conversion and storage devices such as fuel cells, electrolyzer cells and advanced batteries. Here, we have prepared and studied ether-free polyfluorenes tethered with N,N-dimethylpiperidinium (DMP) and 6-azonia-spiro[5.5]undecane (ASU) cations, respectively, attached through heteroatom-free alkyl spacers. By employing alkyl–alkyl Suzuki cross-coupling, these alicyclic quaternary ammonium cations are attached at the 4-position to impede ionic loss. Thus, all the β-hydrogens sensitive to elimination reactions are placed in strain-free rings able to fully relax by the spacer flexibility. Consequently, the AEM carrying DMP cations shows a very high alkaline and thermal stability, retaining more than 91% of the cations after 2400 h immersion in 2 M NaOH at 90 °C. Compared with corresponding AEM functionalized with N-alkyl-N-methylpiperidinium (AMP) cations [conventionally tethered via the 1(N)-position], the ionic loss by β-elimination is successfully reduced by up to 92%. The AEM functionalized with DMP also reaches a high hydroxide conductivity of 124 mS cm−1 at 80 °C. Consequently, tethering piperidine-based cations via the 4-position instead of the 1(N)-position results in AEMs with substantially improved thermal and alkaline stability, combined with high hydroxide conductivity.
1. Introduction
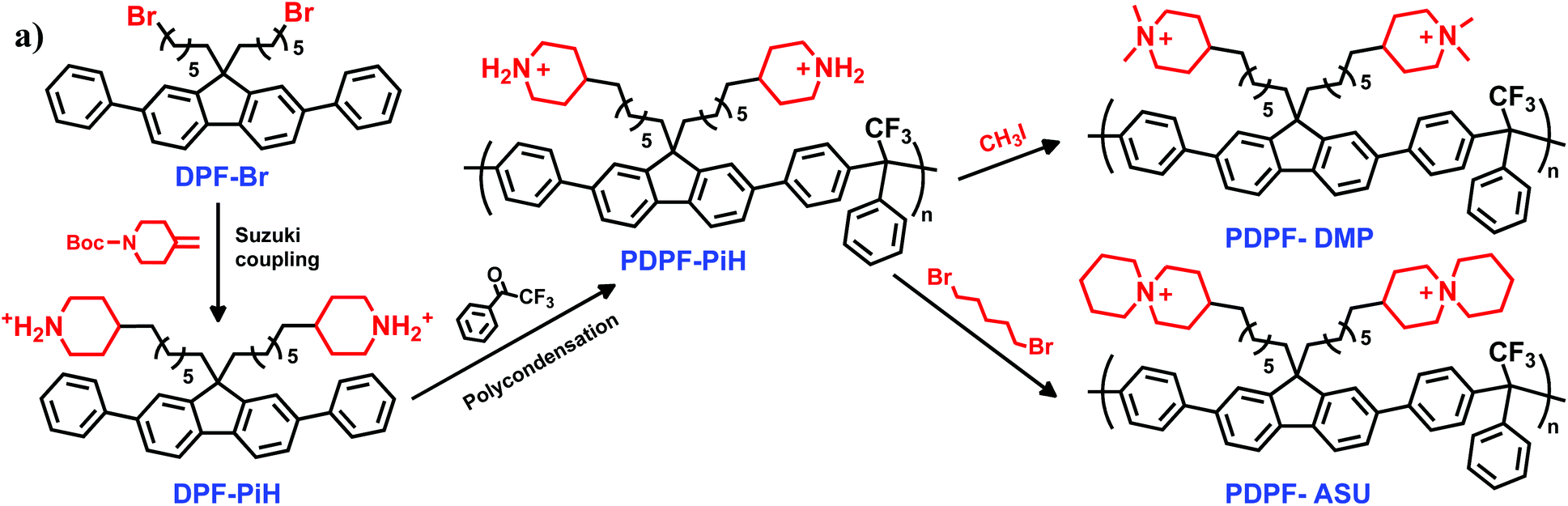
Durable cationic polymers (ionomers) and anion exchange membranes (AEMs) are critical materials for alkaline electrochemical energy technologies such as fuel cells, water electrolyzers, and advanced batteries.1–8 These devices present a promising alternative to acidic proton exchange membrane applications which require platinum group metal electrocatalysts. Potentially, alkaline devices can operate with lower cost catalysts (e.g., cobalt, silver, and nickel), benefit from faster oxygen reduction and oxygen evolution reactions compared with the acidic environment, and may exploit new electrochemical reactions.1,9–12 However, the development has been significantly hampered by a lack of AEMs that show high performance in combination with sufficient alkaline stability. Both the backbones and the cations of the polymers in AEMs are susceptible to nucleophilic attack by hydroxide ions. This leads to reduced mechanical strength and losses in ion-exchange capacity (IEC) and conductivity, and hence to irreversible losses in performance and lifetime of the devices.Polymer backbone degradation in AEMs has primarily been observed to occur via aryl ether bonds cleavage,13–18 motivating a recent change in research focus towards aryl-ether free backbones19 such as polynorbornenes,20–24 poly(arylene alkylene)s25–32 and polystyrenes.33–38 A more serious issue is the alkaline stability of the tethered cationic groups. These are generally ammonium cations that can degrade by a large number of different mechanisms, including nucleophilic substitutions at α-carbons, elimination of β-hydrogens (Hofmann elimination) and mechanisms unique to the specific structure and configuration of the cation.1,39–43 In a seminal work, Marino and Kreuer reported on the stability of different model ammonium cations under harsh alkaline conditions (6 M aq. NaOH, 160 °C).39 The most simple ammonium cation, tetramethylammonium, was found to be very stable with a half-life of t1/2 = 62 h, but cannot be tethered to polymers. Tetherable alkyltrimethyl- and tetraalkylammonium cations were much less stable because of the possibility of Hofmann elimination (e.g. hexyltrimethyl- and tetrapropylammonium with t1/2 = 32 and 7.2 h, respectively).39 In contrast, the alicyclic piperidine-based N,N-dimethylpiperidinium (DMP) and 6-azonia-spiro[5.5]undecane (ASU) cations were found to possess an exceptional stability with t1/2 = 87 and 110 h, respectively. The reason for their high stability is ascribed to the strain-free rings and the geometric constraint of the rings on the elimination and substitution transition states, which require unfavorable bond angles and lengths.39 These findings have motivated quite extensive research on AEMs functionalized with piperidine-based cations in recent years.44–50 However, when these cations are directly incorporated in stiff aromatic polymer backbones, the alkaline stability is significantly reduced in relation to that of the model compounds.27,28 This has been explained by a distortion of the alicyclic rings by the stiff backbones, which facilitates degradation by Hofmann elimination.27,28 Hence, in order to benefit from the inherent alkaline stability of the alicyclic cations, these cations must be tethered to the polymer backbone via flexible spacer chains that facilitate ring-relaxation and minimize ring-strain.
