
Recent advances in the development and applications of nonconventional luminescent polymers
Kamal
Bauri†
a,
Biswajit
Saha†
b,
Arnab
Banerjee
b and
Priyadarsi
De
 *b
*b
aDepartment of Chemistry, Raghunathpur College, Raghunathpur - 723133, Purulia, West Bengal, India
bPolymer Research Centre and Centre for Advanced Functional Materials, Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur - 741246, Nadia, West Bengal, India. E-mail: p_de@iiserkol.ac.in
First published on 26th October 2020
Abstract
Traditional luminescent polymers typically contain extended π–π conjugated structures that suffer from associated aggregation-caused quenching (ACQ) and high cytotoxicity issues. Therefore, there is a huge demand for stand-alone luminescent polymers that can circumvent these drawbacks and can be employed in diverse applications. In view of that, nonconventional luminescent polymers (NLPs) are being developed and studied extensively for their distinctive characteristics and potential applications in wide-ranging areas, and for an in-depth understanding of photophysical processes in science. These polymers do not contain classical chromophores, but often involve isolated benzene rings or only electron-rich moieties (e.g. amine, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –OH, ether, imide, and heteroatoms N, O, P, S, etc.). This review article highlights the recent progress in the field of NLPs, including their synthesis, underlying emission mechanisms, and applications in materials and biological sciences. Besides, it is identified to be the foremost review that extensively emphasizes on both the fluorescence and phosphorescence features of NLPs. Finally, some prospects have been suggested for further exploration in this flourishing field of research.
O, –OH, ether, imide, and heteroatoms N, O, P, S, etc.). This review article highlights the recent progress in the field of NLPs, including their synthesis, underlying emission mechanisms, and applications in materials and biological sciences. Besides, it is identified to be the foremost review that extensively emphasizes on both the fluorescence and phosphorescence features of NLPs. Finally, some prospects have been suggested for further exploration in this flourishing field of research.
1. Introduction
Conventional luminescent polymers are classified as polymers integrated with large π-aromatic building blocks. On account of their strong emission properties, these materials have been the center of great attention for a variety of applications in organic lasers,1,2 organic light-emitting diodes (OLEDs),3,4 solar cells,5 sensors,6,7 bioimaging,8etc. Despite their wide utility, complex synthesis and toxicity of large conjugated systems, poor miscibility in water medium and low resistance to photobleaching restrict further improvement and applications of such luminogens. Additionally, they are eminently luminescent in dilute solutions but exhibit weaker/no emission in their concentrated or aggregated state due to the aggregation-caused quenching (ACQ) effect.9 The aforesaid bottlenecks have prompted researchers to contrive a new class of photoluminescent materials devoid of large π–π conjugated structures, referred to as nonconventional luminescent polymers (NLPs).10 This new category of luminescent polymers often consists of isolated benzene rings or involves different subgroups (–OH, –NH2, CC, and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and electron-rich heteroatoms (N, O, P, and S). Unlike traditional luminogens, their emission phenomenon shows certain unique characteristics including the following: they (i) do not require any excitation of delocalized electrons and (ii) act as collective molecular entities rather than a single or independent unit leading to substantial emission in their concentrated/aggregated states.11 Moreover, the advantages of these photoluminescent polymers over conventional ones are easy preparation, low cost, environmental friendliness, good processability, excellent biocompatibility, and low toxicity or even complete non-toxicity, thereby enabling them to be molded into potential materials for desired applications.12,13 Because of such intriguing properties, hitherto diverse luminogens have been demonstrated aiming at the identification of emission mechanisms, and unfortunately, it remains disputed. Clustering-triggered emission (CTE) and/or aggregation-induced emission (AIE) have been proposed to rationalize the emission properties of these nonconventional luminogens. Nevertheless, the emergence of such materials in the areas of luminescent polymers has led to immense leap-frogging from the perspective of applications based on their inexplicable luminescence properties. Eventually, the past decade realization of rational synthetic strategies have offered numerous nonconventional luminescent polymers and actuated their implementation in a myriad of applications namely light-emitting diodes (LEDs), light-harvesting systems, chemical sensing, drug delivery, intracellular temperature measurements, cell imaging, ink-free anti-counterfeiting printing, data encryption, and so on. Broadly, the majority of these luminescent materials have been found to be phosphorescence inactive due to the lack of a higher fluorescence lifetime until recently. Now, researchers have witnessed not only the cryogenic long-lived phosphorescence but also room temperature phosphorescence (RTP) from these unorthodox luminogens. Certainly, this fact further lightens up their importance, and thus the catalog of such materials is ever-escalating.
N) and electron-rich heteroatoms (N, O, P, and S). Unlike traditional luminogens, their emission phenomenon shows certain unique characteristics including the following: they (i) do not require any excitation of delocalized electrons and (ii) act as collective molecular entities rather than a single or independent unit leading to substantial emission in their concentrated/aggregated states.11 Moreover, the advantages of these photoluminescent polymers over conventional ones are easy preparation, low cost, environmental friendliness, good processability, excellent biocompatibility, and low toxicity or even complete non-toxicity, thereby enabling them to be molded into potential materials for desired applications.12,13 Because of such intriguing properties, hitherto diverse luminogens have been demonstrated aiming at the identification of emission mechanisms, and unfortunately, it remains disputed. Clustering-triggered emission (CTE) and/or aggregation-induced emission (AIE) have been proposed to rationalize the emission properties of these nonconventional luminogens. Nevertheless, the emergence of such materials in the areas of luminescent polymers has led to immense leap-frogging from the perspective of applications based on their inexplicable luminescence properties. Eventually, the past decade realization of rational synthetic strategies have offered numerous nonconventional luminescent polymers and actuated their implementation in a myriad of applications namely light-emitting diodes (LEDs), light-harvesting systems, chemical sensing, drug delivery, intracellular temperature measurements, cell imaging, ink-free anti-counterfeiting printing, data encryption, and so on. Broadly, the majority of these luminescent materials have been found to be phosphorescence inactive due to the lack of a higher fluorescence lifetime until recently. Now, researchers have witnessed not only the cryogenic long-lived phosphorescence but also room temperature phosphorescence (RTP) from these unorthodox luminogens. Certainly, this fact further lightens up their importance, and thus the catalog of such materials is ever-escalating.
To date, limited reviews have amply outlined and discussed the synthesis and properties of NLPs. In 2016, Yuan et al.10 reported the progress in the development of nonconventional macromolecular luminogens and proposed a clustering-triggered emission mechanism to rationalize the photophysical behavior of most of the systems, based on the existing literature focused at that time. A year later, Tomalia et al.11 provided a comprehensive overview of the history and evolution of non-traditional intrinsic luminescence (NTIL) in the form of dendrimers, small/macromolecular architectures, elemental compositions and assemblies. They also illustrated the mechanism of NTIL phenomena and handful of applications of dendrimers/hyperbranched polymers. Very recently, Tang and co-workers14 highlighted the intrinsic luminescence behavior of metal clusters, natural and synthetic polymers, and small molecules.
In the last few years, there has been an astonishing evolution of nonconventional polymers featuring afterglow, dual-emission (fluorescence and phosphorescence) behaviors, and RTP, and their various applications have been explored at a rapid pace. Yet, there is no such review that summarizes these fascinating aspects of such materials in a single article. While advances in this field have been continuing, it should be noted that there are challenges in understanding the exact emission mechanism. But as the nonconventional fluorescent polymer library is getting enriched day by day, we think that giving an overview of the developments in the current field reported to date will help the readers think in a more logical way about the underlying mechanism of this inexplicable emission behavior. Our objectives in this review are therefore to assemble state-of-the-art nonconventional luminescent materials and concurrently highlight the associated significant photophysical characteristics and advanced applications in a more systematic and comprehensive way (Fig. 1).15 We firmly believe that by doing so, we will provide thorough and unambiguous insight into each class of nonconventional luminescent polymers to both experienced and new researchers in the scientific world. Finally, several remaining challenges and some promising future directions have been suggested.
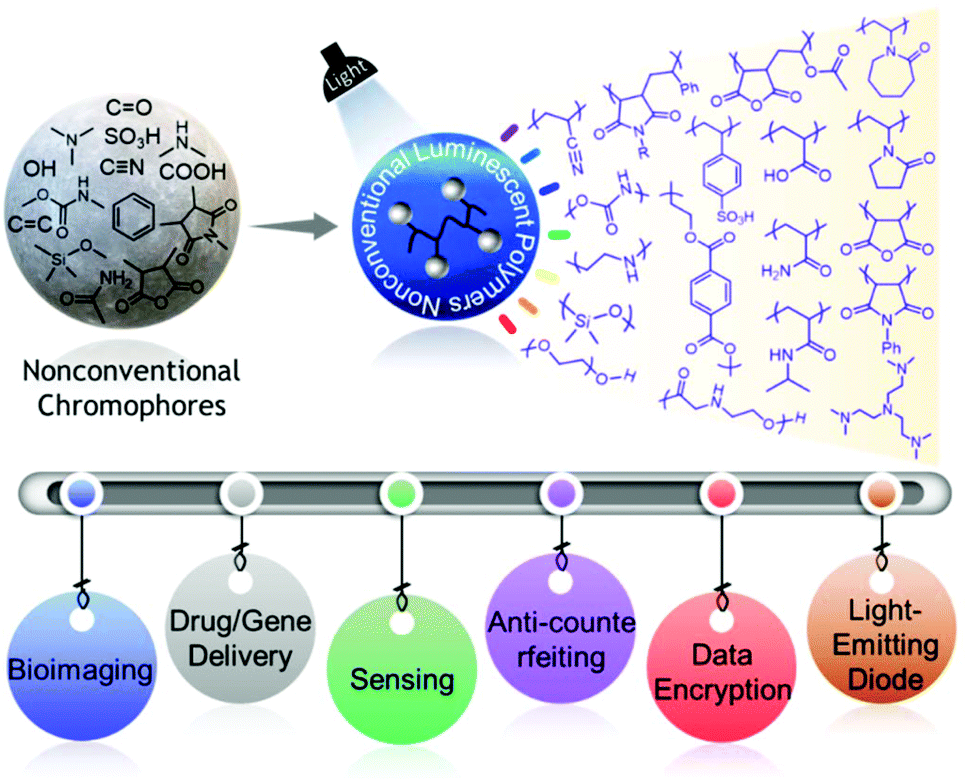 | ||
| Fig. 1 Schematic representation demonstrating different classes of NLPs fabricated from nonconventional chromophores and their applications. | ||
2. Structural diversity of nonconventional luminescent polymers
2.1. Biological macromolecules
In 2013, Tang and coworkers observed the intriguing bright blue emission from common rice upon irradiation with 365 nm UV light. To gain a clear idea about the mechanism of this unexpected autofluorescence, the photophysical properties of the major components of rice, namely starch and other natural products, were further examined. At room temperature, an aqueous solution of starch was non-emissive but after cooling with liquid nitrogen (77 K) it became highly emissive. Moreover, cellulose and the globular protein bovine serum albumin (BSA) exhibited similar emission behavior to starch both in the solution and solid states. As all the three natural products did not show any fluorescence in solution and amorphous states but exhibited strong emission in the crystalline state, it was obvious for the authors to conclude that crystallization is helpful in such unusual emission. Other natural macromolecules viz. chitosan, dextran, and glycogen, and some small sugar molecules such as glucose, xylose and galactose were also found to display similar emission phenomena. In fact, clustering of electron-rich groups with lone pair electrons was believed to be responsible for such emission and was termed ‘clustering-triggered emission’ (CTE).16In another study, the photophysical properties of polyhydroxy polymers, including microcrystalline cellulose (MCC) and its derivatives, namely hydroxypropyl cellulose (HPC), cellulose acetate (CA) and 2-hydroxyethyl cellulose (HEC), were investigated and compared. Except for the CA, all three polymers HPC, HEC and MCC have shown bright emissions with distinct RTP. Such intrinsic emission could be ascribed to the CTE and conformation rigidification.17
Yuan et al. unveiled the clustering-triggered emission and persistent room-temperature phosphorescence (p-RTP) of sodium alginate (SA), which is a naturally occurring anionic polysaccharide composed of guluronic acid (G) and mannuronic acid (M). Dilute solutions of SA were virtually non-emissive even after cooling at 77 K but they became highly fluorescent in concentrated solutions, films and solid powders. Unlike pure organic luminophores, amorphous SA solids possess the p-RTP feature. Cross-linking with Ca2+ boosted the film emission because of the existence of strong chain–chain association and junction zone formation between G residues and Ca2+. Rheological measurements proved that the entanglement of chains and their overlap in concentrated counterparts are invariant with the enhanced emissions, thus certifying the CTE mechanism. Moreover, SA was utilized for endosome specific intracellular imaging of HeLa cells, which has a great significance in biomedical research and clinical diagnostic applications.18
Amino acids are the constituent components of peptides and proteins.19 Earlier it was generally believed that protein emission occurred primarily because of three aromatic amino acids, namely phenylalanine, tyrosine, and tryptophan. But, Ye et al. showed that oligopeptides and polypeptides based on nonaromatic amino acids emitted fluorescence in the solid state.20 It was observed that those oligopeptides adopted ordered conformations such as β-sheets, α-helixes and turns stabilized through inter/intramolecular hydrogen bonds. It was speculated that these abundant hydrogen bonds bring the highly polarized amide group into close proximity, leading to the through-space interaction of amide groups’ electrons resembling those of the aromatic structure based on the through-space π electron interaction. In 2018, Zhang and coworkers found that peptides could not only emit but also their basic building blocks aliphatic amino acids could generate noticeable visible light emission in concentrated solutions, and particularly in the solid state.21 In addition to prompt fluorescence, persistent RTP was observed in powders of poly(amino acids) of ε-poly-L-lysine (ε-PLL) after ceasing UV illumination. In concentrated solutions, amino acid molecules may form diverse nanoaggregates approaching each other with the aid of intermolecular hydrogen bonding interactions; however, in dilute solutions they exist as molecularly dissolved individuals. Furthermore, NH2, CO, and OH subunits afforded through space electronic communications among different π and n (lone pair) electrons, resulting in electron cloud overlap and thus extended electron delocalization together with simultaneously rigidified conformations. These clustered chromospheres are responsible for the unexpected emission.
