
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding structure–property relationships of main chain cyclopropane in linear polyesters†
Connor J.
Stubbs
and
Andrew P.
Dove
 *
*
School of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: a.dove@bham.ac.uk
First published on 28th August 2020
Abstract
Rigid ring structures have gained increasing interest in the polymer materials community as an effective means to manipulate bulk properties. Despite this, little work has focussed on the smallest ring: cyclopropane. Herein, we report a polymerisation that enables incorporation of stereopure cis and trans 1,2-cyclopropanedimethanol through the thiol-Michael “click” reaction between dithiol and diacrylate monomers. Polyesters containing a cyclopropane backbone were found to be amorphous, whilst comparable polymers with a 1,4-butanediol backbone were semi-crystalline. By copolymerising cyclopropane monomers at varying ratios with 1,4-butanediol monomers, the crystalinity of the resulting polymer could be effectively tuned. Successfully adjusting the resulting crystallinity resulted in control over the ultimate tensile strength (UTS) and the Young's modulus (E) of the material. Generally, it was found that increasing cyclopropane content led to a decreased UTS and E as a result of lower polymer crystalinity. Effects of cyclopropane stereochemistry on thermomechanical properties were found to be minimal at low cyclopropane backbone ratios.
Introduction
Conformationally-locked ring structures have recently garnered considerable attention in the design of new thermoplastic materials.1–3 This derives from the ability to manipulate and predict bulk properties from their incorporation into linear polymer systems. Typically, property manipulation is achieved by altering functionalities within a polymer, which can require synthetically demanding steps. Conformationally locked rings offer simple and convenient control of bulk properties in linear polymers. Hence, continual understanding into the influence of rigid ring structures on the thermomechanical properties of the resultant polymers is vital to designing the next generation of high-performance materials.Oil-sourced cyclohexylene rings have been extensively investigated in various thermoplastic systems, including polyesters, polyamides and polycarbonates.3–6 By incorporating and controlling the ratios of cyclohexane units in polymers, numerous bulk properties can be manipulated. For example, the ratio of 1,4-cyclohexanedimethanol in polymer materials has been reported to impact bulk properties such as glass transition temperature (Tg), bulk crystallinity, tensile strength and gas permeability.7–9 More recently bio-based bicyclic diols have gained significant attention in thermoplastic polymers.10–13 Wang and co-workers reported that incorporation of isosorbide, a simple isohexide unit, in polyesters led to a higher Tg and lower polymer crystallinity.14 Increasing isosorbide content also correlated with the speed at which hydrolytic degradation occurred.15
An additional structural feature to these ring systems is stereochemistry. Most rigid rings, including isohexides and cyclohexylenes, are geometrically locked which gives rise to multiple stereoisomers. There has been a recent surge in interest in leveraging stereochemistry in linear polymers to alter bulk properties.16 The stereochemical contribution of isohexides and cyclohexylene in polymer backbones has already been briefly investigated.8,17 Nevertheless, further investigation in the influence of stereochemistry on the physical and mechanical properties of the resultant materials remains a requirement for future polymer systems containing ring structures.
Despite the recent attention into cyclic structures in polymer materials, there lacks considerable investigation into the smallest cycle, cyclopropane. Previous works have focussed on cyclopropane within the polymer side chains.18 This is typically achieved via post-polymerisation modification of unsaturated polymers with diazo species.19 Mecking and co-workers have demonstrated that an increase in the side chain cyclopropane content within polybutadiene led to a higher Tg and a decrease in bulk crystallinity.20 Whilst this effectively manipulated thermal properties, the focus was on side chain functionality. As a result of this, the architectural and stereochemical influence that could have resulted from main chain cyclopropane was not examined.
Few studies have focussed on cyclopropane in the main chain of the polymer. One notable example from Takao and co-workers demonstrated the synthesis of a range of polyesters, polyamides and polyurethanes containing cyclopropane in the main chain.21 Di-functional 1,2-cyclopropanes were synthesised and subsequently polymerised; initial observations suggested that these polymers could be compressed into free-standing films. However, no further analysis was carried out. An extensive study into cyclopropane-containing polymers is needed to further our understanding of the 3-membered structure.
Herein, we present a study into the thermomechanical properties of polyesters containing main chain cyclopropane. Polymers are synthesised in step-growth fashion using mild thiol-Michael addition between multiple dithiols and cyclopropane diacrylate monomers. The effect of stereochemistry in the chain is also investigated.