Another factor that has a significant effect on the alkaline stability of cationic groups in AEMs is the position at which they are attached to the polymer structure.39 Most studies on AEMs functionalized with piperidinium cations have been carried out on N-alkyl-N-methylpiperidinium (AMP) tethered to the backbone in the 1(N)-position via an alkyl chain, (Fig. 1).49,51,52 This functionalization is readily achieved in Menshutkin reactions of bromoalkylated polymers with N-methylpiperidine. Very recently, we have employed this approach to tether AMP to an aryl-ether free polyfluorene (PDPF) backbone.53 However, this molecular design opens up for Hofmann elimination in the linear alkyl tether. Alternatively, the tethering at the 3- or 4-position instead of the 1(N)-position places all the sensitive β-hydrogens in strain-free alicyclic rings, can be expected to significantly reduce Hofmann elimination.
 | ||
| Fig. 1 Cations tethered to polymers via alkyl spacers: (a) alkyltrimethylammonium (ATMA), (b) N-alkyl-N-methylpiperidinium (AMP), (c) 4-alkyl-DMP, and (d) 4-alkyl-ASU. Red circles indicate β-hydrogens with a varied sensitivity towards elimination reactions. | ||
In the present work, we have developed and explored a rational molecular design in which DMP and ASU cations, respectively, are tethered in the 4-positions of the respective rings to PDPF backbones via heptyl spacer chains (Scheme 1). Hence, we combine several beneficial molecular design features, including the use of an ether-free backbone and the attachment of piperidine-based cations on flexible spacers which allow full relaxation of the ring strain. The impact of this approach on the thermal and alkaline stability, as well as water uptake and ion conductivity, of the resulting AEMs was subsequently investigated.
 | ||
| Scheme 1 Synthetic pathway to PDPF-DMP and PDPF-ASU. | ||
2. Results and discussion
2.1. Polymer synthesis
Attaching piperidine-based cations in the 4-position is challenging due to the lack of straightforward synthetic pathways and commercially available building blocks. In the current work this was achieved by attaching 4-methylenepiperidine to a dibromoalkylated diphenylfluorene via Suzuki coupling to produce monomer DPF-PiH (Scheme 1). After modification of a published procedure,54 the synthesis was performed in three steps; hydroboration of N-Boc-4-methylene-piperidine with 9-BBN, Suzuki coupling with 9,9-bis(6-bromohexyl)-2,7-diphenyl-9H-fluorene (DPF-Br) and finally deprotection in aq. HCl. The key step here is the alkyl–alkyl Suzuki cross-coupling which is a rarely reported reaction, most probably because of the slow oxidative addition of alkyl halides to palladium and facile β-hydride elimination.54 In order to achieve an efficient Suzuki cross-coupling with DPF-Br, a catalyst system consisting of the bulky and electron-rich phosphine ligand PCy3, the catalyst Pd(OAc)2 and the base K3PO4·H2O was employed. The successful synthesis of DPF-PiH was confirmed by 1H NMR spectroscopy (Fig. 2a). As expected, the chemical shifts of the aromatic protons (h–m) and the methylene protons in the alkyl spacers (e, f, g) remained unchanged. Concurrently, the signals originating from the –CH2CH2Br and –CH2CH2Br protons were replaced by those of the methylene and methine protons of the piperidinium moieties (b, c, d). In addition, the amine protons (a) generated two signals above 8 ppm. Notably, the clear difference in chemical shifts between equatorial and axial protons at N-, α- and β-positions (a, b, c) indicated limited conformational change of the ring. | ||
| Fig. 2 1H NMR spectra of (a) DPF-PiH, (b) PDPF-PiH, (c) PDPF-DMP and (d) PDPF-ASU. | ||
