
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacromolecular design and preparation of polymersomes
James
Lefley
,
Christopher
Waldron
and
C. Remzi
Becer
 *
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: remzi.becer@warwick.ac.uk; Web: http://www.becergroup.com Web: http://www.twitter.com/remzibecer
First published on 19th October 2020
Abstract
From drug delivery to nanoreactors and protocells, polymersomes have gained considerable interest from researchers due to their novel applications. However, one of the main challenges is the selection of the most appropriate synthetic route. It is crucial to consider factors such as desired application, functionalisation, and environment of the polymersome when designing a synthetic route. This review will explore the current scope of polymersome synthesis and preparation methods, and will conclude by highlighting the most recent reports on polymersome related systems such as virus-like nanoparticles. From the choice of monomers, to polymerisation techniques, and to preparation methods used, we aim for this review to be utilised as a tutorial guide for the synthesis of a range of polymersomes, each with varying characteristics and applications.
1. Introduction
Compartmentalisation is crucial for life to occur.1 Almost all cells, be it eukaryotic or prokaryotic, consist of an outer protective vesicle containing the vital organelles needed to carry out a specific role in the body. Compartmentalisation provides a secure environment to facilitate essential biological processes such as DNA replication and aerobic respiration.2 Billions of years of evolution have allowed eukaryotic and prokaryotic cells to perfect the art of cellular compartmentalisation. As a result, such systems have a highly complex and specialised structure, with each organelle existing as its own entity within the cell, e.g. nucleus, mitochondria, lysosomes etc. In 1960s, artificial vesicular cell mimics consisting of a phospholipid bilayer, known as liposomes (Scheme 1), were first discovered.3,4 These vesicles contain an aqueous solution core and can be used as a mode of transport for small hydrophilic/hydrophobic molecules. The liposome phospholipid bilayer can fuse with cell membranes and subsequently release its aqueous payload into a cell. This discovery was promising as new methods were being developed for the delivery of drugs and other biological molecules to cells within the body. However, liposomes are prone to stability and permeability issues.5 | ||
| Scheme 1 2D and 3D representations of a liposome and a polymersome made from their respective subunits.9 Adapted from E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 40. | ||
In 1999, the term polymersome was first used to describe a vesicle formed from the self-assembly of an amphiphilic block copolymer by the Discher group.6 However, polymer vesicles have been known for some time. With pioneering work being conducted by the Eisenberg7 and Meijer groups8 in 1995. Polymersomes are similar to liposomes in terms of function and morphology but differ in chemistry. The vesicle bilayer is formed from hydrophilic and hydrophobic blocks and encapsulates an aqueous solution core like liposomes. However, by using synthetic monomer building blocks, characteristics such as stability, biodegradability and type of release mechanism can be controlled.10–12 Furthermore, surface modification of polymersomes with certain sugars/proteins can direct the vesicle towards specific cells and targeted drug delivery can be achieved.13 As mentioned previously, drug delivery is the most widely researched application of polymersomes.14 But polymersomes have been synthesised for use in gene delivery in biological applications as well as being used as nanoreactors in enzyme catalysis and cascade reactions.15 More recently, polymersomes have found significant importance in the establishment of cellular biomimetics. Here, polymersomes are utilised as artificial organelles and/or cells. Their functions as cellular biomimetics include the uptake and release of specific cargoes, mimicking living cellular function.16–18
It is worth mentioning the theory behind the formation of a polymersome from its corresponding block copolymer. Essentially, the amphiphilic nature of block copolymers enables self-assembly of the linear chains into nanoparticles of varying morphologies. In almost all cases, a macroinitiator consisting of a pre-synthesised hydrophilic polymer chain is polymerised with a hydrophobic monomer. As the hydrophobic block increases in the length, the overall amphiphilicity of the block copolymer changes as does the morphology of the nanoparticle (Scheme 2). Micelles, worm-like, rod-like and vesicles are achieved with an increasing DP of the hydrophobic block. The relative volume fractions of the hydrophilic and hydrophobic block are described by the packing parameter P.20
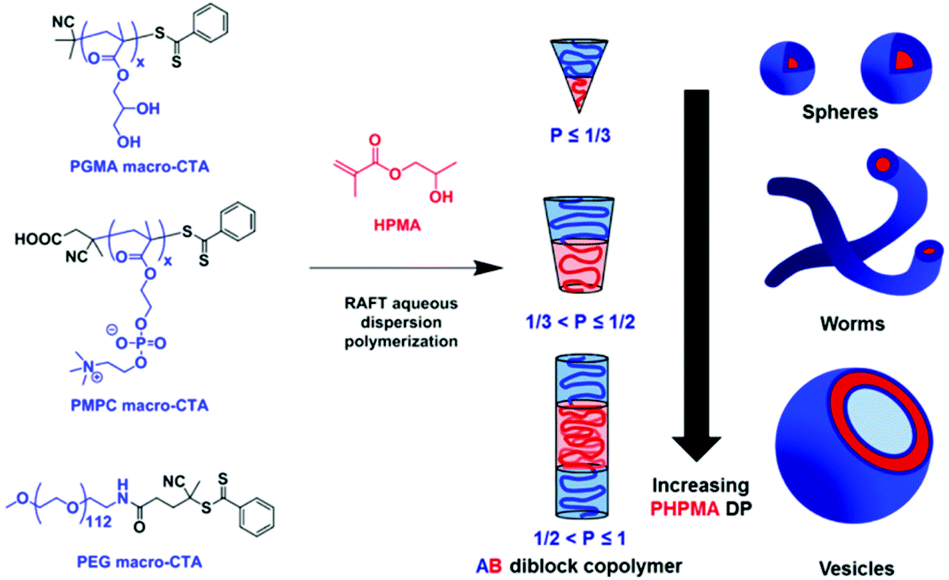 | ||
| Scheme 2 The RAFT dispersion polymerisation of 2-hydroxypropyl methacrylate (HPMA) as described by Armes and coworkers. Here, a variety of RAFT macroinitiators illustrate how the varying lengths of the PHPMA block, and therefore packing parameter P, dictate the morphology of the nanoparticle.19 Adapted from S. Armes, J. Am. Chem. Soc., 2014, 136, 10174. | ||
Many key review articles focusing on the applications of smart polymersomes have been published in the past decade. Notably, those published by Meier and van Hest have done well to highlight the current research of the biological and biomedical applications.5,21 However, after conducting an extensive literature search, it was found that there is a lack of review articles highlighting current research in the synthesis and preparation of polymersomes. Therefore, we present a review that aims to provide an overview of the chemistry and processing of polymersomes. This review will introduce the choice and variety of monomers that can be used for the synthesis of degradable and non-degradable polymersomes. Next, the types of polymerisation and preparation techniques are discussed with detailed examples. The review will conclude by exploring other nanoparticle-like systems similar to polymersomes such as virus-like nanoparticles (VLNPs). From the combination of monomers to the polymerisation and preparation methods used (Scheme 3), this review will serve as a comprehensive guide in synthesising a variety of polymersomes for multiple applications.
