
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ε-caprolactone-derived 2-oxazoline inimer for the synthesis of graft copolymers†
Jungyeon
Kim
 a,
Christopher
Waldron
a,
Beatrice
Cattoz
b and
C. Remzi
Becer
*a
a,
Christopher
Waldron
a,
Beatrice
Cattoz
b and
C. Remzi
Becer
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Remzi.Becer@warwick.ac.uk
bMilton Hill Business & Technology Centre, Infineum UK Ltd, Abingdon, Oxfordshire OX13 6BB, UK
First published on 8th October 2020
Abstract
An inimer approach is presented here for the formation of bottlebrush copolymers consisting of a poly(2-oxazoline) backbone and acrylate branches. A hydroxyl group containing 2-oxazoline (2-n-pentanol-2-oxazoline) was synthesized from ε-caprolactone, which is a sustainable starting material. 2-n-Pentanol-2-oxazoline was then further functionalized with a bromoisobutyrate group, which was used to initiate a Cu(0)-mediated reversible-deactivation radical polymerisation (RDRP) of acrylates. This compound is termed an “inimer” because it contains both a monomer (2-oxazoline) and an initiator (the RDRP initiator in this study). Herein, we report the use of an inimer to form 2-oxazoline-based polymer backbones through cationic ring opening polymerisation, and further polymerisation via the Cu(0)-mediated RDRP of acrylates was achieved by initiating from the RDRP-initiator sites embedded within the poly(2-oxazoline) backbone. Furthermore, statistical copolymers of 2-ethyl 2-oxazoline and the inimer were also formed leading to statistically distributed radical initiating sites, which were then used to form graft copolymers of varying densities.
Introduction
Poly(2-oxazoline)s (POx) have been widely studied for their versatile applications including their use as antifouling agents,1 biomaterials,2 and a means for drug delivery.3,4 They are becoming of increasing importance, especially in the biomedical field, displaying biocompatibility and stealth behavior.1,4,5 Through cationic ring opening polymerisation (CROP), 2-oxazolines can be polymerized in a “living” manner and depending on the chain length and/or functional group attached to the oxazoline ring, a wide range of hydrophobic, hydrophilic, thermoresponsive polymers can be synthesized. Although 2-oxazoline-based copolymers have been investigated for these various properties6 and self-assembly behaviors,7 the combination of 2-oxazolines and other monomers, such as acrylates, is also of great interest. By combining two different types of monomers through CROP and controlled radical polymerisation techniques, multiple functionalities can be incorporated, resulting in a wider range of functional monomer combinations than when using one polymerisation technique.Graft copolymers comprise a polymeric backbone with side chains (branches), and in the case of bottlebrush copolymers, the branches are very densely grafted onto the backbone. In biology, these macromolecular structures are often present, with examples including lubricin8–10 in joint lubrication, and proteoglycans.11–13 Bottlebrush copolymers have been used for drug delivery where it has been reported that they display multivalency as well as extended in vivo circulation time in comparison with their linear analogues. Bottlebrush copolymers, therefore, show advantageous features and so they have been reported extensively.8,14–18 However, the combination of 2-oxazolines and acrylates as a graft copolymer has seldom been reported, with even fewer reports on the 2-oxazoline backbone and acrylate brush combination.19–23
Graft copolymers can be synthesized from three synthetic routes: grafting to, grafting through, and grafting from. While each method is associated with its own advantages and disadvantages,24–26 the grafting from approach, the chosen synthetic route in this research, can give superior control over the grafting density.
In this study, we demonstrate a synthetic approach to a novel inimer structure from ε-caprolactone, a deviation from the conventional 2-oxazoline synthetic routes (Scheme 1). Lactones are a class of cyclic carboxylic esters that can also undergo ring opening polymerisation.27,28 Poly(caprolactone)s are both biocompatible and biodegradable and have garnered much interest as a means for controlled drug delivery.29 However, ε-caprolactone, or other members of the lactone family, has not been previously investigated for the formation of inimer structures. Based on previous reports,30,31 a hydroxyl functional group containing oxazoline was designed from a commercially available and sustainable starting material, namely ε-caprolactone. To the best of our knowledge, this is the first example of the use of a caprolactone-derived hydroxyl oxazoline to form a functional inimer. From this, highly dense ridged bottlebrush copolymers as well as flexible brushes were formed through the copolymerisation of the inimer with non-initiating group containing 2-oxazoline monomers.