DPF-PiH was subsequently employed in a Friedel–Crafts type polyhydroxyalkylation reaction with 2,2,2-trifluoroacetophenone (TFAp) to produce the precursor homopolymer PDPF-PiH (Scheme 1). Trifluoromethanesulfonic (TFSA) was used as a co-solvent together with dichloromethane to generate the superacidic medium necessary for the polycondensation.27,55,56 The use of the rather large DPF monomer enables the preparation of well-defined and rigid ether-free PDPF AEMs with the target IEC-range (2.0–2.2 mequiv. g−1) to reach high conductivity without excessive water uptake, thus avoiding the complications of copolymerization. In comparison with for example a backbone based on non-phenylated fluorene, the higher rigidity of the PDPF backbone facilitates film-formation and the mechanical strength of the AEM. Furthermore, DPF-Br is more reactive than the corresponding non-phenylated fluorene monomer in the Friedel–Crafts type polyhydroxyalkylations. In the present case, the high polymerization rate was indicated by the rapid increase in viscosity of the reaction mixture, despite the low temperature (0 °C). After only 1 h, the viscosity became too high for magnetic stirring and the reaction was stopped. The resulting polymer formed transparent and flexible membranes after casting from DMSO solution at 85 °C and the structure of the polymer was verified by 1H NMR spectroscopy (Fig. 2b). Moreover, PDPF-PiH displayed a high thermal stability and decomposed only above Td,95 = 403 °C (5% weight loss) as determined by thermogravimetric analysis (TGA).
Following our previously reported procedures,27,28,57 the DMP and ASU cations were introduced by quaternizing PDPF-PiH using methyl iodide and 1,5-dibromopentane, respectively, to produce the cationic polymers PDPF-DMP and PDPF-ASU (Scheme 1). In order to avoid potential branching/crosslinking reactions during the cyclo-quaternization in the synthesis of PDPF-ASU, a solution of PDPF-PiH was added drop-wise into a dilute solution of 1,5-dibromopentane. No sign of gelation or particle formation was noted throughout the reaction and the resulting PDPF-ASU had an excellent solubility in DMSO. The formation of both PDPF-DMP and PDPF-ASU from PDPF-PiH was confirmed by NMR spectroscopy. 1H NMR spectra of these samples (Fig. 2c and d) did not show any signal from protonated amine hydrogens (a), hence confirming the complete conversion of the secondary piperidine groups of DPF-PiH. The signals from the piperidinium ring (b, c, d) were shifted downfield, while the signals from the polymer backbone and spacer (e, f, g) remained the same. The two methyl groups in the DMP cation (o) gave rise to two singlets at ∼3.04 and 2.93 ppm (Fig. 2c). For the ASU cation, the signals originating from the methylene units of the pendant piperidine ring (p, q, r) appeared at lower chemical shift than the corresponding signals from the piperidine ring attached to the spacer (b, c, d) (Fig. 2d). Due to the restricted conformational change, the shifts of both the α- and β-protons in the pendant rings (p, q) split into two distinct signals in the same manner as the corresponding protons from the piperidine ring attached to the spacer (b, c).
2.2. Membrane characterization
 | ||
| Fig. 3 Photographic images showing folded AEMs cast from (a) PDPF-DMP and (b) PDPF-ASU. | ||
| AEM | IECBr [mequiv. g−1] | IECOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [mequiv. g−1] a [mequiv. g−1] |
WUc [wt%] | SWin-planec [%] |
SWthrough-planec [%] |
λ | σ [mS cm−1] |
T
d,95d [°C] |
|
|---|---|---|---|---|---|---|---|---|---|
| Theoreticala | Titratedb | ||||||||
| a Calculated from the chemical structure of the polymers. b Determined by Mohr's titration. c Measured at 80 °C in hydroxide form, under fully hydrated conditions (immersed). d Measured by TGA under N2 at 10 °C min−1. | |||||||||
| PDPF-DMP | 1.90 | 1.95 ± 0.03 | 2.15 | 111 | 30 | 30 | 29 | 124 | 273 |
| PDPF-ASU | 1.76 | 1.62 ± 0.05 | 1.98 | 45 | 15 | 15 | 12 | 76 | 332 |
Sufficient water uptake is crucial for the formation of a percolating hydrated phase domain, thus directly affects the hydroxide conductivity. After ion-exchange to the hydroxide form, the water uptake of PDPF-ASU and PDPF-DMP at 20 °C was 28 and 58 wt%, respectively (Fig. 4a, Table 1). At 80 °C the values increased to 45 and 111 wt%, respectively (Fig. 4a, Table 1). We have previously observed a significantly lower water uptake of different poly(arylene alkylene)-based AEMs functionalized with ASU cations compared with corresponding membranes carrying DMP cations.57 In the present case, this observation is probably a combined effect of the lower IEC and the more bulky ASU cations. The swelling (SW) is another important factor that affects the applicability of AEMs. The swelling of both PDPF-ASU and PDPF-DMP in the hydroxide form was seemingly isotropic in water. At room temperature, the in- and through-plane swelling were 8 and 10%, respectively, for PDPF-ASU. For PDPF-DMP, these values were 20 and 22%, respectively. At 80 °C, the in- and through-plane swelling were the same for each AEM and reached 15 and 30% for PDPF-ASU and PDPF-DMP, respectively (Table 1).