Results and discussion
In order to fully understand how cyclopropane units in the polymer main chain affects bulk properties, both cis and trans stereoisomers need to be examined independently to elucidate the stereochemical contribution. Stereopure cis and trans 1,2-cyclopropane were obtained from established literature procedures (Fig. S1†).22 The diols were functionalised with acryloyl chloride to afford diacrylate-containing monomers: Cy(trans) and Cy(cis) (Fig. S1†). An additional commercially available diacrylate monomer, 1,4-butanedioldiacrylate (BD), was also used to serve as a linear comparison to the cyclopropane-containing polymers. Nucleophilic thiol-Michael addition was selected because the mild and efficient conditions can afford high molecular weight polymers without causing isomerisation or other potential side reactions.23,24 A range of polyesters were synthesised from the diacrylate monomers through a phosphine catalysed polyaddition with commercially available dithiols (Scheme 1, Table 1).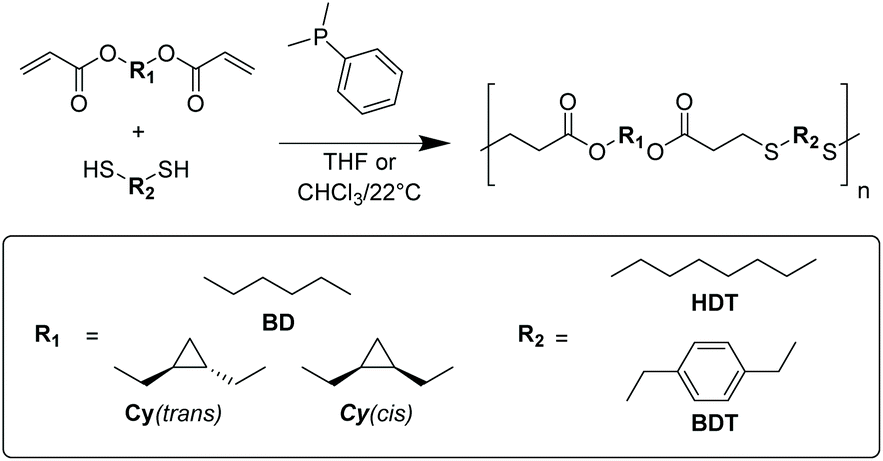 | ||
| Scheme 1 General scheme for linear polyesters synthesised from phosphine-catalysed thiol-Michael addition of diacrylate and dithiol monomers. | ||
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kDa) a (kDa) |
Đ M |
T
gb (°C) |
ΔHmb (J g−1) |
T
d, 5%c (°C) |
|
|---|---|---|---|---|---|
| a M w and ĐM were determined by size exclusion chromatography (THF, 2% v/v NEt3) analysis against polystyrene (PS) standards. b Glass transition temperature obtained from second heating scan. Total enthalpy of melting calculated from integration of all melt transitions. c Temperature after 5% weight loss obtained from thermogravimetric analysis. | |||||
| Cy(trans)-co-HDT | 38.0 | 4.30 | −52 | — | 276 |
| Cy(cis)-co-HDT | 37.4 | 4.08 | −47 | — | 293 |
| BD- co -HDT | 52.7 | 3.13 | −66 | −64.0 | 315 |
| Cy(trans)-co-BDT | 23.5 | 6.58 | −17 | — | 256 |
| BD-co-BDT | 22.0 | 7.78 | −27 | −58.7 | 273 |
Initial polymerisations used 1,6-hexanedithiol (HDT) with the three diacrylate monomers (BD, Cy(cis) and Cy(trans)) to produce comparable homopolymers. Relatively high molecular weights were achieved (35 kDa < Mw) that possessed modest dispersities (4.5 > ĐM, Table 1). 1H NMR spectroscopy confirmed the reaction conditions had not impacted the stereochemistry or cyclopropane structure of the resulting polymers (Fig. 1a). Most noticeably, the polymers displayed vastly different physical appearances. Both cyclopropane polyesters (Cy(trans)-co-HDT and Cy(cis)-co-HDT) were translucent and possessed a tacky consistency, even after aging for multiple months at benchtop conditions. Whilst the linear control (BD-co-HDT) was an opaque white solid. This observation indicated that the cyclopropane structure had significantly impacted bulk properties: crystallinity in particular.
 | ||
| Fig. 1 (a) 1H NMR spectra of polymers synthesised using HDT in CDCl3 (400 MHz, 298 K) (b) normalised DSC thermograms of polymers synthesised using HDT at a heating and cooling rate of 10 K min−1 (solid line = heating run, dashed line = cooling run). | ||
To quantify the thermal properties and gain insight into the crystallinity, differential scanning calorimetry (DSC) was performed (Fig. 1b). The Tgs of all the polymers were well below ambient temperature (−66 to −47 °C). There was a small deviation in the Tg observed between the cis and trans cyclopropane polyesters, where Cy(cis)-co-HDT had a Tg 5 °C higher than Cy(trans)-co-HDT. This could be ascribed to a higher free volume in Cy(trans)-co-HDT caused by the trans isomer. Copolymer BD-co-HDT had the lowest Tg which is attributed to more efficient chain packing of the butyl-group in comparison to the cyclopropane ring. However, the most striking observation from the DSC thermograms was the lack of melt transitions (Tm). Both cyclopropane-containing polyesters lacked a melt transition, while BD-co-HDT possessed a sharp melting peak at 52 °C. This indicates that the cyclopropane polymers are amorphous whereas BD-co-HDT is crystalline, which is in agreement with the observed physical differences. It was clear that the irregularity of the cyclopropane structure had inhibited chain alignment and, therefore, negated any possible crystallisation that would have otherwise occurred. Consequently, the polymers produced were excessively tacky and unable to produce free-standing films.