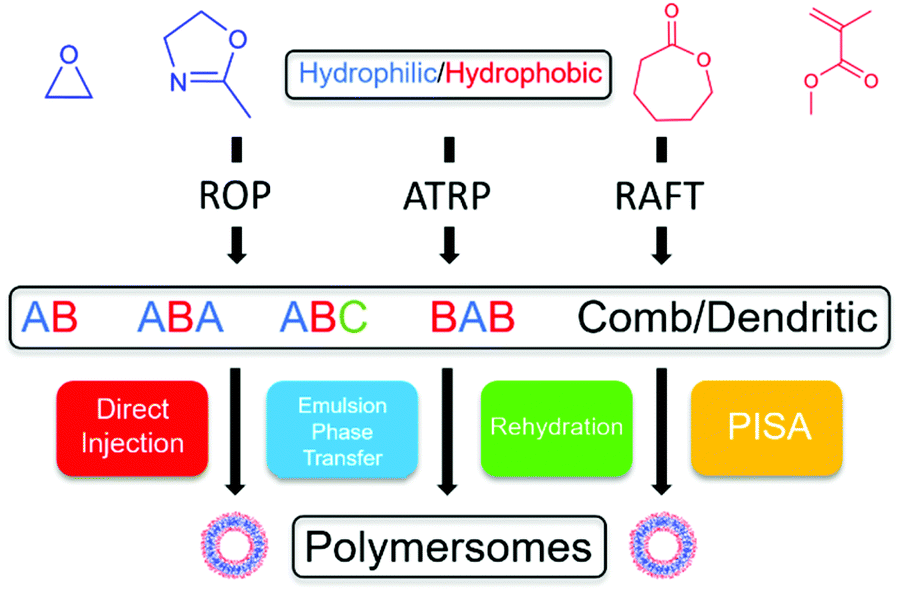 | ||
| Scheme 3 A flowchart showing the various combinations of polymerisation and preparation techniques to synthesise polymersomes. | ||
2. The chemistry of polymersomes
2.1 The choice of monomers
The main characteristic that defines polymersomes is the amphiphilic block copolymer that forms the bilayer of the vesicle. This is achieved by incorporating, at a minimum, a hydrophilic homopolymer block and a hydrophobic homopolymer block. By controlling properties such as degrees of polymerisation (DP), composition and ratio of the hydrophilic to hydrophobic blocks, the desired vesicle morphology can be obtained.In general, polymersomes can be split into two categories. Degradable and non-degradable polymersomes. The degree of degradability of a polymersome can be dictated by the choice of monomers used. Non-degradable polymersomes typically include conjugated/aromatic hydrocarbon blocks such as poly(ethylethylene) (PEE),6 poly(butadiene) (PBD)22 and poly(styrene) (PSt)22–26 as well as poly(dimethylsiloxane) (PDMS).27–29 These polymers form the hydrophobic block of the copolymers but lack any bonds susceptible to hydrolytic cleavage. As a result, polymersomes synthesised with any of these monomers are considered non-degradable and are not biocompatible. Research on non-biocompatible polymersomes for biomedical applications has largely been disregarded as biocompatible vesicles are needed.
Polymersomes made from water soluble monomers including poly(ethylene glycol) (PEG),30–34 poly(2-methyloxazoline) (PMOXA),35 poly(acrylic acid) (PAA),36 poly(ε-caprolactone) (PCL)32–34 and poly(lactic acid) (PLA)30,31,37 are regarded as degradable. In these examples, the ester and carbonate bonds within the polymer chains can be hydrolytically cleaved by the addition of a small molecule, usually water. PEG, PMOXA and PAA homopolymers form the hydrophilic blocks in degradable polymersomes whereas PCL and PLA form the hydrophobic blocks. PEG is the most common and well researched monomer used in synthesising polymersomes due to its proven biocompatibility, water solubility and very low toxicity.38–40
Current research on degradable polymersomes has led to the production of a catalogue of polypeptide-based polymersomes.41,42 Advantages in using amino acid-based monomers include complete biocompatibility, almost zero toxicity and easier functionalisation due to the variety of amino acids functional groups in the polymer chain. The review by Iatrou et al. in 2018, describes the properties and characteristics of polypeptide-based polymersomes and provides an excellent summary of some recent examples of these polymersomes.43
Examples of degradable and non-degradable copolymers are summarised in Table 1.
| Polymers | Degradable | Polymerisation technique | Preparation method | Encapsulated payload | Ref. |
|---|---|---|---|---|---|
| PEG-b-PEE | No | Anionic/ hydrogenation | Film rehydration | N/A | 6 |
| iPSt-b-PBD | No | Anionic | Self-assembly | N/A | 22 |
| PSt-b-PEG | No | Anionic | Phase inversion | N/A | 23 |
| PSt-b-NIPAM-co-SPO | No | RAFT | Double emulsion (w/o/w) | N/A | 24 |
| PVBC-b-PS-b-PVBC | No | RAFT | Self-assembly | N/A | 25 |
| PSt-b-Dex | No | ATRP | Self-assembly | N/A | 26 |
| PDMS-b-PMOXA | No | Cationic | Film rehydration | Paclitaxel, carboxyfluorescein | 27–29 |
| pPEGMA-S-S-PLA | Yes | ATRP | Nanoprecipitation | Doxorubicin | 30 |
| PEG-b-PLA | Yes | Ring opening | Film rehydration | BSA and ASNase | 31 |
| PEG-b-PCL | Yes | Ring opening | Film rehydration | Quercetin | 32 |
| PMOXA-b-PDMS-b-PMOXA | Yes | Ring opening | Film rehydration | Neurotrophins and Curcumin | 35 |
| PLA-b-PEG-b-PLA | Yes | Ring opening | Double emulsion (w/o/w) | Lisonopril and atorvastatin | 34 |
| PEG-b-PLeu-b-PLGA | Yes | Ring opening | Film rehydration | Doxorubicin | 41 |
| PLGA-b-PPhe | Yes | Ring opening | Phase inversion | N/A | 42 |
2.2 Polymerisation techniques
Living polymerisation techniques are characterised by the absence of chain termination and chain transfer reactions during polymerisation, as well as having a much larger rate of initiation compared to the rate of propagation.44 As a result, all chains grow at a near constant rate owing to very little distribution in the molecular weights of the polymer chains. Living polymerisation techniques are mainly employed in the synthesis of block copolymers as the lack of chain termination in these reactions allow polymer blocks to be synthesised in stages by the sequential addition of different monomers. Living polymerisation techniques such as ROP, RAFT and ATRP allow extensive control over parameters such as the composition and block lengths of the hydrophilic and hydrophobic chains.44,45 Seen as the assembly of block copolymers into polymersomes vesicles is entirely dictated by these parameters, living polymerisation is paramount in the synthesis of well-defined amphiphilic block copolymers. The advantages and challenges of each technique will be explored in this section.In 2020, Meier et al. reported the synthesis of a new biocompatible amphiphilic block copolymer poly(ethylene oxide)-block-poly(2-(3-ethylheptyl)-2oxazoline) (PEO-b-PEHOx) via an optimised microwave-assisted CROP procedure.49 Meier and co-workers opted for a nosylated PEO macroinitiator as small molecule nosylates (Nos) show faster initiation and better reactivity compared to tosylates.50 The final optimised procedure used Nos-PEO in chlorobenzene at 140 °C as these conditions gave the lowest dispersity and highest conversion. The two different preparation methods used gave complex nanoparticle morphologies. Film rehydration of PEO-b-PEHOx gave multi-compartment micelles (MCMs) with increasing PEHHOx block length and the solvent switch method gave the morphologies of pseudo-vesicles and yolk/shell nanoparticles at large PEHOx block lengths. In 2019, van Hest et al. reported the step-wise ROP synthesis of poly (ethylene glycol)-b-poly(caprolactone-g-trimethylene carbonate) (PEG-b-PCL-g-PTMC) yielding ionically-induced nanoworms as asymmetric therapeutic carriers.51 This is a perfect example of sequential addition of different monomers producing a well-defined triblock copolymer. Each step produced an ever growing macroinitiator until finally reacted with the final monomer, TMC-Q (Scheme 4).
 | ||
| Scheme 4 How molecular programming of the terpolymer achieves the ionically-induced morphology switch from micelles to nanoworms. (a) the structure of the (PEG-b-PCL-g-PTMC) terpolymer and the morphology change by increasing the ionic strength of the solution with NaCl. (b) The electrostatic interaction and membrane insertion between the terpolymer and cancer cells.51 Adapted from J. C. M. van Hest, Small, 2019, 15, 8. | ||
The inclusion of the quaternary ammonium-TMC (PTMC-Q) chain end allows an ionically-induced morphology change from micelles to nanoworms by varying the ionic strength of the solution with NaCl. The morphology change is ultimately driven by the adjustment of the interchain interactions thus altering the packing parameter of the block copolymer. These nanoworms can be loaded with chemotherapeutics such as doxorubicin (DOX) and can be used for targeted drug delivery. The cationic nature of the terpolymer facilitates greater adhesion and membrane penetration with negatively charged cancer cells.