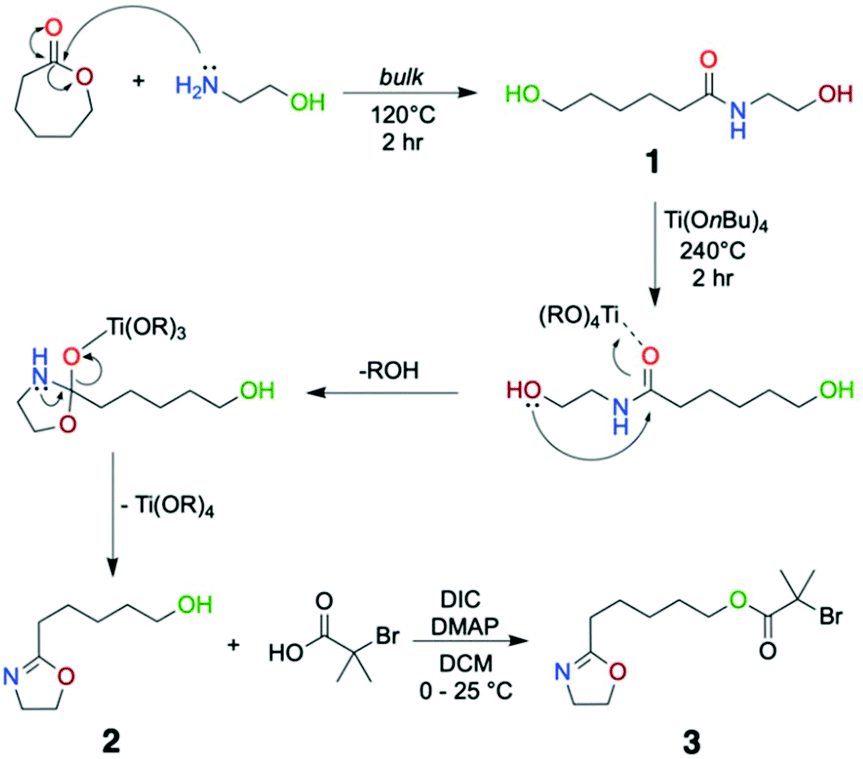 | ||
| Scheme 1 Three-step synthetic approach to the Cu(0)-mediated RDRP inimer where structure 1 is the amide precursor, 2 is the hydroxyl oxazoline and 3 is the desired inimer structure. | ||
Results and discussion
Synthesis of the inimer from ε-caprolactone
A three-step synthetic strategy was developed to prepare the inimer structure. As shown in Scheme 1, the first step is amidation of ε-caprolactone through a nucleophilic attack of 2-aminoethanol which occurs under bulk conditions (Structure 1 in Scheme 1). In 2 hours, based on the 1H NMR analysis, we observed a quantitative conversion to an amide structure (Fig. 1, top). Subsequent addition of catalytic amounts of titanium(IV) butoxide at 240 °C led to ring closing around the amide according to the mechanism outlined in Scheme 1, where coordination between the carbonyl group and the titanium complex assists in the ring closure reaction. Distillation of the product yielded the purified hydroxyl oxazoline monomer (Fig. 1, middle). This two-step synthetic route to the functional oxazoline can be performed in the absence of any solvent and with readily available starting materials in a moderately scalable manner (>30 g). However, the multiple distillations required (in this case four) to remove residual impurities and by-products (butanol and water) significantly lower the overall yield through the course of the purification procedure (18.2% yield overall). Although, multiple distillations are not necessarily required, we have decided to obtain 2-n-pentanol-2-oxazoline at its highest purity as any impurities could lead to further unwanted side reactions in the third step. Compared to the thioether-linked oxazoline, as previously demonstrated by our group,20 it is a less toxic and easily scalable procedure owing to the cheaper and non-toxic nature of the starting material, namely caprolactone. | ||
| Fig. 1 1H NMR spectra of structure 1 (amide intermediate; top), structure 2 (2-n-pentanol-2-oxazoline; middle), and structure 3 (inimer; bottom). EA denotes ethyl acetate as a solvent. | ||
The final step of the synthesis is a Steglich esterification reaction using α-bromoisobutyric acid and 4-N,N′-diisopropylcarbodiimide (DIC) as the coupling reagent. It should be noted that the esterification step directly using α-bromoisobutyryl bromide causes ring opening of the 2-oxazoline as well as ester formation. Therefore, a DMAP-catalyzed acid–alcohol coupling reaction was performed in order to obtain the desired product 3, which was finally purified by column chromatography (Fig. 1, bottom). This three-step synthetic route to the inimer from caprolactone was scaled up to the 20 g scale.