Recently, Yuan and coworkers reinvestigated the photoluminescence of natural protein taking BSA as the model protein under different conditions (Fig. 2A). Like polysaccharide SA, BSA showed AIE and concentration enhanced emission characteristics. Intriguingly, BSA exhibited distinct p-RTP in the form of amorphous powder or a tablet form under vacuum even under ambient conditions (Fig. 2B). This fact may be ascribed to the conformation rigidification caused by the cluster formation among the peptide backbone and pendant groups, which resulted in through-space electronic communications. Some other natural proteins, notably human serum albumin (HAS) and ovalbumin (OVA), showed similar emission characteristics to BSA.22
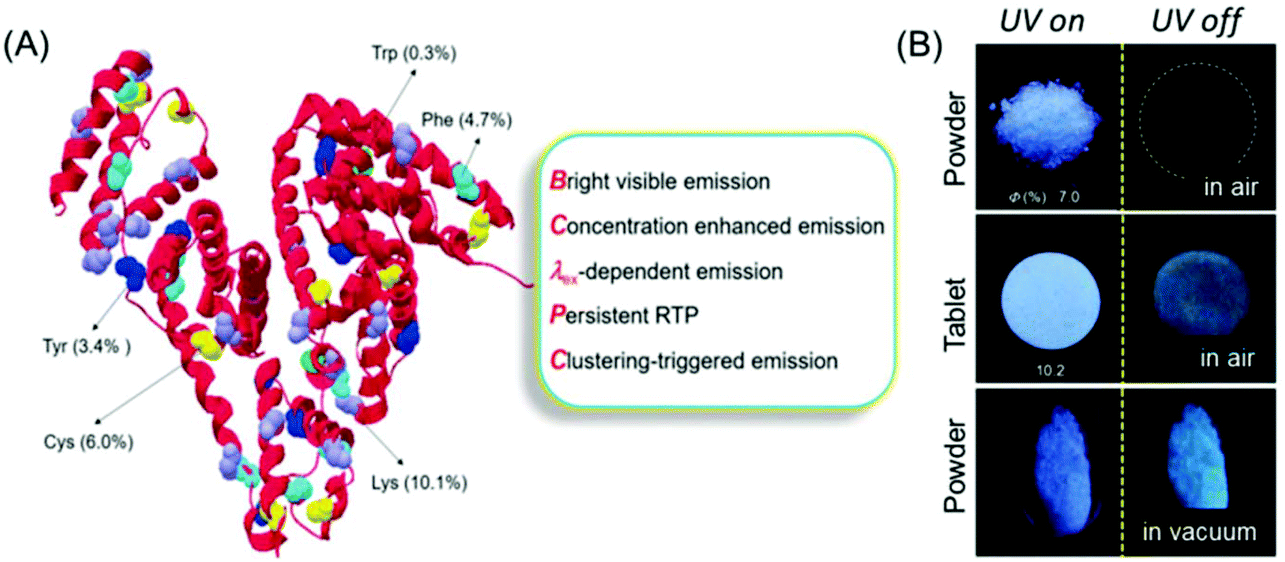 | ||
| Fig. 2 (A) BSA structure with nonaromatic (purple and yellow) and aromatic (green, cyan and blue) residues. (B) Photographic images of the solid powder and tablet of BSA taken under 312 nm UV excitation in air or under vacuum. Reprinted with permission from ref. 22. Copyright (2019), WILEY-VCH. | ||
2.2. Synthetic macromolecules
In 2009, Pan's research group synthesized a novel disulfide-functionalized biodegradable HPAMAM by the Michael addition polymerization of N,N′-cystaminebisacrylamide (CBA) and 1-(2-aminoethyl) piperazine (AEPZ) (Fig. 3A). The resultant HPAMAMs displayed an absorption band at 360 nm in the UV-Vis spectrum and excitation wavelength-dependent bright wide-band fluorescence (Fig. 3B). In contrast, an aqueous solution of the linear PAMAMs did not display the UV-Vis absorption band, and only a weak emission was found. It was believed that the tertiary nitrogen with lone pair electrons inside the hyperbranched structure acted as the emissive species. This was verified by converting the tertiary amine into a quaternary ammonium group upon the reaction of the polymers with CH3I. The absorption band at 360 nm disappeared almost completely, and a drastic decrease in the photoluminescence intensity at around 460 nm was observed in the case of quaternized HPAMAM. Such disulfide-functionalized polymers were easily cleaved under mild conditions by several stimulus reagents, such as 2-mercaptoethanol or glutathione via disulfide/thiol chemistry.24 Chu et al. demonstrated the use of intrinsically fluorescent PAMAM dendrimers as an efficient nonviral gene carrier that could perform multiple functions such as bioimaging, delivery and transfection at the same time. To this end, NH2-terminated generation 4 PAMAM dendrimers were readily treated with an oxidizing agent to yield blue fluorescence with a moderate quantum yield (QY). Three negatively charged antisense oligonucleotides NGF2, NGF1 and (AS-ODN) p75 were electrostatically associated with the positively charged PAMAM dendrimer to produce fluorescent complexes at various nitrogen-to-phosphorus (N/P) ratios. Similar to the hyperbranched polyethyleneimine-mediated gene transfection, these complexes were also found to be capable of knocking down the specific protein expression to a certain level, confirmed by western blot analysis. The intrinsic fluorescence properties of PAMAM dendrimers enabled direct analysis of cellular internalization behavior by fluorescence microscopy and flow cytometry.25
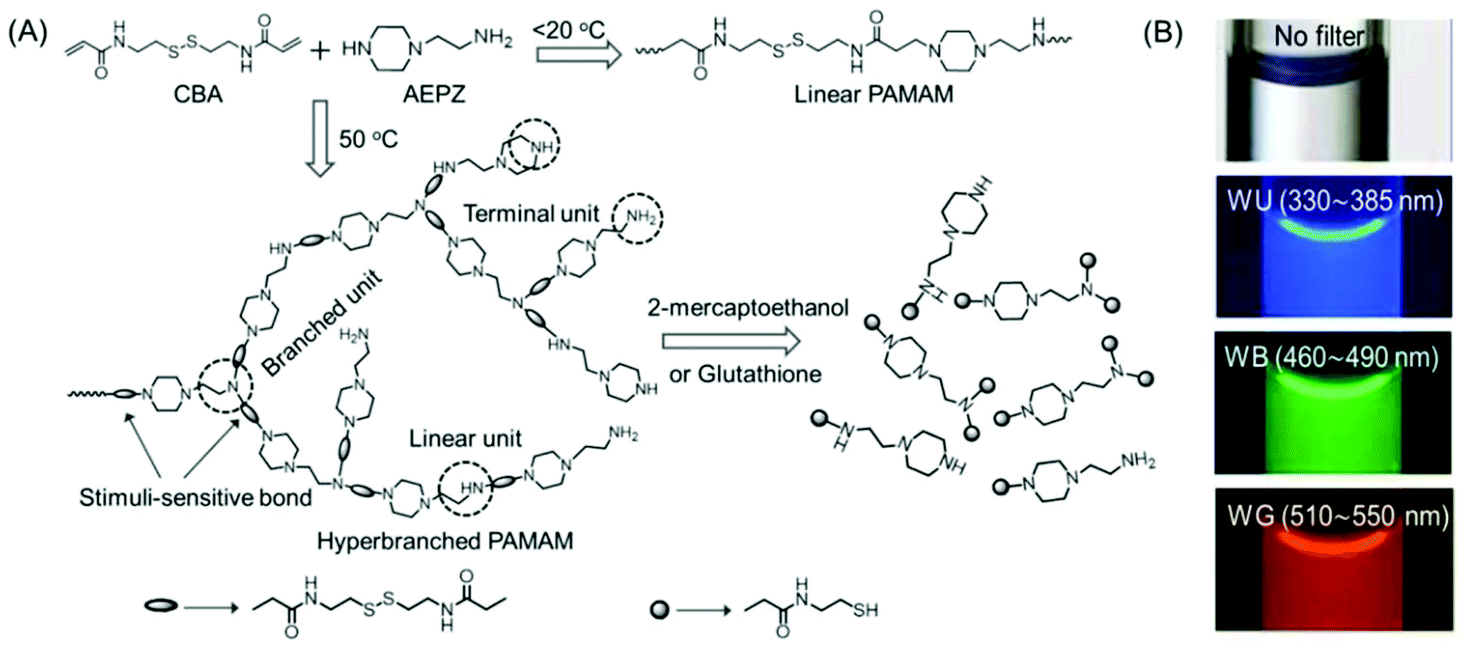 | ||
| Fig. 3 (A) Synthetic strategy adopted for the preparation of linear and hyperbranched PAMAMs, and stimuli-induced degradation of the hyperbranched analog. (B) Fluorescence microscopy images of aqueous solutions of hyperbranched PMAMAMs in a capillary acquired with 40× magnification. Reprinted with permission from ref. 24. Copyright (2009), WILEY-VCH. | ||
In another report, biodegradable less toxic β-cyclodextrin (β-CD) was incorporated into HPAMAM by a simple and efficient one-step Michael addition copolymerization reaction to improve biocompatibility and enhance photoluminescence. The amplified fluorescence of the resultant β-CD containing HPAMAM (HPAMAM-CD) was assumed to be generated from the restricted rotational motion of terminal chains due to the presence of large β-CD units, which suppressed collisional relaxation and a self-quenching phenomenon. The HPAMAM-CD could condense plasmid DNA (pDNA) very efficiently. The intrinsic luminescence of HPAMAM-CD aided in the study of the cellular uptake and gene transfection processes of the complexes through confocal laser scanning microscopy (CLSM) and flow cytometry, without any additional fluorescence labeling.26
Wang and coworkers developed a novel conventional chromophore-free photoluminescent supramolecular hyperbranched polymer (SHP) via inclusion complexation between diethylenetriamine (guest) and α-cyclodextrin (host). The SHP displayed excitation wavelength-dependent wide-band fluorescence. The inclusion complex prevented the chain movement and increased the rigidity of the SHP, which eventually suppressed the non-radiative decay and amplified the fluorescence.27
A series of photoluminescent HPAMAMs with dual thermal and pH sensitivity, and reducible degradability were successfully prepared by the Michael addition mediated copolymerization of tris(2-aminoethyl) amine and two bisacrylamide monomers CBA and N,N′-hexamethylenebisacrylamide, and the subsequent modification with isobutyric anhydride at room temperature. The fluorescence intensity was enhanced by lowering the pH and oxidizing the polymer by air. The tertiary amine oxide was assumed to be the fluorescent species.28
Shi and coworkers reported the synthesis of a label-free green fluorescent and biocompatible PAMAM dendrimer derivative via surface modification of pristine PAMAM with acetaldehyde for traceable and controlled drug delivery. Further PEGylation enables the fluorescent PAMAM to demonstrate excellent intracellular tracking in human melanoma SKMEL28 cells. The encapsulation of a model anticancer drug, doxorubicin (DOX), into the new nanocarriers helps this nanocomposite achieve higher drug loading capability and better therapeutic efficiency compared to the hydrophobic DOX and the original DOX loaded PAMAM nanocomposite.29
Hao's research group unveiled HPAMAMs as novel mechano-responsive fluorescent polymers where the luminescence intensity of HPAMAM films increased upon stretching the sample and the fluorescence intensity quickly returned to the nearly original value automatically once the strain was relaxed. HPAMAMs exhibited sensitivity for the entire strain range from 0 to 250% or even up to 350%. Besides, they showed good recyclability in terms of fluorescence responses.30
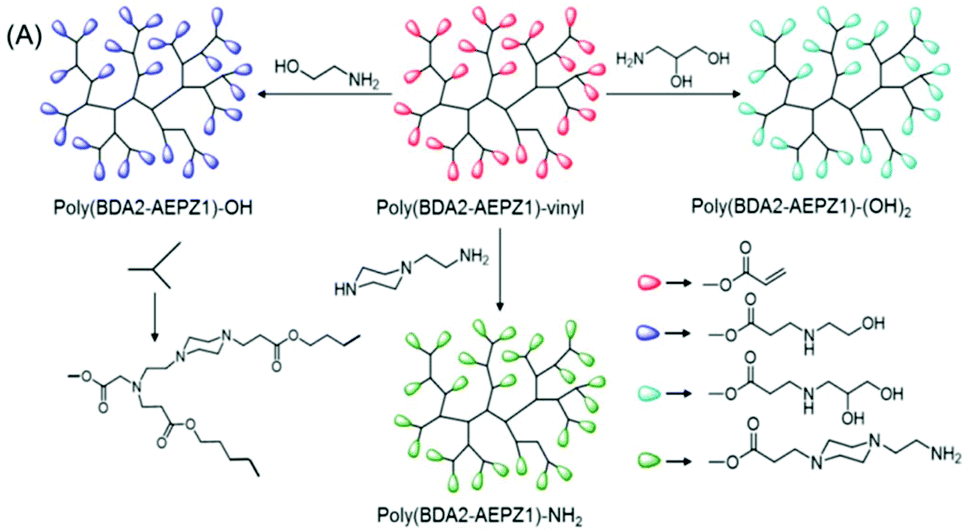 | ||
| Fig. 4 Synthetic strategy for the preparation of HPAE with diverse terminal functional groups: monohydroxy, diol and amine. Reproduced with permission from ref. 31. Copyright (2005), American Chemical Society. | ||
The first report of degradable dual pH- and temperature-responsive dendrimers displaying photoluminescence was provided by Shen et al. This dual-stimuli responsive dendrimeric polymer was proved to be a perfect drug-delivery carrier. DOX was incorporated into the dendrimers at low temperatures without using organic solvents. The loaded DOX release was very slow under physiological conditions, but very fast at acidic pH, such as the lysosomal pH (pH 4–5) which caused quenching of the autofluorescence of the dendrimer. Moreover, the fluorescence intensity decreased linearly when the DOX content was increased from the range of 1–16 wt%. Hence, the encapsulation and release of DOX can be monitored from the dendrimer's autofluorescence activity.32
Yan et al. reported melt polycondensation of triethanolamine and itaconic anhydride for facile preparation of water-soluble poly(amino ester) polyols. The synthesized polymers displayed unexpected bright blue emission which originated due to the clusters of carbonyl (CO) and oxidized tertiary amine functionalities. These fluorescent polymers have been employed for consecutive detection of unlabeled Fe3+ ions and L-cysteine via “on–off–on” fluorescence switching.33
An unprecedented multicolor photoluminescent HPAE containing non-conventional chromophores was synthesized via one-step A2 + B3 melt polycondensation using N-methyldiethanolamine and citric acid. The as-synthesized polymers exhibited a full-spectrum of bright photoluminescence including blue, cyan, green and red fluorescence under different excitation wavelengths. This unique emissive characteristic of a non-conjugated polymer is caused by the heterogeneity and wide size distribution of the polymer aggregates. Besides, its fluorescence was very sensitive to Fe3+ ions.34
Recently, another HPAE has been reported from the polycondensation reaction between maleic acid and triethanolamine by Yan et al. The as-prepared HPAE did not display multicolor fluorescence; rather it emitted only blue light on excitation with different wavelengths, which was assigned to the homogeneous electron delocalization. Intriguingly, the HPAE exhibited a multi-stimuli-responsive photoluminescence feature against solvent, pH, temperature and Fe3+, enabling it to act as a multifunctional macromolecular probe towards the sensing of solvent polarity, temperature, pH and Fe3+.35
Water-soluble fluorescent hyperbranched poly(amido acids) with terminal –CO2H functionalities were synthesized using straightforward self-condensation of N-(3-aminopropyl) diethanosuccinate amine (AB2 monomer). The amphiphilic poly(amido acids) formed nanoscopic globular micelle structures with a size around 30–50 nm in water and showed strong blue emissions with a QY of 23%. Moreover, their photoluminescence intensity was highly influenced by solution pH.36
A series of hyperbranched graft copolymers (HPEI-g-HPG) were synthesized through the ‘grafting from’ method. These copolymers contain HPEI as the core and hyperbranched polyglycerol (HPG) as a shell with different numbers of glycidol units. HPEI-g-HPGs emitted blue fluorescence centered at ca. 470 nm albeit it did not contain any traditional fluorophores. The fluorescence intensity was found to be enhanced by a combination of factors such as increasing the shell thickness, oxidation of the polymer by air at 95 °C, adding tetrahydrofuran (THF) into the solvent in an appropriate ratio, and lowering the pH of the solution below 4. It was believed that tertiary amine oxide acted as a primary fluorescence center.38
A water-soluble amphiphilic polyethyleneimine–poly(D,L-lactide) (PEI–PDLLA) copolymer was synthesized which emitted intrinsic blue light in both the solid-state and concentrated solution under a 365 nm UV lamp (Fig. 5A). Further transformation of the amphiphilic copolymer into polymeric nanoprobes (PEI–PDLLA NPs) was attained by a facile nanoprecipitation method. Interestingly, the synthesized NPs exhibited concentration enhanced fluorescence properties and a significant excitation-dependent multi-wavelength emission characteristic (Fig. 5B). The in vitro and in vivo imaging techniques corroborated tumor-targeted imaging and lysosome-specific staining capability of the PEI–PDLLA NPs with good biocompatibility and excellent photostability (Fig. 5C).39
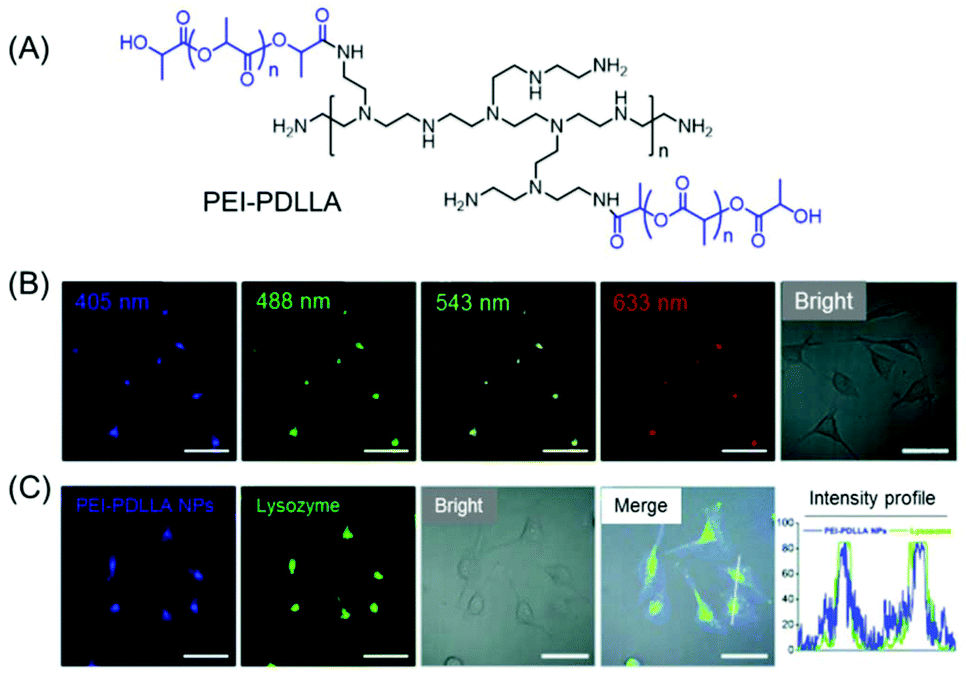 | ||
| Fig. 5 (A) Molecular structure of an intrinsically fluorescent PEI–PDLLA polymer. (B) Excitation wavelength-dependent multiemission cellular imaging of MCF-7 cells incubated with PEI–PDLLA NPs. Scale bars = 50 μm. (C) CLSM images of PEI–PDLLA (blue fluorescence) treated MCF-7 cells followed by staining with LysoTracker Red (green fluorescence). The intensity profiles indicated the polymer's ability to function as an alternative dye for lysosome imaging. Scale bar is 50 μm. Reprinted with permission from ref. 39. Copyright (2019), Royal Society of Chemistry. | ||
Self-monitoring and triple-collaborative nanotheranostics (NPICS) was successfully constructed from the positively charged amphiphilic autofluorescent copolymer PEI–polylactide (PEI–PLA) which can simultaneously load NIR dye IR825 and hydrophobic antiangiogenesis agent combretastatin A4 (CA4), and interact with the negatively charged heat shock protein 70 (HSP70) inhibitor (siRNA against HSP70). The NPICS was applied for dual-modal imaging-guided cancer anti-angiogenesis as well as gene silencing enhanced photothermal therapy. Importantly, the inherent luminescence of PEI–PLA assisted it in not only serving as the drug carrier but also self-monitoring of the biodistribution and tumor accumulation in a real-time manner by virtue of fluorescence microscopy imaging.40