As mentioned previously, ROP is an effective method in synthesising block copolymers that incorporate heteroatoms in the polymer backbone. In 2020, Wang et al. synthesised a dual stimuli-responsive, polyorganophosphazene (POP) diblock copolymer poly((mPEG-SSamino)(N,N-diisopropylethylene diamino)phosphazene) (PPDP) via thermal ring opening polymerisation to encapsulate and achieve stimuli-responsive release of DOX.52 The hydrophobic block of this polymer consists of an entirely heteroatom backbone of nitrogen and phosphorus atoms. A PEG-S-S-NH2 macroinitiator was first synthesised in a two-step process. Using this macroinitiator, the thermal ring opening of Hexachlorocyclotriphosphazene (six membered nitrogen and phosphorus cyclic ring) proceeds at 250 °C with catalytic amounts of AlCl3. The S–S disulphide bond is redox-responsive whereas the diisopropylethylenediamino (DPA) moiety accounts for the pH-responsiveness. POPs have gathered a lot of attention recently for drug delivery due to their excellent biocompatibility and degradability into non-toxic nitrogen/phosphorus-based products.53,54
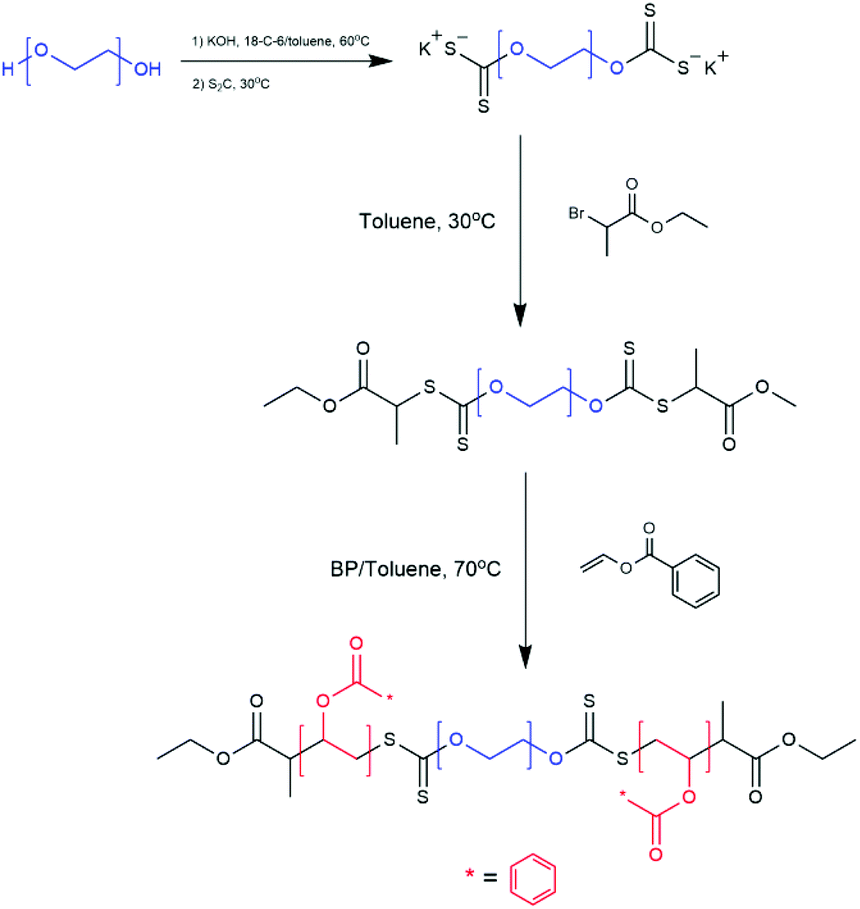 | ||
| Scheme 5 Synthetic Route of PVBz-b-PEG-b-PVBz via RAFT dispersion polymerisation.61 Adapted from M. S. Cortizo et al., J. Nanopart. Res., 2018, 20, 67. | ||
Using RAFT dispersion polymerisation, Cheng et al. synthesised a self-assembling diblock copolymer poly(ethylene oxide)-b-poly(styrene) PEO-b-PSt which produces ultra-thick membrane, surfactant-resistant polymersomes.58 A PEO-CTA macroinitiator was used to polymerise the hydrophobic PSt block. Methanol was chosen as the choice of solvent for the reaction as it solubilises the PEO-CTA macroinitiator and Styrene well but solubilises the PSt block poorly. The poor solubility of PSt is the driving force for self-assembly. Low PSt block lengths gave micellar and worm-like morphologies whereas larger block lengths gave the desired vesicle morphology for polymersomes. The large hydrophobic PSt block lengths also gave the polymersomes ultra-thick membranes. As a result, the resistance to surfactant solubilisation of the polymersomes was greatly improved, reporting less than 7% leakage of the encapsulated cargo after 30 days in a surfactant solution.
Mantovani and coworkers also used RAFT dispersion polymerisation to produce two well define amphiphilic block copolymers using a PEG acrylate macroinitiator and 2-(acryl-oyloxy)ethyl-3-chloro-4-hydroxybenzoate (ACH) or 2-(3-chloro-4-hydroxybenzamido)ethyl acrylate(CHB).59 Parameters such as choice of solvent, CTA and temperature were carefully selected to ensure the ‘livingness’ of the growing polymer chains. The one-pot RAFT dispersion polymerisation method, developed by Perrier et al.,60 achieved high monomer conversions of ≥94% with vesicle morphology being obtained at ACH and CHB hydrophobic block lengths of DP = 34 (f = 35% ± 10). The vesicles were loaded with terbinafine and cyanocobalamin to determine their potential as nanocarriers of hydrophilic and hydrophobic drugs.
Cortizo et al. synthesised a triblock copolymer poly(vinylbenzoate)-b-poly(ethylene glycol)-b-poly(vinyl benzoate) (PVBz-b-PEG-b-PVBz) via RAFT polymerisation.61 A PEG macroinitiator was synthesised bearing two thiocarbonylthio functional groups at either end of the hydrophilic PEG block. Vinyl benzoate was then polymerised from both ends of the macroinitiator producing a BAB triblock copolymer as seen in Scheme 5.61 The formation of vesicles via self-assembly in THF/water solution was reported when the triblock copolymer had a hydrophilic PEG weight fraction f, of f = 30–40%.
Theato et al. have developed a unique ‘breathable’ polymersome from the triblock copolymer poly[(ethylene glycol)methyl ether]-b-poly(N,N-dimethylamino ethyl methacrylate-co-2,2,2-trifluoroethyl methacrylate)-b-poly(4-(4-methoxy-phenylazo)phenoxy methacrylate) (PEG-b-P(DMAEMA-co-TFEMA)-b-PMEPPMA) via step-wise ATRP.64 A PEGylated ATRP initiator was used to polymerise DMAEMA and TFEMA to form the first statistical polymer block. The resulting diblock was used as a macroinitiator to polymerise MEPPMA to form the triblock copolymer. The amphiphilic nature of the polymer allowed self-assembly into vesicles which exhibited triple stimuli responsive character to CO2, O2 and light (Scheme 6). The membrane permeability of the vesicle can be fine-tuned by changing a single stimulus or a combination of stimuli.