Polymerisation of the oxazoline backbone via CROP
An initial CROP of inimer 3 (In) alone using the initiator methyl tosylate was carried out. Alkyl halides are known initiators of CROP,32 and it was also reported by our group that a similar inimer structure can self-initiate even at 80 °C, which is significantly lower than the standard CROP temperature of 140 °C.20 This could be associated with the electrophilicity of the tertiary alkyl halide also present in the inimer structure in this research, which could cause the initiation of the oxazoline and lead to unwanted hyperbranched structures. Therefore, a lower reaction temperature of 60 °C was chosen for the polymerisation of the backbone by CROP. A short DP of 10 was initially targeted and resulted in P1 (DP = 10) with a dispersity Đ = 1.32 (Mn, SEC. = 2200 Da). The detailed kinetics of this reaction showed that this polymerisation followed first-order kinetics, and the calculated kapp was determined to be 1.231 × 10−4 s−1. Subsequent attempts at increasing the length of the backbone (P2, P3) resulted in larger molecular weight polymers but as higher chain lengths were targeted (in the case of P3, DP = 100, Đ = 2.20) (Fig. S1†), it resulted in broader dispersity, due to the extended reaction time.Consequently, higher molecular weight polymers were investigated through copolymerisation with a comonomer that did not contain other functionalities such as 2-ethyl-2-oxazoline (EtOx). The thioether linker containing inimer structure we previously reported showed narrow dispersity for block copolymers of EtOx and the inimer, but statistical copolymers of the two led to undesirable polymerisation reactions leading to poorly defined polymers.20 In that report, it was concluded that the susceptibility of EtOx to the tertiary alkyl bromide of the inimer led to the polymerisation of the monomers, both by the initiator and the tertiary alkyl bromide of the inimer. However, upon copolymerisation of the inimer presented in this research (In) with EtOx at a ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 (P5), well-defined polymers were synthesized even at higher molecular weights (Fig. 2f). The main difference between the two inimer structures is that of the methyl group adjacent to the oxazoline ring and the thioether linker connecting the oxazoline ring and the radical initiating group, both of which are not present in the inimer structure reported here. These results led to the conclusion that the poor polymers shown using the thioether linker containing inimer structure were due to the methyl group adjacent to the oxazoline ring, which can change the nucleophilicity of the monomer and hence change the reactivity.33 In addition, the sulfur could act as a nucleophile, attacking the oxazolium group, leading to unwanted termination reactions.34 Therefore, In can be considered to be the more desirable inimer structure because of the less toxic starting materials required for its synthesis, better stability (thioether bonds are prone to oxidation35), and statistical copolymerisation with other 2-oxazoline monomers.