O groups caused by N → Si coordination bonds.41 This emission intensity was found to decrease with increasing temperature and quench rapidly in the presence of Fe3+ and Hg2+ ions.42
In this context, Yan et al. introduced two novel water-soluble hyperbranched polysiloxanes (WHPSs) simultaneously carrying primary amine and –OH groups via melt polycondensation, that is, transesterification reaction between the ethoxyl groups of (3-aminopropyl)triethoxysilane and –OH groups of dihydric alcohols such as 2-methyl-1,3-propanediol (MPD) and neopentylglycol (NPG) using a one-step process under solvent-free and catalyst-free conditions. Both the WHPSs exhibited bright blue luminescence. However, the emission intensity was much intense for the NPG moiety containing compound compared to that of the other, plausibly due to the more easily restricted intramolecular rotations of the former than that of the later. They showed that the terminal OH functionalities instead of primary amine groups in WHPSs were mainly responsible for producing the blue-luminescent species. Moreover, the photoluminescence intensities of the two unhydrolyzed polymers were enhanced after hydrolysis.43
The same group has also synthesized fluorescent hyperbranched polysiloxanes (HBPSi) comprising epoxy and –OH groups, and observed that the aggregation of oxygen-rich heteroatoms from both the –OH and epoxy groups in the polymers played a vital role in the formation of fluorescent species.44 It is noteworthy that these hyperbranched polymers displayed reinforced photoluminescence in water after polyetherification by 20 wt% polyether amine. However, the fluorescence intensity of the modified polymer in water diminished on further increase of the polyether content from 20 to 40 wt%, owing to its increased dissolubility that obstructs the aggregation of the polymer and as a result an incompact texture was formed.45
Using a similar synthetic strategy they constructed luminescent HBPSi, composed of plenty of –OH groups and non-conjugated CC bonds. With increasing molecular weight and concentration of the polymer, the fluorescence intensity was found to be dramatically strengthened, and even the brightest light emission was observed in the solid-state. The authors concluded that both the –OH groups and CC bonds play a pivotal role in creating the emissive centers, and their persistent aggregation was responsible for the glow.46
For the first time, carbosiloxane dendrimers bearing CC bonds were fabricated from a monomer containing two kinds of CC and a dithiol via orthogonal thiol–ene click chemistry. Generation 1 dendrimers showed a stronger blue glow than the monomer containing two types of CC bonds, whereas polydimethylsiloxane (PDMS) showed no glow at all under similar conditions. In comparison with the non-luminescent PDMS, the authors speculated that the introduction of sulfur is accountable for such unusual emission; however, they mentioned that more experimental data are required to understand the mechanism.47 Again, OH-terminated HBPSi containing aliphatic tertiary amines have been reported to show blue fluorescence both in solution (water) and in the bulk state, and the emission intensities in water increased progressively with concentration. It was revealed that the –OH group, rather than the aliphatic tertiary amine, is crucial for the formation of blue-luminescent species. Additionally, Fe3+ ions acted as a highly selective fluorescence quencher of the polymers.48
Zhang and coworkers unprecedentedly constructed hydrophilic diamond-structured organosilica nanocrystals (ca. 2–6 nm) with finely tunable fluorescence (460–625 nm) from nonfluorescent precursors utilizing the classic Stöber strategy. The synthesized nanocrystals exhibited size-dependent fluorescence emission originating from the π–π interaction of orderly stacked vinyl groups throughout the particle.49
A series of water-soluble intrinsically luminescent comb polysiloxanes containing various ratios of mercaptopropyl groups and polyether were synthesized via thiol–ene click reaction by Feng's research group. It was presumed that the unprecedented fluorescence is generated because of the formation of S → Si coordination bonds that facilitate the through-space charge transfer of the lone pair electron in sulfur to the 3d empty orbital of silicon. However, in response to Hg2+ and Fe3+, the S–Si interaction was broken and eventually quenching of fluorescence occurred.50
Novel imidazole functionalized polysiloxanes were synthesized for the consecutive detection of Fe3+ and cysteine by fluorescence quenching and a lighting mechanism. Here, imidazole groups and the whole polysiloxane structure contributed to the fluorescence. The present system could be useful for monitoring the Fe3+/cysteine cycle in situ in living HeLa cells.51
The HBPSi or common unconventional luminescent polymers discussed above suffer from low fluorescence intensity and efficiency; in particular the QY is low in comparison with those of the traditional conjugated luminescent materials. Recently, Feng et al. showed how the fluorescence QYs of HBPSi could be improved. They synthesized HBPSi with both CO and vinyl groups, which exhibited a nontraditional intrinsic luminescence QY up to 43.9%, the highest value obtained to date among the reported silica-containing hyperbranched fluorescent polymers. To elucidate the fluorescence mechanism, the results were compared with those of two reference oligomers, which contain either CO groups or vinyl groups (Fig. 6A). The absolute fluorescence QYs for only CO groups and only vinyl groups bearing polymers have been reported to be 16.3% and 10.5%, respectively. Thus, the high fluorescence QY of the designed hyperbranched polymer was attributed to the synergism of vinyl and CO groups along with the Si–O groups which facilitated the through-space conjugation (Fig. 6B).52
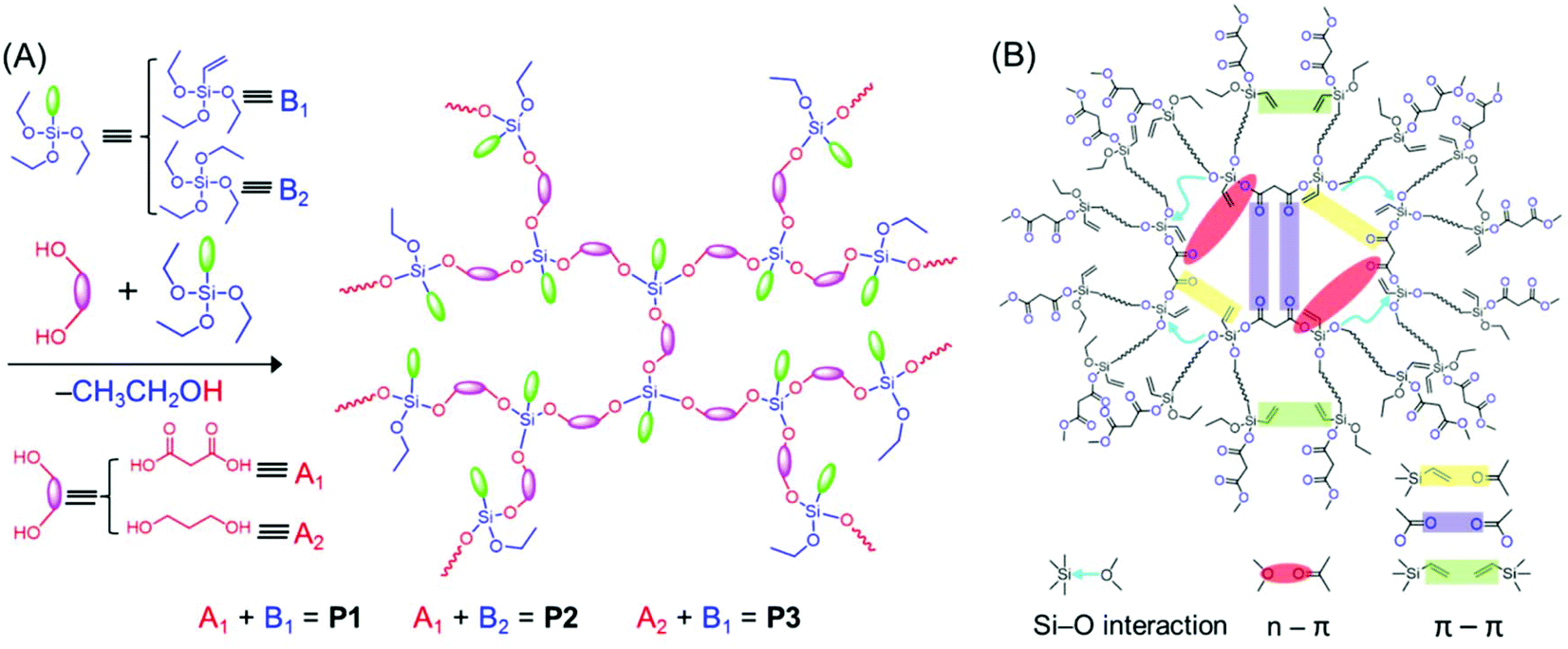 | ||
| Fig. 6 (A) One-step synthetic method for the preparation of P1–P3. (B) Schematic diagram of different interactions between the functional groups, responsible for the bright-blue emission of P1. Reprinted with permission from ref. 52. Copyright (2019), American Chemical Society. | ||
Although the QYs of such nontraditional luminescent polysiloxanes substantially improved, their bioapplications are still associated with a major shortcoming viz, low water-solubility.
Accordingly, to enhance the fluorescence intensity and water solubility of HBPSi, β-CD, a rigid biodegradable molecule with plenty of hydroxyl groups was decorated onto the surface of the polymer architecture. The β-CD decorated HBPSi showed promise not only as a drug delivery vehicle that can achieve high loading capacity and pH-responsive drug release performance, but also in simultaneous cell imaging.53
Very recently, a novel multicolor emissive HBPSi with adjacent CO and CC groups was designed and developed by Tang et al. (Fig. 7A). To explore the multicolor emission mechanism, a reference linear polymer with the same chemical components was synthesized (Fig. 7B), and its photophysical properties were compared with a more compact hyperbranched one. The hyperbranched analog showed higher QY (7.71%) and luminescence lifetime (1.0 ns) compared to the linear one (QY = 1.12%, lifetime = 0.57 ns). Notably, the hyperbranched polymer displayed multicolor luminescence at different excitation wavelengths (Fig. 7C) in contrast to the linear polymer which emitted only weak blue fluorescence. An innovative luminescence mechanism namely “multiring-induced multicolor emission” was proposed based on the results obtained from density functional theory (DFT) calculations (Fig. 7D). Furthermore, on account of their quenching effect in response to Fe3+, the application of the hyperbranched polymer in data encryption was accomplished by using disodium ethylenediaminetetraacetate dehydrate (Na2EDTA) as the “ink” and iron(III) chloride (FeCl3) as a “key” to decode the encrypted information on the security paper (Fig. 7E).54
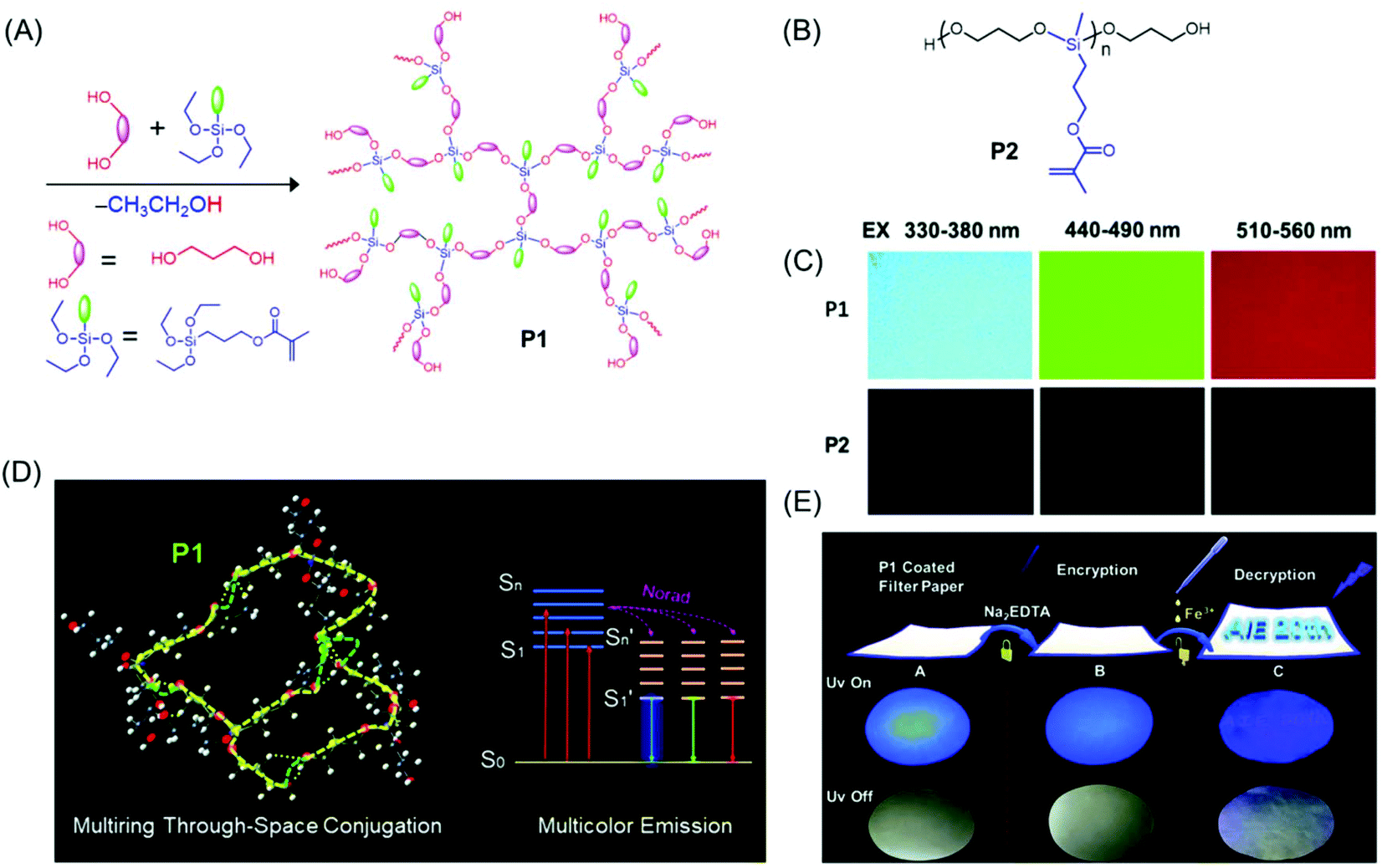 | ||
| Fig. 7 (A) Synthetic route employed for the preparation of P1. (B) Chemical structure of P2. (C) Fluorescence microscopy images of hyperbranched (P1) and linear (P2) polysiloxanes acquired under different UV filters. (D) Occurrence of multiring through-space conjugation through a silicon-bridge in P1 and schematic representation of its luminescence mechanism. (E) Chronological steps involved in the data encryption process with P1 coated security paper and their photographs under both natural and UV light (λ = 365 nm). Reprinted with permission from ref. 54. Copyright (2020), Royal Society of Chemistry. | ||
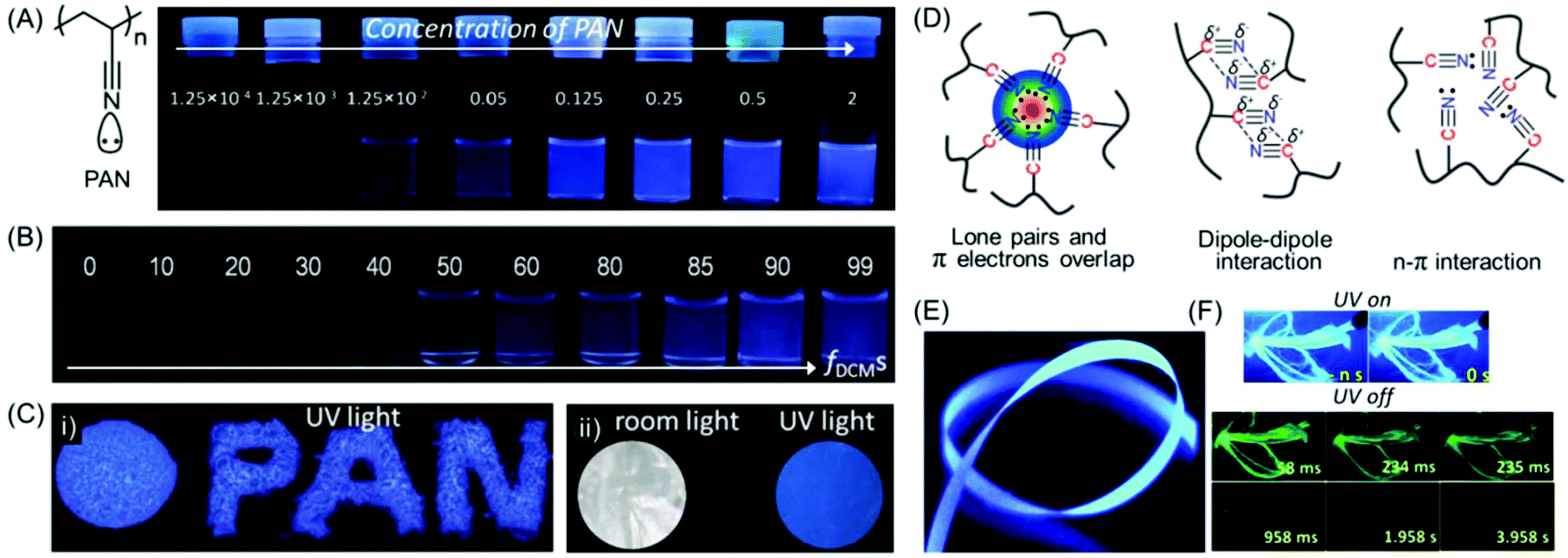 | ||
| Fig. 8 (A) Chemical structure of intrinsically emissive PAN. Photographs of PAN in only DMF and (B) a DCM/DMF mixture with various fDCMs obtained under a 365 nm UV lamp. (C) Luminescence images of (i) solid powders and (ii) a film of PAN on exposure to 365 nm UV excitation. (D) Schematic illustration of plausible intra- and inter-molecular interactions present within nitrile clusters in the aggregated state. (E) Photoluminescence image of SR6 ENRs taken under 365 nm UV irradiation. (F) Photographic images of ENRs at 77 K in UV lamp-on/lamp-off mode at different time courses. Figures A, B, C and D are reprinted with permission from ref. 55. Copyright (2016), John Wiley and Sons. Figures E and F are reprinted with permission from ref. 57. Copyright (2020), Royal Society of Chemistry. | ||
Surface-grafted PAN brushes were synthesized by copper (Cu)-mediated photoinduced atom transfer radical polymerization (ATRP) using α-bromophenyl acetic acid-based initiator-modified silicon wafers. The AIE behavior of PAN was validated by making polymer brushes with various film thicknesses, and it depends on the molecular weight of the grafted chains and grafting density. It was observed that, as the film thickness increases the emission is more pronounced, which is attributed to the formation of larger fluorescent clusters.56
In a recent work, the non-conjugated PAN was implemented for the fabrication of electrospun nanofiber ribbons (ENRs) by electrospinning and high heat stretching above the glass transition temperature (Tg). The resultant mesoscopically ordered ENRs were light-weight and found to exhibit tremendous mechanical strength and toughness. It is noteworthy that upon excitation at 340 nm, the ribbons showed polarized deep blue luminescence with anisotropy and QY values of 0.37 and 31%, respectively. Along with the prompt luminescence (Fig. 8E), delayed deep blue and green phosphorescence was also observed at room temperature (Fig. 8F). In a nutshell, the authors have exemplified a very fascinating material with the combination of the unique properties of emission, anisotropy and mechanical behavior.57
An unprecedented aggregation-induced long-persistent phosphorescence (>1 s) in a non-conjugated PU system at 77 K was reported for the first time by Bryce et al. It was proposed that the appearance of such unusual behavior is because of the formation of intra- and/or inter-molecular CO clusters at low temperature that facilitates spatial electronic overlap between π electrons and lone pairs. Interestingly, the incorporation of conjugated aromatic units into the non-conjugated PU backbone has been shown to increase the lifetime of long-persistent phosphorescence, revealed that the intersystem crossing rate was enhanced through the inter- or intra-molecular n–π* transition from the electron-rich CO groups to the conjugated aromatic units.59 In the follow-up work, they prepared a series of fluorescent non-conjugated PU derivatives by incorporating classical π-aromatic chromophores, and applied them for highly selective and sensitive recognition of PA both in solution and in the solid state. The sensing mechanism was ascribed to the combined effect of Förster resonance energy transfer (FRET), photoinduced electron transfer (PET) and strong IFE.60