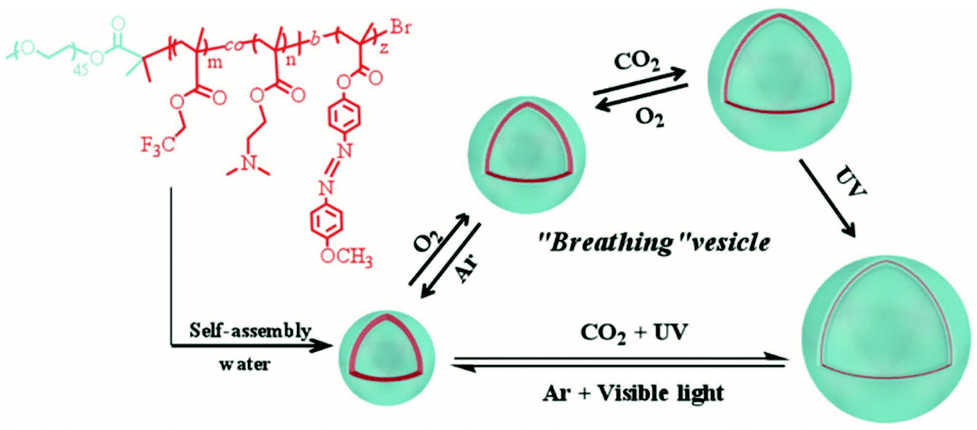 | ||
| Scheme 6 The structure of the triblock copolymer (PEG-b-P(DMAEMA-co-TFEMA)-b-PMEPPMA) showing self-assembly and its tri-stimuli-responsive ‘breathing’ behaviour.64 Adapted from P. Theato et al., Macromol. Rapid Commun., 2018, 39, 6. | ||
In 2020, Liu et al. reported the synthesis of novel cationic nanoparticles (cNPs) from the coself-assembly of poly(lactic-co-gly-colic acid)-b-poly(2-(dimethylamino)ethyl methacrylate) (PLGA-b-PDMA) and poly(lactic-co-glycolic acid)-b-poly(ethylene glycol) (PLGA-b-PEG) block copolymers via ATRP.65 The use of a PLGA macroinitiator was employed in the homopolymerisation of the PDMA and PEG blocks separately. The two diblock copolymers coself-assemble to form cNPs that bind to cell-free DNA (cfDNA) within inflamed joints and inhibits rheumatoid arthritis. PDMA blocks provides the cationic nature of the polymeric nanoparticles which binds to the negatively charged DNA. Meanwhile, PEG blocks increase accumulation and the retention time of cNPs in the joints thus lowering the administration frequency.
Lee et al. used a disulphide bridge-containing bifunctional initiator to produce a triblock copolymer poly(oligo(ethylene glycol)methyl ether methacrylate)-b-poly(2-(diisopropyl amino)ethyl methacrylate)-b-poly(2(methacryloyloxy)ethyl phosphorylcholine) POEGMA-b-PDPA-b-PMPC for DOX encapsulation and release.66 The POEGMA, PDPA and PMPC blocks were synthesised by polymerisation of the respective monomers to full conversion then sequential addition of the next monomer in the sequence. The pH-sensitive nature of PDPA allows pH-responsive release of DOX.
2.3 Block copolymer architectures
To further add diversity to the synthetic route, a vast choice of block copolymer architectures has been employed in the formation of polymersomes. To recap, the main feature of a polymersome is the amphiphilicity of the bilayer membrane. The copolymers assemble so that the hydrophilic blocks form the exterior and interior layers whilst the hydrophobic blocks form the middle layer. These well-defined hydrophilic and hydrophobic regions are utilised for the encapsulation and transportation of various hydrophilic and hydrophobic drugs. AB, ABA, BAB, ABC block copolymers are the main architectures that are synthesised in literature. With blocks A and C being hydrophilic and block B being hydrophobic. Other alternative architectures such as comb and dendrimersomes exist too but are less studied. | ||
| Scheme 7 The four main architectures of block copolymers that form polymersomes. Hydrophilic blocks are blue and green whilst hydrophobic blocks are coloured in red.70 Adapted from J. S. Lee and J. Feijen, J. Control. Release, 2012, 161, 473–483. | ||
Dendrimersomes are a unique class of synthetic vesicles with the term being coined by Percec in 2005.86 They are formed from the self-assembly of low generation (1st or 2nd) amphiphilic Janus dendrimers consisting of a hydrophilic and hydrophobic side (Scheme 8). Percec and co-workers are considered the pioneers of dendrimersomes and have contributed immensely to the field with a number of seminal papers and reviews being published on them.87–89 In one of his more recent publications, Percec et al. have developed amphiphilic dendrimersomes that engulf bacteria (Pseudomonas aeruginosa) via endocytosis mimicking the role of macrophages of the body. The development of these synthetic protocells that are capable of carrying out complex biological processes in collaboration with living matter is another step closer to the development of fully synthetic and artificial cells.
 | ||
| Scheme 8 A diagram illustrating the formation and structure of dendrimersomes. Blue dendrons denote hydrophilic blocks whilst green dendrons denote hydrophobic blocks. Hydrophilic and/or hydrophobic cargo can be encapsulated in the respective blocks of the dendrimersomes.89 Adapted from Percec et al., Chem. Rev., 2017, 117, 6538–6631. | ||
3. Preparation of polymersomes
The next stage in the formation of polymersomes is choosing which preparation method to use. Many have been reported over the years but essentially, they can be classified into two distinct categories, solvent-free and solvent displacement. Solvent free methods include film rehydration, PISA and electroformation where the amphiphilic block copolymers are formed in aqueous solution. Solution displacement methods such as direct injection, emulsion phase transfer and microfluidics involve the use of organic solvent to dissolve the block copolymers. The addition of this solvent-polymer solution in aqueous solution and subsequent removal of the organic solvent produces polymersomes. Properties such as hydrodynamic radius and size distribution can be controlled using these methods. In recent times, it has been found that the shape of polymersomes can also be controlled very effectively. Polymersomes were long thought of as spherical entities but in the 2019 review by Stenzel and co-workers, they highlight many different examples of non-spherical polymersomes, including preparation methods and applications.903.1 Rehydration
Rehydration of thin polymer films is the most common preparation technique used due to it being an inexpensive process that is relatively easy to perform.91,92 A sample of a block copolymer is dissolved in an organic solvent. The selected solvent must solvate the hydrophilic and hydrophobic blocks of the copolymer well. This polymer-solvent solution is then placed on a solid surface, such as a metal or glass plate, and the solvent is evaporated. Remaining on the surface is a thin dehydrated polymer film. Upon the addition of an aqueous solution, water molecules permeate through small defects in the polymer film. The accumulation of aqueous solution causes bulging and eventual separation of the vesicles from the polymer film is achieved (Scheme 9). This technique produces a fairly broad distribution of vesicles sizes. To narrow the distribution, vesicles are extruded through a filter with set pore diameters.93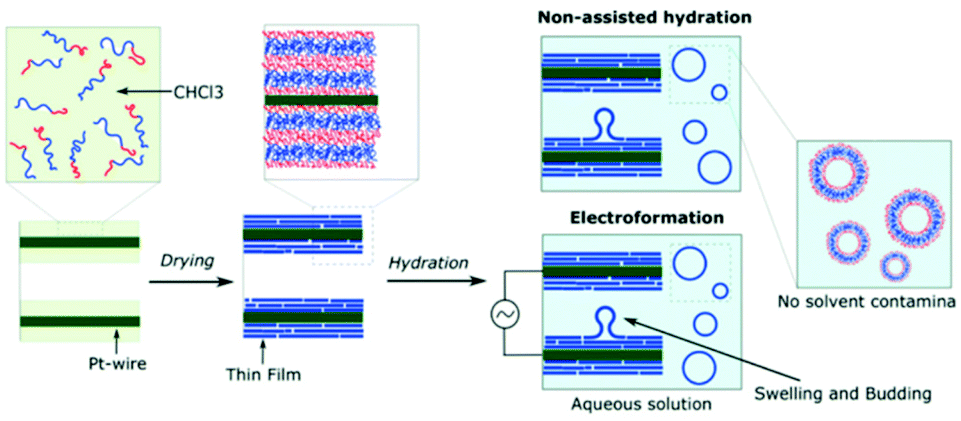 | ||
| Scheme 9 A diagram showing how polymersomes are formed via the film rehydration and electroformation methods.79 Adapted from E. Rideau et al., Polym. Chem., 2018, 9, 5385–5394. | ||
Direct hydration using low molecular weight PEG can also be a very effective polymersome preparation method. In 2015, Sui et al. reported a modified direct hydration method to produce PEG-b-PCL polymersomes.94 The PEG-b-PCL block copolymer with an appropriate PEG block ratio was dissolved in PEG 550 at a relatively high concentration. The addition of water to this solution drove the rapid formation of highly concentrated polymersomes. This method yields polymersomes of high quality, sufficient size and good control over membrane thickness. Moreover, this methods does not involve the use of organic solvent or any agitation methods that could otherwise effect the quality of the polymersomes.