:25 (P5), well-defined polymers were synthesized even at higher molecular weights (Fig. 2f). The main difference between the two inimer structures is that of the methyl group adjacent to the oxazoline ring and the thioether linker connecting the oxazoline ring and the radical initiating group, both of which are not present in the inimer structure reported here. These results led to the conclusion that the poor polymers shown using the thioether linker containing inimer structure were due to the methyl group adjacent to the oxazoline ring, which can change the nucleophilicity of the monomer and hence change the reactivity.33 In addition, the sulfur could act as a nucleophile, attacking the oxazolium group, leading to unwanted termination reactions.34 Therefore, In can be considered to be the more desirable inimer structure because of the less toxic starting materials required for its synthesis, better stability (thioether bonds are prone to oxidation35), and statistical copolymerisation with other 2-oxazoline monomers.
 | ||
| Fig. 2 CROP of In: (a) kinetic studies of P1 and P4 at 60 °C, (b) Mn,SEC values and the corresponding dispersity values of P1 and P4, (c) SEC traces of P1 (PIn10) and P4 (PEtOx10), (d) kinetics of each component in P5, (e) Mn, SEC values and the corresponding dispersity values, and (f) the SEC trace of copolymer P5. | ||
The differing reaction rates of EtOx and In at 60 °C, with the inimer polymerizing more slowly than EtOx (P4, Fig. 2a), suggested that the copolymerisation of these two monomers could form a gradient copolymer structure. Further investigation of the reaction kinetics of each component through 1H NMR of P5 (PEtOx25-r-PIn25) showed similar rates of reaction during copolymerization (P(EtOx25-r-In25), kapp,EtOx = 2.404 × 10−5 s−1, kapp,In = 2.304 × 10−5 s−1) (Fig. 2d). These similar reactivity rates would indicate that the inimer is statistically distributed along the backbone rather than in a gradient or block copolymer form. Different ratios of In and EtOx were polymerized into P6 (P(EtOx40-r-In10), kapp,EtOx = 2.562 × 10−5 s−1, kapp,In = 2.588 × 10−5 s−1) (Fig. S6†) and have suggested that the reaction rates of EtOx and In were very similar in each case.
Brush synthesis via Cu(0)-mediated RDRP
After establishing the scope of the polymerisation of In through a systematic investigation of the backbone length and brush initiator density, graft copolymers were synthesized using various acrylates (Scheme 2). Under a well-established Cu(0)-mediated RDRP system in organic media (where [monomer]/[In]/[Me6Tren]/[CuBr2] = DP/1/0.18/0.05, and 5 cm of pre-activated copper wire), 2-ethyl hexyl acrylate (EHA) was primarily used for brush formation, first using P1 as the brush initiator. With a Cu(0)-RDRP initiating group on every repeating unit of the backbone, a very short, dense graft copolymer of EHA, B1, was formed.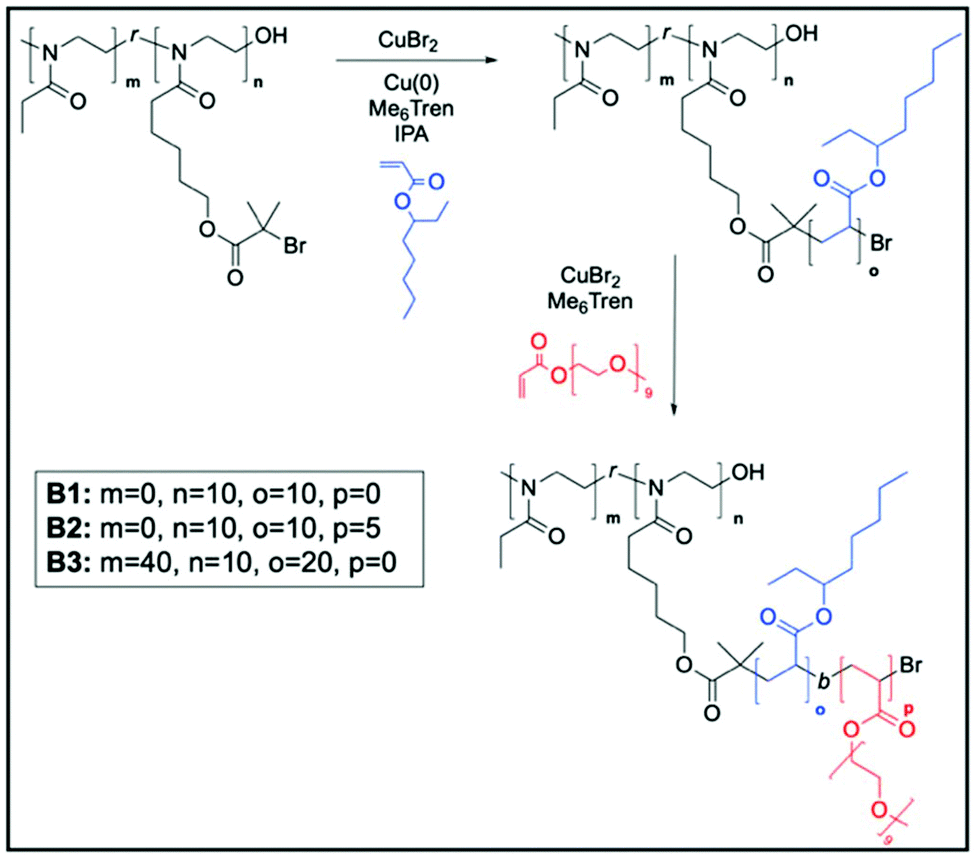 | ||
| Scheme 2 Polymerisation scheme of 2-ethyl hexyl acrylate using brush initiators P1 and P6 for B1 and B2 and for B3, respectively. m denotes the number of non-initiating monomers, n is the number of initiating sites, and o and p are brush lengths. | ||