Zhao and coworkers reported NLPs based on dynamic covalently cross-linked polyhydroxyurethane (PHU2), synthesized from amines and aliphatic multifunctional cyclic carbonates avoiding the use of toxic isocyanates (Fig. 9A). It was observed that the fluorescence intensity of PHU2 progressively became stronger without any sign of quenching on increasing the curing time from 0 to 8 h (Fig. 9B). It is noteworthy that it displayed long persistent phosphorescence under cryogenic conditions. In addition to the unique luminescence properties, the new NLP possessed excellent mechanical strength along with smart characteristics including shape memory (Fig. 9C) and self-healing (Fig. 9D and E). The efficient transcarbamoylation dynamic exchange reaction between carbamate and –OH groups was responsible for the good shape memory property and self-healing behavior of PHU2. Utilizing these alluring characteristics of the synthesized NLP, “lightassisted ink-free screen printing” was established to prepare anti-counterfeiting materials, which eliminated the use of expensive security inks.61
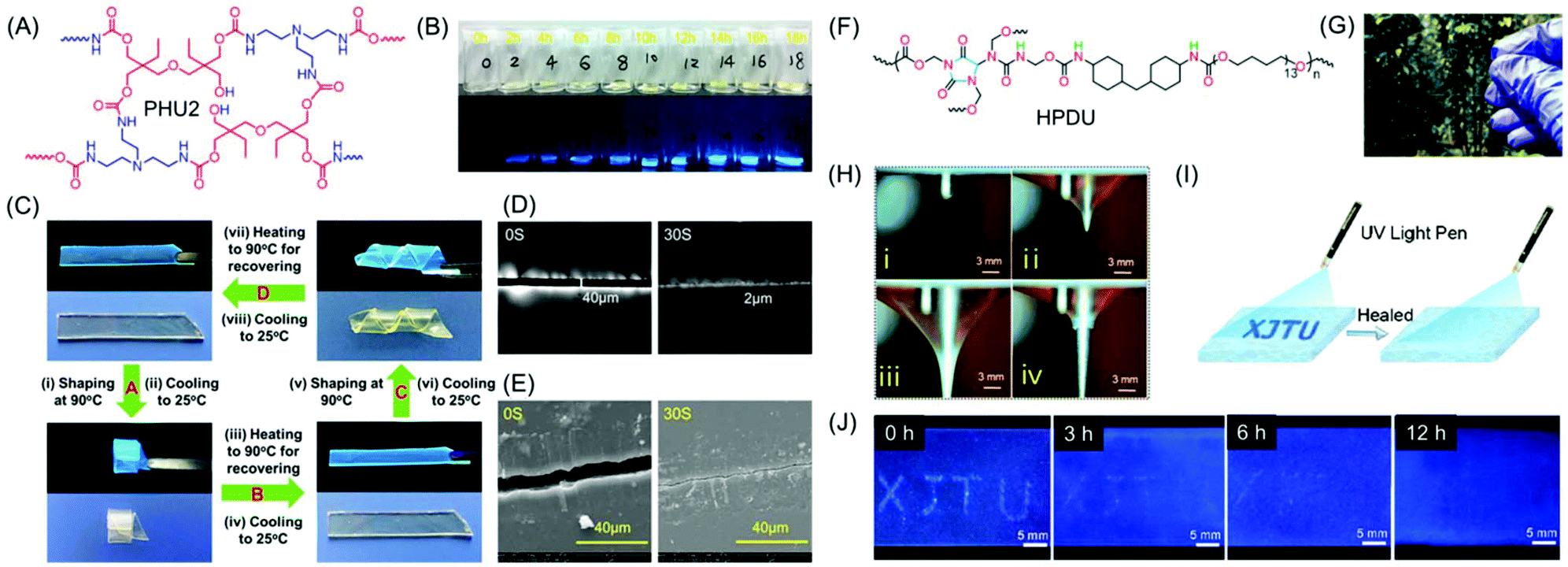 | ||
| Fig. 9 (A) Chemical structure of PHU2 and (B) its fluorescence images taken under daylight and UV excitation (λ = 365 nm) at various reaction time periods at 90 °C. (C) The shape memory properties of a PHU2 sheet and its self-healing behaviour monitored at 160 °C using (D) a polarizing microscope and (E) a SEM. (F) Chemical structure of HPDU. (G) Transparent photograph of a HPDU4 film. (H) Illustration of puncture resistant properties of the HPDU10 film. (I) Schematic view of a crack shaped as XJTU with bright-blue fluorescence on the film surface. (J) Photographs of crack XJTU on the HPDU4 film surface when exposed to UV excitation after being healed for different time intervals at 90 °C. Figures A, B, C, D and E are reprinted with permission from ref. 58. Copyright (2020), American Chemical Society. Figures F, G, H, I and J are reprinted with permission from ref. 59. Copyright (2020), American Chemical Society. | ||
Very recently, a series of novel hyperbranched polyurethane (HPDU) elastomers have been developed by Zhang et al. using a simple polycondensation reaction among commercially available poly(tetramethylene) glycol, methylene-bis(4-cyclohexyl isocyanate) and diazolidinyl urea by following the A2 + B4 strategy (Fig. 9F). The resultant PUs were transparent (Fig. 9G) and showed super-tough, puncture-resistant (Fig. 9H) and photoluminescence elastomeric behavior with crack self-diagnosis and healing tracking ability. Upon irradiation with 365 nm UV light, the fracture surface furnished strongest emission intensity as compared to the entire film, which was found to diminish gradually with self-healing. Consequently, the change in the emission intensity on the crack guides in the diagnosis of the crack location and concomitant tracking of the progress of the self-healing process (Fig. 9I and J).62
Carbon dioxide (CO2)-derived siloxane (Si–O–Si)-containing linear PHU was designed and synthesized and it possessed strong photoluminescence (QY = 23.6%), high photostability, and broad absorption and emission characteristics both in solution and bulk states. The hydrophobicity and flexibility of the siloxane linkages in PHU led to strong fluorescence through the significant aggregation of hydroxyurethane chromophores along with H-bonding interactions. To demonstrate a practical application, a low voltage cool white LED was fabricated with a color rendering index (CRI) of 83 by combining the polymer as a single phosphor with a commercially available UV chip.63
Recently, a white LED with a high CRI of 95 has been reported from a non-conjugated PHU microsphere (PHUM) prepared via the crosslinking of 1,6-hexanediamine and trimethylolpropane tri(cyclic carbonate) ether. The high degree of crosslinking as well as various H-bonding structures prompted the construction of carbamate clusters with different sizes and broad distributions, enabling a wide-range emission spectrum (white light emission).64 Another report used a similar synthetic strategy to prepare non-traditional fluorescent PHU. The collective results of the methylthiazolyldiphenyl-tetrazolium bromide (MTT), hemolysis and cytokine release assays for PHU certified its much lower cytotoxicity and immunotoxicity along with good blood compatibility with a <5% hemolysis rate in comparison with the traditional isocyanate-derived PU. This polymer was applied for fluorometric detection of Fe3+, and the LOD was calculated to be 4.56 μM.65
O groups.66 They also described the unprecedented strong blue photoluminescence characteristics of non-conjugated hyperbranched polycarbonates (HBPCs). The hyperbranched polycarbonates were synthesized via catalyst or solvent-free one-pot A2 + B3 transesterification reaction between diethyl carbonate and triol (glycerol67 or trimethylolpropane68). The resultant HBPC exhibited excitation wavelength-dependent and concentration-dependent emission behaviors in ethanol. The authors have envisioned that the intermolecular H-bond probably formed between the oxygen atom of the carbonate group and the unreacted –OH group suppressed the non-radiative decay processes through rigidifying the polymer conformation and thus furnished bright emission.
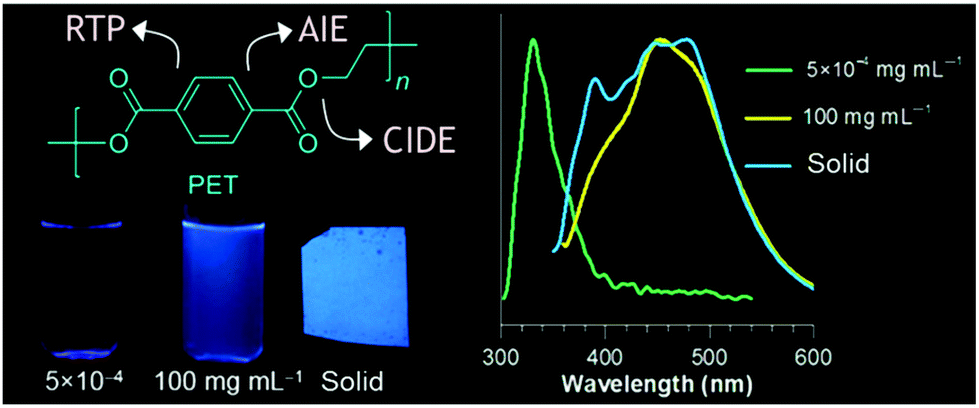 | ||
| Fig. 10 Overall emission properties of PET at different states. Reprinted with permission from ref. 69. Copyright (2018), American Chemical Society. | ||
Li et al. demonstrated a new class of polymers based on a non-conjugated methacrylate backbone appended with a single switchable boron chromophore (Fig. 11A). Impressively, the homopolymers displayed rare multicolor emission in solution which was found to be dependent on the degree of polymerization (DP). Moreover, these homopolymers exhibit solvent and temperature-tunable fluorescence color changes (Fig. 11B and C).
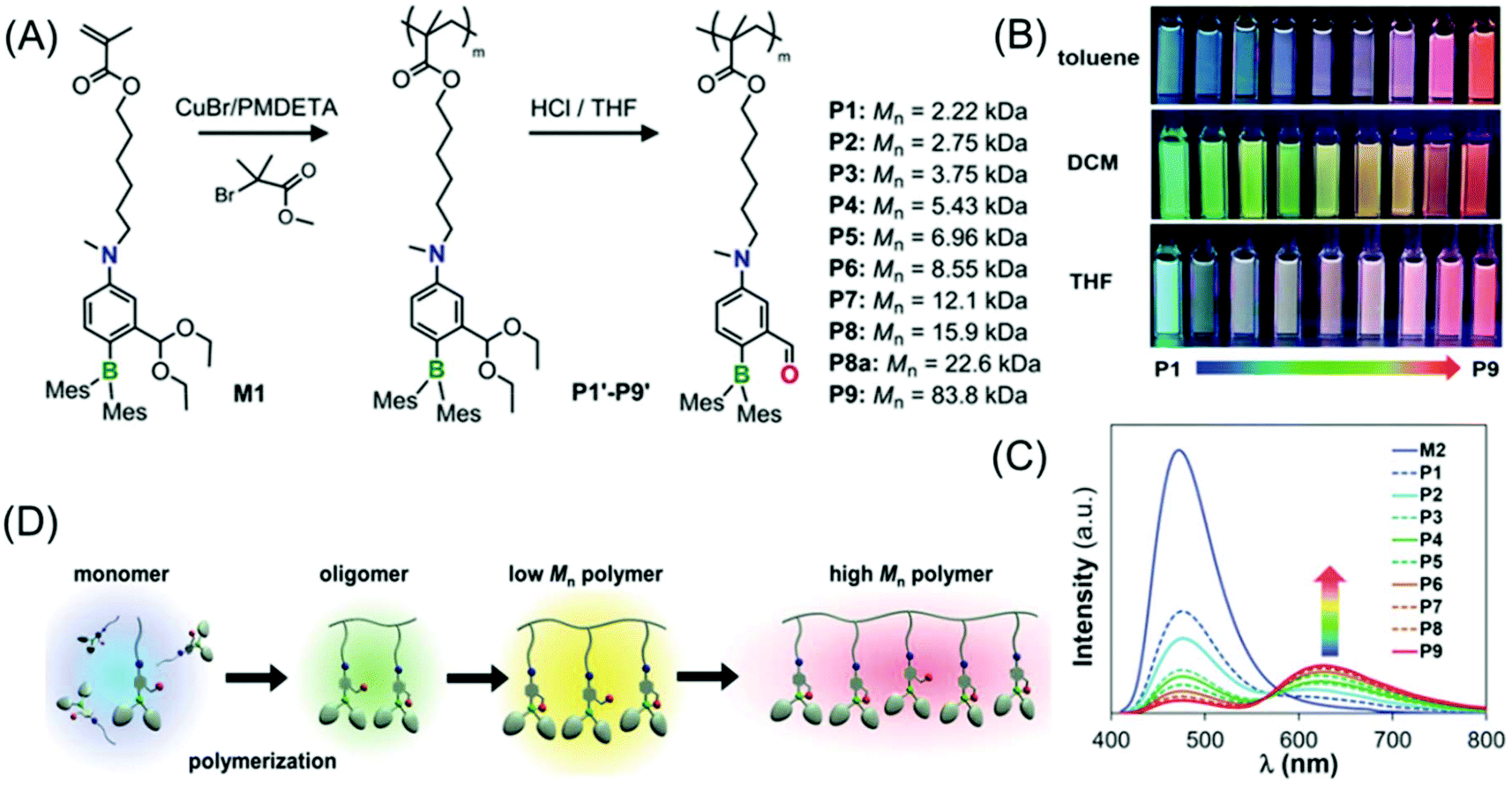 | ||
| Fig. 11 (A) Synthesis of methacrylate-based polymers containing pendant switchable boron chromophores. (B) Photographs of P1–P9 showing multicolour emission in toluene, DCM and THF acquired under UV illumination of 365 nm. (C) Emission spectra of P1–P9 in toluene. (D) Schematic view of the DP-dependent boron chromophore structural change and fluorescent colors of the methacrylate polymers. Reprinted with permission from ref. 70. Copyright (2019), WILEY-VCH. | ||
The color-switching mechanism is associated with the structural transition from an open form (tricoordinate boron chromophore, blue-emitting) to a closed-form (tetracoordinate boron chromophore, red-emitting). In a nutshell, the solvent, temperature and DP (molecular weight of the polymer) have immense effects on the open-to-closed form ratios of the attached pendant boron chromophore (Fig. 11D). Notably, a pure white light emission was attained in solution by finely adjusting the homopolymer molecular weight, and also in a thin-film/solid-state by copolymerizing the luminescent boron monomer with non-fluorescent monomers in an appropriate ratio.70
Long-lived room temperature phosphorescence (LRTP) has emerged as an appealing optical feature in the area of organic photonics and electronics.71 Amorphous polymer poly(styrene sulfonic acid) (PSS) showed green phosphorescence in air (dry solid-state) with a maximum lifetime up to 1.22 s. The RTP lifetime was greatly influenced by the molecular weight of PSS and percentage of sulfonic acid groups in PSS incorporated through sulfonation reaction. It was presumed that the inter-/intra-chain H-bonding network amongst the pendant sulfonic acid groups suppressed non-radiative transition processes via restricting the motion of the chromophores, enabling ultralong RTP lifetimes. However, exposure to water caused RTP quenching of solid-state PSS, and restoration occurred by removal of water. This reversible “off–on” RTP switching of PSS powder upon exposure and expulsion of water was realized in lifetime-encoding application simply by injecting water vapor within the PSS film using a mask.72
Huang et al. reported the achievement of the longest lifetime (RTP lifetime is around 2.1 s) of organic phosphorescence in polymer luminogens by a facile ionic bonding cross-linking strategy. Instead of PSS, poly(sodium 4-styrenesulfonate) (PSSNa) as an ionic polymer model was taken to prove the hypothesis. Similar steady-state fluorescence and phosphorescence spectra were observed after the replacement of Na+ with different cations (Li+, K+, Rb+ and NH4+). The universality of the ionic interlocking for LRTP was verified by introducing the ionic bonding cross-linking into nonaromatic polymers. So a set of totally four ionic polymers, namely PSSNa, poly(acrylic acid sodium salt) (PAANa), poly(maleic acid sodium salt) (PMANa), and poly(maleic acid-co-4-styrenesulfonic acid sodium salt) (PSSNa-co-PMANa), were designed and synthesized. On chromophore variations, the LRTP emission colors were successfully transformed from yellow to blue. It is noteworthy that both aromatic and nonaromatic polymers exhibited ultralong phosphorescence on varying the excitation wavelength under ambient conditions.73 The delay time-dependent RTP emission was first reported in the case of poly(4-styrenesulfonic acid-co-maleic acid) salt by Gong et al. The existence of the free volume that provided space for altering the conformation in order to relax the excited states was primarily responsible for the observed phenomenon.74
On the other hand, amorphous purely organic phosphorescent polymer materials with color-tunable and long-lived emission by a concise chemical ionization strategy have been reported. Poly(4-vinylpyridine) (PVP) was chosen as an ionization model. Upon ionization with 1,4-butanesultone, the PVP-based ionization product poly(4-vinylpyridine)butane-1-sulfonate (PVP-S) (Fig. 12A) showed distinct phosphorescence behavior with an emission lifetime of 578.36 ms, which is 525 times longer than that of the PVP polymer under ambient conditions. Intriguingly, the phosphorescence emission color could be precisely tuned over the full-spectrum of the visible region from blue to red, by changing the excitation wavelength (Fig. 12B), whereas, the PVP polymer did not show any excitation-dependent phosphorescence at room temperature, even when cooled to 77 K. It was proposed that the strong ionic interactions between PVP and sulfonic acid help in generating ultralong phosphorescence in the PVP-S polymer by improving the local rigidity and suppressing the non-radiative decay of the excited triplet state through the restriction of the vibration and rotation of the molecules (Fig. 12C).75
 | ||
| Fig. 12 (A) Synthetic route to an amorphous PVP-S polymer. (B) Phosphorescence spectra of PVP-S acquired at 77 K at various excitation wavelengths. Inset shows the afterglow multicolor phosphorescence emission on excitation with a wide range of wavelengths from 280 to 500 nm at 77 K. (C) Schematic illustration of ionization-induced ultralong phosphorescence of PVP-S. Reprinted with permission from ref. 75. Copyright (2019), WILEY-VCH. (D) Graphical representation of the RTP polymer involving boron–nitrogen dative bonds and their fluorescence images under sunlight and UV “on–off” conditions. Reprinted with permission from ref. 76. Copyright (2020), American Chemical Society. | ||
The development of the foremost instance of the amorphous RTP polymer containing nonmetal dative bonds has been reported. In the copolymer of vinylphenylboronic acid and acrylamide, nonmetal dative bonds formed between boron and nitrogen/oxygen atoms enhanced the probability of an intersystem crossing (ISC) process to populate the triplet excitons; the charge transfer and subsequent inhibition of the motions of the phosphors resulted in an effective RTP lifetime in air (Fig. 12D). Additionally, the dynamic dative bonds and H-bonds endowed the material with self-healing and outstanding anti-counterfeiting capabilities.76
Jin's research group recently reported non-conjugated RTP phosphoramidic acid oligomers consisting of a phosphoryl group having dπ–pπ bonds and a diamino group as a H-bond donor with ultralong phosphorescence lifetimes and phosphorescence QY up to 776.9 ms and 4.6–10.5%, respectively. The system was easily synthesized by the polycondensation reaction of H3PO4 (phosphoric acid) and variable diamines including 1,3-bis(aminomethyl)cyclohexane (BAC), norbornane dimethylamine (ND) and m-xylylenediamine (XY). The ultralong phosphorescence lifetime was attributed to the H-bond trapping excited electrons, albeit it was not fully understood.77