3.2 Electroformation
Electroformation is another solvent-free preparation method and can be considered as an aided-film rehydration method.6 Here, block copolymers are deposited onto electrodes (platinum, gold or indium-titanium oxide (ITO) glass) via the same process as film rehydration (Scheme 9). By applying an alternating current to the electrodes, the rate of water diffusion across the polymer film can be controlled. As a result, the degree of bulging and separation of the vesicles can be controlled to give a narrow distribution of polymersome sizes.95,963.3 Polymerisation-induced self-assembly (PISA)
One of the more recent methods of preparation that has been extensively used in research is polymerisation-induced self-assembly. This method exploits the differences between the solubility of the monomer and polymer in solution. Like the example in Scheme 2, as a hydrophobic monomer is polymerised from a hydrophilic chain macroinitiator, the resulting block copolymer becomes increasingly hydrophobic.97 The amphiphile will self-assemble so that the interfacial tension between the water and hydrophobic block is minimised. Recently, more information regarding the proposed mechanism of self-assembly has been published by Ianaro et al. in 2019.98 It was found that liquid–liquid phase separation precedes self-assembly of the amphiphile. Liquid–liquid phase separation is responsible for determining the size, membrane thickness and other structural properties of polymersomes. The morphology of the nanoparticle is related to the packing parameter P of the amphiphile which, in turn, is related to the DP of the hydrophobic block. Initially, self-assembly does not occur at very low DP. However, the DP of the hydrophobic block will reach a critical micelle concentration (CMC) upon which micelles start to form. Worm-like, rod-like and vesicle morphologies are achieved at higher DPs.19 Therefore, a specific morphology can be obtained by fine-tuning the weight fractions of the hydrophilic and hydrophobic blocks or controlling the DP of the hydrophobic block. PISA is a popular method in producing nanoreactors with selective membrane permeability as the permeability is dictated by the f ratios of the differing blocks.99–101 PISA has become one of the most popular preparation methods today as polymersome assembly occurs in situ with no post polymerisation processing needed to be done, thus saving time and resources.102 In addition to PISA, self-assembly of block copolymers into polymersomes can be driven by a change in stimuli such as pH, temperature etc. In 2018, Battaglia et al. reported the self-assembly of amphiphilic copolymers poly(2-(methacryloyloxy) ethyl phosphorylcholine)-b-poly(2-(diisopropylamino) ethyl methacrylate) (PMPC-b-PDPA) into polymersomes driven by a change in pH or temperature.103 It was found that an increase in temperature (from 5 °C–60 °C) and pH (from pH 2.0–7.0+) leads to an increase in deprotonation of the PDPA block. Deprotonation leads to an increase in hydrophobicity of the block and therefore the amphiphile self-assembles to form a vesicle. Temperature and pH can then be varied to fine-tune the morphology of the polymersome.3.4 Direct injection
The most common of the solvent displacement methods, direct injection exploits the differences in solubility of the polymer in organic solvent and in aqueous solution.104 Like film rehydration, direct injection is an easy and inexpensive process to perform hence its popularity in literature. An organic solvent which solvates all blocks of the amphiphile is selected to dissolve the block copolymer. Next, the polymer-solvent solution is injected into an aqueous solution where the hydrophobic block becomes insoluble due to an increase of interfacial tension between the block and surrounding water molecules. This interaction triggers the self-assembly of vesicles (Scheme 10). The reverse process can also produce polymersomes where water is injected into the polymer-solvent solution to trigger self-assembly.105 Direct injection can produce a broad distribution of vesicle sizes. However, the size distribution can be homogenised by extrusion as well as changing the solvent and concentration of solvent too.93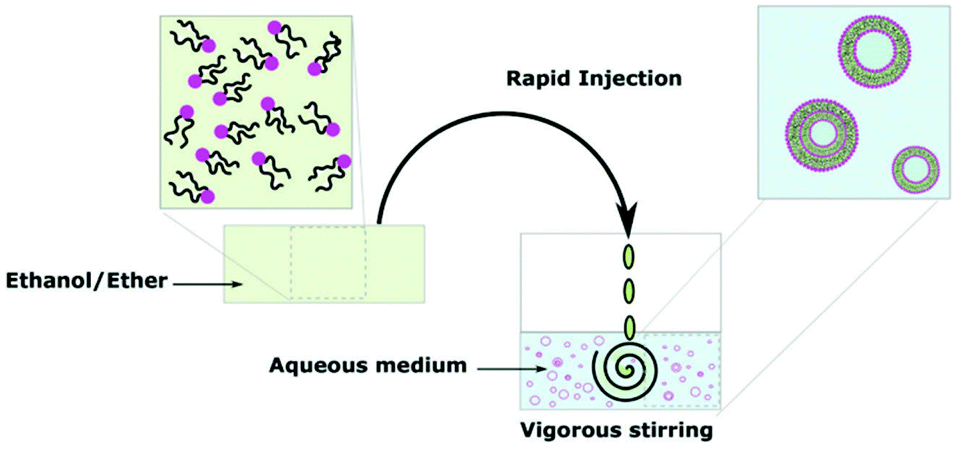 | ||
| Scheme 10 A diagram showing the formation of polymersomes via the direct injection method.9 Adapted from E. Rideau et al., Chem. Soc. Rev., 2018, 47, 40. | ||
3.5 Emulsion phase transfer
Emulsion phase transfer takes the direct injection method and adds more control, producing a homogenous sample of uniform vesicle sizes.24,37 As highlighted in Scheme 11, an emulsion of water-in-oil droplets are produced by vigorous agitation of a small amount of aqueous solution in a solution of polymer in organic solvent. In a separate vessel, an oil-in water biphasic system is set up, with a layer of amphiphiles sitting on the oil/water interface. The emulsion is then poured into the biphasic system. Due to density differences between the oil and water, the oil-coated water droplets sink to the aqueous phase and are coated in another layer of amphiphiles. The final product is a water-in-oil-in-water (w/o/w) double emulsion. The production of vesicles by w/o/w double emulsion allows the encapsulation of hydrophilic in the aqueous core and/or hydrophobic drugs in the oil layer.106,107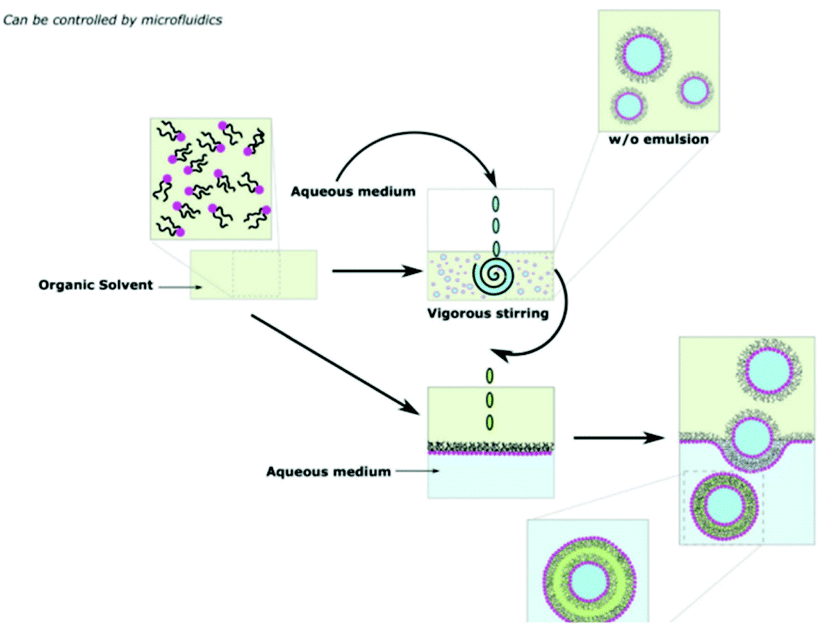 | ||
| Scheme 11 A diagram showing how polymersomes are formed via the water-in-oil–water (w/o/w) double emulsion phase transfer method.9 Adapted from E. Rideau et al., Chem. Soc. Rev., 2018, 47, 40. | ||
3.6 Microfluidics
Emulsion phase transfer can be taken one step further to give almost complete control over the production and size distribution of polymersomes.108 This method is known as microfluidics and is perhaps the most recent and up-to date advancement in the polymersomes preparation field.109,110 From Scheme 12, a jet of organic solvent containing dissolved amphiphiles (middle fluid) is injected through the 1st junction into an aqueous solution (inner fluid) to produce amphiphile stabilised water-in-oil droplets. These droplets are then injected through a 2nd junction into an amphiphile-coated aqueous phase (outer fluid) to produce w/o/w double emulsion vesicles. By varying the flow rates through each junction, the size of the resulting polymersome can be controlled.108,110 | ||
| Scheme 12 A diagram showing the formation of polymersomes via a simplified microfluidic device.111 Adapted from J. H. Jang et al., Sens. Actuator B-Chem., 2017, 241, 636–643. | ||
4. Beyond polymersomes