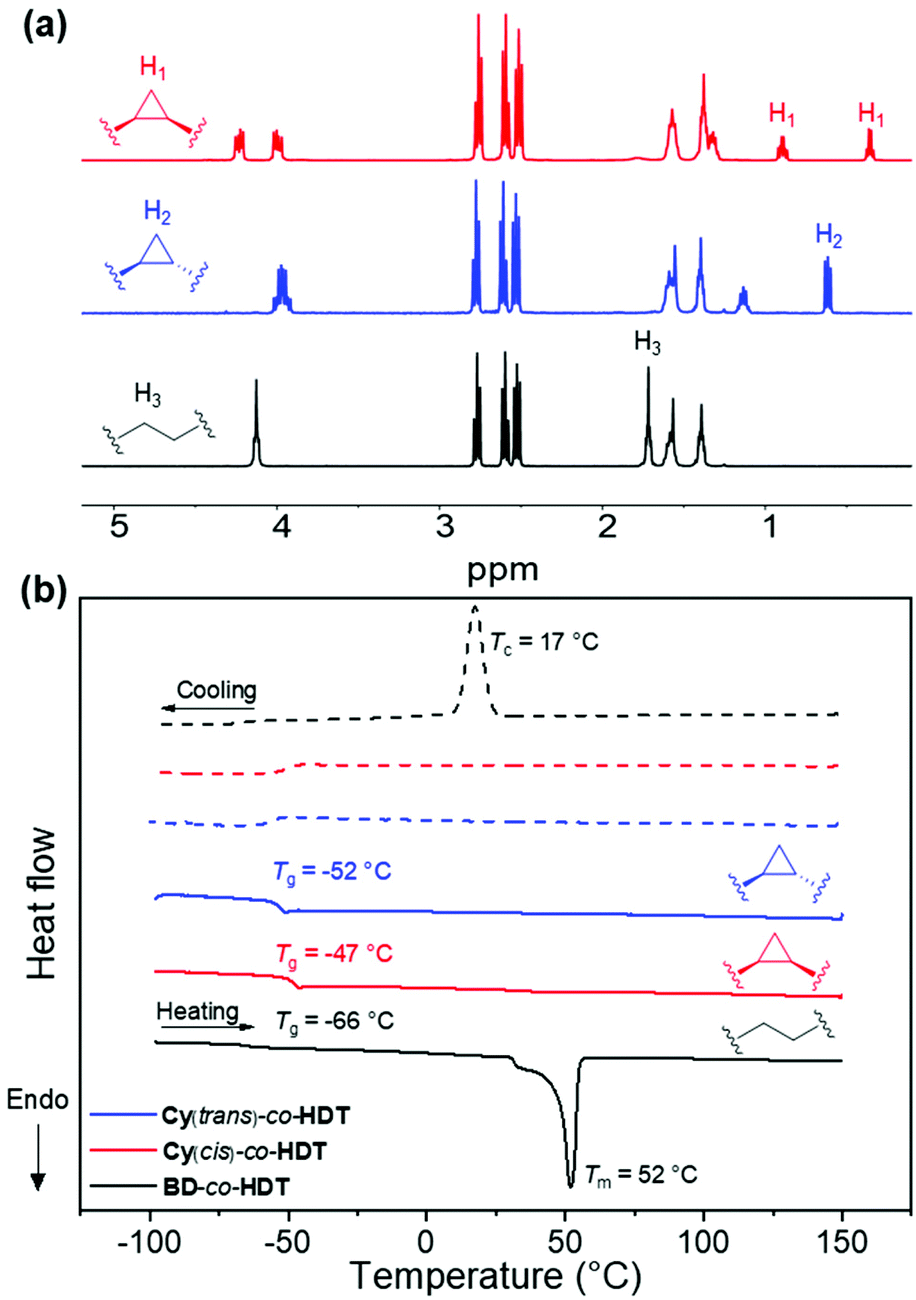
In attempts to produce a polymer suitable for heat-pressing and forming a stable film, a rigid aromatic dithiol was used as comonomer. 1,4-Benzenedimethanethiol (BDT) was reacted with the diacrylate monomers, Cy(trans) and BD. The polymerisation produced polyesters of a notably lower Mw and higher ĐM than the previously synthesised polymers. A similar physical difference was observed in this aromatic system in comparison to the HDT polymers: Cy(trans)-co-BDT produces a tacky texture whilst BD-co-BDT system produces an opaque white solid (Fig. 2a). It was observed that the BD-co-BDT visibly changed over the course of ∼12 h at room temperature after melting which was hypothesised to be slow crystallisation. In efforts to produce comparable data, both polymers were melted until liquid-like flow was achieved (∼100 °C) then cooled to room temperature (22 °C) and annealed in a thermostated incubator at 22 °C for 48 h. DSC was then performed on the annealed polymers to elucidate the thermal properties (Fig. 2b).
 | ||
| Fig. 2 (a) Polyester architecture with variable structure from diacrylate monomer with BDT. Image of annealed polyesters illustrating differences in physical appearance (b) normalised DSC thermograms of 2 day-annealed polymers made using BDT at a heating and cooling rate of 10 K min−1 (solid line = heating run, dashed line = cooling run). | ||
Both Cy(trans)-co-BDT and BD-co-BDT polymers possessed a higher Tg (−17 and −27 °C respectively) than the previous HDT system, which was expected as a consequence of increased planarity and rigidity. It was also evident that Cy(trans)-co-BDT had a higher Tg than BD-co-BDT resulting from the increased rigidity of the cyclopropane ring. The physical appearances of these polymers were further corroborated by the DSC thermograms. BD-co-BDT possessed multiple melt transitions which indicates that the material has multiple crystalline domains.25 Interestingly there was also an absence of a crystallisation peak (Tc) in BD-co-BDT, which we attributed to inefficient crystallisation under cooling run conditions. This corroborates the slow physical change to a white solid observed after melting. In contrast, the Cy(trans)-co-BDT does not display a Tm peak, indicative of an amorphous polymer. Despite the higher chain ordering promoted by BDT's planarity, Cy(trans)-co-BDT was unable to produce a free-standing film. This has further demonstrated the lack of chain-packing, and subsequent crystallisation in polymers that contain the cyclopropane structure. We postulate that the geometry of 1,2-cyclopropanes can prevent packing in two ways: (i) the rigidity of the ring prevents the rotation and flexibility that aids chain packing and (ii) the 1,2-substitution of the rigid cyclopropane geometry induces a “kink” in the linear chain that further disadvantages facile chain packing and hence crystallisation.
In order to create cyclopropane-containing polymers that could be processed into a flexible free-standing film, we leveraged the efficient thiol–ene polyaddition to introduce cyclopropane units into BD-containing copolymers. By polymerising BD and Cy monomers with HDT, random copolymers could be accessed. Additionally, this method allowed total control over the backbone content through varying monomer ratios within the feedstock (Scheme 2).
 | ||
| Scheme 2 Scheme illustrating the co-polymerisation of cyclopropane diacrylates and 1,4-butanediol diacrylate (BD) with 1,6-hexanedithiol (HDT) to create random co-polymers. | ||
The three copolymerised polyesters were of comparable Mw (63.5 to 91.8 kDa) and ĐM (4.28 to 4.62, Table 2). 1H NMR spectroscopic analysis indicated that the initial feed ratio of the monomers had been successfully translated into the resulting polymers (ESI†). By keeping the cyclopropane content in the polymer low (<30%), the polymers could be heat-pressed to produce non-tacky free-standing films. It was noted that some of the films visibly became more opaque and stiffer at room temperature (22 °C) a few hours after heat-pressing; particularly polymers containing cyclopropane. This was attributed to slow crystallisation. Therefore, to ensure the polymers had reached thermal equilibrium and were comparable, they were heat-pressed and annealed for 2 days at 22 °C in a thermostated incubator. These samples were then characterised by uniaxial tensile testing until breakage (Fig. 3).