 | ||
| Fig. 4 (a) Water uptake of fully hydrated (immersed) AEMs in the hydroxide form as a function of temperature, (b) hydroxide conductivity of fully hydrated AEMs as a function of T−1 and (c) hydroxide conductivity of fully hydrated AEMs as a function of water uptake. | ||
As expected, the hydroxide conductivity of the AEMs in the fully hydrated (immersed) state increased with the temperature and water uptake (Fig. 4b and c, Table 1). At 20 and 80 °C, PDPF-ASU showed a conductivity (σ) of 31 and 76 mS cm−1, respectively. PDPF-DMP reached a very high conductivity, 53 and 124 mS cm−1 under the same conditions. Hence, the conductivity of PDPF-DMP was significantly higher than PDPF-ASU at 80 °C. The conductivity results are in good agreement with previous findings on AEMs with DMP and ASU cations directly attached to the poly(arylene alkylene)s.57 Beside the higher IEC and water uptake, the less bulky DMP cations might also facilitate ion clustering during casting, thus promoting phase separation and ion transport. The conductivity of PDPF-DMP was comparable or significantly higher than with that of AEMs with similar or higher IEC (Table 2),46–48,53,58 demonstrating the advantages of the present molecular design on the hydroxide conductivity of AEMs.
| AEM | IECa [mequiv. g−1] | WUb [wt%] | σ [mS cm−1] | Structure | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Backbone | Cation | Spacer | Attachment position | |||||
| a Calculated from the chemical structure of the polymers in hydroxide form. b Measured at 80 °C in hydroxide form under fully hydrated conditions (immersed). c Determined by acid–base titration. | ||||||||
| PDPF-DMP | 2.15 | 111 | 124 | PDPF-based | DMP | Alkyl | 4 | Present |
| PDPF-ASU | 1.98 | 45 | 76 | PDPF-based | ASU | Alkyl | 4 | Present |
| PDPF-TMA | 2.24 | 178 | 107 | PDPF-based | ATMA | Alkyl | 1(N) | 53 |
| PFF+ | 2.45 | 26 | 48 | Fluorene-based | ATMA | Alkyl | 1(N) | 58 |
| PBF2 | 2.19 | 160 | 86 | Fluorene-based | AMP | Alkyl | 1(N) | 47 |
| PBPipQ66Ap | 2.24 | 141 | 118 | Biphenyl-based | DMP | None | 4 | 59 |
| QPES | 1.89 | 20–25 | 59.8 | Polysulfone | DMP | None | 4 | 48 |
| PPO-DMP | 1.98c | — | 71.8 | Poly(phenylene oxide) | DMP | Triazolium | 4 | 46 |
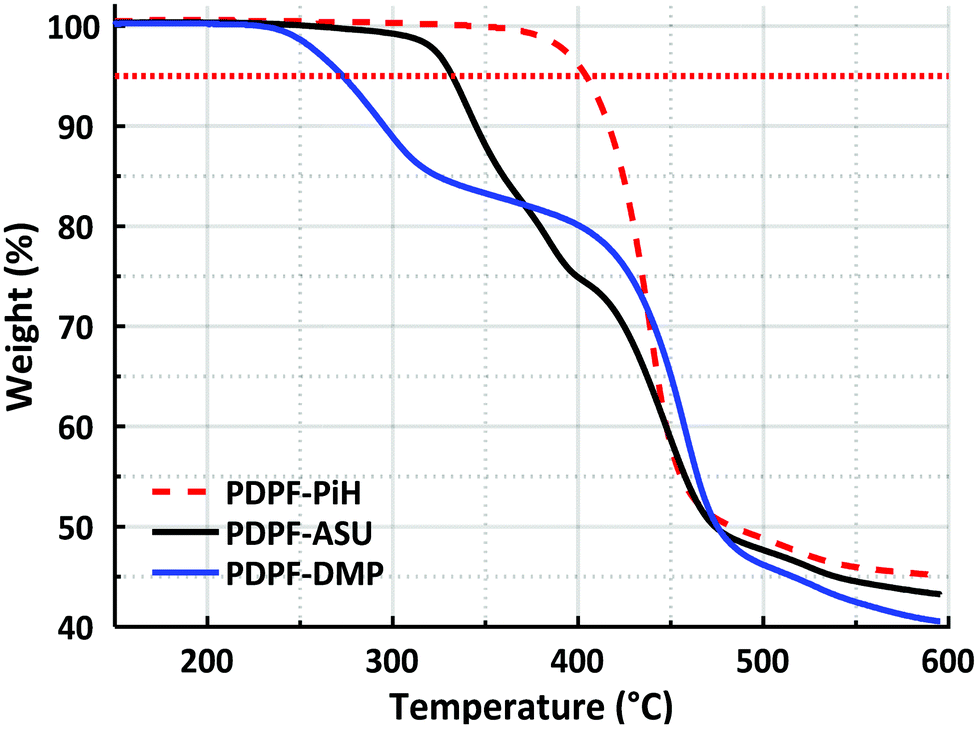 | ||
| Fig. 5 TGA profiles of PDPF-PiH, PDPF-ASU and PDPF-DMP (dotted horizontal line indicates the 95 wt% limit). | ||
 | ||
| Fig. 6 1H-NMR spectra of (a) PDPF-ASU and (b) PDPF-DMP before and after immersion in 2 M aq. NaOH solutions at 90 °C. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm (no data available for PDPF-ASU after 2400 h because of insolubility). | ||