Since the first use of the term polymersome in 1999, research in this area of polymer chemistry has come a very long way since. Starting from the simplest diblock copolymer of PEO-b-PEE, we arrive at multi stimuli-responsive multiblock copolymers and complex architectures in today's research. This rapid progression driven by intense research in the field has led many academic groups to explore alternative systems related to polymersomes with the same applications and goals in mind. In recent years, researchers have been inspired by some of the oldest organisms in the world to achieve targeted drug delivery. Viruses have long been regarded as excellent vectors for intracellular delivery due to their size and ability to change morphology to traverse the body of its host on a cellular level.112 By designing polymeric systems that mimic viruses, researchers have managed to synthesise a variety of highly specialised and efficient nanoparticles for intracellular delivery of many cargoes.113,114Armes et al. have recently reported a Dengue virus-mimicking, pH-responsive, framboidal ABC triblock copolymer vesicles that target breast cancer cells.115 poly(glycerol monomethacrylate)-b-poly(hydroxypropyl methacrylate)-b-poly(2-(diisopropylamino)ethyl methacrylate) PGMA-b-PHPMA-b-PDPA vesicles were synthesised via RAFT emulsion polymerisation and were prepared by PISA (Scheme 13). A small fraction (3%) of the hydrophilic PGMA chains were functionalised with phosphorylcholine (PMPC). This targeting ligand for the SR-B1 receptor is crucial for intracellular uptake by triple negative breast cancer cells in which this receptor is overexpressed. The pH-responsive nature of the nanoparticles allows for release of a payload (protein, gene or drug) inside these cancer cells.
 | ||
| Scheme 13 Synthetic route of mPGMA58−P(HPMA300-stat-ERh1)–PDPA48 framboidal vesicles (left) where m = 100 and mPGMA58-nPMPC60−P(HPMA300-stat-ERh1)–PDPA52 framboidal vesicles (right) where m = 97 and n = 3. Corresponding cartoon illustrations of each vesicle are included, where PGMA = red, PMPC = green, P(HPMA-stat-ERh1) = blue and PDPA = black. As reported by Armes et al.115 | ||
Wang et al. also reported the synthesis of a pH-responsive virus-like nanoparticle consisting of an acetylated dextran-based diblock copolymer of poly(methyl methacrylate)-b-poly(lauryl methacrylate) (MMA-b-LMA).116 The MMA-b-LMA block copolymer mimics the capsid protein for endosomal escape and acetylated dextran resembles the viral core for encapsulating payloads. It was found that conjugating the tumour penetrating peptide TT1 to the virus-like nanoparticle increases its cellular uptake in colon carcinoma cells.
As well as synthetic polymeric systems, a variety of peptide-based virus-like nanoparticles have been reported.117–119 These nanoparticles take the protein structure of a virus and are functionalised to improve its physiological properties inside the body. Functionalisation strategies include PEGylation for increased biocompatibility and retention in the body. As well as sugar/peptide conjugation for specific receptor targeting. Viruses such as the physalis mottle, cowpea mosaic, tobacco mosaic and hepatitis B have all been studied as vectors for drug delivery and vaccinations.120–122
5. Conclusion and outlook
In this review, we have seen that polymersomes are diverse in synthesis and preparation as there is no standard synthetic route in their formation. The amphiphilic block copolymers are synthesised via one of three main living polymerisation techniques. ROP is used for the polymerisation of cyclic monomers whereas RAFT and ATRP polymerise vinyl monomers with the use of a hydrophilic or hydrophobic macroinitiator with mono- or di-functionality. Depending on the chemistry of the amphiphiles, polymersomes can be prepared via solvent-free or solvent dissolution methods. Solvent-dissolution methods such as microfluidics offer finer control over polymersome size and size distribution. However, for quick and easy preparation of polymersomes with less emphasise on size uniformity, solvent-free methods followed by extrusion are perfectly viable. Evidently, the synthetic route is entirely dictated by factors such as the monomers used and the eventual application of the polymersome.Drug delivery still remains the main application of polymersomes as both hydrophilic and hydrophobic cargoes can be encapsulated within the nanoparticle. Furthermore, stimuli-responsive release of the encapsulated cargoes can be achieved by incorporating stimuli-responsive polymer blocks within the amphiphile. However, there is growing interest in utilising polymersomes as nanoreactors for biological cascade reactions where compartmentalisation is key for these biological processes to occur. Research interest has also grown in synthesising virus-like nanoparticles as vectors for intracellular cargo delivery too. These specialised nanoparticles have the ability to change morphology to pass through cellular membranes and barriers. Sugars and/or peptides can be conjugated to the virus head for targeted delivery to a desired destination.
Considering their significant contribution towards targeted drug delivery, why have we not seen polymersomes reach clinical trials or the commercial market yet. Unfortunately, there are a number of drawbacks that have made the use of these drug delivery vectors not viable yet. Firstly, and quite possibly most importantly, only a handful of polymers have been approved for use in pharmaceuticals by government health and safety agencies such as the MHRA and FDA. These include PEG, PLA, PCL, and PMMA. Any of the more unique, less reported stimuli-responsive polymers will need to be extensively researched, tested and approved by such agencies first. With only a limited number of building blocks to choose from, very few combinations of polymersomes can be produced for actual clinical use. The preparation of polymersomes also presents a few challenges holding back polymersomes for commercial use. During the preparation, any presence of residual cytotoxic organic solvent or monomers in the polymersomes renders them non-biocompatible regardless whether the amphiphiles are biocompatible themselves. Poor drug loading efficiencies, lack of absolute control over the targeting and release of drugs and the issue of scalability for production on a commercial level presents further problems to be addressed. For polymersomes to be incorporated in pharmaceuticals in the future, we believe cytotoxicity, drug loading efficiencies and long-term stability of polymersomes must be extensively researched and understood. Furthermore, new methods to allow the facile and cost-effective production of polymersomes on an industrial scale must also be explored.
Conflicts of interest
There are no conflicts to declare.References
- X. Zhang, P. Tanner, A. Graff, C. G. Palivan and W. Meier, J. Polym. Sci., Polym. Chem., 2012, 50, 2293–2318 CrossRef CAS.
- M. C. M. van Oers, F. Rutjes and J. C. M. van Hest, Curr. Opin. Biotechnol., 2014, 28, 10–16 CrossRef CAS.
- A. D. Bangham, R. W. Horne, A. M. Glauert, J. T. Dingle and J. A. Lucy, Nature, 1962, 196, 952–955 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660–668 CrossRef CAS.
- H. L. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS.
- J. C. M. van Hest, D. A. P. Delnoye, M. W. P. L. Baars, M. H. P. van Genderen and E. W. Meijer, Science, 1995, 268, 1592 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 40 RSC.