Comparing the SEC traces of the backbone (P1) to the graft copolymer, there was a significant shift from P1 and B1, showing a molecular weight of 14 kDa (Fig. 3). The end group fidelity, or “livingness” of the chain end, was investigated through bottlebrush block copolymerisation with oligo(ethylene glycol)acrylate (OEGA, Mn = 480 Da) (B2). P1 was used as a brush initiator and as was the case for B1 (PIn10-brush-PEHA10). EHA was polymerized under standard Cu(0)-mediated RDRP conditions in IPA. Once near-full conversion of EHA was confirmed, the second brush block was polymerized using OEGA. In order to maintain a high conversion of the second block,36 additional portions of the deactivator (Cu2Br) and ligand (Me6Tren) were added. This led to near-full conversion of the second monomer (B2, Mn, SEC = 28 kDa, Đ = 1.25) (Fig. S9†).
 | ||
| Fig. 3 Analysis of the bottlebrush copolymer PIn10-brush-PEHA10 (B1): (a) SEC traces of the bottlebrush copolymer compared to the linear backbone, (b) evolving SEC traces of B1 over time, (c) kinetics of B1, and (d) Mn, SEC and the corresponding dispersity values. | ||
The inimer backbone structure was further investigated by grafting from a copolymer of EtOx and In. We have established that copolymerisation of EtOx and In results in well-defined copolymers with the radical initiating sites distributed along the polymer backbone. Hence, polymer P6 was used to form graft copolymers, again by Cu(0)-mediated RDRP in organic media. This resulted in longer, less dense brush-like structures compared to those when using the homopolymer of the inimer as the brush initiator. The study of the reaction kinetics, however, showed an induction period of around 2 hours before the reaction showed any conversion. Although Cu(0)-mediated RDRP is known to have some induction period,37 the much longer induction period observed in B3 compared to B1 or B2 could be due to the relatively lower solubility of P6 in the solvent, namely IPA (Fig. S10†). Nevertheless, the polymerisation led to 77% acrylate monomer conversion after 5 hours, and SEC traces of the resulting graft copolymer showed the residual oxazoline backbone polymer (Fig. 4). Based on the GPC traces in Fig. 4b, there is an indication that some polymer chains cannot initiate the brush polymerisation. For B3, only 20% of the backbone has brush initiating sites, whereas in B1, 100% of the backbone was brush initiating. Therefore in B3, where P6 was used as the backbone (DP = 50, EtOx = 40, In = 10), it is statistically possible that there might be some chains where there are less than 10 initiating sites on the backbone, or in some cases, none. Although we showed that because of the similar rates of reaction between EtOx and In, the brush initiating sites were statistically distributed along the backbone, there is still a limit to the precise control over the monomer location within the polymer.38 Because of this statistical distribution of brush initiating and non-brush initiating monomers, it is difficult to have a uniform distribution of the brush polymer since the backbone itself is a distribution of linear polymers containing ten or less brush initiating sites. There is another possibility that may result in the formation of the low molar mass peak, which is the presence of small initiating groups such as 2-bromoisobutyrate. This compound may form upon hydrolysis of the ester group in the inimer structure or might arise during the synthesis of the inimer; however, the latter is not possible as that would have stopped the cationic ring opening polymerisation. We have looked at the 3–4 ppm region in the 1H NMR spectra of P6, which would show two different shifts of the –CH2 peak adjacent to the ester group if hydrolysis had taken place, one associated with the inimer structure and the other associated with 2-n-pentanol-2-oxazoline. However, we could not identify a second peak that may belong to a hydrolyzed 2-bromoisobutyrate compound.