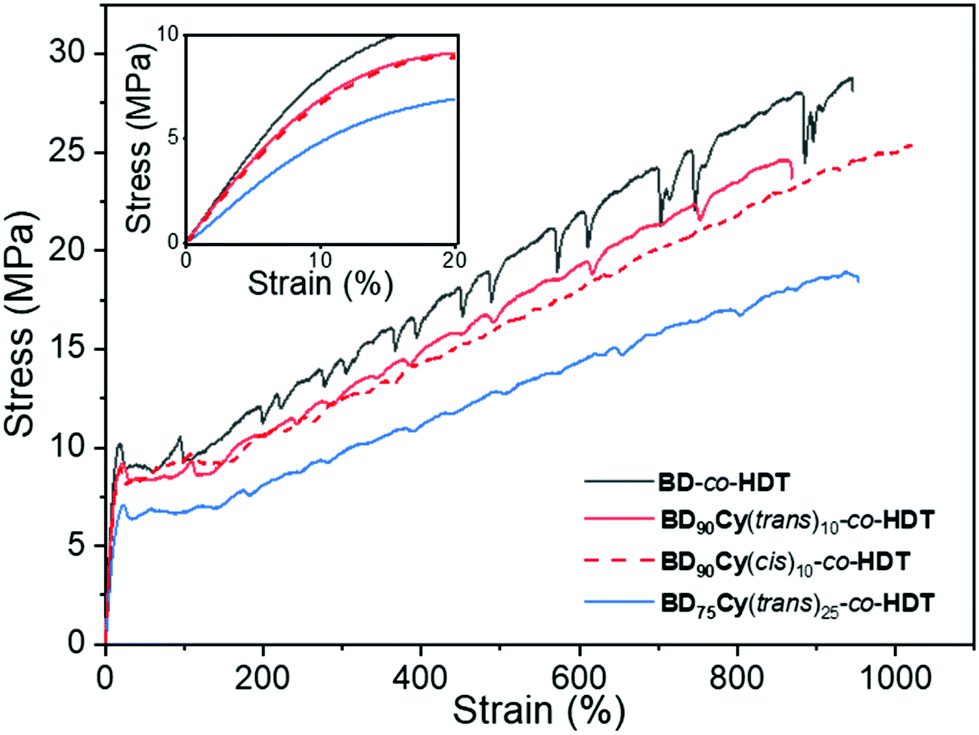 | ||
| Fig. 3 Representative stress vs. strain curves of the random copolymers Inset data between 0 and 20% strain illustrating the difference in Young's modulus. | ||
|
M
wa (kDa) |
Đ M |
T
gb (°C) |
T
d, 5%c (°C) |
|
|---|---|---|---|---|
| a M w and ĐM were determined by size exclusion chromatography (THF, 2% v/v NEt3) analysis against polystyrene (PS) standards. b Glass transition temperature obtained from second heating scan. c Temperature after 5% weight loss obtained from thermogravimetric analysis. | ||||
| BD90Cy(trans)10-co-HDT | 63.5 | 4.28 | −64 | 315 |
| BD90Cy(cis)10-co-HDT | 76.9 | 4.62 | −63 | 314 |
| BD75Cy(trans)25-co-HDT | 91.8 | 4.54 | −62 | 313 |
The newly synthesised random co-polymers were compared alongside BD-co-HDT to assess the impact cyclopropane had on tensile properties. All the samples displayed tensile curves that are consistent with a tough thermoplastic and possessed a similar strain at break (Table S1†). All samples also displayed periodic drops in stress which was most apparent in BD-co-HDT. This is characteristic of stress-oscillation behaviour observed in numerous polymer systems, however origins of this phenomenon are often disputed.26,27 A possible explanation is the formation of “voids” in the amorphous regions during elongation which leads to a stress drop and the re-orientation of polymer crystallites leading to a stress increase.28 Remarkably, this behaviour was considerably reduced in samples containing cyclopropane. This is attributed to a reduction in chain packing caused by the cyclopropane geometry, and subsequent impact on the size and spacing between the crystallites which are typically responsible for stress-oscillation behaviour.
BD-co-HDT displayed the highest strength at break (UTS) and Young's Modulus (E). Upon incorporation of either cyclopropane isomer: the tensile strength and Young's modulus slightly decreased. Reduction of strength and stiffness is expected as a result of cyclopropane inhibiting chain crystallisation which ultimately leads to a weaker material. Interestingly, there appeared to be negligible tensile difference between the cis and trans cyclopropane isomers at 10% backbone content (Table S1†). Whilst this is a contrast to several previously reported cis/trans polymer systems, the low cyclopropane content within this system could inhibit the influence of stereochemistry.29,30 Further increase of the trans-cyclopropane content to 25% led to a more flexible and weaker material. BD75Cy(trans)25-co-HDT possessed an average Young's modulus of 60 MPa in comparison to BD-co-HDT which was 105 MPa. A similar reduction in UTS was also observed between these two polymers (19.2 MPa from 29.7 MPa). This clear manipulation of tensile properties as a result of cyclopropane indicates that efforts to control polymer crystallinity were successful.
To quantify the impact of cyclopropane incorporation on polymer crystallinity in these random co-polymers, DSC analysis was performed (Fig. 4a). Incorporation of cyclopropane led to slight increases in the co-polymer Tg (Table 2). Unlike Cy(trans)-co-HDT and Cy(cis)-co-BDT, the butyl/cyclopropane random co-polymers all possessed a Tm and a Tc intuitive of a semi-crystalline material. This indicates that the large BD content was able to drive chain crystallisation within the polymer material. To quantify the extent of crystallinity, the melt transitions were normalised and integrated to obtain the total enthalpy of melting (ΔHm). A higher value for ΔHm indicates more extensive polymer crystallinity. As hypothesised, increasing the content of cyclopropane led to a lower ΔHm indicating a loss of crystallinity. The other striking observation of these DSC thermograms is the shift seen in the Tc to lower temperatures with an increasing cyclopropane content. Both of these observations corroborate the amorphous nature of the cyclopropane structure and its ability to impede crystallinity when implemented into highly ordered polymers. This loss in crystallinity could be directly correlated with strength and Young's modulus of the resulting copolymer (Fig. 4b). Thus, the tensile properties of the polymers can not only be modulated but can be predicted from the thermal transitions.