- Y. L. Lin, H. Y. Chang, Y. J. Sheng and H. K. Tsao, Soft Matter, 2013, 9, 4802–4814 RSC.
- H. C. Shum, J. W. Kim and D. A. Weitz, J. Am. Chem. Soc., 2008, 130, 9543–9549 CrossRef CAS.
- U. Kauscher, M. N. Holme, M. Bjornmalm and M. M. Stevens, Adv. Drug Delivery Rev., 2019, 138, 259–275 CrossRef CAS.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- F. H. Meng, Z. Y. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS.
- A. F. Mason, N. A. Yewdall, P. L. W. Welzen, J. X. Shao, M. van Stevendaal, J. C. M. van Hest, D. S. Williams and L. K. Abdelmohsen, ACS Cent. Sci., 2019, 5, 1360 CrossRef CAS.
- X. Liu, P. Formanek, B. Voit and D. Appelhans, Angew. Chem., Int. Ed., 2017, 56, 16233 CrossRef CAS.
- A. Belluati, S. Thamboo, A. Najer, V. Maffeis, P. von Claudio, I. Craciun, C. G. Palivan and W. Meier, Adv. Funct. Mater., 2020, 30, 2002949 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174 CrossRef CAS.
- R. Nagarajan, Langmuir, 2002, 18(1), 31–38 CrossRef CAS.
- C. G. Palivan, R. Goers, A. Najer, X. Y. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- H. Q. Liang, Q. H. Zhou, Y. J. Long, W. C. Wei, S. Feng, G. D. Liang and F. M. Zhu, RSC Adv., 2018, 8, 12752–12759 RSC.
- S. So and T. P. Lodge, Langmuir, 2015, 31, 594–601 CrossRef CAS.
- M. R. Kim and I. W. Cheong, Langmuir, 2016, 32, 9223–9228 CrossRef CAS.
- J. Jonikaite-Svegzdiene, A. Kudresova, S. Paukstis, M. Skapas and R. Makuska, Polym. Chem., 2017, 8, 5621–5632 RSC.
- C. Houga, J. Giermanska, S. Lecommandoux, R. Borsali, D. Taton, Y. Gnanou and J. F. Le Meins, Biomacromolecules, 2009, 10, 32–40 CrossRef CAS.
- K. Kiene, S. H. Schenk, F. Porta, A. Ernst, D. Witzigmann, P. Grossen and J. Huwyler, Eur. J. Pharm. Biopharm., 2017, 119, 322–332 CrossRef CAS.
- F. Porta, D. Ehrsam, C. Lengerke and H. E. M. zu Schwabedissen, Mol. Pharm., 2018, 15, 4884–4897 CrossRef CAS.
- L. Martin, P. Gurnani, J. L. Zhang, M. Hartlieb, N. R. Cameron, A. M. Eissa and S. Perrier, Biomacromolecules, 2019, 20, 1297–1307 CrossRef CAS.
- C. Nehate, A. A. M. Raynold, V. Haridas and V. Koul, Biomacromolecules, 2018, 19, 2549–2566 CrossRef CAS.
- A. C. Apolinario, M. S. Magon, A. Pessoa and C. D. Rangel-Yagui, Nanomaterials, 2018, 8, 16 CrossRef.
- F. Lu, Z. Y. Pang, J. J. Zhao, K. Jin, H. C. Li, Q. Pang, L. Zhang and Z. Q. Pang, Int. J. Nanomed., 2017, 12, 2117–2127 CrossRef CAS.
- E. Konishcheva and W. Meier, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 2016, 252, 2 Search PubMed.
- A. A. Asl and S. Rahmani, Int. J. Polym. Mater. Polym. Biomater., 2019, 68, 540–550 CrossRef.
- A. Moquin, J. Ji, K. Neibert, F. M. Winnik and D. Maysinger, ACS Omega, 2018, 3, 13882–13893 CrossRef CAS.
- N. A. N. Mendez, E. B. N. Rodriguez, R. H. Najera and A. E. C. Castellanos, Int. J. Polym. Mater. Polym. Biomater., 2014, 63, 143–148 CrossRef.
- H. Danafar, K. Rostamizadeh, S. Davaran and M. Hamidi, Drug Dev. Ind. Pharm., 2014, 40, 1411–1420 CrossRef CAS.
- K. J. Zhu, X. Z. Lin and S. L. Yang, J. Appl. Polym. Sci., 1990, 39, 1–9 CrossRef CAS.
- A. Beletsi, Z. Panagi and K. Avgoustakis, Int. J. Pharm., 2005, 298, 233–241 CrossRef CAS.
- E. Locatelli and M. C. Franchini, J. Nanopart. Res., 2012, 14, 17 CrossRef.
- P. P. Chen, C. Deng, F. H. Meng, J. Zhang, R. Cheng and Z. Y. Zhong, J. Controlled Release, 2015, 213, E87–E88 CrossRef.
- E. Vlakh, A. Ananyan, N. Zashikhina, A. Hubina, A. Pogodaev, M. Volokitina, V. Sharoyko and T. Tennikova, Polymers, 2016, 8, 14 CrossRef.
- E. Liarou, S. Varlas, D. Skoulas, C. Tsimblouli, E. Sereti, K. Dimas and H. Iatrou, Prog. Polym. Sci., 2018, 83, 28–78 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- C. Jerome and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
- I. Conejos-Sanchez, A. Duro-Castano, A. Birke, M. Barz and M. J. Vicent, Polym. Chem., 2013, 4, 3182 RSC.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373 RSC.
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC.
- M. Glassner, D. R. D'Hooge, J. Y. Park, P. H. M. Van Steenberge, B. D. Monnery, M. F. Reyniers and R. Hoogenboom, Eur. Polym. J., 2015, 65, 298–304 CrossRef CAS.
- S. P. Cao, J. X. Shao, Y. F. Xia, H. L. Che, Z. Y. Zhong, F. H. Meng, J. C. M. van Hest, L. Abdelmohsen and D. S. Williams, Small, 2019, 15, 8 Search PubMed.
- R. U. Khan, H. Yu, L. Wang, Q. Zhang, W. Xiong, Z. ul-Abdin, A. Nazir, S. Fahad, X. Chen and T. Elsharaarani, J. Mater. Sci., 2020, 55, 8264–8284 CrossRef CAS.
- A. P. Martinez, B. Qamar, T. R. Fuerst, S. Muro and A. K. Andrianov, Biomacromolecules, 2017, 18, 2000–2011 CrossRef CAS.
- K. Schulze, T. Ebensen, L. A. Babiuk, V. Gerdts and C. A. Guzman, Nanomedicine, 2017, 13, 2169–2178 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312–325 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- Z. Y. Song, Y. Z. Huang, V. Prasad, R. Baumgartner, S. Y. Zhang, K. Harris, J. S. Katz and J. J. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 17033–17037 CrossRef CAS.
- F. Mastrotto, A. F. Breen, G. Sicilia, S. Murdan, A. D. Johnstone, G. E. Marsh, C. Grainger-Boultby, N. A. Russell, C. Alexander and G. Mantovani, Polym. Chem., 2016, 7, 6714–6724 RSC.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- L. N. Besada, P. Peruzzo, A. M. Cortizo and M. S. Cortizo, J. Nanopart. Res., 2018, 20, 67 CrossRef.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- S. T. U. Borman, Chem. Eng. News Archive, 2006, 84, 40–41 CrossRef.
- S. J. Lin, J. J. Shang, X. X. Zhang and P. Theato, Macromol. Rapid Commun., 2018, 39, 6 Search PubMed.
- J. J. Wu, H. Y. Liang, Y. C. Li, Y. Shi, M. Bottini, Y. M. Chen and L. X. Liu, Adv. Funct. Mater., 2020 DOI:10.1002/adfm.202000391.
- S. Huber and S. Mecking, Macromolecules, 2019, 52, 5917–5924 CrossRef CAS.