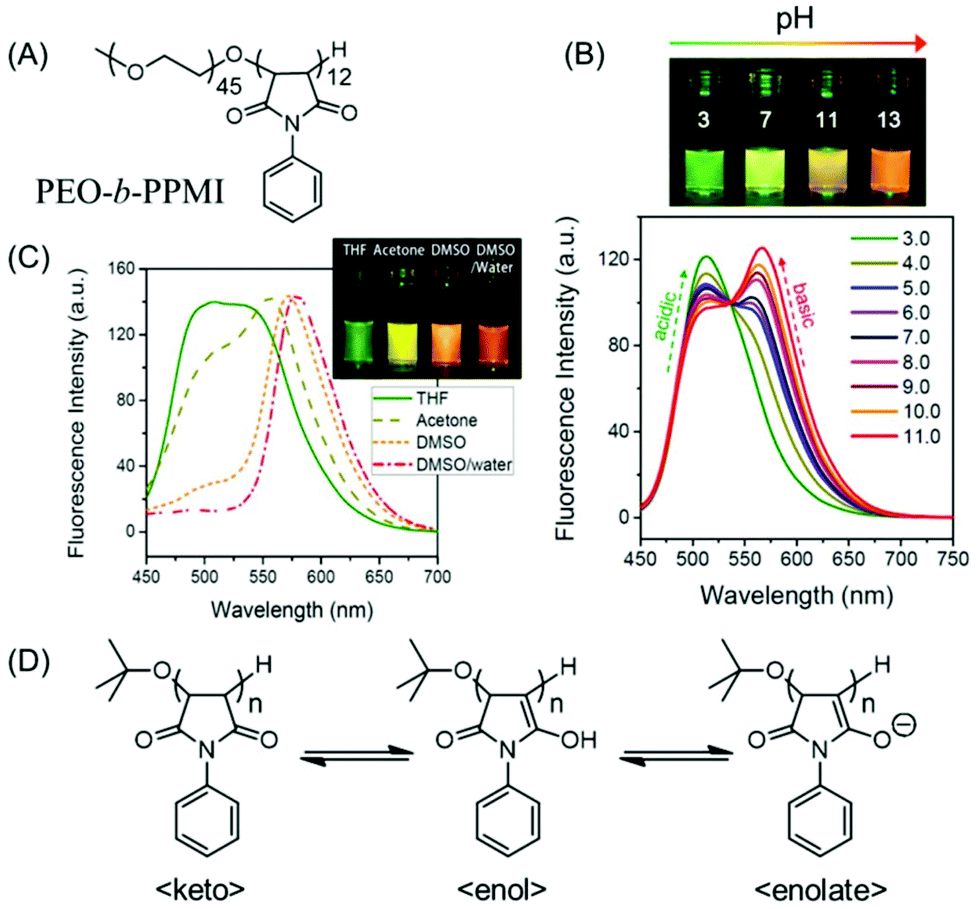 | ||
| Fig. 13 (A) Chemical structure of poly(ethylene oxide-b-N-phenylmaleimide) (PEO-b-PPMI). (B) Ratiometric fluorescence changes and multiple emission colors of PEO-b-PPMI at different pH values in water on excitation at 445 nm. (C) Emission profiles (λex = 445 nm) of PPMI in different solvents. (D) Tautomeric structures of PPMI, responsible for pH-tunable emission and solvatochromism phenomena. Reprinted with permission from ref. 78. Copyright (2015), American Chemical Society. | ||
Tang and coworkers showed how a pure oxygenic non-conjugated polymer based on poly[(maleic anhydride)-alt-(vinyl acetate)] (PMV) could emit strong blue-light upon UV illumination (Fig. 14A). Moreover, its photophysical behavior could be manipulated by varying the solvent (Fig. 14B). Because of the rigid conformation of succinimide-based polymers, a heterodox cluster by the collection of many CO groups is generated. It was believed that the clusters formed within the locked CO groups by interacting in a through-space manner generated the actual chromophore, leading to bright emission upon UV light irradiation. Again, the existence of the polymer/solvent complex is accountable for the solvatochromism.79
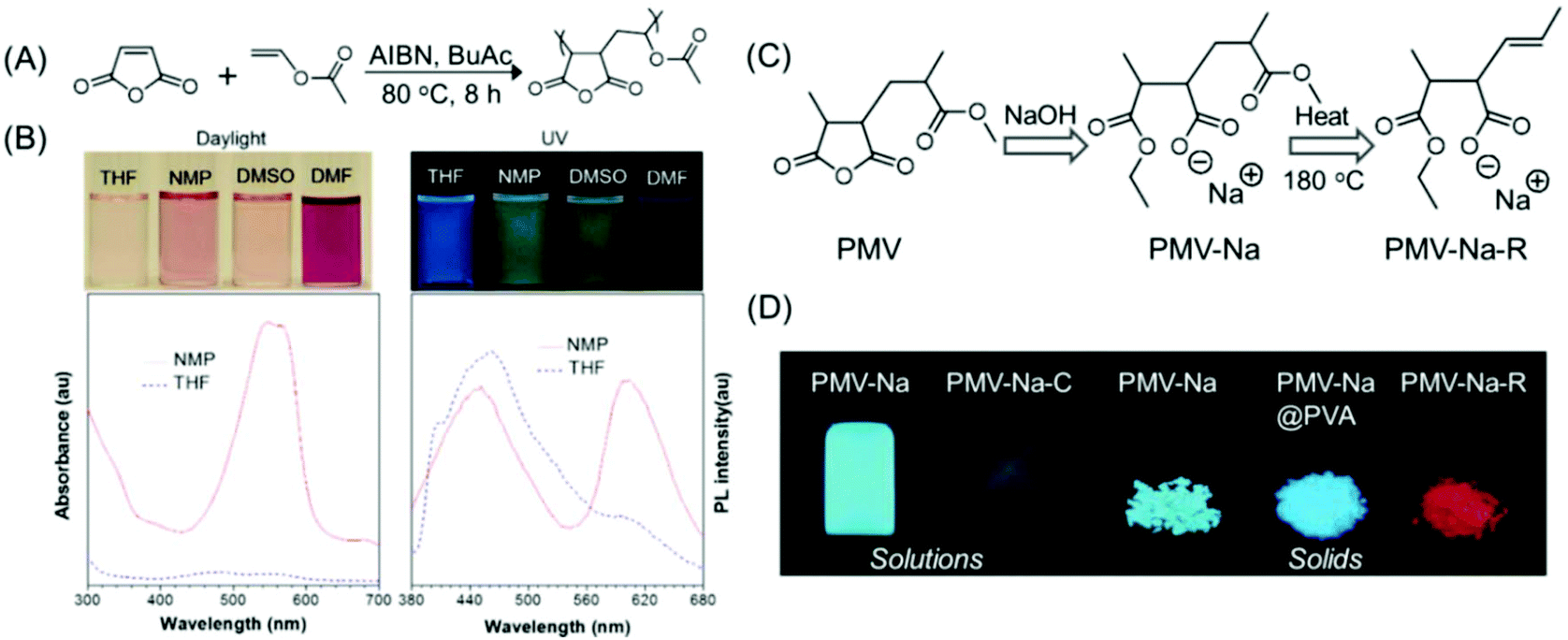 | ||
| Fig. 14 (A) Synthesis of PMV. (B) Photographs of PMV solutions under daylight/365 nm UV illumination in different organic solvents and corresponding UV and fluorescence spectra (λex = 330 nm) in NMP and THF. (C) The synthetic route to PMV-Na and PMV-Na-R. (D) Photoluminescence images of PMV-derived polymers in solution and in the solid state upon UV excitation at 365 nm. Figures A and B are reprinted with permission from ref. 79. Copyright (2015), American Chemical Society. Figures C and D are reprinted with permission from ref. 80. Copyright (2017), WILEY-VCH. | ||
Qiao and coworkers have demonstrated how the emission intensity/QY of PMV could be enhanced, and how the emission color could be altered from common blue to red. Hydrolysis and esterification reaction of PMV with a strongly alkaline sodium hydroxide solution afforded PMV-Na, which emitted high-intensity blue-green light in solution and solid-states. The calculated QYs of the PMV-Na solution and solid were found to be as high as 35% and 66%, respectively. By thermally treating at around 170 °C for 5 min, blue-emissive PMV-Na solids could be converted into a red emission agent. It was recommended that the construction of isolated CC bonds is responsible for the unusual red emission from such non-conjugated PMV polymers (Fig. 14C and D).80 In the subsequent year, a new kind of PMV derivative was synthesized. The new red-emissive PMVs were synthesized by utilizing large alcohol molecules such as diacetone alcohol produced from acetone under basic catalysis. Two types of CO chromophores were present in the newly synthesized PMV derivatives: a blue light-emitting sodium carboxylate group and a red light-emitting bulky carboxylic ester group, and both exhibited excitation-dependent luminescence. Also, such non-conjugated photoluminescence polymers having dual chromophores showed AIE characteristics.81
Novel multicolored AIE-active photoluminescent polymers were developed by mixing the PMV solid with basic aqueous solutions of variable pH in suitable ratios at room temperature. The PMV/alkaline sodium hydroxide ratio played a key role in tuning the emission color from cyan to red.82 The mechanism behind the above phenomenon was not fully elucidated. However, it has been assumed that the addition of NaOH altered the clustering state of the carbonyl chromophores present in PMV.
Wang's research group utilized a conventional free radical precipitation copolymerization technique to synthesize a series of poly(maleic anhydride-alt-vinyl pyrrolidone) (PMVP) copolymers with variable molecular weights. The resultant PMVPs emitted strong light in both DMSO solvent and solid-state, demonstrating their AIE characteristics. Besides, the PMVPs exhibited distinct excitation-dependent fluorescence (EDF) and molecular weight-dependent fluorescence (MWDF). More importantly, their emissions were significantly red-shifted in comparison with those of the homopolymers poly(maleic anhydride) (PMAh) and poly(N-vinyl-2-pyrrolidone). They suggested that the copolymerization of N-vinyl-2-pyrrolidone with MAh increased the flexibility of polymer chains and as a result the rigidity of the polymer chain was compromised, which facilitated the stronger and more extended intra- and/or inter-chain n–π* and π–π* interactions between the anhydride and pyrrolidone groups in the aggregation state, leading to the strong and red-shifted emission of PMVPs.83
To prove the previous hypothesis that the polymer chain flexibility plays an imperative role in enhancing the photoluminescence of NLPs, they synthesized poly(itaconic anhydride) (PITA) and poly[(1-octene)-co-(itaconic anhydride)] (POITA) by using 1-octene and itaconic anhydride (ITA) as the comonomers.84 The photophysical properties of these as-synthesized copolymers were compared with those of PMAh and poly[(maleic anhydride)-alt-(2,4,4-trimethyl-1-pentene)] (PMP) to elucidate the mechanism in terms of chain flexibility. In comparison with PITA, POITA exhibited red-shifted and stronger luminescence both in solution and in the solid state. The finding is completely different from the result of PMP, where the bulky 2,4,4-trimethyl-1-pentene (TMP) segregated the MAh moieties and thus obstructed their intra-chain interactions, leading to very weak emission. Thus, the increased chain flexibility enabled the functional groups to adopt proper conformations to brighten up the emissions.
The photoluminescence characteristics of poly(itaconic anhydride-co-vinyl caprolactam) (PIVC) and poly(itaconic anhydride-co-vinyl pyrrolidone) (PIVP) were recently highlighted keeping in mind that in some cases the chain flexibility is beneficial for boosting the fluorescence of NLPs.85 ITA was chosen, as it has a cyclic acid anhydride ring that is attached to the polymeric structure via only one carbon atom; therefore these structures possess much higher flexibility in comparison with PMAh. It was found that the emissions of both the copolymers are significantly red-shifted (emission maxima in the orange-red region) in solutions. Interestingly, PIVP and PIVC displayed bright white and orange-red emission in the solid-state upon 365 nm UV illumination, respectively. Both the polymers emitted blue fluorescence at a low concentration in DMSO, but surprisingly the emissions changed to orange as the concentration was increased. It was speculated that two distinct chromophores were present causing the blue and orange emissions, and the number of chromophores corresponding to the orange emission increased with the increase of the polymer concentration.
The reversible addition–fragmentation chain transfer (RAFT) method was employed to prepare alternating copolymers based on the poly(styrene-alt-maleimide) skeleton by the sequence-defined copolymerization of para-methoxydiethylene glycol-substituted styrenes and N-(2-salicylaldehyde-aminoethyl)maleimides. The as-synthesized alternating copolymers exhibited a fast and highly sensitive “off–on” fluorometric response toward Zn2+ with a LOD value of 0.25 μM by inhibiting the PET process. Apart from this, a notable green fluorescence “off–on” switching (230-fold) was arrested while the pH changed from 11 to 12.86
Polyhedral oligomeric silsesquioxane (POSS)-containing homopolymer poly(maleimide isobutyl POSS) and various alternating copolymers namely poly(styrene-alt-maleimide isobutyl POSS), poly(4-acetoxystyrene-alt-maleimide isobutyl POSS) and poly(4-hydroxystyrene-alt-maleimide isobutyl POSS) were synthesized through free radical polymerization and subsequent post-polymerization modification reactions. Although the POSS-containing polymer does not contain any common fluorescent moiety, it exhibited emission at high concentration and in the solid-state. Moreover, the POSS-appending homopolymer displayed stronger photoluminescence emission in the bulk state with a much higher φ compared to those of the POSS-integrated alternating copolymers, presumably due to the former's crystallinity and aggregation of locked CO groups of five-membered rings.87
Qiao and coworkers developed a new family of photoluminescent alternating copolymers by the copolymerization of MAh and diverse olefin derivatives. These fluorescent polymers were totally transparent and exhibited thermoplastic behavior. Additionally, the copolymers prepared from styrene or its derivatives and MAh were found to be phosphorescent.88
Concurrently, we reported the development of dual pH- and thermo-responsive luminescent alternating copolymers based on the poly(styrene-alt-maleimide) skeleton (Fig. 15A). To this end, an N-substituted maleimide monomer bearing a diethylene oxide side-chain, (N-(methoxydiethyleneglycol) maleimide) and a tert-butylcarbamate (Boc)-protected leucine appended styrenic monomer was copolymerized at an equimolar feed ratio to obtain well-defined copolymers with perfectly alternating sequences of these two monomers. The as-prepared Boc-protected copolymers showed bright-blue emission in organic solvents, whereas the deprotected copolymers retained their luminescence behavior in organic solvents and also exhibited dual pH/temperature-tunable fluorescence activity in water.89 This conventional fluorophore-free water-soluble luminescent copolymer was applied as a turn-off fluorescent sensor for selective, sensitive and instantaneous detection of the nitro explosive PA in 100% water. Besides, this polymeric sensor showed the ability to detect PA in both solid and vapor phases (Fig. 15B). The ground-state electrostatic complex formation with concomitant ground-state electron transfer from the analyte (picrate anion) to the polymeric probe was proposed as the fluorescence quenching mechanism.90
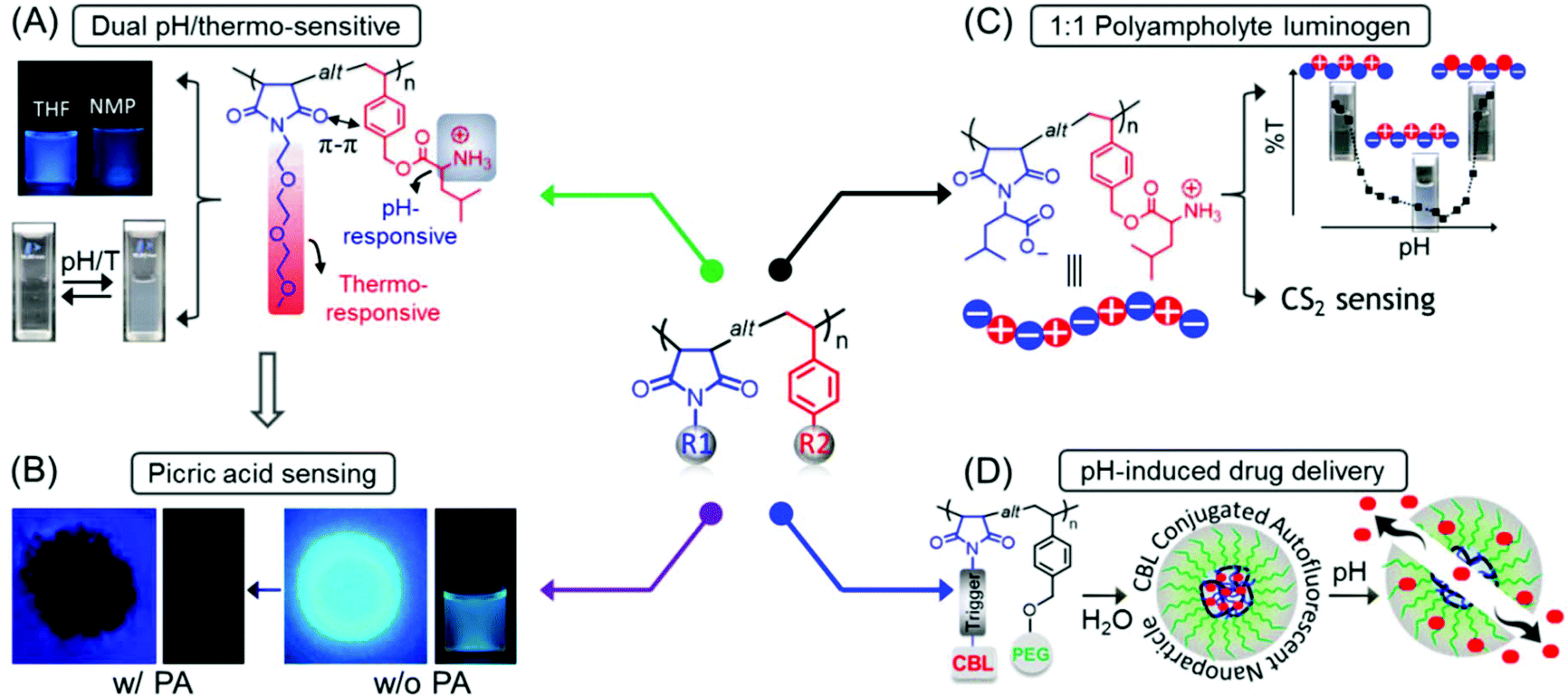 | ||
Fig. 15 General structure of a poly(styrene-alt-maleimide) copolymer with variable pendants R1 and R2. (A) Solution properties and unusual photoluminescence behaviour of a dual pH and thermo-responsive copolymer and (B) its PA sensing application. (C) Chemical structure of leucine-derived alternating 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 polyampholyte luminogen and its implementation in CS2 sensing in both solution and vapour phases. (D) Schematic representation demonstrating pH-induced drug delivery application using such an alternating copolymer. Figure A reprinted with permission from ref. 89. Copyright (2016), Royal Society of Chemistry. Figure B reprinted with permission from ref. 90. Copyright (2017), Royal Society of Chemistry. Figure C reprinted with permission from ref. 91. Copyright (2019), Royal Society of Chemistry. Figure D reprinted with permission from ref. 92. Copyright (2019), American Chemical Society. :1 polyampholyte luminogen and its implementation in CS2 sensing in both solution and vapour phases. (D) Schematic representation demonstrating pH-induced drug delivery application using such an alternating copolymer. Figure A reprinted with permission from ref. 89. Copyright (2016), Royal Society of Chemistry. Figure B reprinted with permission from ref. 90. Copyright (2017), Royal Society of Chemistry. Figure C reprinted with permission from ref. 91. Copyright (2019), Royal Society of Chemistry. Figure D reprinted with permission from ref. 92. Copyright (2019), American Chemical Society. | ||
In another report, we engineered an alternating polyampholyte based on the same polymeric building block by copolymerization of two rationally designed monomers: L-leucine-derived functional group protected maleimide and styrene-based monomers via RAFT polymerization. The expulsion of protection groups resulted in the formation of a charge-neutral polyampholyte having an anionic and a cationic functional entity placed in a perfectly alternating fashion. Interestingly, the as-synthesized polyampholyte displayed emission behavior in aqueous media and was applied for the specific recognition of carbon disulfide (CS2) amongst volatile organic compounds (VOCs) in pure water (Fig. 15C). In addition, a colorimetric “naked-eye” detection of CS2 vapor with significant sensitivity and selectivity under ambient conditions was accomplished with this polymeric probe.91