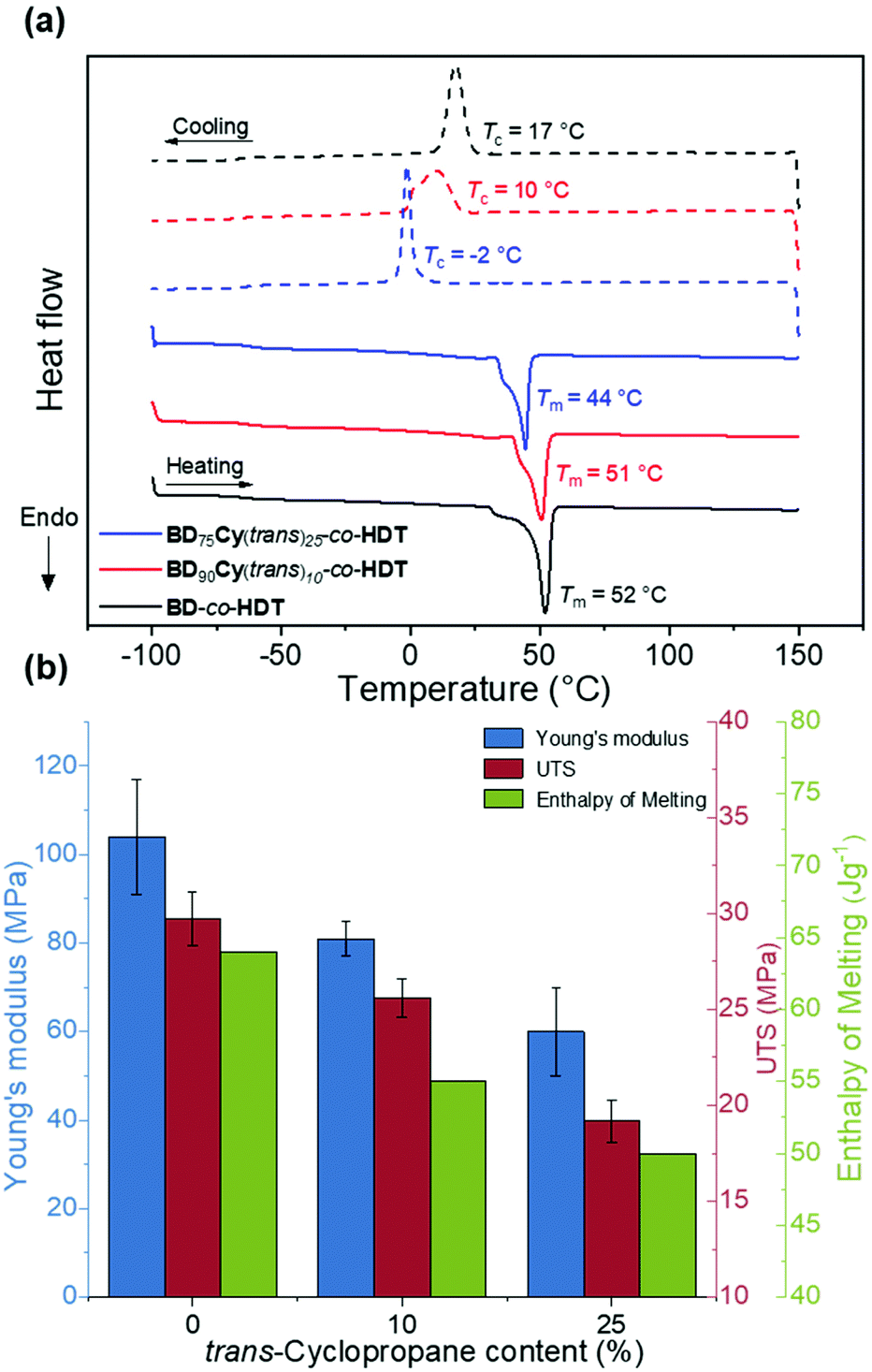 | ||
| Fig. 4 (a) Normalised DSC thermograms of 2 day annealed polymers synthesised using different feed ratios of trans-cyclopropane Cy(trans) at a heating and cooling rate of 10 K min−1 (solid line = heating run, dashed line = cooling run). (b) Bar chart of Young's modulus (E), ultimate tensile strength (UTS) and Enthalpy of Melting vs. trans-cyclopropane content (%) in polyester backbone. | ||
There were minimal differences in Tg, Tm and ΔHm between the cis and trans stereochemistry at 10% backbone content (Table S1†). This was expected given the comparable tensile properties of BD90Cy(trans)10-co-HDT and BD90Cy(cis)10-co-HDT. However, previous investigations report that changing the cis/trans content of linear polymers led to a control of bulk crystallinity, and hence, tensile properties could be tuned.29,31 The lack of crystallinity in the initial cyclopropane-containing polymers demonstrates that neither stereochemistry promotes crystallisation. Therefore, we observe minimal differences in tensile properties between the cis and trans stereochemistry in this cyclopropane system. It was clear that controlling the content of either cyclopropane isomer in a polymer backbone led to manipulation of thermomechanical properties. Future investigation is required to understand the full extent main chain cyclopropane has on further bulk properties.
Conclusion
A polymer system has been designed to incorporate both isomers of 1,2-cyclopropane into the main chain of a polyester using nucleophilic thiol–ene addition chemistry. Cyclopropane backbones were found to be unusually amorphous. However, used as a comonomer, cyclopropane could effectively manipulate polymer crystallinity. Through the control of polymer crystallinity, it was demonstrated that Young's modulus and UTS could be decreased by ∼40% with as little as 25% cyclopropane content. This demonstrates an effective and simple means to control polymer crystallinity. Typically, elucidation and design of polymers with specific properties is a laborious experimental task. But through detailed understanding of structure–property relationships, design of polymers that satisfy diverse applications can be straightforward. Further study into cyclopropanes in polymers and networks are still required to understand its future potential in the synthesis of high-performance materials.Experimental
Materials
1,6-Hexanedithiol was distilled under vacuum before use and stored under N2 in a Schlenk-flask. All other chemicals were used as received.Measurements
All NMR experiments were performed at 298 K on a Bruker DPX-400 NMR instrument equipped operating at 400 MHz for 1H (100.57 MHz for 13C). 1H NMR spectra are referenced to residual proton solvent (δ 7.26 for CDCl3δ 2.50 for DMSO) and 13C NMR spectra are referenced to the solvent signal (δ 77.2 for CDCl3δ 39.52 for DMSO). The resonance multiplicities are described as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet).Mass spectrometry was performed by University of Birmingham school of Chemistry on a Waters GCT Premier for compounds 2 and 3. Mass spectrometry was performed on compounds 4 and 5 using Waters Xevo G2-XS QTof.
Size Exclusion Chromatography (SEC) in THF was performed on a system composed of an Agilent 1260 Infinity II LC system equipped with an Agilent guard column (PLGel 5 μM, 50 × 7.5 mm) and two Agilent Mixed-C columns (PLGel 5 μM, 300 × 7.5 mm). The mobile phase used was THF (HPLC grade) containing 2% v/v NEt3 at 40 °C at flow rate of 1.0 mL min−1 (polystyrene (PS) standards used for calibration). Number average molecular weights (Mn), weight average molecular weights (Mw) and dispersities (ĐM = Mw/Mn) were determined using Agilent SEC software.
Determination of the thermal characteristics of the polymers were carried out using a STARe system DSC3 with auto-sampler (Mettler Toledo, Switzerland). Samples were added to 40 μL aluminum pans. Thermograms obtained with a heating rate of 10 K min−1 were recorded from −100–150 °C with a 10 K min−1 heating and cooling rate over two cycles. The glass transition temperature (Tg) was determined by the minimum of the first derivative in the second heating cycle. Total enthalpy of melting (ΔHm) was calculated from integration and normalization of all endothermic peaks present using the STARe software.
Thermal degradation was quantified using a Q550 Thermogravimetric analyser (TA instruments). Thermograms were recorded under an N2 atmosphere at a heating rate of 10 K min−1 from 25–600 °C. Decomposition temperatures were reported at the 5% weight loss temperature (Td, 5%).
Thin polymer films were fabricated using a Specac Atlas™ Manual Hydraulic Press 15 T fitted with Specac heated plates. Films with a thickness of 0.3 ± 0.1 mm were prepared by melt compressing the polymers under ca. 5 kN of force in a hot press followed by cooling to ambient temperature in the press whilst under compression. Polymer samples were in an aluminium rectangular spacer (20 × 40 × 0.5 mm) between PTFE sheets and placed between heated plates at 90 °C unless stated otherwise and then cooled to ambient temperature maintaining 5 kN of force. Film samples were visually inspected for deformation and bubbles before annealing for 2 days at 25 °C. Dogbones ASTM Type IV were cut out of the films and subjected to uniaxial tensile testing on a Testometric M350-5CT fitted with a load cell of 10 kN. Each specimen was clamped into the tensile holders and subjected to an elongation rate of 10 mm min−1 until failure (n = 3). The data was averaged, and the standard deviation is posed as the uncertainty.
Synthesis of racemic diethyl-1,2-cyclopropanedicarboxylate (1)
A dry 3-necked 1000 mL round bottom flask fitted with a Suba-Seal®, dropping funnel and gas adapter with light nitrogen flow and was then charged with sodium hydride 60% dispersion in mineral oil (24 g, 0.6 mol, 1.5 equiv.) and cooled 0 °C with an ice bath. The sodium hydride was then suspended in dry toluene (400 mL). Ethyl acrylate (40 g, 0.4 mol, 1 equiv.) and ethyl chloroacetate (49 g, 0.4 mol, 1 equiv.) were mixed in a beaker and charged to the dropping funnel. Approximately 5 mL of this mixture was added to the cooled sodium hydride solution. Nitrogen flow was increased whilst ethanol (2 mL) was charged via needle to the reaction whilst maintaining vessel at 0 °C with an ice bath. Once bubbling had ceased, the remaining ethyl acrylate and ethyl chloroacetate mixture was added dropwise through the dropping funnel over the course of 3 h to maintain the reaction temperature at 0 °C. The reaction was stirred and allowed to warm to room temperature over ca. 16 h. The reaction was quenched over crushed ice (∼400 mL) then extracted with ethyl acetate (3 × 200 mL) before removal of solvent in vacuo. The remaining orange oil was distilled under vacuum to obtain a crude mixture containing title compound (15% trans isomer and 85% cis isomer calculated by 1H NMR spectroscopy) as a colourless oil (31.8 g, 170 mmol, 42% overall yield). This was used as obtained in further reactions (crude NMR shown Fig. S2†). Spectral data consistent with previous work.32Synthesis of cis-1,2-cyclopropanedimethanol (2)