- C. Lebleu, L. Rodrigues, J. M. Guigner, A. Brulet, E. Garanger and S. Lecommandoux, Langmuir, 2019, 35, 13364–13374 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- H. O. Kim, S. H. Lee, W. Na, J. W. Lim, G. Park, C. Park, H. Lee, A. Kang, S. Haam, I. Choi, J. T. Kang and D. Song, J. Mater. Chem. B, 2020, 8, 2476–2482 RSC.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS.
- R. Ghasemi, M. Abdollahi, E. E. Zadeh, K. Khodabakhshi, A. Badeli, H. Bagheri and S. Hosseinkhani, Sci. Rep., 2018, 8, 13 CrossRef.
- W. Y. Ning, P. Shang, J. Wu, X. Y. Shi and S. X. Liu, Polymers, 2018, 10, 18 Search PubMed.
- S. K. Kozawa, K. Matsumoto, A. Suzuki, M. Sawamoto and T. Terashima, J. Polym. Sci., Polym. Chem., 2019, 57, 313–321 CrossRef CAS.
- P. Biais, P. Beaunier, F. Stoffelbach and J. Rieger, Polym. Chem., 2018, 9, 4483–4491 RSC.
- A. Zahoranova, M. Mrlik, K. Tomanova, J. Kronek and R. Luxenhofer, Macromol. Chem. Phys., 2017, 218, 12 CrossRef.
- J. Gaitzsch, S. Hirschi, S. Freimann, D. Fotiadis and W. Meier, Nano Lett., 2019, 19, 2503–2508 CrossRef CAS.
- E. V. Konishcheva, D. Daubian, S. Rigo and W. P. Meier, Chem. Commun., 2019, 55, 1148–1151 RSC.
- X. R. Wang, C. Z. Yao, G. Y. Zhang and S. Y. Liu, Nat. Commun., 2020, 11, 13 CrossRef.
- O. F. Mutaf, Y. Anraku, A. Kishimura and K. Kataoka, Polymer, 2017, 133, 1–7 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS.
- Q. Qu, G. Liu, X. Lv, B. Zhang and Z. An, ACS Macro Lett., 2016, 5, 316–320 CrossRef CAS.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS.
- C. Liao, Y. Chen, Y. Yao, S. Zhang, Z. Gu and X. Yu, Chem. Mater., 2016, 28, 7757–7764 CrossRef CAS.
- Y. L. Dong, J. H. Zhou, C. R. Wang, Y. P. Wang, L. D. Deng, A. H. Zhang and A. J. Dong, Macromol. Biosci., 2020 DOI:10.1002/mabi.202000143.
- J. J. Nie, W. Y. Zhao, H. Hu, B. R. Yu and F. J. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 8376–8385 CrossRef CAS.
- V. Percec, M. R. Imam, T. K. Bera, V. S. K. Balagurusamy, M. Peterca and P. A. Heiney, Angew. Chem., Int. Ed., 2005, 44, 4739–4745 CrossRef CAS.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS.
- S. E. Sherman, Q. Xiao and V. Percec, Chem. Rev., 2017, 117, 6538–6631 CrossRef CAS.
- C. K. Wong, M. H. Stenzel and P. Thordarson, Chem. Soc. Rev., 2019, 48, 4019–4035 RSC.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Sci. Rep., 2016, 6, 33491 CrossRef.
- E. Rideau, F. R. Wurm and K. Landfester, Polym. Chem., 2018, 9, 5385–5394 RSC.
- J. E. Bartenstein, J. Robertson, G. Battaglia and W. H. Briscoe, Colloids Surf., A, 2016, 506, 739–746 CrossRef CAS.
- X. Sui, P. Kujala, G. Janssen, E. de Jong, I. S. Zuhorn and J. C. M. van Hest, Polym. Chem., 2015, 6, 691–696 RSC.
- E. Ibarboure, M. Fauquignon and J.-F. Le Meins, JoVE, 2020, e60199, DOI:10.3791/60199.
- C. Kunzler, S. Handschuh-Wang, M. Roesener and H. Schönherr, Macromol. Biosci., 2020, 20, 2000014 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- A. Ianiro, H. Wu, M. M. J. van Rijt, M. P. Vena, A. D. A. Keizer, A. C. C. Esteves, R. Tuiner, H. Friedrich, N. A. J. M. Sommerdijk and J. P. Patterson, Nat. Chem., 2019, 11, 320 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- J. He, J. Cao, Y. Chen, L. Zhang and J. Tan, ACS Macro Lett., 2020, 9, 533–539 CrossRef CAS.
- A. N. Albertsen, J. K. Szymański and J. Pérez-Mercader, Sci. Rep., 2017, 7, 41534 CrossRef CAS.
- C. Contini, R. Pearson, L. Wang, L. Messager, J. Gaitzsch, L. Rizzello, L. Ruiz-Perez and G. Battaglia, iScience, 2018, 7, 132–144 CrossRef CAS.
- K. Vijayakrishna, D. Mecerreyes, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5167–5174 CrossRef CAS.
- I. A. B. Pijpers, F. H. Meng, J. C. M. van Hest and L. Abdelmohsen, Polym. Chem., 2020, 11, 275–280 RSC.
- N. Soomherun, N. Kreua-ongarjnukool, S. Chumnanvej and S. Thumsing, Int. J. Biomater., 2017, 2017, 1743765 Search PubMed.
- J. S. Winkler, M. Barai and M. S. Tomassone, Exp. Biol. Med., 2019, 244, 1162–1177 CrossRef CAS.
- A. S. Utada, E. Lorenceau, D. R. Link, P. D. Kaplan, H. A. Stone and D. A. Weitz, Science, 2005, 308, 537–541 CrossRef CAS.
- J. Ahn, J. Ko, S. Lee, J. Yu, Y. Kim and N. L. Jeon, Adv. Drug Delivery Rev., 2018, 128, 29–53 CrossRef CAS.
- C. B. Roces, D. Christensen and Y. Perrie, Drug Delivery Transl. Res., 2020, 10, 582–593 CrossRef CAS.
- J. H. Jang and S. Y. Park, Sens. Actuators, B, 2017, 241, 636–643 CrossRef CAS.
- M. Elsabahy and K. L. Wooley, J. Polym. Sci., Polym. Chem., 2012, 50, 1869–1880 CrossRef CAS.
- M. J. Rohovie, M. Nagasawa and J. R. Swartz, Bioeng. Transl. Med., 2017, 2, 43–57 CrossRef CAS.
- Y. Zou, M. Zheng, W. Yang, F. Meng, K. Miyata, H. J. Kim, K. Kataoka and Z. Zhong, Adv. Mater., 2017, 29, 1703285 CrossRef.
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. U. Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 RSC.
- S. Wannasarit, S. Q. Wang, P. Figueiredo, C. Trujillo, F. Eburnea, L. Simon-Gracia, A. Correia, Y. P. Ding, T. Teesalu, D. F. Liu, R. Wiwattanapatapee, H. A. Santos and W. Li, Adv. Funct. Mater., 2019, 29, 14 Search PubMed.
- R. Biabanikhankahdani, N. B. M. Alitheen, K. L. Ho and W. S. Tan, Sci. Rep., 2016, 6, 13 CrossRef.
- J. A. Finbloom, I. L. Aanei, J. M. Bernard, S. H. Klass, S. K. Elledge, K. Han, T. Ozawa, T. P. Nicolaides, M. S. Berger and M. B. Francis, Nanomaterials, 2018, 8, 12 CrossRef.
- C. Pretto and J. C. M. van Hest, Bioconjugate Chem., 2019, 30, 3069–3077 CrossRef CAS.
- H. Hu, H. Masarapu, Y. N. Gu, Y. F. Zhang, X. Yu and N. F. Steinmetz, ACS Appl. Mater. Interfaces, 2019, 11, 18213–18223 CrossRef CAS.
- E. Alemzadeh, A. Dehshahri, K. Izadpanah and F. Ahmadi, Colloids Surf., B, 2018, 167, 20–27 CrossRef CAS.
- M. Perotti and L. Perez, Viruses, 2020, 12, 17 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |