
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solubility-governed architectural design of polyhydroxyurethane-graft-poly(ε-caprolactone) copolymers†
Charalampos
Pronoitis
 ,
Minna
Hakkarainen
and
Karin
Odelius
*
,
Minna
Hakkarainen
and
Karin
Odelius
*
Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, 100 44 Stockholm, Sweden. E-mail: hoem@kth.se
First published on 9th November 2020
Abstract
Polyhydroxyurethanes (PHUs) are strong candidates for replacing conventional isocyanate-based polyurethanes (PUs), yet analogous structural diversity and subsequent properties have so far not been reached for PHUs. Here, we demonstrate that by rational design of the PHU microstructure and utilization of solubility as an enabling tool, complex architectures can be realized with benign reagents and atom-economical reactions, without harsh reaction conditions and solvents. Polyhydroxyurethane-graft-poly(ε-caprolactone), PHU-graft-PCL, copolymers were synthesized through graft ring-opening polymerization of ε-caprolactone (CL) from the pendent –OH groups of a PHU. The structure of the PHU was carefully designed to allow its dissolution in CL and enable bulk polymerization conditions. Solubility tests of different PHUs in various lactones corroborated that the PHU-lactone structure pair was critical to achieve dissolution. The optimum reaction conditions were kinetically determined, the copolymers’ structure was confirmed by 1H, DOSY and 31P NMR spectroscopy and their thermal properties were evaluated by DSC and TGA analysis. Our work demonstrated the effectiveness of utilizing solubility to achieve complex architectures, expanding the synthetic intricacy for PHUs and could expand their future applications and opportunities.
Introduction
Significant effort is undertaken to develop solvent-free, atom-economical synthetic procedures, utilizing benign reagents and mild conditions as alternatives for the production of conventional polymers. There is also an ongoing quest to decrease the oil dependence of the chemical industry and therefore, great interest lies in exploiting renewable resources to obtain platform chemicals that can serve as precursors to functional monomers and polymers. One such “greener” class of polymers is polyhydroxyurethanes (PHUs). PHUs have emerged as a safer alternative to conventional thermoplastic and thermoset polyurethanes (PUs) that utilize toxic isocyanates for their production.1 The most common synthesis route to create PHUs is step-growth polymerization between diamines and cyclic dicarbonates, a highly attractive method from a sustainability point of view. It is atom-economical, it can be performed under bulk conditions and can be combined with extrusion techniques2,3 to shorten the polymerization time needed and improve the final characteristics of the PHUs. Structural diversity in the linear and crosslinked PHUs can also be achieved by utilizing one of the numerous dicarbonate precursors in combination with one or more of the several diamines available. These building blocks can be derived from renewable resources,4,5 enabling biobased PHUs with tailored microstructure, designed to reach the properties needed for the intended application. A plethora of cyclic carbonates can be accessed by incorporating CO2 to oxiranes6,7 and oxetanes8 or benign carbonylating agents such as dimethyl carbonate to diols9,10 thereby avoiding toxic phosgene and contributing to the fixation of CO2 for materials’ production.11Much fewer examples exist, however, of PHUs combined with other classes of polymers, creating more complex structures such as block, star or graft copolymers. This could be due to the inherent challenge of synthetic control over step-growth polymerization, the low molar masses achieved and the limited solubility of PHUs caused by the hydrogen bonding (H-bonding).12,13 Carbonate, ester and ether,14 thioether,15 sulfone,16 siloxane,17 amide18 and tertiary amine19 groups or longer segments of oligo-esters, -ethers and -carbonates,20,21 have for instance been reported along the backbone of linear PHUs, yielding diverse microstructures. Hydroxyurethane moieties have also been incorporated as side-groups in fluorinated, alternating copolymers22 and polyester-graft-poly(ethylene glycol) brush copolymers,23 and recently biobased ester-urethane star polymers based on glycerol and dimethyl-2,5-furan dicarboxylate were reported.24 Hence, PHUs with diverse microstructures can be achieved and hydroxyurethane functionalities can be used to decorate various polymer backbones, but copolymers with a PHU block or more complex architectures are lacking, especially when considering synthesis conditions devoid of solvents and hazardous reagents.
To truly promote sustainable polymers, in addition to utilizing benign reactants and mild reaction conditions, it is critical to prove that they are not limited to linear or crosslinked structures, but that they can be utilized for the synthesis of functional copolymers with complex architecture even for ‘hard to work with’ systems. We hypothesized that the flexibility of the PHU chemistry could enable this. By designing a biobased PHU with a microstructure that would allow its dissolution in ε-caprolactone (CL), a brush-like copolymer could be created by graft ring-opening polymerization (ROP) of CL from the pendent hydroxyl groups of the PHU and the copolymer characteristics could be simply controlled by adjusting the monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator ratio. 2,2′-(Ethylenedioxy)bis(ethylamine) (EDOBA) and diglycerol carbonate (DGC) were selected as monomers for the PHU. This selection had twofold importance. First, DGC can be easily synthesized from renewable, benign and inexpensive diglycerol and dimethyl carbonate, thereby, the sustainability requirements are met. Second, it bears an ether bridge which in combination with the ethylene glycol moiety of EDOBA should favor the dissolution of the PHU in CL allowing for bulk polymerization conditions and achieving an overall more sustainable synthesis.
:initiator ratio. 2,2′-(Ethylenedioxy)bis(ethylamine) (EDOBA) and diglycerol carbonate (DGC) were selected as monomers for the PHU. This selection had twofold importance. First, DGC can be easily synthesized from renewable, benign and inexpensive diglycerol and dimethyl carbonate, thereby, the sustainability requirements are met. Second, it bears an ether bridge which in combination with the ethylene glycol moiety of EDOBA should favor the dissolution of the PHU in CL allowing for bulk polymerization conditions and achieving an overall more sustainable synthesis.
Experimental
Materials
All reagents were used as received without further purification, unless stated otherwise. Moisture- and air-sensitive reagents were stored and used under inert N2 atmosphere in a MBraun glovebox. Diglycerol (DG, mixture of isomers, >80.0% with respect to the α,α′-isomer) was purchased from TCI Europe. Dimethyl carbonate (DMC, 99%) was purchased from Alfa Aesar. Calcium hydride (CaH2, ≥90%), potassium carbonate (K2CO3, ≥99.0%), tin(II) 2-ethylhexanoate (Sn(Oct)2, 92.5–100%), ε-caprolactone (CL, 97%), 2,2′-(ethylenedioxy)bis(ethylamine) (EDOBA, 98%), methanesulfonic acid (MSA, ≥99.0%), 1,5,7-triazabicyclo[4.4.0]-dec-5-ene (TBD, 98%), chromium(III) acetylacetonate (Cr(acac)3, 97%), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP, 95%), N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (HNOB, 97%), toluene (anhydrous, 99.8%), tetrahydrofuran (THF, ≥99.0%), ethyl acetate (EtOAc, ≥99.5%), butyl acetate (BuOAc, ≥99.5%), δ-valerolactone (δVL, technical grade), γ-valerolactone (γVL, 99%), ω-pentadecalactone (ωPDL, ≥98%), hexadecanolide (HDL, 97%), ethylene brassylate (EB, ≥95%), δ-decalactone (δDL, ≥98%), ε-decalactone (εDL, ≥99%) and α-bromo-γ-butyrolactone (αBr-γBL, 97%) were purchased from Sigma-Aldrich. Pyridine (≥99.7%), methanol (MeOH, ≥98.5%), n-heptane (≥99%), acetone (≥99%), chloroform-d (CDCl3, 99.8% D) and dimethyl sulfoxide-d6 (DMSO-d6, 99.8% D) were purchased from VWR. Chloroform (CHCl3, ≥99.8%) was purchased from Fisher Scientific. CL was stirred over CaH2 for 24 h, distilled under vacuum and stored over activated 4 Å molecular sieves under inert atmosphere before use. Sn(Oct)2 was stored over activated 4 Å molecular sieves under inert atmosphere before use.:1:0.02 of CL:OH:Sn(Oct)2 ratio is described as a representative example. Inside the glovebox, 3.25 g of CL (100 CL monomers per −OH) and 50 mg of PHU (0.285 mmol of −OH) and a stirring bar were added in a 25 mL Schlenk flask and sealed with a glass stop. The flask was taken out of the glovebox, connected to a Schlenk line and placed in a thermostated oil bath at 110 °C under stirring. A N2 gas flow was applied and kept during the entire reaction. A solution of Sn(Oct)2 (2.3 mg, 0.0057 mmol, 0.02 eq. per −OH) in a minimum amount of anhydrous toluene was added to the flask after the PHU had completely dissolved in the CL. Time zero was set as the time when all of the catalyst had been added. At regular time intervals, samples were withdrawn with oven-dried, glass Pasteur pipettes and directly dissolved in CDCl3 for further 1H NMR analysis. Once the reaction was completed, the crude product was cooled down to room temperature and purified by two successive precipitations from CHCl3, first in a 10-fold excess of cold MeOH, and then in cold heptane. The solvent was filtered out and the copolymer was dried under vacuum at room temperature and stored. For the MSA-catalyzed reactions, the same procedure was followed, only MSA was directly added in the flask with an automatic pipette, and a CDCl3 with 0.1 M pyridine solution was employed to quench the catalyst. Table S1† summarizes the masses of the reactants for all the kinetic reactions.
The same procedure was followed for the synthesis and purification of the copolymers under the optimized conditions with Sn(Oct)2 as catalyst, but crimp-top glass vials sealed with butyl/PTFE septum caps were employed instead of Schlenk flasks. The masses of the reactants for these reactions are summarized in Table S2.†
Characterization
000000 g mol−1. PSS WinGPC Unity v. 7.2 software was used to process the data. For the PHU-graft-PCL copolymers, a Malvern GPCMAX instrument was employed equipped with a PLgel 5 μm guard column (7.5 × 50 mm), two PLgel 5 μm MIXED-D (300 × 7.5 mm) columns and a Viscotek VE3580 RI detector. The mobile phase was CHCl3 at 35 °C, at a flow rate of 0.5 mL min−1 and toluene was used as internal standard for flow rate fluctuation corrections. The system was calibrated with narrow poly(styrene) standards with molar mass range from 1200 to 940000 g mol−1. The data were processed by OmniSEC v. 5.1 software.
 | (1) |
Results and discussion
Graft copolymers, PHU-graft-PCL, were successfully designed and synthesized implementing a “grafting from” approach through bulk, graft ROP of ε-caprolactone (CL) from the pendent –OH groups of a polyhydroxyurethane (PHU). As PHUs are very versatile with respect to the chemical structure of their building blocks and thereby their microstructure, the predefined choice of 2,2′-(ethylenedioxy)bis(ethylamine) (EDOBA) and diglycerol carbonate (DGC) allowed tailoring a PHU that was soluble in the monomer CL, thereby enabling bulk polymerization, Scheme 1.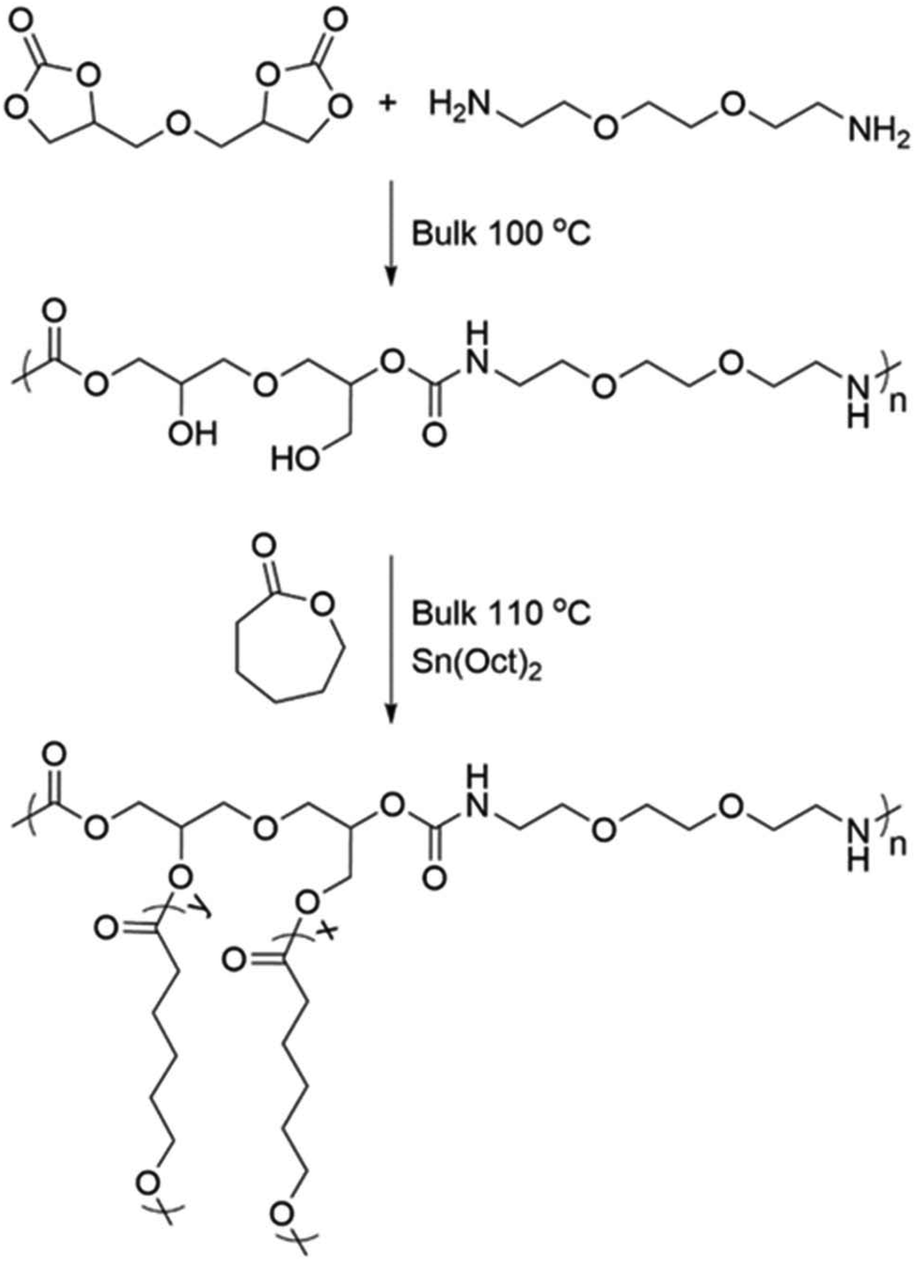 | ||
| Scheme 1 Synthesis of the EDOBA PHU and the PHU-graft-PCL copolymers. | ||
Polyhydroxyurethane (PHU) synthesis and characterization