Next, to explore the biomedical applications of such intrinsically emissive alternating copolymers, an autofluorescent amphiphilic brush copolymer was fabricated as a pH-sensitive drug delivery nanocarrier for the intracellular administration of the fluorescent inactive aromatic nitrogen mustard chlorambucil (CBL) drug into the cancer cells. To serve the purpose, a CBL functionalized N-substituted maleimide monomer was copolymerized with a poly(ethylene glycol) (PEG) attached styrenic macromonomer, which provided a well-defined luminescent alternating copolymer. These copolymers were self-assembled into nanoparticles in aqueous medium where the CBL drug was enclosed into the core of the self-assembled nanostructures. At endolysosomal pH 5.0, 90% of the CBL was released from the nanocarrier by the cleavage of pH-responsive aliphatic ester linkages, connecting CBL to the polymeric chain. The unique luminescence property assisted intracellular monitoring of CBL-incorporated nanocarriers by virtue of fluorescence microscope imaging is noteworthy (Fig. 15D).92
The generation of fully controlled 1:1 alternating D- and L-alanine containing sequence-regulated polymers based on the poly(styrene-alt-maleimide) backbone using an N-substituted maleimide monomer bearing Boc-L-alanine and a styrene conjugated Boc-D-alanine monomer via the RAFT polymerization process has been reported. It is noteworthy that these copolymers displayed fluorescence in various organic solvents and retained their fluorescence activity even after deprotection as observed in organic and aqueous media.93 Recently, we employed an alanyl–alanine tethered maleimide monomer and a glycyl–glycine functionalized styrenic monomer to make these two dipeptides containing 1:1 alternating copolymers on the same polymer skeleton. Unusual luminescence characteristics in solution and in the solid state were observed, where only blue emission occurred in solution but in the solid-state tri-color emission (blue, green and red) was observed.94
Yang et al. observed that although succinimides act as common fluorescence quenchers of proteins, and common succinimide monomers are nonfluorescent but succinimide-based compounds and also succinimide-containing polymers became fluorescent only after functionalization with dithiol/amino substituents without introducing any conjugated fluorescent entity. The unexpected fluorescence of dithio/amino-succinimides originated from the spatial separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), realized from DFT calculations. Compared with monomers, the 2-amino-succinimide-based polymers showed considerable enhancement in emission intensity (up to ∼200-fold) owing to the isolation effect where the non-radiative decay occurring among succinimide units was significantly suppressed.95
Poly(amide-imide)s (PAIs) belong to a class of outstanding engineering materials which express the combined properties of both polyamides (PAs) and polyimides (PIs) viz. superior thermal stabilities, high mechanical strength, chemical resistance and facile processability.96,97 Broadly, PAIs are nonemissive, and do not exhibit fluorescence except when they are totally aromatic or semi-aromatically connected with emissive species. Therefore, the construction of fluorescent aliphatic PAIs has been challenging and has rarely been reported. Meanwhile, PAIs mostly suffer from their solubility issue in common organic solvents and water, restricting their potential roles in biological applications. However, Yang et al. demonstrated the synthesis of luminescent aliphatic PAI with excellent solubility in both organic and aqueous media. To this end, initially, a functional thiolactone–maleimide monomer was synthesized using a Cu(I)-catalyzed click reaction. An efficient polycondensation between the thiolactone–maleimide and an aliphatic diamine afforded fluorescent aliphatic PAIs containing pendant thiols and 2-aminosuccinimide fluorophores through the combination of the ring-opening of thiolactones and amine–maleimide Michael addition reaction. Furthermore, the released thiol was employed for PEGylation via a thiol–methacrylate Michael addition that significantly resolved the solubility issues of PAIs in both common organic solvents and aqueous media.98 Using similar types of chemical reactions, they also fabricated hyperbranched aliphatic PAI with fluorescence behavior. The as-prepared polymer was found to be soluble in various solvents and showed solvent-dependent emission with moderate QYs.99
Again, by using a similar synthetic route, non-conjugated PEGylated PAI was prepared with isolated benzene rings. The authors serendipitously discovered that this amphiphilic PEGylated PAI self-assembled into nanovesicles with unexpected red fluorescence in water. It was believed that the synergistic effects of π–π stacking interactions and H-bonding endowed non-conjugated PAI with a significant bathochromic shift (>100 nm) from the blue-green fluorescence of the aminosuccinimide fluorophore to red emission. Moreover, the self-assembled structure shielded PAIs from the interaction with water and suppressed the H-bond formation between aminosuccinimide fluorophores and water, prohibiting the quenching of fluorescence in protic solvents and as a consequence the PAIs were found to be emissive in water. Finally, the graft copolymer showed excellent biostability and photostability and a promising cell imaging ability.100
Tang et al. developed an easy and effective multicomponent tandem polymerization method to afford a series of NTIL poly(aminomaleimide)s (PAMIs) with well-defined structures and moderate molecular weights in excellent yields from alkynes and diamines. The as-synthesized PAMIs were employed as macromonomers to carry out further polymerization which resulted in PAMIs that barely contained simple and single maleimide moieties in the polymer main-chain. However, they exhibited strong blue-green emission in DMSO/ethyl acetate (EA) mixtures. Interestingly, the emission color and intensity did not change with the addition of different EA fractions (fEA) in the DMSO/EA solvent mixture, although EA is a bad solvent for PAMI and it aggregated in solvent mixtures with a high fEA. PAMIs showed outstanding biocompatibility, and were therefore advantageous for bioimaging applications.101
Although Tang's group proposed that the unexpected photoluminescence behavior of maleimide or MAh-based homooligomers and alternating copolymers is a result of through-space electronic communication among the locked CO groups of MAh or maleimide units, the mode of interaction of these non-aromatic groups was not known; and therefore further investigation was required. To prove the hypothesis they synthesized the homooligomer of oligo(maleic anhydride)s (OMAhs) and an alternating polymer of PMP as well as their copolymers, and their systematic structure–property relationships were investigated. Instead of a small vinyl acetate comonomer, they have used more bulky TMP as a comonomer partner to isolate the MAh units, thereby inhibiting their intra-chain through-space interactions, and this assists in the examination of the effect of the in-between distance on the fluorescence characteristics in condensed phases. OMAhs showed distinct absorption at ca. 365 and 458 nm, whereas PMP did not absorb above 300 nm. Furthermore, under UV irradiation (365 nm), OMAh4 showed intense emissions, but PMP emitted very weakly both in solution and in the solid state (Fig. 16). It was anticipated that bulky t-butyl groups segregated MAh repeating units in PMP. Again simulation results revealed that the distance between two adjacent MAh units of OMAhs is in the range of 2.84–3.18 Å, whereas for PMP, the distance was 4.90–5.37 Å. It was further unveiled that the existence of the intra- and inter-chain π–π* interactions of the CO groups of MAh units present in OMAhs is responsible for unexpected light emission. Detailed experimental results and theoretical simulation study led to the coining of the term CTE or clusteroluminescence mechanism for rationalizing the inexplicable luminescence of such non-aromatic polymers.102
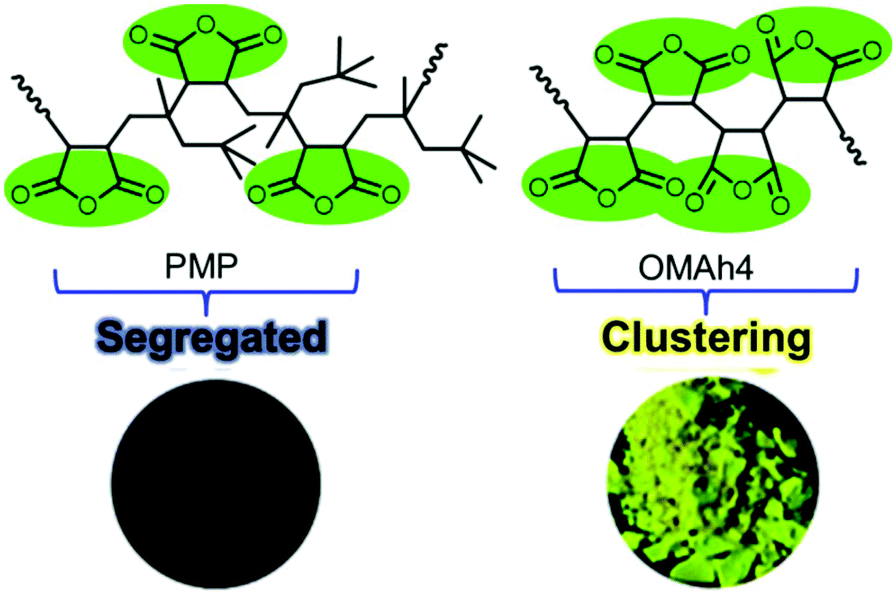 | ||
| Fig. 16 Chemical structures of PMP and OMAh4 and corresponding luminescence photographs of their solids when exposed to 365 nm UV illumination. Reprinted with permission from ref. 102. Copyright (2017), Royal Society of Chemistry. | ||
Turner's research group synthesized tert-butyl carboxylate functionalized four alternating copolymers: two poly(stilbene-alt-maleic anhydride) and two poly(styrene-alt-maleic anhydride), and their fluorescence properties were studied in detail. The stilbene-containing copolymers I and II exhibited strong fluorescence with high QYs of 71.2% and 65.9%, respectively, whereas both of the styrene-containing copolymers III and IV (Fig. 17) exhibited weak fluorescence with low QYs of 2.6% and 3.1%, respectively. The fluorescence remained unaltered even after the removal of tert-butyl groups which makes the resulting copolymer completely water-soluble. The origin of the fluorescent band with high QY was assigned to the extra phenyl groups and “through space” π–π interactions between the phenyl rings of stilbene and CO groups from the succinimides.103
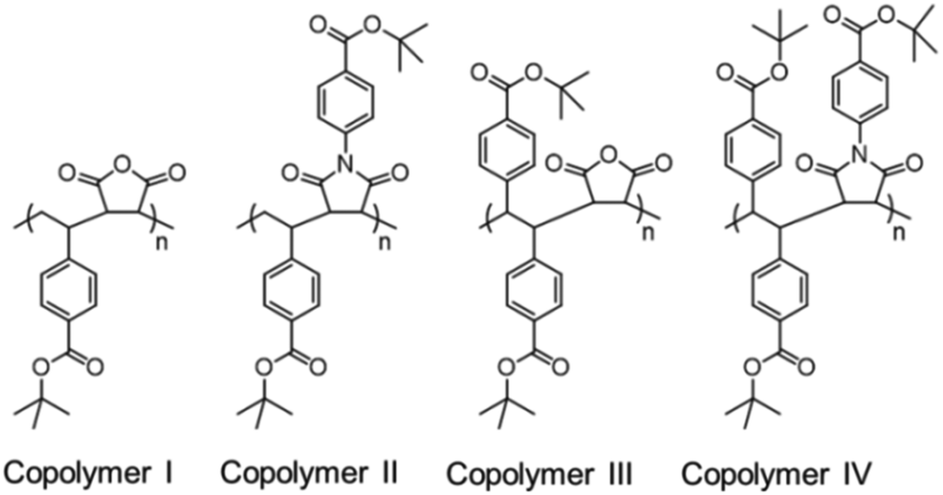 | ||
| Fig. 17 Molecular structures of stilbene-containing luminescent alternating copolymers. Reprinted with permission from ref. 103. Copyright (2018), WILEY-VCH. | ||
O unit of NIPAM and the benzene ring of CTA present in the polymeric pendant and chain end, respectively. Furthermore, the investigators synthesized a well-defined luminescent multiblock copolymer comprising both PNIPAM and poly(oligoethylene glycol acrylate) (POEGA) using the same polymerization technique and used it for cell imaging purposes.104
Tang and coworkers attempted to understand the chromophore responsible for the intrinsic fluorescence of non-aromatic polymers such as PNIPAM and poly(N-tert-butylacrylamide) (PNtBA). By further investigating the emission features of a group of non-aromatic bio-based and synthetic peptides, they proposed that the fluorescence in the aggregation state/condensed phase in such non-conventional fluorescent polymers is attributed to the electronic interaction amongst the amide groups, where H-bonds play a decisive role in bringing these functionalities in the immediate vicinity.105
Autoluminescent micelles were fabricated through the self-assembly of amphiphilic polymer molecules having an intrinsic unconventional fluorophore and AIE-active polyetheramide (PEA). A one-pot synthetic route was employed to prepare the PEA molecule consisting of a hydrophilic poly(ethylene oxide) (PEO) backbone and two hydrophobic chains on both the ends from palmitic acid and commercially obtainable polyetheramine (Jeffamine ED2003). The AIE behavior of PEA emerged from the structural rigidification caused by the restriction of intramolecular rotation by virtue of multiple H-bonding interactions within the amide groups rather than from the clusters of oxygen atoms with lone pair electrons of PEO segments as the aggregation of the PEO–PPO–PEO triblock copolymer (Pluronic F127) was found to be non-emissive.106
Wang et al. identified a new series of nonconventional fluorescent polynorbornenes bearing aminosuccinimide side groups. Under UV illumination, these polymeric materials showed bright-blue emission (470–478 nm) with large Stokes shifts of over 110 nm.107 The same research group also discovered the intrinsic fluorescence of a common non-conjugated compound, poly(N-vinylpyrrolidone) (PVP), a linear polymer with side-chain pyrrolidone rings (a lactam) connected to a simple polyethylene backbone. Although the monomer N-vinyl-2-pyrrolidone (NVP) was very weakly emissive, its polymer version PVP exhibited strong intrinsic fluorescence. Surprisingly, the oxidized hydrolysate of PVP showed much stronger fluorescence, about 1000 times stronger than those of unhydrolyzed compounds (Fig. 18). The increased fluorescence intensity was attributed to the formation of secondary amine oxides.108
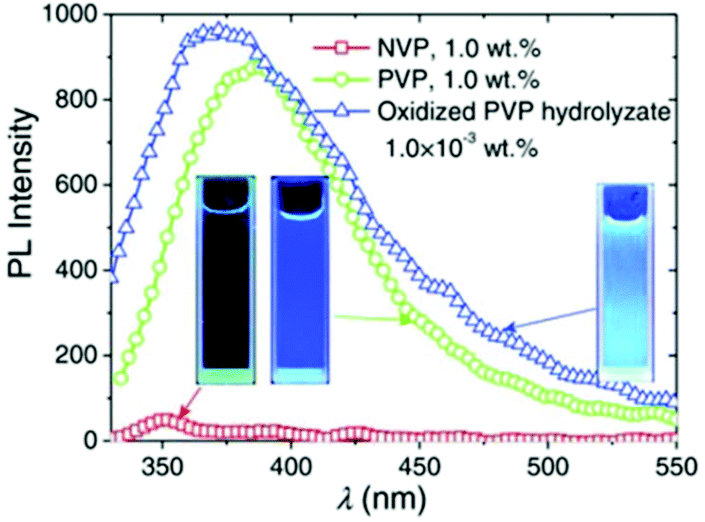 | ||