A dry 3-necked 500 mL round bottom flask fitted with a condenser and gas adapter with light nitrogen flow was then charged with LiAlH4 (7.1 g, 186 mmol, 1.5 equiv.). The vessel was cooled to 0 °C and dry THF (∼200 mL) was added and stirred. A mixture of 1 (23.3 g, 125 mmol, 1.0 equiv.) in THF (∼40 mL) was added dropwise over 30 minutes. The reaction was then heated at reflux for 2 h and allowed to cool to ambient temperature and stirred for ca. 16 h. The reaction was then quenched with saturated NH4Cl solution (42 mL) and diluted with EtOAc (42 mL). Remaining salts are removed by filtration and the filtrate was concentrated yielding a crude orange oil. The oil was further purified by column chromatography (100% EtOAc) yielding title compound as a colourless oil (7.3 g, 71 mmol, 57% yield). 1H NMR (400 MHz; 298 K; DMSO-d6) δ 4.46 (dd, J = 6.1, 4.6 Hz, 2H), 3.56 (m, 2H), 3.33–3.22 (m, 2H), 1.13–0.97 (m, 2H), 0.62 (td, J = 8.2, 4.5 Hz, 1H), 0.11 (m, 1H) 13C NMR (101 MHz; 298 K; DMSO-d6) δ 61.0, 17.8, 8.2. MS (CI-TOF) calculated for C5H10O2NH4 120.10; found 120.03.Synthesis of trans-1,2-cyclopropanedimethanol (3)
A dry 3-necked 500 mL round bottom flask fitted with a condenser and gas adapter with light nitrogen flow was then charged with LiAlH4 (3.1 g, 81 mmol, 1.5 equiv.). The vessel was cooled to 0 °C and dry THF (∼60 mL) was added and stirred. trans-Diethyl-1,2-cyclopropanedicarboxylate (10 g, 54 mmol, 1 equiv.) in THF (∼20 mL) was added dropwise over 30 minutes. The reaction was then heated at reflux for 2 h and allowed to cool to ambient temperature and stirred for ca. 16 h. The reaction was then quenched with saturated NH4Cl solution (20 mL) and diluted with EtOAc (20 mL). Remaining salts are removed by filtration and the filtrate is concentrated yielding a crude orange oil. The oil was further purified by column chromatography (100% EtOAc) to yield title compound as colourless oil (3.47 g, 34 mmol, 63% yield) 1H NMR (400 MHz, DMSO-d6) δ 4.37 (d, J = 11.1 Hz, 1H), 3.35–3.23 (m, 2H), 3.26–3.14 (m, 2H), 2.08 (s, 1H), 0.86–0.72 (m, 2H), 0.33–0.25 (m, 2H). 13C NMR (101 MHz; 298 K; DMSO-d6) δ 64.0, 18.9, 7.7. Spectral data consistent with previous work.33 MS (CI-TOF) calculated for C5H10O2NH4 120.1025; found 120.005.Synthesis of cis-Cy diacrylate (4)
A 2-necked flask 100 mL round bottom flask fitted with a gas adapter and a dropping funnel was charged with 2 (2.0 g, 19 mmol, 1 equiv.), dry THF (∼30 mL) and acryloyl chloride (4.4 g, 49 mmol, 2.5 equiv.). Triethylamine (5.9 g, 59 mmol, 3 equiv.) was added dropwise slowly over the course of 1 h at 0 °C whilst stirring. The reaction was allowed to warm to ambient temperature and stirred for ca. 16 h. The reaction was then quenched with water (∼50 mL) and was extracted with EtOAc (3 × 30 mL). The organic layer was dried with Mg2SO4 and concentrated to an orange oil. The oil was subject to further purification by column chromatography (3:1 hexanes to ethyl acetate) yielding title compound as a colourless oil (2.3 g,11 mmol, 58% yield). 1H NMR (400 MHz, CDCl3) δ 6.41 (dd, J = 17.3, 1.5 Hz, 2H), 6.11 (dd, J = 17.3, 10.4 Hz, 2H), 5.82 (dd, J = 10.4, 1.5 Hz, 2H), 4.45–4.28 (m, 2H), 4.13–3.96 (m, 2H), 1.50–1.30 (m, 2H), 0.94 (td, J = 8.4, 5.3 Hz, 1H), 0.40 (q, J = 5.6 Hz, 1H). 13C NMR (101 MHz; 298 K; CDCl3) δ 166.3, 131.0, 128.6, 64.7, 14.9, 8.9. MS (CI-TOF) calculated for C11H14O4NH4 228.1236; found 228.1241.
Synthesis of trans-Cy diacrylate (5)
A 2-necked flask 100 mL round bottom flask fitted with a gas adapter and a dropping funnel was charged with 3 (2.0 g, 19 mmol, 1 equiv.), dry THF (∼30 mL) and Acryloyl chloride (4.4 g, 49 mmol, 2.5 equiv.). Triethylamine(5.9 g, 59 mmol, 3 equiv.) was added dropwise slowly over the course of 1 h at 0 °C whilst stirring. The reaction was allowed to warm to ambient temperature and stirred for ca. 16 h. The reaction was then quenched with water (∼50 mL) and was extracted with EtOAc (3 × 30 mL). The organic layer was dried with Mg2SO4 and concentrated to an orange oil. The oil was subject to further purification by column chromatography (3:1 hexanes to ethyl acetate) yielding title compound as a colourless oil (2.1 g, 10 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 6.40 (dd, J = 17.3, 1.5 Hz, 2H), 6.19–6.04 (m, 2H), 5.82 (dd, J = 10.4, 1.5 Hz, 2H), 4.14–4.00 (m, 2H), 4.05–3.91 (m, 2H), 1.24–1.18 (m, 2H), 0.70–0.59 (m, 2H). 13C NMR (101 MHz; 298 K; DMSO-d6) δ 166.4, 130.9, 128.6, 67.7, 16.3, 9.1. MS (CI-TOF) calculated for C11H14O4H 211.0970; found 211.0978.