As a first step towards the PHU-graft-PCL copolymers, the PHU main chain was synthesized under rather mild reaction conditions, 100 °C, in bulk, without the addition of catalyst and with stoichiometric balance between DGC and EDOBA.Thanks to the comparably mild reaction conditions, the undesired side-reactions, mainly urea formation, common during PHUs’ synthesis,7,12,13,29,30 were circumvented and only <2% urea was formed. The chemical structure of the PHU was confirmed by 1H NMR spectroscopy and all signals are attributed to the desired PHU structure. An overlap of the –CHCH2O– and –CHCH2OH protons of DGC with the –OCH2CH2O– protons of the diamine was observed, giving a sharp peak at 3.49 ppm, Fig. 1. The ratio of primary to secondary hydroxyl groups, pOH:sOH was 27:73, in agreement with other PHUs reported in the literature.10,31–35 The pOH:sOH ratio is especially important in this system for the second step of the graft ROP, where pOH is considered a more active initiator than sOH.36,37 The urea byproducts (<2%) were identified by the characteristic –NHCONH– protons at 5.98 ppm,38Fig. 1. The synthesized PHU was water-soluble and completely amorphous with glass transition temperature (Tg) of 9.6 ± 1.6 °C. The number-average molar mass (Mn) and dispersity (Đ) were 5300 g mol−1 and 1.9, respectively. The total −OH concentration of the PHU was 5.7 mmol g−1, calculated through 31P NMR spectroscopy after phosphitylation of the hydroxyl groups with Cl-TMDP,26,27 Fig. S2.† Additionally, the pOH:sOH ratio was 32:68 which is in good agreement with the ratio determined from 1H NMR spectroscopy.
 | ||
| Fig. 1 1H NMR spectrum (400 MHz, DMSO-d6) of the EDOBA PHU. | ||
Designing the graft ROP of CL from the PHU
To achieve a complex brush-like architecture in a sustainable manner, the inherent and extensive H-bonding in the PHU13 needs to be overcome. Protic or polar aprotic solvents such as MeOH, DMF and DMSO disrupt the intermolecular H-bonding between the pendent hydroxyl groups, acting as Lewis acid or base,39 respectively, allowing the dissolution even in less polar CHCl3.13,32 The carbonyl oxygen of CL could potentially act as a H-bonding acceptor, similar to a solvent with an available lone electron pair, facilitating the disruption of the intermolecular H-bonding of the PHU. Combined with elevated temperature, which further contributes to the disruption of the H-bonding, the PHU could ultimately be dissolved in CL. This would create appropriate conditions for a robust, solvent-free, ring-opening polymerization of CL. To test this hypothesis, the EDOBA PHU (10 wt%) was mixed with CL at 90 °C and a slow dissolution was observed, further improved by increasing the temperature to 100 and 110 °C. At 110 °C, the PHU dissolved completely in CL and a clear solution was retained for several hours at that temperature. Upon cooling, the PHU gradually precipitated out. At low PHU concentrations (1 wt%), no precipitation was observed during cooling even after one week at room temperature.To prove the contribution of CL's structure to the dissolution of the PHU, the solubility of the PHU was investigated in the linear equivalent of CL, butyl acetate (BuOAc) at 110 °C. Visually, the PHU was insoluble in BuOAc. The differences in solubility parameters (Δδ) between EDOBA PHU, CL and BuOAc were calculated by Hoy's method of group contribution, providing a theoretical explanation for the observed behavior, Table S3.†40–43 The ΔδPHU–CL is 7.3 (MJ m−3)1/2 and the ΔδPHU–BuOAc is 11.4 (MJ m−3)1/2. Both values are higher than the 5 (MJ m−3)1/2 threshold below which the polymer is soluble in a solvent,43 but clearly, dissolution in CL is favored over BuOAc. The high temperature applied (110 °C) has probably a beneficial effect towards dissolution, but the discrepancy could be further related to the structure of the solvent. The linear structure of BuOAc makes the alkyl groups more accessible and the polar interactions between the PHU and the CL's carbonyl oxygen may be hindered. In contrast, the ring structure of CL could be responsible for the adoption of a more favorable conformation that suppresses the freedom of movement of the alkyl groups and allows for more interactions between the carbonyl oxygen of CL and the H-bonding donor sites of the PHU, Fig. S3.† Additionally, although the ester groups of both CL and BuOAc are described as of moderate H-bonding capability,44 lactones are more basic than linear esters due to conformational differences.45 This increased basicity could result in slightly stronger H-bonding of CL compared with BuOAc and therefore, the ability to dissolve the PHU would be influenced as well. Ethyl acetate (EtOAc) was also evaluated near its boiling point (77 °C) due to its structural similarity with CL46 and with the possibility that the shorter ethyl group could facilitate dissolution. The PHU was, however, again insoluble, suggesting that either the lower temperature prevents dissolution or that the same effect as for BuOAc occurs.
To further evaluate whether the microstructure of the PHU or the elevated temperature was the sole reason for dissolution in CL, we compared the solubility of a PHU with an aromatic backbone in CL. The aromatic PHU was synthesized under the same polymerization conditions as DGC and EDOBA but m-xylylenediamine (mXDA) was employed instead of EDOBA (Mn = 3500 g mol−1, Đ = 2.5, Tg = 37 ± 1.1 °C). Even after prolonged heating at 130 °C in CL, a considerable part of the mXDA PHU was not dissolved, as visually observed, confirming that high temperature is substantially contributing to the dissolution of the PHU but the chemical structure is equally important. The polar ethylene glycol moiety of EDOBA probably favored the interactions between the EDOBA PHU and CL in contrast to the hydrocarbon structure of mXDA. mXDA PHU was also not dissolved in boiling toluene, confirming that the structural similarity between the PHU and the solvent is contributing to, yet not exclusively governing, the dissolution of the PHU and that the H-bonding has a central role in it.
To gain further insight into the dissolution of the PHU, a series of lactones with varying size and functionality was evaluated for their ability to dissolve the EDOBA PHU at 110 °C, Fig. 2. Lactones with long aliphatic segments, either in the main ring (ωPDL, HDL, EB) or as side chains (δDL, εDL), were unable to dissolve the EDOBA PHU. This is most probably due to unfavorable interactions between the polar groups of the PHU and the apolar hydrocarbon moieties of the lactones, similarly to the BuOAc effect. The insolubility in α-bromo-γ-butyrolactone (αBr-γBL) should be associated with the Br-group which can act as an additional H-bonding acceptor site, but it also makes the molecule more hydrophobic. As a 6-membered ring, δ-valerolactone (δVL) could, similarly to CL, also adopt a favoring conformation that resulted in complete dissolution of the PHU after heating. However, the solution seemed less stable compared to CL and faster precipitation of the PHU occurred upon cooling, as determined by an increased haziness of the solution. The effect of the smaller ring size on dissolution was evaluated through γ-valerolactone (γVL). Low solubility was observed, with the exo-methyl substituent contributing to this behavior. An identical solubility test was also performed for mXDA PHU, which presented similar or inferior dissolution for all lactones, again highlighting the pivotal role of the PHU's microstructure.
 | ||
| Fig. 2 Solubility of EDOBA PHU in various lactones at 110 °C. γ-valerolactone (γVL), δ-valerolactone (δVL), ε-caprolactone (CL), ω-pentadecalactone (ωPDL), hexadecanolide (HDL), ethylene brassylate (EB), δ-decalactone (δDL), ε-decalactone (εDL), α-bromo-γ-butyrolactone (αBr-γBL). | ||
Catalyst selection for the graft ROP of CL
To screen catalysts that enable a fast and efficient polymerization of CL, but hinder simultaneous PHU scrambling and thereby the formation of e.g. urea byproducts, initial trials were performed by adding catalysts to the PHU and keeping the system at 120 °C. A plethora of catalysts have been reported to efficiently catalyze the ROP of CL, ranging from metal based catalysts, such as tin- and zinc alkoxides, and pure organocatalysts, both acidic and basic such as sulfonic acids and guanidines.47,48 To screen broadly, one of the most efficient and well-studied catalysts, tin(II) 2-ethylhexanoate (Sn(Oct)2)49 was selected along with two organocatalysts, methanesulfonic acid (MSA)50 and 1,5,7-triazabicylco[4.4.0]-dec-5-ene (TBD).51 The scrambling effect was evaluated by 13C NMR spectroscopy monitoring changes in the carbonyl region, as it is the clearest and most direct way to determine if scrambling reactions occur. The results were compared to an uncatalyzed PHU sample heated to the same temperature, Fig. 3. | ||
| Fig. 3 Carbonyl region of 13C NMR spectra (100 MHz, DMSO-d6) of the EDOBA PHU tested with different catalysts at 120 °C. | ||
Three chemical shifts are observed for the PHU, the urethane's carbonyl carbon at 156.3 and 156.0 ppm and a small signal at 157.9 ppm corresponding to the carbonyl carbon of the urea byproduct. The –NHCOO– signals of the sample with no catalyst, Sn(Oct)2 and MSA were completely intact after heating. In the MSA sample a new signal at 163.05 ppm appeared, most likely representing a second type of urea byproduct. This was surprising as the strong acid MSA should protonate the –OH and –NH2 groups thereby, it should not allow side reactions to occur. It is possible that due to the slight basicity of DMSO, a reaction between DMSO and MSA takes place producing the corresponding salt46 and affecting the chemical shifts of the urea byproducts that were already present. The spectrum of the TBD sample was vastly changed. The urethane signals disappeared while the 157.9 ppm signal was enhanced and a new peak at 167.6 ppm appeared. This was explained by the ability of TBD to catalyze side reactions involving the deprotonation of amine species concurrently with activation of the urethane's carbonyl and subsequent attack on the urethane bonds producing urea linkages.38 Therefore, Sn(Oct)2 and MSA were chosen as catalysts for the ROP of CL.
Monitoring of the graft ROP of CL from the PHU
The kinetics of the graft ROP of CL from the PHU main chain were monitored to evaluate the efficacy of the catalysts and establish the optimum polymerization conditions for the system, varying the catalyst loading and CL loading. Initially, the CL:OH ratio was fixed at 100:1 and the catalyst loading was varied for both MSA and Sn(Oct)2. The loadings of the catalysts were chosen based on conventional loadings of each catalyst49,50 and to reflect the effect of higher catalyst content on the reaction rate. The MSA catalyzed polymerization of CL has been shown to occur significantly faster when the MSA:OH ratio is increased from 1:1 to 3:1, at room temperature.50 Here, the polymerization was carried out at 110 °C and a 1:0.05 OH:MSA ratio was included to evaluate the influence of high temperature at a low MSA loading. Four different OH:Sn(Oct)2 ratios were evaluated, 1:0.01, 1:0.02, 1:0.05 and 1:1 probing the effect of both low and high catalyst loading. A reaction temperature of 110 °C was considered ideal for the reaction to ensure full dissolution of the PHU in CL and high activity of the catalyst. The reactions were monitored by 1H NMR spectroscopy and SEC until high conversions were reached. The samples are named after the CL and catalyst eq. per −OH group, for instance CL100MSA1 denotes that the reaction was catalyzed by MSA and the CL:OH and OH:MSA molar ratio was 100:1 and 1:1, respectively. The reaction conditions and characteristics of the samples are summarized in Table 1 and the conversion, Mn and Đ vs. time profiles are presented in Fig. 4.
 | ||
| Fig. 4 Graft ROP of CL from the EDOBA PHU with varying catalyst and catalyst loading. Conversion, Mn and Đ vs. time plots of the MSA (a, b and c) and Sn(Oct)2 (d, e and f) catalyzed reactions. The CL:OH ratio was 100:1 and each line corresponds to the eq. of catalyst per −OH group. For clarity, the 5–60 min points of conversion and Mnvs. time are shown in the inset graphs. | ||
| Sample name | Catalyst | CL:OH:Cat. |
CL conversionc (%) |
M
nd (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a Total reaction time was 24 h. b Conversion after 72 h. c Calculated through I4.06/(I4.06 + I4.24–4.22), where I4.06 and I4.24–4.22 are the integrals of the –COOCH2− signals of PCL and CL, at 4.06 and 4.24–4.22 ppm respectively, in the 1H NMR spectra. d Values of purified samples, calculated by SEC in CHCl3 against narrow PS standards. Mn values are rounded to the closest multiple of hundred. e The value corresponds to the crude product. f Sample gelled, the values in the brackets correspond to the crude fraction obtained after swelling and extraction in CHCl3. | |||||
| CL100 | — | 100:1:— |
4.7 | — | — |
| CL100MSA0.05 | MSA | 100:1:0.05 |
9 (96)b | 5300e | 6.0e |
| CL100MSA1 | MSA | 100:1:1 |
98 | 36200 |
2.2 |
| CL100MSA3 | MSA | 100:1:3 |
98 | 32500 |
1.8 |
| CL100Sn0.01 | Sn(Oct)2 | 100:1:0.01 |
39 (98)b | 39500 |
1.7 |
| CL100Sn0.02 | Sn(Oct)2 | 100:1:0.02 |
99 | 61600 |
6.5 |
| CL100Sn0.05 | Sn(Oct)2 | 100:1:0.05 |
96 | 62800 |
7.5 |
| CL100Sn1 | Sn(Oct)2 | 100:1:1 |
97 | 57700 |
3.7 |
| CL10Sn0.02 | Sn(Oct)2 | 10:1:0.02 |
96 | Gelled (4300)f | Gelled (8.9)f |
| CL25Sn0.02 | Sn(Oct)2 | 25:1:0.02 |
96 | Gelled (8200)f | Gelled (33.2)f |
| CL50Sn0.02 | Sn(Oct)2 | 50:1:0.02 |
98 | 71600 |
8.9 |
The reactions presented similar features; as expected increasing the catalyst loading resulted in a faster CL conversion, while in the absence of any catalyst (CL100), thermal polymerization of CL took place to a small extent and after a long reaction time (4.7% conversion after 8 h), Fig. S4.† Transesterification reactions took place in the PCL blocks as indicated by the increase in Đ, and CL homopolymer was formed as seen from the bimodal SEC traces. Naturally, Đ values less than ∼2 are not possible as the PHU backbone already had Đ = 1.9. Both catalysts, at all loadings, reached high conversion at the end of the reaction, Table 1, but overall, CL's conversion was faster for the MSA than the Sn(Oct)2 catalyzed reactions as seen from the conversion vs. time profiles in Fig. 4. CL100MSA1 and CL100MSA3 reached 98% conversion within 45 and 5 min, respectively, whereas the CL100Sn0.02, CL100Sn0.05 and CL100Sn1 samples achieved conversion >95% within 24, 8 and 2 h, respectively. The reason is most likely the different activation mechanisms for the two catalysts. MSA catalyzes an activated monomer mechanism by protonating the CL's carbonyl and activating the hydroxyl group by H-bonding. The reaction proceeds in two steps (nucleophilic attack and ring-opening) through a tetrahedral intermediate.52 Instead, Sn(Oct)2 follows a coordination–insertion mechanism.49 This includes three distinct steps before the active tin alkoxide species, responsible for propagation, are generated and therefore, slower kinetics are observed. At low catalyst loading (CL100MSA0.05 and CL100Sn0.01), MSA seemed initially faster but after 24 h merely 9% conversion was reached, Table 1. In contrast, for Sn(Oct)2 (CL100Sn0.01) a conversion of 39% was reached. The induction period observed is likely due to more protonation instead of activation of the −OH in the case of MSA,50 and due to the equilibrium between the dormant chain end species and the active tin alkoxide species in the case of Sn(Oct)2.49
The formation of the active species is favored at higher Sn(Oct)2 loadings leading to faster initiation, as confirmed by CL100Sn0.02 and CL100Sn0.05. Their induction period was approximately 60 and 30 min, while the induction period of CL100Sn1 was ≤5 min due to the high catalyst amount, Fig. 4.
In both systems, after full conversion was reached, Mn decreased concomitantly with an increase of Đ. This indicated the existence of side reactions, most likely inter- and/or intramolecular transesterification, Fig. 4b, c, e and f. Transesterification is a prominent reaction in the ROP of lactones and it is largely dependent on the nature of the catalyst, reaction temperature and time. Sn-based catalysts, and specifically Sn(Oct)2, are well known to induce such side reactions,53 in linear49,54–56 and more complex architectures such as star57 or graft (co)polymers.58,59 Similarly, transesterification has been observed in some sulfonic acid-catalyzed polymerizations of CL and other lactones towards linear and star-based homo- or copolymers.60–64 Due to the abundance of hydroxyl groups and at high catalyst loadings (CL100MSA3, CL100Sn1), transesterification may take place already from the early stages of the reaction, but at a lower rate compared to the polymerization rate. In other words, the competition between transesterification and polymerization is negligible when copious CL monomer is available, but it becomes significant once high monomer conversion has been reached. This is why the low catalyst loading reactions, CL100MSA0.05 and CL100Sn0.01, did not present analogous behavior. Although prolonged reaction times open up for side reactions to occur, the conversion of those samples was low until 24 h due to the slow polymerization rate.