| Fig. 18 Fluorescence spectra of NVP, PVP and oxidized PVP hydrolysate in water. Reprinted with permission from ref. 108. Copyright (2015), WILEY-VCH. | ||
We unveiled the unprecedented luminescence, AIE activity and heat-induced AIE characteristics of well-known non-conjugated thermoresponsive poly(N-vinylcaprolactam) (PNVCL), a higher homolog of PVP. The PNVCL film displayed multicolor emission (blue, green and red) upon illumination with different excitation wavelengths, suggesting the coexistence of multiple emissive species with different energy levels. Impressively, the PNVCL (concentration = 0.1 wt% in water) exhibited a bright blue emission upon heating above its lower critical solution temperature (LCST) (37.5 °C), while it was nonfluorescent below this temperature (Fig. 19A). This fact was ascribed to the heat-assisted AIE phenomenon. Subsequently, these fascinating properties, temperature-assisted aggregation and AIE characteristics, were ingeniously utilized to apply PNVCL as a suitable fluorescent polymeric thermometer to determine the intracellular temperature (Fig. 19B).109
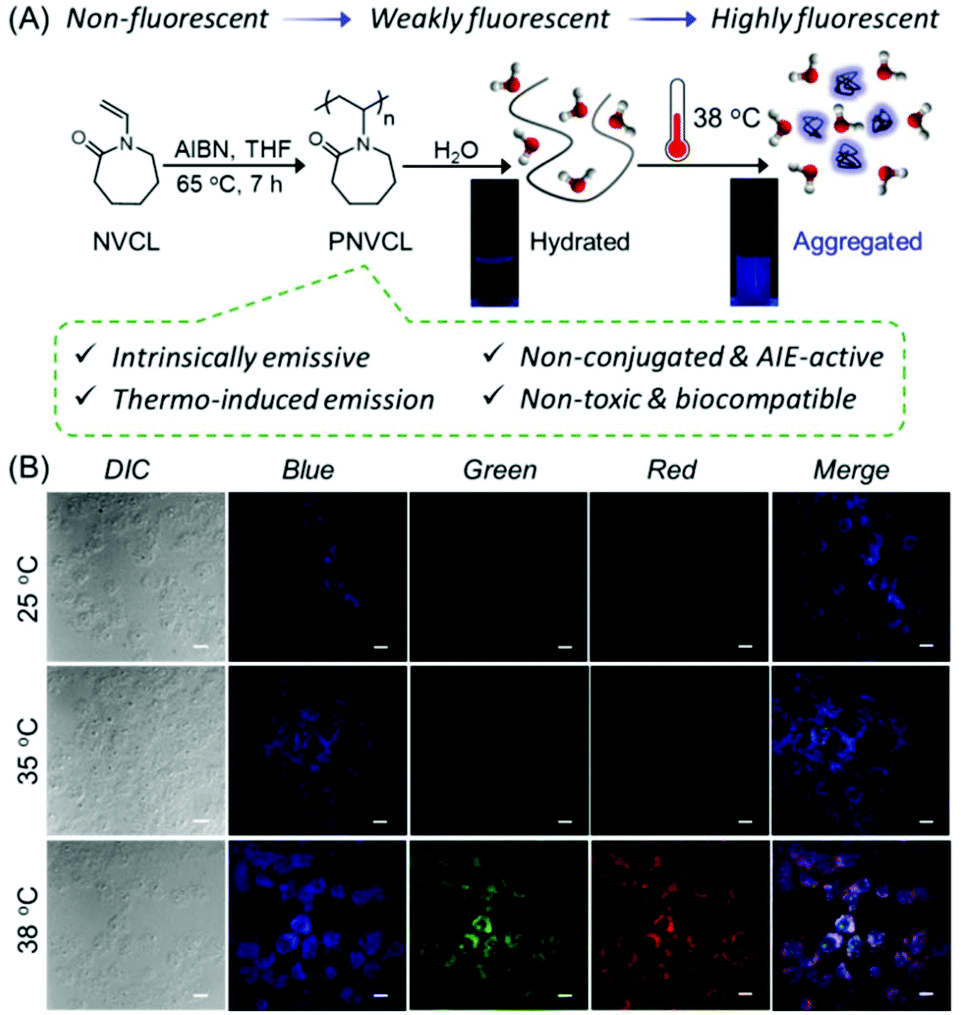 | ||
| Fig. 19 (A) Synthetic route to PNVCL and its thermo-induced fluorescence photographs on exposure to a 365 nm UV lamp in an aqueous medium. (B) CLSM images of PNVCL treated MCF-7 cells at 25, 35 and 38 °C. Scale bar is 10 μm. Reprinted with permission from ref. 109. Copyright (2020), Royal Society of Chemistry. | ||
Yuan et al. showed that the CTE hypothesis to rationalize the inexplicable fluorescence properties of nonconventional luminogens is also applicable to those compounds with merely oxygen moieties (i.e., ether and –OH). Impressively, in solid-state xylitol, PEG and F127 displayed cryogenic persistent phosphorescence sustaining for almost 12 s, and even persistent RTP emission was observed from xylitol crystals.110
Wang and coworkers observed autofluorescence of hydrogels made of polyacrylamide (PAM) or polyacrylic acid (PAA) and their derivatives devoid of any traditional fluorophore. This autofluorescence was successfully applied for self-monitoring the gelation process, as the internal structural changes of the gels strongly correlated with the fluorescence response. It was found that with the decrease of water content in the as-prepared gels, the fluorescence intensity gradually increased. When the water content was >40%, a very weak fluorescence was observed, while the emission intensity sharply increased once the water content decreased from 40% to 20%. Moreover, the gel exhibited dazzling fluorescence on further diminishing the water content to 10%. However, the quenching of fluorescence takes place with some particular metal ion namely Fe3+, the erasure. Accordingly, information rewriting in the presence of visible and UV light, respectively, was successfully realized by using an ethanol solution containing Fe3+ as a “writer” and ethanol as a “remover”.111
Yuan et al. reported the preparation of new sulfur-containing polythioether via a Michael polyaddition between 1,4-butanedithiol and 1,4-butanedioldiacrylate. The synthesized polythioether was found to be emissive under UV irradiation in aggregates, despite the absence of remarkable conjugations. Furthermore, boosting of the emission efficiency was realized by the oxidation of polythioether to polysulfoxide and polysulfone both in the solid state and in concentrated solutions. The clustering of sulfur-containing functional groups (OSO or SO, S) and ester moieties (COO) assisted in the “through-space” electronic interaction and concurrently rigidified the molecular conformations, making the clusters fluorescent active under UV light irradiation.112
They also reported the unique intrinsic luminescence and additionally p-RTP from a group of non-conjugated and aromatic-ring-free amorphous polymers PAA, PAM and PNIPAM (Fig. 20A). The polymers exhibited no emission in dilute solutions, but became highly luminescent in solid powders/films, nanosuspensions and concentrated solutions. Interestingly, PAA and PAM solids showed distinct p-RTP in air, while PNIPAM solids did so under vacuum or nitrogen (Fig. 20B and C). Furthermore, ionization-induced increased efficiency and prolonged p-RTP were realized in the case of PAANa and PAMHCl powders resulting from the neutralization of PAA and PAM with NaOH or fuming HCl, respectively. The p-RTP of PNIPAM was also modulated by pressurization (Fig. 20D).113
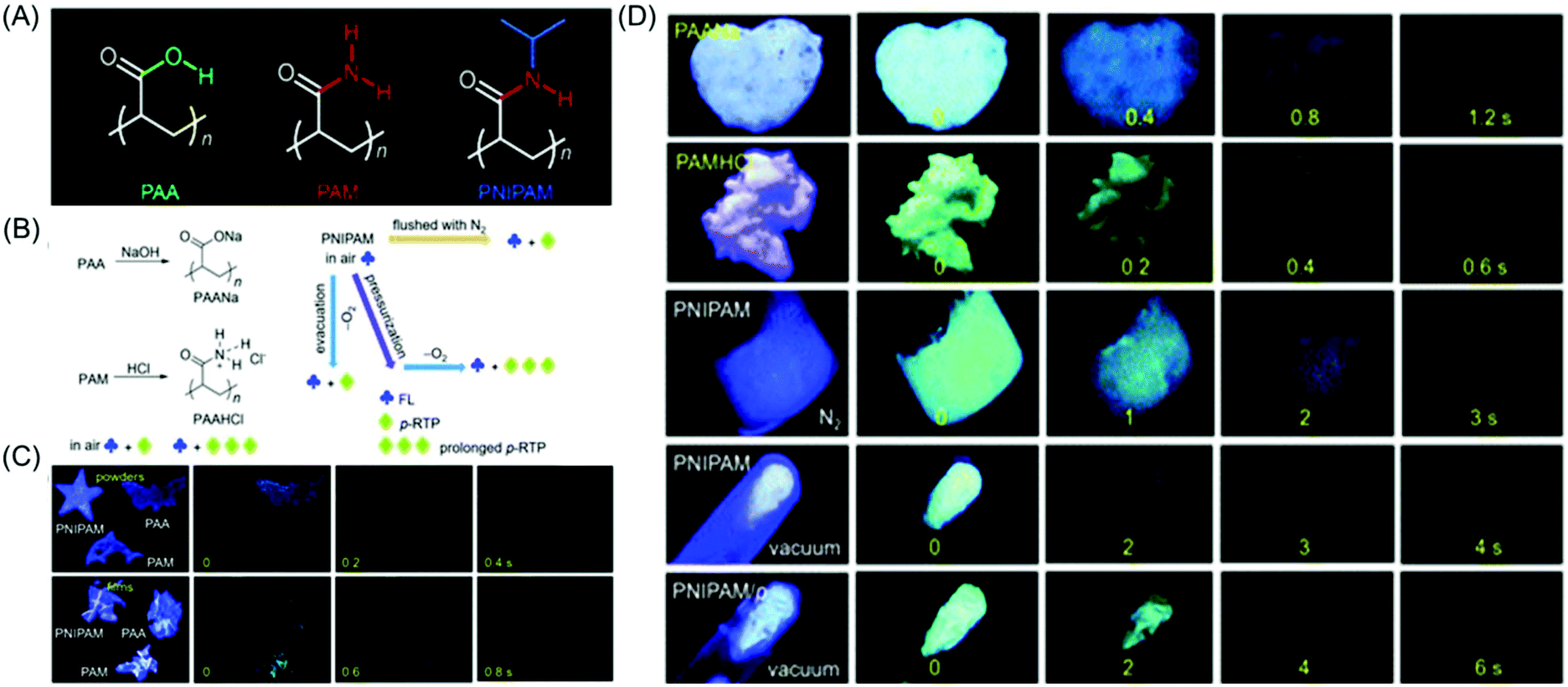 | ||
| Fig. 20 (A) Molecular structures of PAA, PAM and PNIPAM. (B) Schematic diagram of the variation of p-RTP features of the mentioned polymers. (C and D) Luminescence pictures of different films and powders before and after ceasing the UV excitation at 312 nm and room temperature. PNIPAM/p means PNIPAM powders are pressed at 2000 kg cm−2 pressure for one min. Reprinted with permission from ref. 113. Copyright (2019), Royal Society of Chemistry. | ||
Tang and coworkers reported the synthesis of luminescent well-defined polythioamide via catalyst-free atom economical multicomponent polymerization (MCP) reaction using elemental sulphur, aliphatic diamines, and aromatic diynes as monomers. The synthesized polythioamides were found to be emissive both in solution and in the powder state upon UV irradiation. They envisioned that the H-bonding between the secondary amine and thiocarbonyl group along with the n → π* interaction among thioamides causes the formation of “heterodox clusters” which may behave as an actual luminogen.114
Recently, Ling et al. achieved colorful ultralong clusterization-triggered phosphorescent (CTP) PAMs by controlling the polymerization requisites (Fig. 21A). Two reaction parameters, specifically initiators and polymerization temperatures, impose immense effects on the emission intensity and color of the CTP PAMs. It was found that water-soluble initiators ((NH4)2S2O8, and Na2SO3/(NH4)2S2O8) can cause quite high molecular weights of PAMs and strong persistent CTP than oil-soluble 2,2′-azobis(2-methylpropionitrile) (AIBN) initiated PAM (PAM1) and there was a correlation between the afterglow color and polymerization temperature. PAMs initiated at high temperature (PAM2 and PAM3) or aged at 100 °C (PAM5) displayed green ultralong RTP by the formation of side-by-side small clusters through intra-chain H-bonding among acrylamide (AM) units, and that PAM4 obtained at low polymerization temperature without heat treatment exhibited a yellow color afterglow via the formation of head-to-head large clusters mainly through inter-chain hydrogen bonding (Fig. 21B).115
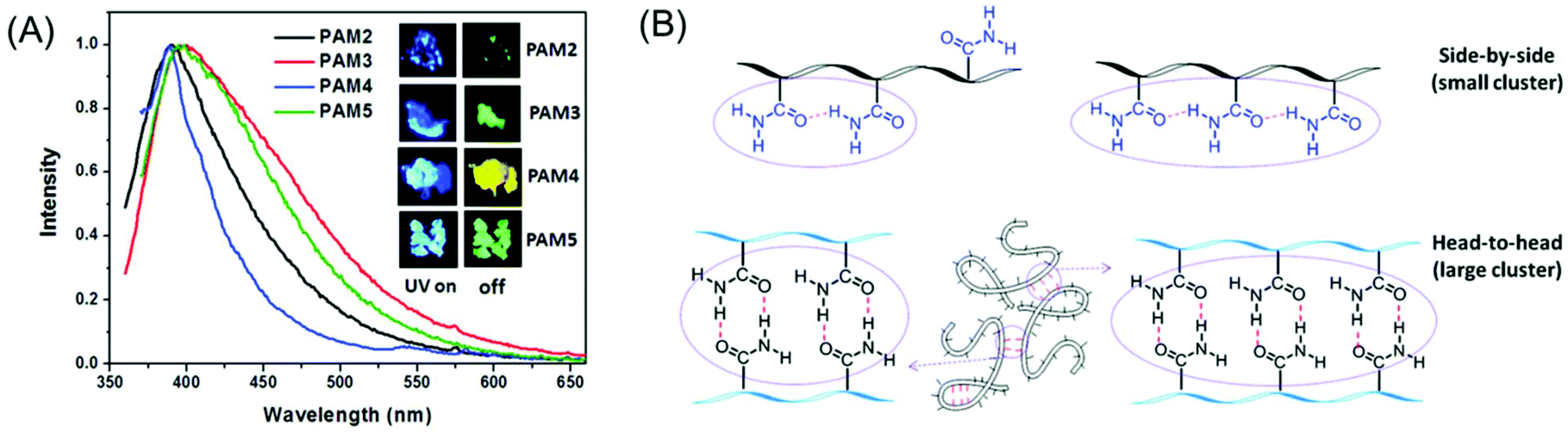 | ||
| Fig. 21 (A) Prompt fluorescence (λex = 347 nm) response of polyacrylamides (PAM2–PAM5). The inset shows their afterglow images while the 365 nm UV source was switched off. (B) Schematic representation demonstrating two different orientations of the polymer chain of PAMs: small and large clusters were formed by intra- and inter-molecular hydrogen bonding, respectively. Reprinted with permission from ref. 115. Copyright (2020), Royal Society of Chemistry. | ||
3. Conclusions and outlook
Intrinsic luminescence was serendipitously discovered from a few nonaromatic biomacromolecules and subsequently further systematic investigation was performed in some other natural polymers. These biopolymers include a few polysaccharides such as starch, cellulose, chitosan, dextran, glycogen, sodium alginate, etc., and some proteins such as BSA, OVA and HSA. Apart from natural polymers, other synthetic nonaromatic polymers, for example, PAMAMs, PAE, PEIs, PEA, PU, polyester, polycarbonate, PAN, PEG, PAA, PAM, PNIPAM, PNVP, and PNVCL lacking traditional aromatic chromophores also furnish intrinsic fluorescence. So the above libraries of luminescent polymers strongly indicate that the aromatic entities are not a key requirement to acquire light emission behavior from such polymers. In addition, a few polymers with isolated benzene ring structures, such as PET, PSS, PVP-S and PVP, showed exceptional fluorescence and phosphorescence behavior.The CTE mechanism was envisaged to justify the unorthodox photophysical characteristics of nonconventional luminogens. That is, the electron cloud overlap of nonconventional chromophores bearing lone-paired (n) electrons of O, N, P and S atoms and π orbitals of double and/or triple bonds or CO groups in the cluster with concurrent conformational rigidification can account for the unprecedented luminescence behavior of nontraditional luminophores. However, most of the reported NLPs normally emit excitation-independent blue luminescence upon irradiation with UV light and excitation-dependent multicolor emission with relatively low QYs at longer wavelength ranges. The aforementioned drawbacks would limit their application possibilities, particularly in the area of bioimaging, due to strong biological autofluorescence in the blue region and potential tissue damage on exposure to UV light.116 As the underlying photoluminescence mechanism of NLPs is not yet crystal clear, only very few reports could be made on the successful preparation of NLPs with long-wavelength emissions. Therefore, achieving intrinsic long-wavelength emission from NLPs upon excitation with visible light of higher QYs is highly desirable for unequivocal understanding of their luminescence mechanism/sources and for widening their practical enforcement.
Researchers before 2017 mostly focused on the fluorescence processes of the NLPs, but their fascinating phosphorescence behavior, particularly room temperature phosphorescence (RTP), was almost overlooked. However, in the last 3 years, special attention has been paid to glorious developments of RTP materials. Various strategies such as hydrogen bonding, ionic cross-linking, strong ionic interactions, nonmetal dative bond formation, etc. have been employed for designing many fascinating polymer-based RTP materials and strengthening their emission characteristics. It is noteworthy that another review article highlighting the phosphorescence properties of NLPs was published by Bryce et al.117 when this manuscript was in the evaluation stage. Generally, nonconventional polymers exhibited cryogenic phosphorescence or RTP mostly in the solid-state, so it is a great challenge to generate phosphorescence in the solution phase especially in aqueous medium.
The discovery of the cryogenic phosphorescence and RTP phenomenon in NLPs is a huge breakthrough and one of the most recent advancements. This will include another dimension within the field of bioimaging in the foreseeable future due to the longer excitation lifetime of phosphorescence relative to fluorescence, and therefore more sophisticated instruments could be potentially used or invented to look inside the cells and subsequently perceive the dynamics of life processes. To date, phosphorescent NLPs have been utilized mainly for information encryption. But owing to their high biocompatibility and less toxicity, these RTP polymers could be beneficial as in vitro and in vivo bioprobes to improve the detection sensitivity by diminishing the signal-to-noise ratio. Furthermore, organelle targeted (such as the nuclei, lysosomes, and mitochondria) bioimaging could be a research hotspot for low cytotoxicity NLPs. By highlighting the fluorescence and phosphorescence behavior of NLPs in this article, we firmly believe that this manuscript will help new and experienced researchers from broad research areas to advance their knowledge and to come up with new possibilities for expanding the horizon of these NLPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BS acknowledges the Council of Scientific and Industrial Research (CSIR), Government of India, for the senior research fellowship (SRF).Notes and references
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS.
- M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 348, 352–352 CrossRef.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- A. Mishra, C. Uhrich, E. Reinold, M. Pfeiffer and P. Bäuerle, Adv. Energy Mater., 2011, 1, 265–273 CrossRef CAS.
- H. Zhou, J. Mei, Y.-A. Chen, C.-L. Chen, W. Chen, Z. Zhang, J. Su, P.-T. Chou and H. Tian, Small, 2016, 12, 6542–6546 CrossRef CAS.
- X. Lou, L. Zhang, J. Qin and Z. Li, Chem. Commun., 2008, 5848–5850 RSC.
- Y. Yuan, S. Xu, X. Cheng, X. Cai and B. Liu, Angew. Chem., Int. Ed., 2016, 55, 6457–6461 CrossRef CAS.
- Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
- W. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 560–574 CrossRef.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- R. B. Restani, P. I. Morgado, M. P. Ribeiro, I. J. Correia, A. Aguiar-Ricardo and V. D. B. Bonifacio, Angew. Chem., Int. Ed., 2012, 51, 5162–5165 CrossRef CAS.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626–14637 CrossRef CAS.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- The review articles reported by Yuan et al., Tomalia et al., and Tang et al. mainly highlight the history and evolution of non-traditional luminescence, photophysical properties of metal clusters, natural/synthetic polymers and small molecules and their handful applications based on the existing literature focused at that time. Moreover, the above review articles did not discuss the phosphorescence properties of the NLPs as they have been developed very recently. Hence, in the present review article, we have provided a comprehensive and extensive discussion on the newly discovered photophysical properties including dual-emission (fluorescence and phosphorescence) of NLPs along with their underlying mechanisms and advanced applications which are transpiring at a rapid pace nowadays.
- Y. Y. Gong, Y. Q. Tan, J. Mei, Y. R. Zhang, W. Z. Yuan, Y. M. Zhang, J. Z. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1178–1181 CrossRef CAS.
- L.-L. Du, B.-L. Jiang, X.-H. Chen, Y.-Z. Wang, L.-M. Zou, Y.-L. Liu, Y.-Y. Gong, C. Wei and W.-Z. Yuan, Chin. J. Polym. Sci, 2019, 37, 409–415 CrossRef CAS.
- X. Dou, Q. Zhou, X. Chen, Y. Tan, X. He, P. Lu, K. Sui, B. Z. Tang, Y. Zhang and W. Z. Yuan, Biomacromolecules, 2018, 19, 2014–2022 CrossRef CAS.
- K. Bauri, M. Nandi and P. De, Polym. Chem., 2018, 9, 1257–1287 RSC.
- R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard III and B. Z. Tang, Polym. Chem., 2017, 8, 1722–1727 RSC.
- X. Chen, W. Luo, H. Ma, Q. Peng, W. Z. Yuan and Y. Zhang, Sci. China: Chem., 2018, 61, 351–359 CrossRef CAS.
- Q. Wang, X. Dou, X. Chen, Z. Zhao, S. Wang, Y. Wang, K. Sui, Y. Tan, Y. Gong, Y. Zhang and W. Z. Yuan, Angew. Chem., Int. Ed., 2019, 58, 12667–12673 CrossRef CAS.
- C. L. Larson and S. A. Tucker, Appl. Spectrosc., 2001, 55, 679–683 CrossRef CAS.
- W. Yang and C.-Y. Pan, Macromol. Rapid Commun., 2009, 30, 2096–2101 CrossRef CAS.
- Y.-J. Tsai, C.-C. Hu, C.-C. Chu and T. Imae, Biomacromolecules, 2011, 12, 4283–4290 CrossRef CAS.
- Y. Chen, L. Zhou, Y. Pang, W. Huang, F. Qiu, X. Jiang, X. Zhu, D. Yan and Q. Chen, Bioconjugate Chem., 2011, 22, 1162–1170 CrossRef.
- W. Li, J. Qu, J. Du, K. Ren, Y. Wang, J. Sun and Q. Hu, Chem. Commun., 2014, 50, 9584–9587 RSC.
- C. Zhan, X.-B. Fu, Y. Yao, H.-J. Liu and Y. Chen, RSC Adv., 2017, 7, 5863–5871 RSC.
- G. Wang, L. Fu, A. Walker, X. Chen, D. B. Lovejoy, M. Hao, A. Lee, R. Chung, H. Rizos, M. Irvine, M. Zheng, X. Liu, Y. Lu and B. Shi, Biomacromolecules, 2019, 20, 2148–2158 CrossRef CAS.
- W. Yang, S. Wang, R. Li, J. Xu and W. Hao, React. Funct. Polym., 2018, 133, 57–65 CrossRef CAS.
- D. Wu, Y. Liu, C. He and S. H. Goh, Macromolecules, 2005, 38, 9906–9909 CrossRef CAS.
- Y. Shen, X. Ma, B. Zhang, Z. Zhou, Q. Sun, E. Jin, M. Sui, J. Tang, J. Wang and M. Fan, Chem. – Eur. J., 2011, 17, 5319–5326 CrossRef CAS.
- H. Chen, W. Dai, J. Huang, S. Chen and X. Yan, J. Mater. Sci., 2018, 53, 15717–15725 CrossRef CAS.
- L. Yuan, H. Yan, L. Bai, T. Bai, Y. Zhao, L. Wang and Y. Feng, Macromol. Rapid Commun., 2019, 40, 1800658 CrossRef.
- L. Bai, H. Yan, L. Wang, T. Bai, L. Yuan, Y. Zhao and W. Feng, Macromol. Mater. Eng., 2020, 305, 2000126 CrossRef CAS.
- S.-F. Shiau, T.-Y. Juang, H.-W. Chou and M. Liang, Polymer, 2013, 54, 623–630 CrossRef CAS.
- L. Pastor-Perez, Y. Chen, Z. Shen, A. Lahoz and S.-E. Stiriba, Macromol. Rapid Commun., 2007, 28, 1404–1409 CrossRef CAS.
- Y. Fan, Y.-Q. Cai, X.-B. Fu, Y. Yao and Y. Chen, Polymer, 2016, 107, 154–162 CrossRef CAS.
- L. Shao, K. Wan, H. Wang, Y. Cui, C. Zhao, J. Lu, X. Li, L. Chen, X. Cui, X. Wang, X. Deng, X. Shi and Y. Wu, Biomater. Sci., 2019, 7, 3016–3024 RSC.
- L. Shao, Q. Li, C. Zhao, J. Lu, X. Li, L. Chen, X. Deng, G. Ge and Y. Wu, Biomaterials, 2019, 194, 105–116 CrossRef CAS.
- H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476–482 CrossRef CAS.
- H. Lu, J. Zhang and S. Feng, Phys. Chem. Chem. Phys., 2015, 17, 26783–26789 RSC.
- S. Niu, H. Yan, Z. Chen, L. Yuan, T. Liu and C. Liu, Macromol. Rapid Commun., 2016, 37, 136–142 CrossRef CAS.
- S. Niu, H. Yan, S. Li, C. Tang, Z. Chen, X. Zhi and P. Xu, J. Mater. Chem. C, 2016, 4, 6881–6893 RSC.
- S. Niu, H. Yan, S. Li, P. Xu, X. Zhi and T. Li, Macromol. Chem. Phys., 2016, 217, 1185–1190 CrossRef CAS.
- S. Niu, H. Yan, Z. Chen, S. Li, P. Xu and X. Zhi, Polym. Chem., 2016, 7, 3747–3755 RSC.
- Z. Zhang, S. Feng and J. Zhang, Macromol. Rapid Commun., 2016, 37, 318–322 CrossRef CAS.
- S. Niu, H. Yan, Z. Chen, Y. Du, W. Huang, L. Bai and Q. Lv, RSC Adv., 2016, 6, 106742–106753 RSC.
- L. Yang, L. Wang, C. Cui, J. Lei and J. Zhang, Chem. Commun., 2016, 52, 6154–6157 RSC.
- J. Cao, Y. Zuo, H. Lu, Y. Yang and S. Feng, J. Photochem. Photobiol., A, 2018, 350, 152–163 CrossRef CAS.
- Y. Zuo, Y. Zhang, Z. Gou and W. Lin, Sens. Actuators, B, 2019, 291, 235–242 CrossRef CAS.
- Y. Feng, T. Bai, H. Yan, F. Ding, L. Bai and W. Feng, Macromolecules, 2019, 52, 3075–3082 CrossRef CAS.
- L. Bai, H. Yan, T. Bai, Y. Feng, Y. Zhao, Y. Ji, W. Feng, T. Lu and Y. Nie, Biomacromolecules, 2019, 20, 4230–4240 CrossRef CAS.
- Y. Feng, H. Yan, F. Ding, T. Bai, Y. Nie, Y. Zhao, W. Feng and B. Z. Tang, Mater. Chem. Front., 2020, 4, 1375–1382 RSC.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS.
- M. Kopeć, M. Pikiel and G. J. Vancso, Polym. Chem., 2020, 11, 669–674 RSC.
- X. Liao, F.-J. Kahle, B. Liu, H. Bässler, X. Zhang, A. Köhler and A. Greiner, Mater. Horiz., 2020, 7, 1605–1612 RSC.
- X. Chen, X. Liu, J. Lei, L. Xu, Z. Zhao, F. Kausar, X. Xie, X. Zhu, Y. Zhang and W. Z. Yuan, Mol. Syst. Des. Eng., 2018, 3, 364–375 RSC.
- N. Jiang, G.-F. Li, B.-H. Zhang, D.-X. Zhu, Z.-M. Su and M. R. Bryce, Macromolecules, 2018, 51, 4178–4184 CrossRef CAS.
- N. Jiang, G. Li, W. Che, D. Zhu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2018, 6, 11287–11291 RSC.
- Z. Feng, W. Zhao, Z. Liang, Y. Lv, F. Xiang, D. Sun, C. Xiong, C. Duan, L. Dai and Y. Ni, ACS Appl. Mater. Interfaces, 2020, 12, 11005–11015 CrossRef.
- X. Chen, Q. Zhong, C. Cui, L. Ma, S. Liu, Q. Zhang, Y. Wu, L. An, Y. Cheng, S. Ye, X. Chen, Z. Dong, Q. Chen and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 30847–30855 CrossRef CAS.
- B. Liu, Y.-L. Wang, W. Bai, J.-T. Xu, Z.-K. Xu, K. Yang, Y.-Z. Yang, X.-H. Zhang and B.-Y. Du, J. Mater. Chem. C, 2017, 5, 4892–4898 RSC.
- B. Liu, B. Chu, Y.-L. Wang, Z. Chen and X.-H. Zhang, Adv. Opt. Mater., 2020, 8, 1902176 CrossRef CAS.
- W. Xu, B. Liu, X. Cai, M. Zhang, X.-H. Zhang and P. Yu, J. Appl. Polym. Sci., 2018, 135, 46723 CrossRef.
- Y. Du, H. Yan, W. Huang, F. Chai and S. Niu, ACS Sustainable Chem. Eng., 2017, 5, 6139–6147 CrossRef CAS.
- W. Huang, H. Yan, S. Niu, Y. Du and L. Yuan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3690–3696 CrossRef CAS.
- Y. Du, Y. Feng, H. Yan, W. Huang, L. Yuan and L. Bai, J. Photochem. Photobiol., A, 2018, 364, 415–423 CrossRef CAS.
- X. Chen, Z. He, F. Kausar, G. Chen, Y. Zhang and W. Z. Yuan, Macromolecules, 2018, 51, 9035–9042 CrossRef CAS.
- J. Wang, N. Wang, G. Wu, S. Wang and X. Li, Angew. Chem., Int. Ed., 2019, 58, 3082–3086 CrossRef CAS.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- T. Ogoshi, H. Tsuchida, T. Kakuta, T. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- S. Cai, H. Ma, H. Shi, H. Wang, X. Wang, L. Xiao, W. Ye, K. Huang, X. Cao, N. Gan, C. Ma, M. Gu, L. Song, H. Xu, Y. Tao, C. Zhang, W. Yao, Z. An and W. Huang, Nat. Commun., 2019, 10, 4247 CrossRef.
- L. Zou, X. Qin, H. Sun, S. Wang, W. Ding, Y. Liu, C. Wei, B. Jiang and Y. Gong, RSC Adv., 2019, 9, 36287–36292 RSC.
- H. Wang, H. Shi, W. Ye, X. Yao, Q. Wang, C. Dong, W. Jia, H. Ma, S. Cai, K. Huang, L. Fu, Y. Zhang, J. Zhi, L. Gu, Y. Zhao, Z. An and W. Huang, Angew. Chem., 2019, 58, 18776–18782 CrossRef CAS.
- Q. Wu, H. Xiong, Y. Zhu, X. Ren, L.-L. Chu, Ye.-F. Yao, G. Huang and J. Wu, ACS Appl. Polym. Mater., 2020, 2, 699–705 CrossRef CAS.
- Z.-F. Liu, X. Chen and W. J. Jin, J. Mater. Chem. C, 2020, 8, 7330–7335 RSC.
- H. Ahn, J. Hong, S. Y. Kim, I. Choi and M. J. Park, ACS Appl. Mater. Interfaces, 2015, 7, 704–712 CrossRef CAS.
- E. Zhao, J. W. Y. Lam, L. Meng, Y. Hong, H. Deng, G. Bai, X. Huang, J. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64–71 CrossRef CAS.
- Z. Guo, Y. Ru, W. Song, Z. Liu, X. Zhang and J. Qiao, Macromol. Rapid Commun., 2017, 38, 1700099 CrossRef.
- C. Hu, Z. Guo, Y. Ru, W. Song, Z. Liu, X. Zhang and J. Qiao, Macromol. Rapid Commun., 2018, 39, 1800035 CrossRef.
- C. Hu, Y. Ru, Z. Guo, Z. Liu, J. Song, W. Song, X. Zhang and J. Qiao, J. Mater. Chem. C, 2019, 7, 387–393 RSC.
- C. Shang, N. Wei, H. Zhuo, Y. Shao, Q. Zhang, Z. Zhang and H. Wang, J. Mater. Chem. C, 2017, 5, 8082–8090 RSC.
- C. Shang, Y. Zhao, N. Wei, H. Zhuo, Y. Shao and H. Wang, Macromol. Chem. Phys., 2019, 220, 1900324 CrossRef CAS.
- C. Shang, Y. Zhao, J. Long, Y. Ji and H. Wang, J. Mater. Chem. C, 2020, 8, 1017–1024 RSC.
- S. Wang, B. Wu, F. Liu, Y. Gao and W. Zhang, Polym. Chem., 2015, 6, 1127–1136 RSC.
- M. G. Mohamed, K.-C. Hsu, J.-L. Hong and S.-W. Kuo, Polym. Chem., 2016, 7, 135–145 RSC.
- Y. Ru, X. Zhang, W. Song, Z. Liu, H. Feng, B. Wang, M. Guo, X. Wang, C. Luo, W. Yang, Y. Li and J. Qiao, Polym. Chem., 2016, 7, 6250–6256 RSC.
- B. Saha, K. Bauri, A. Bag, P. K. Ghorai and P. De, Polym. Chem., 2016, 7, 6895–6900 RSC.
- K. Bauri, B. Saha, J. Mahanti and P. De, Polym. Chem., 2017, 8, 7180–7187 RSC.
- B. Saha, N. Choudhury, A. Bhadran, K. Bauri and P. De, Polym. Chem., 2019, 10, 3306–3317 RSC.
- B. Saha, N. Choudhury, S. Seal, B. Ruidas and P. De, Biomacromolecules, 2019, 20, 546–557 CrossRef CAS.
- K. G. Goswami, B. Saha, S. Mete and P. De, Macromol. Chem. Phys., 2018, 219, 1800398 CrossRef.
- K. G. Goswami, B. Saha and P. De, J. Macromol. Sci., Pure Appl. Chem., 2020, 57, 675–683 CrossRef CAS.
- J. Yan, B. Zheng, D. Pan, R. Yang, Y. Xu, L. Wang and M. Yang, Polym. Chem., 2015, 6, 6133–6139 RSC.
- J. K. Fink, in High Performance Polymers, William Andrew Inc., Norwich, NY, 1st edn, 2008, ch. 14, p. 449 Search PubMed.
- H.-M. Wang and S.-H. Hsiao, Polym. Chem., 2010, 1, 1013–1023 RSC.
- J. Yan, R. Wang, D. Pan, R. Yang, Y. Xu, L. Wang and M. Yang, Polym. Chem., 2016, 7, 6241–6249 RSC.
- R.-R. Wang, J.-J. Yan, R.-L. Yang, D.-H. Pan, W. Li, Y.-P. Xu, L.-Z. Wang, X.-Y. Wan and M. Yang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2053–2060 CrossRef CAS.
- J.-J. Yan, X.-Y. Wang, M.-Z. Wang, D.-H. Pan, R.-L. Yang, Y.-P. Xu, L.-Z. Wang and M. Yang, Biomacromolecules, 2019, 20, 1455–1463 CrossRef CAS.
- B. He, J. Zhang, H. Zhang, Z. Liu, H. Zou, R. Hu, A. Qin, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2020, 53, 3756–3764 CrossRef CAS.
- X. Zhou, W. Luo, H. Nie, L. Xu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 4775–4779 RSC.
- J. Huang, X. Geng, C. Peng, T. Z. Grove and S. R. Turner, Macromol. Rapid Commun., 2018, 39, 1700530 CrossRef.
- J.-J. Yan, Z.-K. Wang, X.-S. Lin, C.-Y. Hong, H.-J. Liang, C.-Y. Pan and Y.-Z. You, Adv. Mater., 2012, 24, 5617–5624 CrossRef CAS.
- R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard III and B. Z. Tang, Polym. Chem., 2017, 8, 1722–1727 RSC.
- C. Liu, Q. Cui, J. Wang, Y. Liu and J. Chen, Soft Matter, 2016, 12, 4295–4299 RSC.
- J. Xu, D. Luo, X. Yin, H. Zhang, L. Wang and H. Wang, Macromol. Chem. Phys., 2017, 218, 1700410 CrossRef.
- G. Song, Y. Lin, Z. Zhu, H. Zheng, J. Qiao, C. He and H. Wang, Macromol. Rapid Commun., 2015, 36, 278–285 CrossRef CAS.
- B. Saha, B. Ruidas, S. Mete, C. D. Mukhopadhyay, K. Bauri and P. De, Chem. Sci., 2020, 11, 141–147 RSC.
- Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Macromol. Rapid Commun., 2018, 39, 1800528 CrossRef.
- H.-X. Xu, Y. Tan, D. Wang, X.-L. Wang, W.-L. An, P.-P. Xu, S. Xu and Y.-Z. Wang, Soft Matter, 2019, 15, 3588–3594 RSC.
- Z. Zhao, X. Chen, Q. Wang, T. Yang, Y. Zhang and W. Z. Yuan, Polym. Chem., 2019, 10, 3639–3646 RSC.
- Q. Zhou, Z. Wang, X. Dou, Y. Wang, S. Liu, Y. Zhang and W. Z. Yuan, Mater. Chem. Front., 2019, 3, 257–264 RSC.
- W. Li, X. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Macromolecules, 2015, 48, 7747–7754 CrossRef CAS.
- S. Wang, D. Wu, S. Yang, Z. Lin and Q. Ling, Mater. Chem. Front., 2020, 4, 1198–1205 RSC.
- L. Pan, S. Sun, L. Zhang, K. Jiang and H. Lin, Nanoscale, 2016, 8, 17350–17356 RSC.
- N. Jiang, D. Zhu, Z. Su and M. R. Bryce, Mater. Chem. Front., 2020 10.1039/D0QM00626B.
Footnote |
| † These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2020 |