General polymerisation procedure (BD-co-HDT)
A scintillation vial was charged with 1,4-butanedioldiacrylate (0.1696 g, 0.856 mmol, 1 equiv.) and another scintillation vial was charged with 1,6-hexanedithiol (0.1285 g, 0.856 mmol, 1 equiv.). The contents of both scintillation vials were dissolved in THF (1.8 mL) and quantitatively transferred to a 25 mL scintillation vial and stirred at ambient temperature. Whilst stirring, polymerization was initiated with DMPP (2.4 μL, 2%mol) and left to stir. Initially an exotherm was observed with a visible increase in viscosity. After ca. 16 h the polymer solution was precipitated in MeOH (100 mL) and left to stir for 1 h before decanting the supernatant. Polymer was then dried under vacuum for approximately 2 h. SEC analysis (THF + 2% v/v NEt3) Mw = 52.7, ĐM = 3.13, DSC analysis Tg = −66 °C, 1H NMR (400 MHz, CDCl3) δ 4.21–4.05 (m, 4H), 2.77 (t, J = 7.5 Hz, 4H), 2.60 (t, J = 7.5 Hz, 4H), 2.57–2.49 (m, 4H), 1.77–1.66 (m, 4H), 1.64–1.51 (m, 6H), 1.46–1.34 (m, 4H). 13C NMR (101 MHz; 298 K; CDCl3) δ 172.2, 64.3, 35.0, 32.3, 29.6, 28.6, 27.2, 25.5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme under grant agreement No 681559.Notes and references
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef
.
- D. J. Saxon, A. M. Luke, H. Sajjad, W. B. Tolman and T. M. Reineke, Prog. Polym. Sci., 2020, 101, 101196 CrossRef
- X. Li and A. F. Yee, Macromolecules, 2003, 36, 9421–9429 CrossRef
- C.-W. Chen, C.-W. Lin, Y.-H. Chen, T.-F. Wei, S.-P. Rwei and R. Sasikumar, Polym. Bull., 2020, 77, 235–253 CrossRef
- N. Sánchez-Arrieta, A. M. de Ilarduya, A. Alla and S. Muñoz-Guerra, Eur. Polym. J., 2005, 41, 1493–1501 CrossRef
- X. Li and A. F. Yee, Macromolecules, 2004, 37, 7231–7239 CrossRef
- J. Wang, X. Liu, Y. Zhang, F. Liu and J. Zhu, Polymer, 2016, 103, 1–8 CrossRef
- F. Liu, J. Zhang, J. Wang, X. Liu, R. Zhang, G. Hu, H. Na and J. Zhu, J. Mater. Chem. A, 2015, 3, 13637–13641 RSC
- A. Celli, P. Marchese, L. Sisti, D. Dumand, S. Sullalti and G. Totaro, Polym. Int., 2013, 62, 1210–1217 CrossRef
- S.-A. Park, J. Choi, S. Ju, J. Jegal, K. M. Lee, S. Y. Hwang, D. X. Oh and J. Park, Polymer, 2017, 116, 153–159 CrossRef
- C. Lavilla and S. Muñoz-Guerra, Green Chem., 2013, 15, 144–151 RSC
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Polym. Chem., 2017, 8, 3746–3754 RSC
- N. G. Moon, F. Mazzini, A. M. Pekkanen, E. M. Wilts and T. E. Long, Macromol. Chem. Phys., 2018, 219, 1800177 CrossRef
- D.-D. Xing, Y.-W. Jia, D.-F. Li, X.-L. Wang and Y.-Z. Wang, ACS Sustainable Chem. Eng., 2017, 5, 7040–7051 CrossRef CAS
- J. Qi, J. Wu, J. Chen and H. Wang, Polym. Degrad. Stab., 2019, 160, 229–241 CrossRef CAS
- J. C. Worch, H. Prydderch, S. Jimaja, P. Bexis, M. L. Becker and A. P. Dove, Nat. Rev. Chem., 2019, 3, 514–535 CrossRef
- R. J. Kieber, S. A. Silver and J. G. Kennemur, Polym. Chem., 2017, 8, 4822–4829 RSC
- A. B. Glazyrin, M. I. Abdullin, E. R. Atnabaeva, R. M. Sultanova, V. P. Volodina and V. A. Dokichev, Polym. Bull., 2019, 76, 3643–3657 CrossRef
- Á. Beltrán, B. P. Gómez-Emeterio, C. Marco, G. Ellis, M. D. Parellada, M. M. Díaz-Requejo, S. Corona-Galván and P. J. Pérez, Macromolecules, 2012, 45, 9267–9274 CrossRef
- J. Urbano, B. Korthals, M. M. Díaz-Requejo, P. J. Pérez and S. Mecking, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4439–4444 CrossRef CAS
- R. Oda, T. Shono, A. Oku and H. Takao, Makromol. Chem., 1963, 67, 124–131 CrossRef CAS
- L. L. McCoy, J. Am. Chem. Soc., 1958, 80, 6568–6572 CrossRef CAS
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS
- J. Vandenbergh, K. Ranieri and T. Junkers, Macromol. Chem. Phys., 2012, 213, 2611–2617 CrossRef CAS
- A. Frick and A. Rochman, Polym. Test., 2004, 23, 413–417 CrossRef CAS
- H. Ebener, B. Pleuger and J. Petermann, J. Appl. Polym. Sci., 1999, 71, 813–817 CrossRef CAS
- K. K. Childress, M. D. Alim, J. J. Hernandez, J. W. Stansbury and C. N. Bowman, Polym. Chem., 2020, 11, 39–46 RSC
- C. Wan, E. L. Heeley, Y. Zhou, S. Wang, C. T. Cafolla, E. M. Crabb and D. J. Hughes, Soft Matter, 2018, 14, 9175–9184 RSC
- C. J. Stubbs, J. C. Worch, H. Prydderch, M. L. Becker and A. P. Dove, Macromolecules, 2020, 53, 174–181 CrossRef CAS
- Y. Yu, Z. Wei, X. Leng and Y. Li, Polym. Chem., 2018, 9, 5426–5441 RSC
- C. A. Bell, J. Yu, I. A. Barker, V. X. Truong, Z. Cao, A. V. Dobrinyin, M. L. Becker and A. P. Dove, Angew. Chem., Int. Ed., 2016, 55, 13076–13080 CrossRef
- R. Csuk and Y. von Scholz, Tetrahedron, 1994, 50, 10431–10442 CrossRef
- W. S. McDonald, C. A. Verbicky and C. K. Zercher, J. Org. Chem., 1997, 62, 1215–1222 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and thermomechanical characterisation data included. See DOI: 10.1039/d0py01004a |
| This journal is © The Royal Society of Chemistry 2020 |