The achieved Mn was independent of the catalyst loading, Table 1, although the products of the MSA catalyzed reactions exhibited lower Mn values compared to the Sn(Oct)2 catalyzed reactions. This was explained by the bimodal molar mass distribution observed in the SEC chromatograms, which was more pronounced for the MSA than the Sn(Oct)2 catalyzed reactions and notably, it was the lowest for the CL100Sn0.02, Fig. S5 and S6.† The bimodality indicated the existence of a second species, apart from the PHU-graft-PCL copolymer, which could be a CL homopolymer initiated by adventitious H2O or by 6-hydroxyhexanoic acid resulting from hydrolysis of CL monomer catalyzed by MSA. In either case, PCL with an α-carboxylic acid group (HOOC-PCL-OH) would be produced. The origin of the two species was determined by fractionating the 30 min sample of CL100MSA1 by SEC and analyzing the fractions by 1H NMR spectroscopy, Fig. S7.† The spectrum of the first peak, i.e. the species with highest molar mass, presented a signal at 12.4 ppm, which is attributed to an acidic proton and thus, it corroborates our hypothesis. In the spectrum recorded for the second peak, the –CH– protons adjacent to the esterification sites are observed along with some PHU backbone signals, confirming that this species is the PHU-graft-PCL copolymer. The –CH– signals can be weakly seen in the first spectrum too, probably due to partial mixing of the two fractions during separation, Fig. S7.†
To evaluate the reactivity of the hydroxyl groups, 31P NMR spectra of the purified products were recorded after –OH phosphitylation, Fig. S8.† A linear PCL synthesized in bulk with Sn(Oct)2 as catalyst (Mn,NMR = 12200 g mol−1) was taken as a reference. In the spectra of the copolymers, the PHU's −OH signals at 147.5 and 146.1 ppm have disappeared and only the terminal −OH from the PCL blocks are detected at 146.9 ppm indicating the successful graft ROP of CL from all the −OH groups. This proves that the system was not limited either by its inherent features such as steric hindrance and high viscosity of the bulk or surprisingly, by the reactivity difference between the secondary and primary hydroxyl groups. The signal at 134.7 ppm was attributed to the acid group of the CL homopolymer. The possibility of this signal belonging to 2-ethylhexanoic acid residue in the Sn(Oct)2 catalyzed reactions, was considered highly unlikely as it should have been removed during precipitation.55
Overall, the kinetics of the MSA and Sn(Oct)2 catalyzed reactions with varying catalyst loading, showed that the copolymers formed under Sn(Oct)2 catalysis exhibited higher molar mass and lower CL homopolymer content, compared to their MSA analogues. Hence, Sn(Oct)2 was selected for the rest of the study. The CL100Sn0.02 sample portrayed monomodal molar mass distribution and reached high conversion (88.5%) and Mn (64300 g mol−1) within a reasonable timeframe (8 h) while the extent of transesterification was comparably low (Đ = 2.2). Therefore, the 1:0.02 OH:Sn(Oct)2 ratio was considered the optimum. Keeping the OH:Sn(Oct)2 ratio fixed at 1:0.02, the reactions were kinetically monitored by varying the CL:OH ratio to 10:1, 25:1, 50:1 and 100:1. The samples were again named according to the CL:OH and OH:Sn(Oct)2 molar ratio. For instance, CL10Sn0.02 denotes a 10:1 CL:OH ratio and a 1:0.02 OH:Sn(Oct)2 ratio. The reaction characteristics are summarized in Table 1 and the conversion, Mn and Đ vs. time plots are presented in Fig. 5. The reactions fall into two groups depending on the CL loading of the samples. The low CL loading samples, CL10Sn0.02 and CL25Sn0.02, exhibited conversions above 90% within 2–3 h and reached their final Mn approximately at the same time. Thereafter, due to transesterification reactions, the Mn decreased continuously followed by quick increase of Đ for CL10Sn0.02 and a more gradual increase for CL25Sn0.02. After 24 h the samples had gelled, similarly to what has been previously observed in graft copolymers of poly(ethylene-co-vinyl alcohol) and CL.58 The samples with higher CL loadings, CL50Sn0.02 and CL100Sn0.02, reached high conversion, ≥85%, after 8 h and the Mn increased continuously during the total 24 h of reaction time. Đ increased more modestly compared to the reactions with a low CL loading, with a slightly higher value for CL50Sn0.02 than CL100Sn0.02. The presence of CL homopolymer is low in all cases, Fig. S9.† Thus, it is shown that the CL loading can be varied to obtain PHU-graft-PCL copolymers with varying molar mass of the PCL grafts and controlled characteristics.
 | ||
| Fig. 5 Graft ROP of CL from the EDOBA PHU with varying CL loading. (a) Conversion, (b) Mn, (c) Đ vs. time plots. x marks the eq. of CL per −OH group. For clarity, the 5–60 min points of conversion vs. time and the 5–120 min points of Mnvs. time are shown in the inset graphs. CL10Sn0.02 and CL25Sn0.02 samples gelled after 24 h. | ||
Synthesis of PHU-graft-PCL copolymers under the optimized conditions
The synthesis of the PHU-graft-PCL copolymers with 1:0.02 OH:Sn(Oct)2 ratio was repeated for the four CL loadings (10, 25, 50, 100 CL monomers per –OH group) and quenched upon high conversion. The final copolymers were named PHU-graft-PCLx where x signifies the theoretical number of CL repeating units per −OH group. The reaction conditions and characteristics of the final copolymers are presented in Table 2.
| x | CL:OH:Sn(Oct)2 |
t (h) | CL conversionb (%) |
M
nc (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a Total reaction time. b Calculated through I4.06/(I4.06 + I4.24–4.22), where I4.06 and I4.24–4.22 are the integrals of the –COOCH2− signals of PCL and CL at 4.06 and 4.24–4.22 ppm, respectively, in the 1H NMR spectra. c Calculated by SEC in CHCl3 against narrow PS standards, average of triplicate measurements. Mn values are rounded to the closest multiple of hundred. | |||||
| 10 | 10:1:0.02 |
3 | 96 | 23200 ± 500 |
2.23 ± 0.01 |
| 25 | 25:1:0.02 |
3 | 96 | 42700 ± 900 |
4.53 ± 0.25 |
| 50 | 50:1:0.02 |
3.5 | 87 | 75700 ± 1100 |
3.50 ± 0.08 |
| 100 | 100:1:0.02 |
6 | 72 | 111600 ± 1800 |
2.73 ± 0.05 |
Side reactions were suppressed as seen from the final Mn and Đ values, Table 2. The successful graft ROP of CL from the PHU was proven by 1H NMR spectroscopy where the signals at 5.16 and 5.04 ppm, corresponding to the methine protons next to the esterified hydroxyls of the PHU, are detected for all the copolymers, Fig. 6. The PHU's backbone signals between 3.60–3.36 ppm and the terminal –CH2OH of PCL at 3.64 ppm are also clearly observed. Partial overlapping of the –OCH2CH2O– protons of the diamine at 3.60 ppm with the terminal –CH2OH protons of PCL at 3.64 ppm did not allow for an estimation of PCL's degree of polymerization. However, DOSY NMR experiments indubitably showed copolymer formation and the absence of copolymer/CL homopolymer blend, as all of the 1H signals are attributed to a single species with the same diffusion coefficient,65Fig. 7. Finally, 31P NMR spectroscopy confirmed that there are no unreacted −OH groups on the PHU after polymerization, as only the terminal −OH of the PCL blocks were identified at 146.96 ppm, Fig. 8.
 | ||
| Fig. 6 Stacked 1H NMR spectra (400 MHz, CDCl3) of the final PHU-graft-PCLx copolymers. | ||
 | ||
| Fig. 7 DOSY NMR spectra (400 MHz, CDCl3) of the PHU-graft-PCL copolymers. | ||
 | ||
| Fig. 8 31P NMR spectra of the final PHU-graft-PCL copolymers. All of the −OH groups of the PHU have reacted and only the terminal −OH of the PCL grafts are present. | ||
Thermal properties of the PHU-graft-PCL copolymers
The thermal properties of the PHU-graft-PCL copolymers were controlled by the molar mass of the PCL grafts. The melting (Tm) and crystallization (Tc) temperature and the respective enthalpies (ΔHm and ΔHc), increased with increased molar mass of the PCL blocks, Table 3, Fig. S10.† The PHU and PCL segments were not miscible as two discrete Tgs were observed for the PHU-graft-PCL10 copolymer, in the vicinity of the Tgs of the corresponding homopolymers. The absence of clear glass transitions in the thermographs of the other copolymers, is most likely associated to their higher degree of crystallinity (Xc) compared to PHU-graft-PCL10. The degree of crystallinity increased with increasing molar mass of the PCL chains, yet it was slightly lower for the PHU-graft-PCL100 copolymer (49%). This is in line with previous observations of PCL displaying lower crystallinity at higher molar masses.66,67 The onset decomposition temperature (Td, 5%) of the copolymers was in the range of 288–300 °C and, as expected, it increased with increasing molar mass of the PCL grafts, Table 3. The grafted PCL was advantageous as it offered greater thermal stability due to the more stable ester bonds compared to the thermally labile urethane linkages of the PHU.| x |
T
g, PHUb (°C) |
T
g, PCLb (°C) |
T
mb (°C) |
ΔHmb (J g−1) |
T
cc (°C) |
ΔHcc (J g−1) |
X
ce (%) |
T
d,5%f (°C) |
|---|---|---|---|---|---|---|---|---|
| a All values are averages of triplicate measurements. b Determined from the second heating scan. c Determined from the cooling scan. d No Tg could be observed for either block. e Calculated through eqn (1). f Determined by TGA analysis at 5% mass loss. | ||||||||
| PHU | 9.6 ± 1.3 | — | — | — | — | — | — | 241 |
| PCL | — | −50.2 ± 1.4 | 54.8 ± 0.1 | 68.3 ± 2.6 | 34.6 ± 0.3 | 70.8 ± 0.5 | 49 | 268 |
| 10 | 7.8 ± 0.3 | −50.6 ± 0.7 | 42.6 ± 0.1 | 63.2 ± 0.5 | 16.3 ± 0.1 | 63.4 ± 0.9 | 45 | 288 |
| 25d | — | — | 50 ± 0.006 | 71.4 ± 1.3 | 25.4 ± 0.2 | 71.5 ± 1.1 | 51 | 291 |
| 50d | — | — | 53.4 ± 0.01 | 72.8 ± 1.3 | 29.5 ± 0.4 | 72.3 ± 1.4 | 52 | 291 |
| 100d | — | — | 55.4 ± 0.2 | 68.7 ± 2.3 | 28.9 ± 0.6 | 67.7 ± 2.0 | 49 | 301 |
Conclusions
Tunable polyhydroxyurethane-graft-poly(ε-caprolactone) copolymers with brush-like architecture were successfully synthesized through a ‘grafting from’ approach. Judicious selection of the PHU monomers, DGC and EDOBA, afforded a PHU that dissolved in CL at 110 °C and therefore enabled the graft ROP of CL in bulk. The system presented several features of sustainable design, solvent-free preparation of both the PHU backbone and the PHU-graft-PCL copolymers, combined with the biobased origin of DGC and mild reaction conditions. The polymerization reaction was robust and the molar mass of the PCL blocks was tuned by simply varying the CL:OH ratio. The thermal properties of the copolymers were controlled by the molar mass of PCL and the copolymerization was beneficial for the thermal stability, which was enhanced compared to the pure PHU. This work demonstrates that complex structures, that would otherwise be challenging to achieve, can be synthesized by utilizing polymer-in-monomer solubility. This enables the design of solvent-free systems and opens new possibilities for sustainable polymer synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Swedish Research Council Formas (2016-00700). Prof. Zoltán Szabò, Department of Chemistry, KTH Royal Institute of Technology, is gratefully acknowledged for all the help with the DOSY NMR experiments.References
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS.
- V. Schimpf, J. B. Max, B. Stolz, B. Heck and R. Mülhaupt, Macromolecules, 2019, 52, 320–331 CrossRef CAS.
- F. Magliozzi, G. Chollet, E. Grau and H. Cramail, ACS Sustainable Chem. Eng., 2019, 7, 17282–17292 CrossRef CAS.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- M. Alves, B. Grignard, A. Boyaval, R. Méreau, J. De Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jérôme, ChemSusChem, 2017, 10, 1128–1138 CrossRef CAS.
- M. Tryznowski, A. Swiderska, Z. Zołek-Tryznowska, T. Gołofit and P. G. Parzuchowski, Polymer, 2015, 80, 228–236 CrossRef CAS.
- J. L. J. van Velthoven, L. Gootjes, D. S. Van Es, B. A. J. Noordover and J. Meuldijk, Eur. Polym. J., 2015, 70, 125–135 CrossRef CAS.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS.
- V. Besse, F. Camara, F. Méchin, E. Fleury, S. Caillol, J. P. Pascault and B. Boutevin, Eur. Polym. J., 2015, 71, 1–11 CrossRef CAS.
- M. Blain, A. Cornille, B. Boutevin, R. Auvergne, D. Benazet, B. Andrioletti and S. Caillol, J. Appl. Polym. Sci., 2017, 44958, 1–13 Search PubMed.