 | ||
| Fig. 4 Analysis of the bottlebrush copolymer B3: (a) SEC traces of the bottlebrush copolymer compared to the linear backbone, (b) evolving SEC traces of B3 over time, (c) kinetics of B3, and (d) Mn, SEC and the corresponding dispersity values. | ||
These demonstrations of graft copolymer synthesis using In have shown that the Cu(0)-RDRP initiating group on the monomer does not interfere with the backbone polymerisation and in the case of B1 can produce well-defined graft copolymers. However, further structural elucidation through microscopy will be needed to confirm their brush-like structure.
Conclusion
To conclude, a synthetic route to a 2-oxazoline inimer was established whereby ε-caprolactone, a commercially available, non-toxic starting material, was used to form the hydroxyl 2-oxazoline precursor to obtain the inimer in a scalable manner. The synthesis of the monomer led to the formation of brush copolymers using the grafting from approach, first using CROP for the backbone and Cu(0)-mediated RDRP for the brushes. Kinetic studies were carried out on the CROP of the new monomer where it showed that polymerisation on its own or with a comonomer occurs in a “living” manner. Based on the monitoring of monomer consumption throughout the reaction, both EtOx and inimer showed similar rates of reaction indicating incorporation of In in a statistical fashion. Cu(0)-mediated RDRP using the inimer containing polymers with various acrylates resulted in well-defined graft copolymers.Author contributions
J. K. has performed all the experimental work. The project was designed, discussed, and written by the contributions of all authors.Conflicts of interest
The authors declare no competing financial interest.References
- R. Konradi, C. Acikgoz and M. Textor, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS
.
- S. Nam, J. Seo, S. Woo, W. H. Kim, H. Kim, D. D. C. Bradley and Y. Kim, Nat. Commun., 2015, 6, 8929 CrossRef CAS
- K. P. Luef, C. Petit, B. Ottersböck, G. Oreski, F. Ehrenfeld, B. Grassl, S. Reynaud and F. Wiesbrock, Eur. Polym. J., 2017, 88, 701–712 CrossRef CAS
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS
- G. Morgese, B. Verbraeken, S. N. Ramakrishna, Y. Gombert, E. Cavalli, J. G. Rosenboom, M. Zenobi-Wong, N. D. Spencer, R. Hoogenboom and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 11667–11672 CrossRef CAS
- L. T. T. Trinh, H. M. L. Lambermont-Thijs, U. S. Schubert, R. Hoogenboom and A. Kjøniksen, Macromolecules, 2012, 45, 4337–4345 CrossRef CAS
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC
- Y. Xia, V. Adibnia, R. Huang, F. Murschel, J. Faivre, G. Xie, M. Olszewski, G. De Crescenzo, W. Qi, Z. He, R. Su, K. Matyjaszewski and X. Banquy, Angew. Chem., Int. Ed., 2019, 58, 1308–1314 CrossRef CAS
- X. Banquy, J. Burdyńska, D. W. Lee, K. Matyjaszewski and J. Israelachvili, J. Am. Chem. Soc., 2014, 136, 6199–6202 CrossRef CAS
- G. Morgese, E. Cavalli, M. Müller, M. Zenobi-Wong and E. M. Benetti, ACS Nano, 2017, 11, 2794–2804 CrossRef CAS
- G. Delaittre, Eur. Polym. J., 2019, 121, 109281 CrossRef CAS
- W. Yan, S. N. Ramakrishna, N. D. Spencer and E. M. Benetti, Langmuir, 2019, 35, 11255–11264 CrossRef CAS
-
B. S. Sumerlin and K. Matyjaszewski, in Macromolecular Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, vol. 2, pp. 1103–1135 Search PubMed
- G. Xie, M. R. Martinez, M. Olszewski, S. S. Sheiko and K. Matyjaszewski, Biomacromolecules, 2019, 20, 27–54 CrossRef CAS
- G. G. Alvaradejo, H. V. T. Nguyen, P. Harvey, N. M. Gallagher, D. Le, M. F. Ottaviani, A. Jasanoff, G. Delaittre and J. A. Johnson, ACS Macro Lett., 2019, 8, 473–478 CrossRef CAS
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146–1149 CrossRef CAS
- S. I. Yamamoto, J. Pietrasik and K. Matyjaszewski, Macromolecules, 2008, 41, 7013–7020 CrossRef CAS
- A. Kerr, M. Hartlieb, J. Sanchis, T. Smith and S. Perrier, Chem. Commun., 2017, 53, 11901–11904 RSC
- N. Zhang, S. Huber, A. Schulz, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 2215–2221 CrossRef CAS
- V. P. Beyer, B. Cattoz, A. Strong, A. Schwarz and C. R. Becer, Macromolecules, 2020, 53, 2950–2958 CrossRef CAS
- B. A. Drain and C. R. Becer, Eur. Polym. J., 2019, 119, 344–351 CrossRef CAS
- E. Shin, C. Lim, U. J. Kang, M. Kim, J. Park, D. Kim, W. Choi, J. Hong, C. Baig, D. W. Lee and B. Kim, Macromolecules, 2020, 53, 3551–3562 CrossRef CAS
- X. Zheng, C. Zhang, L. Bai, S. Liu, L. Tan and Y. Wang, J. Mater. Chem. B, 2015, 3, 1921–1930 RSC
- R. Article, R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405 RSC
-
N. Hadjichristidis, S. Pispas, M. Pitsikalis, H. Iatrou and D. J. Lohse, Graft Copolymers, 2002 Search PubMed
- Y. Tsukahara, S. I. Namba, J. Iwasa, Y. Nakano, K. Kaeriyama and M. Takahashi, Macromolecules, 2001, 34, 2624–2629 CrossRef CAS
- T. Chen, Z. Qin, Y. Qi, T. Deng, X. Ge, J. Wang and X. Hou, Polym. Chem., 2011, 2, 1190–1194 RSC
- O. Haba and H. Itabashi, Polym. J., 2014, 46, 89–93 CrossRef CAS
- T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15–33 CrossRef CAS
-
J. Jansen, A. Dias and P. Tijssen, US Pat., 20010020050A1, 2001 Search PubMed
- M. Litt and M. Levy, J. Polym. Sci., Part A-1: Polym. Chem., 1968, 6, 1883–1894 CrossRef
- R. A. Young, E. L. Malins and C. R. Becer, Aust. J. Chem., 2012, 65, 1132–1138 CrossRef CAS
- H. Goossens, S. Catak, M. Glassner, V. R. de la Rosa, B. D. Monnery, F. De Proft, V. Van Speybroeck and R. Hoogenboom, ACS Macro Lett., 2013, 2, 651–654 CrossRef CAS
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, Polymer, 2009, 48, 1973–1978 Search PubMed
- Y. Zhang, P. He, X. Liu, H. Zhang, H. Yang, C. Xiao and X. Chen, Eur. Polym. J., 2018, 107, 308–314 CrossRef CAS
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS
- Y. Gao, T. Zhao, D. Zhou, U. Greiser and W. Wang, Chem. Commun., 2015, 51, 14435–14438 RSC
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: SEC, NMR, synthesis, materials. See DOI: 10.1039/d0py01092h |
| This journal is © The Royal Society of Chemistry 2020 |