In order to investigate the alkaline stability, the AEMs were immersed in 2, 5, 7 and 10 M aq. NaOH solutions at 90 °C for different periods of time, followed by 1H NMR analysis of any changes in molecular structure. These alkali concentrations correspond to λ ([H2O]/[OH−]) values of approximately 10.6, 7.4 and 5.0, respectively. All AEM samples remained transparent and were seemingly unaffected after the alkaline stability study. In addition, there was no detectable change in the aromatic region of the NMR spectra, confirming the high stability of the ether-free polymer backbone. In contrast, signs of cationic degradation was indicated by the appearance of new small signals outside the aromatic region (Fig. 6 and 7a). The alicyclic DMP and ASU cations degrade mainly via nucleophilic substitution and Hofmann elimination.39 Both degradation pathways lead to formation of tertiary amines, which were protonated upon addition of TFA and gave rise to NMR signals above 8 ppm (Fig. 6 and 7a). By comparing the intensity of these signals with that of the stable aromatic signals, the total ionic loss was estimated. Concurrently, degradation via Hofmann elimination resulted in the formation of alkene groups, which were readily detected by the appearance of distinct vinyl signals between 4.5 and 5.5 ppm. Integration of these signals gave the quantitative ionic loss caused by Hofmann elimination exclusively.
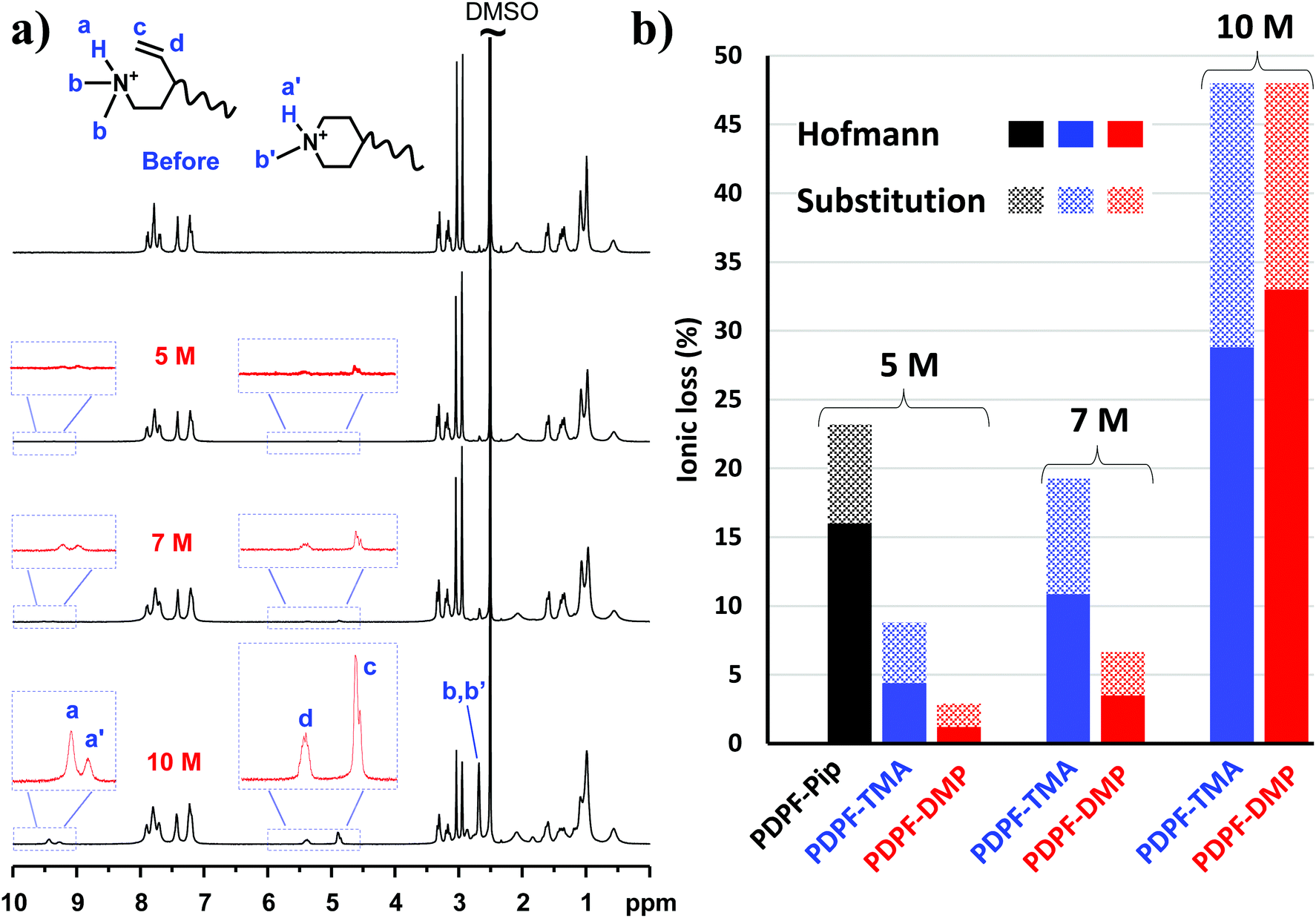 | ||
| Fig. 7 Alkaline stability of AEMs immersed in 5, 7 and 10 M aq. NaOH solutions during 168 h: (a) 1H NMR spectra of PDPF-DMP before and after immersion (TFA was added to shift the water signals originally at ∼3.3 ppm to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm) and (b) the ionic loss of PDPF-Pip, PDPF-TMA and PDPF-DMP via Hofmann elimination and substitution reactions, respectively. | ||
PDPF-ASU showed traces of degradation already after 720 h immersion in 2 M aq. NaOH (Fig. 6a), and the solubility of PDPF-ASU was found to decrease after further alkaline treatment. The solubility decreased both with the treatment time and the alkali concentration which prevented further studies of the alkaline stability of this AEM. The decrease in solubility was most probably due to crosslinking via the degradation products, e.g., through amino alcoholates formed in the ring-opening substitution of ASU. The alcoholates may then react with ASU rings via nucleophilic attack, resulting in ether-containing crosslinks.
PDPF-DMP had a higher alkaline stability compared to PDPF-ASU. After 720 h immersion of PDPF-DMP in aq. NaOH solution at 90 °C, no detectable change was observed in the 1H NMR spectra, suggesting excellent alkaline stability (Fig. 6b). In contrast to PDPF-ASU, PDPF-DMP remained completely soluble in DMSO after the alkaline treatment, suggesting different degradation mechanisms for the two cations. After 2400 h immersion in 2 M aq. NaOH, the total ionic loss of PDPF-DMP remained very low at 8.4%, of which a mere 2.0% was attributed to β-elimination (Fig. 6b, Table 3).
| Treatment | Ionic loss [%] | ||
|---|---|---|---|
| Hofmann elimination | Nucleophilic substitution | Total | |
| 2 M NaOH, 2400 h | 2.0 | 6.4 | 8.4 |
| 5 M NaOH, 168 h | 1.2 | 1.7 | 2.9 |
| 7 M NaOH, 168 h | 3.5 | 3.2 | 6.7 |
| 10 M NaOH, 168 h | 33 | 15 | 48 |
As expected, the rate of degradation increased with the concentration of alkali. Consequently, PDPF-DMP showed signs of degradation after 168 h immersion in 5, 7 and 10 M aq. NaOH solutions at 90 °C (Fig. 7a). Notably, the relative rates of the different degradation pathways varied with the alkaline concentration. With increasing NaOH concentration, the rate of Hofmann elimination accelerated much faster than nucleophilic substitution (Fig. 7b, Table 3). At 10 M, Hofmann elimination became the dominating degradation pathway instead of nucleophilic substitution that dominated at 2 M. Moreover, while PDPF-DMP remained quite stable in 5 M and 7 M aq. NaOH (<10% ionic loss), an increase of alkaline concentration from 7 to 10 M resulted in a sharp increase in the total ionic loss, probably due to increased reactivity of the OH− at this very low λ-value (Fig. 7b, Table 3).60
The positive effect on the alkaline stability by attaching via the 4-position is apparent when comparing the alkaline stability of PDPF-DMP with corresponding AEMs functionalized with AMP (designated PDPF-Pip) and ATMA (designated PDPF-TMA, cations attached at 1(N)-position, Fig. 1).53 The alkaline stability of the two latter AEMs have previously been investigated under conditions similar to those used in the present study (Fig. 7b). After 168 h immersion in 5 M aq. NaOH, the total ionic loss in PDPF-DMP was merely 2.9% (Table 3), nearly 5-fold less than the ionic loss in PDPF-Pip.53 While the loss via nucleophilic substitution was similar for both cations, the Hofmann elimination of the former was reduced by 92%. Furthermore, PDPF-DMP also showed higher alkaline stability than the corresponding AEM functionalized with the benchmark ATMA cation (PDPF-TMA).53 While both polymers showed a similar total ionic loss in 10 M aq. NaOH, PDPF-DMP had ∼3 times less ionic loss than the latter in 5 and 7 M aq. NaOH solutions (Fig. 7b).
3. Conclusions
We have successfully employed alkyl–alkyl Suzuki cross-coupling to attach piperidine-based DMP and ASU cations at the 4-position to ether-free polyfluorenes via alkyl spacers. This approach allowed a molecular design in which all the sensitive β-hydrogens were placed in strain-free alicyclic rings able to fully relax by the flexibility of the spacers. Compared with AEMs conventionally tethered with AMP cations via the 1(N)-position, corresponding AEMs with DMP cations attached via the 4-position showed significantly improved thermal and alkaline stability, while retaining high hydroxide conductivity. After immersion in concentrated (5 M) alkali at 90 °C, the total ionic loss was 80% lower for DMP compared with AMP. While the loss by substitution was approximately the same for both cations, the loss by β-elimination was an order of magnitude lower for DMP compared to AMP. Concurrently, the total ionic loss of DMP was 67% lower than of ATMA. However, in highly concentrated alkali (10 M), the ionic loss of DMP was in the same range as that of ATMA. The findings of the study demonstrate that AEMs with high hydroxide conductivity and substantially improved thermal and alkaline stability are obtained by employing a molecular design where piperidine-based cations are tethered to ether-free aromatic polymers via the 4-position instead of the 1(N)-position.4. Experimental
1-N-Boc-4-methylene-piperidine (96%, Fluorochem), 9-borabicyclo[3.3.1]nonane (9BBN, 0.5 M in THF, Sigma-Aldrich), potassium phosphate (K3PO4, ≥99%, Sigma-Aldrich tetrakis(triphenylphosphine)palladium(0) (99%, Sigma-Aldrich), tricyclohexylphosphine (PCy3, reagent grade, Sigma-Aldrich), HCl (37% in water, VWR), 2,2,2-trifluoroacetophenone (TFAp, 99%, Sigma-Aldrich), 1,1,1-trifluoroacetic acid (TFA, 99%, Acros), triflic acid (TFSA, 99%, Acros), 1,5-dibromopentane (97%, Aldrich), N,N-diisopropylethylamine (DIPEA, ≥99%, Sigma-Aldrich), methyliodide (99%, Sigma-Aldrich), K2CO3 (99%, Sigma-Aldrich), toluene (reagent grade, VWR), isopropanol (IPA, reagent grade, VWR), diethyl ether (Et2O, reagent grade, VWR), N-methyl-2-pyrrolidone (NMP, reagent grade, Acros), dimethyl sulfoxide (DMSO, reagent grade, VWR), ethyl acetate (EtOAc, reagent grade, VWR), heptane (reagent grade, VWR), NaBr (99%, Sigma-Aldrich), ethanol (EtOH, 99.5%, Solveco), NaOH (99% pellets, VWR), KOH (99% pellets, VWR), CDCl3 (99.8 atom% D, Sigma-Aldrich) and DMSO-d6 (99.5 atom% D, Sigma-Aldrich) were all used as received. Dichloromethane was dried using an MBraun dry solvent dispenser system MB-SPS 800. 2,7-Diphenyl-9,9-bis(6-bromohexyl)-fluorene (DPF-Br) was synthesized following a previously published procedure.534.1. Monomer and polymer synthesis
:heptane mixtures (0:100 to 30:70) as eluent to yield a viscous oil. The oil was then stirred with concentrated aq. HCl (7 ml) in a 25 ml round flask overnight. The mixture was evaporated until dryness under reduced pressure and the residue was stirred vigorously in acetone at 0 °C, then filtrated to yield DPF-PiH as a white powder.
The AEM in the in hydroxide form was prepared from bromide form by ion-exchange in 1 M aq. NaOH solution at RT for at least 120 h. After that the membrane was washed thoroughly with degassed deionized water and stored (if necessary) in degassed deionized water under nitrogen for less than 72 h before the measurement.
4.2. Characterization
 | (1) |
 | (2) |
The intrinsic viscosity ([η]) was calculated to be 0.19 dL g−1 by taking the average of the intersections of the linear regressions of ηinh and ηred with the y-axis.
 | (3) |
| mdry = mdry,Br × (1 − 0.0629 × IECBR). | (4) |
Afterward, the AEMs were ion-exchanged to hydroxide form. The weight, length and thickness of the hydrated membranes (mwet, lwet, twet) were obtained after equilibration in DI water at 20, 40, 60 and 80 °C. Their water uptake was calculated as:
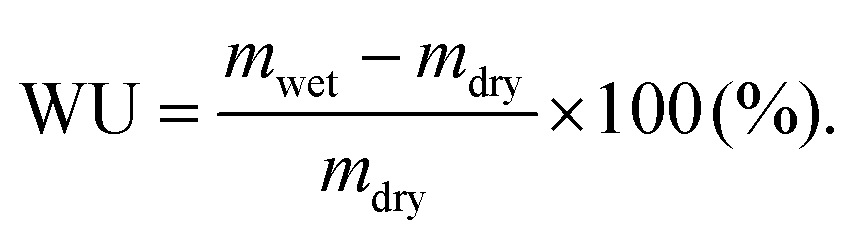 | (5) |
The membrane swelling, i.e., the increase in length and thickness of the membranes, was calculated in similar way to the water uptake. Hence, the in-plane swelling was the average increase in length of the four edges of the quadrangular membranes, and the through-plane swelling was the average increase in thickness at the four corners.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swedish Energy Agency (grants 45057-1 and 37806-3), the Swedish Research Council (grants 45397-1 and 2015-04820) and the Swedish Foundation for Strategic Research, SSF, (grant EM16-0060) for financial support.References
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727–1735 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CAS.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- S. D. Poynton, J. P. Kizewski, R. C. T. Slade and J. R. Varcoe, Solid State Ionics, 2010, 181, 219–222 CrossRef CAS.
- S. Gu, W. Sheng, R. Cai, S. M. Alia, S. Song, K. O. Jensen and Y. Yan, Chem. Commun., 2013, 49, 131–133 RSC.
- T. S. Olson, S. Pylypenko, P. Atanassov, K. Asazawa, K. Yamada and H. Tanaka, J. Phys. Chem. C, 2010, 114, 5049–5059 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- S. Miyanishi and T. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18, 12009–12023 RSC.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- Y.-K. Choe, C. Fujimoto, K.-S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615–F621 CrossRef CAS.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS.
- C. Wang, B. Mo, Z. He, Q. Shao, D. Pan, E. Wujick, J. Guo, X. Xie, X. Xie and Z. Guo, J. Membr. Sci., 2018, 556, 118–125 CrossRef CAS.
- C. Wang, B. Mo, Z. He, X. Xie, C. X. Zhao, L. Zhang, Q. Shao, X. Guo, E. K. Wujcik and Z. Guo, Polymer, 2018, 138, 363–368 CrossRef CAS.
- W. Chen, M. Mandal, G. Huang, X. Wu, G. He and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2458–2468 CrossRef CAS.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- M. Mandal, G. Huang and P. A. Kohl, J. Membr. Sci., 2019, 570–571, 394–402 CrossRef CAS.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- M. Ozawa, T. Kimura, K. Otsuji, R. Akiyama, J. Miyake, M. Uchida, J. Inukai and K. Miyatake, ACS Omega, 2018, 3, 16143–16149 CrossRef CAS.
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. N. Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- S. K. Tuli, A. L. Roy, R. A. Elgammal, T. A. Zawodzinski and T. Fujiwara, Polym. Int., 2018, 67, 1302–1312 CrossRef CAS.
- J. Ponce-González, D. K. Whelligan, L. Wang, R. Bance-Soualhi, Y. Wang, Y. Peng, H. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724–3735 Search PubMed.
- J. Hao, X. Gao, Y. Jiang, H. Zhang, J. Luo, Z. Shao and B. Yi, J. Membr. Sci., 2018, 551, 66–75 CAS.
- Z. Wang, J. Parrondo and V. Ramani, J. Electrochem. Soc., 2017, 164, F1216–F1225 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2020, 53, 4722–4732 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49–59 CrossRef CAS.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS.
- G. Cerichelli, G. Illuminati and C. Lillocci, J. Org. Chem., 1980, 45, 3952–3957 CrossRef CAS.
- A. C. Cope and A. S. Mehta, J. Am. Chem. Soc., 1963, 85, 1949–1952 CAS.
- N. Chen, C. Long, Y. Li, C. Lu and H. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 15720–15732 CAS.
- X. Chu, L. Liu, Y. Huang, M. D. Guiver and N. Li, J. Membr. Sci., 2019, 578, 239–250 CrossRef CAS.
- H. J. Park, X. Chu, S. P. Kim, D. Choi, J. W. Jung, J. Woo, S. Y. Baek, S. J. Yoo, Y.-C. Chung, J. G. Seong, S. Y. Lee, N. Li and Y. M. Lee, J. Membr. Sci., 2020, 608, 118183 CrossRef CAS.
- R. Ren, S. Zhang, H. A. Miller, F. Vizza, J. R. Varcoe and Q. He, ACS Appl. Energy Mater., 2019, 2, 4576–4581 CrossRef CAS.
- F. Wang, B. Xue, S. Zhou, J. Zheng, S. Li, S. Zhang and T. A. Sherazi, J. Membr. Sci., 2019, 591, 117334 CrossRef.
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 RSC.
- M. Niu, C. Zhang, G. He, F. Zhang and X. Wu, Int. J. Hydrogen Energy, 2019, 44, 15482–15493 CrossRef CAS.
- A. Allushi, T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 27164–27174 RSC.
- M. R. Netherton, C. Dai, K. Neuschütz and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 10099–10100 CrossRef CAS.
- D. A. Klumpp, M. Garza, A. Jones and S. Mendoza, J. Org. Chem., 1999, 64, 6702–6705 CrossRef CAS.
- M. J. O'Connor, K. N. Boblak, A. D. Spitzer, P. A. Gucciardo, A. M. Baumann, J. W. Peter, C. Y. Chen, R. Peter, A. A. Mitton and D. A. Klumpp, Tetrahedron Lett., 2010, 51, 4984–4987 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 15895–15906 RSC.
- W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CAS.
| This journal is © The Royal Society of Chemistry 2020 |