- H. Matsukizono and T. Endo, Macromol. Chem. Phys., 2017, 218, 1–11 CrossRef.
- S. Benyahya, M. Desroches, R. Auvergne, S. Carlotti, S. Caillol and B. Boutevin, Polym. Chem., 2011, 2, 2661–2667 RSC.
- O. Lamarzelle, G. Hibert, S. Lecommandoux, E. Grau and H. Cramail, Polym. Chem., 2017, 8, 3438–3447 RSC.
- B. Ochiai, H. Kojima and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1113–1118 CrossRef CAS.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- A. Yuen, A. Bossion, E. Gomez, F. Ruipérez, M. Isik, J. Hedrick, D. Mecerreyes, Y.-Y. Yang and H. Sardon, Polym. Chem., 2016, 7, 2105–2111 RSC.
- M. Helou, J.-F. Carpentier and S. M. Guillaume, Green Chem., 2011, 13, 266–271 RSC.
- L. Annunziata, A. K. Diallo, S. Fouquay, G. Michaud, F. Simon, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Green Chem., 2014, 16, 1947–1956 RSC.
- R. Hamiye, A. Alaaeddine, M. Awada, B. Campagne, S. Caillol, S. M. Guillaume, B. Ameduri and J. F. Carpentier, Polym. Chem., 2014, 5, 5089–5099 RSC.
- Y.-Y. Zhang, Y. Li, X.-J. Zhou, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, Macromol. Rapid Commun., 2015, 36, 852–857 CrossRef CAS.
- B. Quienne, N. Kasmi, R. Dieden, S. Caillol and Y. Habibi, Biomacromolecules, 2020, 21, 1943–1951 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- P. Furtwengler, R. Perrin, A. Redl and L. Avérous, Eur. Polym. J., 2017, 97, 319–327 CrossRef CAS.
- P. Furtwengler and L. Avérous, Sci. Rep., 2018, 8, 1–14 CrossRef CAS.
- V. Crescenzi, G. Manzini, G. Calzolari and C. Borri, Eur. Polym. J., 1972, 8, 449–463 CrossRef CAS.
- L. Maisonneuve, A.-L. Wirotius, C. Alfos, E. Grau and H. Cramail, Polym. Chem., 2014, 5, 6142–6147 RSC.
- O. Lamarzelle, P.-L. Duran, A.-L. Wirotius, G. Chollet, E. Grau and H. Cramail, Polym. Chem., 2016, 7, 1439–1451 RSC.
- M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286–4291 RSC.
- A. Cornille, M. Blain, R. Auvergne, B. Andrioletti, B. Boutevin and S. Caillol, Polym. Chem., 2017, 8, 592–604 RSC.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 162–168 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 851–859 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- F. Yu and R. Zhuo, Polym. J., 2004, 36, 28–33 CrossRef CAS.
- H. Alamri, J. Zhao, D. Pahovnik and N. Hadjichristidis, Polym. Chem., 2014, 5, 5471–5478 RSC.
- A. Bossion, R. H. Aguirresarobe, L. Irusta, D. Taton, H. Cramail, E. Grau, D. Mecerreyes, C. Su, G. Liu, A. J. Müller and H. Sardon, Macromolecules, 2018, 51, 5556–5566 CrossRef CAS.
- Y. Marcus, J. Solution Chem., 1984, 13, 599–624 CrossRef CAS.
- K. L. Hoy, J. Paint Technol., 1970, 42, 76–118 CAS.
- K. L. Hoy, The Hoy Tables of Solubility Parameters, Union Carbide Corporation, Solvents and Coatings Materials Division, South Charleston, WV, 1985 Search PubMed.
- K. L. Hoy, J. Coated Fabr., 1989, 19, 53–67 CrossRef.
- D. W. Van Krevelen and K. Te Nijenhuis, in Properties of Polymers:Their Correlation with Chemical Structure; their Numerical Estimation and Prediction from Additive Group Contributions, Elsevier B.V., New York, 4th edn, 2009, pp. 189–227 Search PubMed.
- E. A. Grulke, in Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, John Wiley & Sons, Inc., 4th edn, 1999, p. 689 Search PubMed.
- K. B. Wiberg and R. F. Waldron, J. Am. Chem. Soc., 1991, 113, 7705–7709 CrossRef CAS.
- P. Olsén, N. Herrera and L. A. Berglund, Biomacromolecules, 2020, 21, 597–603 CrossRef.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- R. F. Storey and J. W. Sherman, Macromolecules, 2002, 35, 1504–1512 CrossRef CAS.
- C. Navarro, B. Martín-Vaca, D. Bourissou, S. Gazeau-Bureau, S. Magnet and D. Delcroix, Macromolecules, 2008, 41, 3782–3784 CrossRef.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581–6587 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- M. Bero, B. Czapla, P. Dobrzyński, H. Janeczek and J. Kasperczyk, Macromol. Chem. Phys., 1999, 200, 911–916 CrossRef CAS.
- R. F. Storey and A. E. Taylor, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35, 723–750 CrossRef.
- A. Kowalski, A. Duda and S. Penczek, Macromol. Rapid Commun., 1998, 19, 567–572 CAS.
- K. Odelius and A. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1249–1264 CrossRef CAS.
- H. Jiang, J. He, J. Liu and Y. Yang, Polym. J., 2002, 34, 682–686 CrossRef CAS.
- Y. Li, J. Nothnagel and T. Kissel, Polymer, 1997, 38, 6197–6206 CrossRef CAS.
- A. Pascual, H. Sardon, A. Veloso, F. Ruipérez and D. Mecerreyes, ACS Macro Lett., 2014, 3, 849–853 CrossRef CAS.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071–7081 CrossRef.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3090–3097 CrossRef.
- J. M. Ren, Q. Fu, A. Blencowe and G. G. Qiao, ACS Macro Lett., 2012, 1, 681–686 CrossRef CAS.
- A. Pascual, J. R. Leiza and D. Mecerreyes, Eur. Polym. J., 2013, 49, 1601–1609 CrossRef CAS.
- P. Groves, Polym. Chem., 2017, 8, 6700–6708 RSC.
- C. G. Pitt, F. I. Chasalow, Y. M. Hibionada, D. M. Klimas and A. Schindler, J. Appl. Polym. Sci., 1981, 26, 3779–3787 CrossRef CAS.
- P. Skoglund and Å. Fransson, J. Appl. Polym. Sci., 1996, 61, 2455–2465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01089h |
| This journal is © The Royal Society of Chemistry 2021 |