
Recent development in halogen-bonding-catalyzed living radical polymerization
Chen-Gang
Wang
 ,
Amerlyn Ming Liing
Chong
,
Houwen Matthew
Pan
,
Jit
Sarkar
,
Xiu Ting
Tay
and
Atsushi
Goto
*
,
Amerlyn Ming Liing
Chong
,
Houwen Matthew
Pan
,
Jit
Sarkar
,
Xiu Ting
Tay
and
Atsushi
Goto
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371 Singapore. E-mail: agoto@ntu.edu.sg
First published on 18th August 2020
Abstract
Halogen bonding (XB) has been used to catalyze organic reactions and polymerizations, which is an emerging research area. Reversible complexation mediated polymerization (RCMP) is an XB-catalyzed living radical polymerization and is one of the most promising examples of the XB catalysis. RCMP utilizes alkyl iodides as initiating dormant species and electro-donating molecules and ions such as amines, iodide anions, and oxyanions as catalysts. Various initiating dormant species and catalysts were developed, enabiling the synthesis of well-defined homopolymers and block copolymers with complex architectures, chain-end functionalization, photo-polymerization, and industrial application. The use of inexpensive non-metallic catalysts and the accessibility to a wide range of polymer structures are attractive features of RCMP. This mini-review summarizes the current research status of RCMP and its uniqueness brought via the XB catalysis.
1. Introduction
Halogen bonding (XB) is a noncovalent bond between an electron-accepting halogen (X) and an electron-donating base (B) (Fig. 1).1 XB is a highly directional bond, and the R–X⋯B angle is close to 180°. The strength of XB increases in the order of X = F ≪ Cl < Br < I (with an increase in the polarizability of X) and with an increase in the electron-withdrawing ability of R. The B group can be electro-donating anions and neutral species such as halide anions, chalcogen- and pnictogen-containing species, and π-electron donors.1,2 XB is widely utilized in crystal engineering,2–4 supramolecular chemistry,5–7 and functional materials such as luminescent crystals, porous frameworks, and conductive organic materials8–12 due to its unique directionality, complementarity, and compatibility with various environments. | ||
| Fig. 1 Halogen bonding. | ||
The application of XB has encompassed the creation of polymer materials. Similar to other non-covalent bonds, XB is used as an efficient driving force to form liquid-crystalline polymers, supramolecular gels, self-healing polymers, conjugated polymers, polymer nano-particles, phase separation structures, and polymer functional surfaces.11–13 Resnati et al. reported a pioneering work on the preparation of liquid-crystalline comb-shaped polymers formed via the XB between poly(4-vinylpyridine) and α,ω-diiodoperfluoroalkanes in 2002.14 XB was also used to form supramolecular polymers using pyridine and iodopentafluorobenzene derivatives as molecular building blocks.15 Steed et al. reported XB-bridged supramolecular gels using bis(pyridylurea) derivatives and 1,4-diiodotetrafluorobenzene.16 Some XB-bridged supramolecular gels were stable in polar media such as aqueous methanol,16 exhibiting the environmental compatibility of XB. Biomolecules bearing X and B moieties were used as building blocks to generate biomedical gels.17 These examples demonstrate the usefulness of XB for the preparation of polymer materials.
XB was also used to form monomer (co)crystals. Polymers were synthesized via solid phase polymerizations of the monomer (co)crystals. Goroff et al. synthesized polyacetylenes via 1,4-addition polymerizations of diiodo-1,3-diacetylene (Fig. 2).18,19 The co-crystal of diiodo-1,3-diacetylene (monomer) and bis(nitrile)oxalamide (XB linker) offered a high regularity of the monomer distance appropriate for topochemical polymerization (Fig. 2). Unlike the solution polymerization, where both 1,2- and 1,4-additions occur to generate branched irregular polymer structures, the topochemical polymerization in the crystal lattices predominantly led to 1,4-addition and afforded ordered polymer structures. Other examples of the solid-phase polymerizations of XB-based monomer (co)crystals are referred to excellent comprehensive reviews.20,21
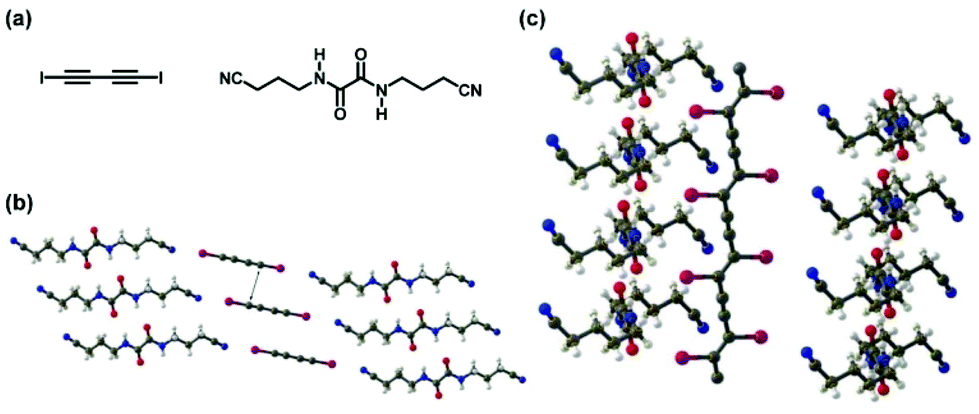 | ||
| Fig. 2 (a) Structures of 1,4-diiodo-1,3-butadiyne (monomer) and bis(4-cyanobutyl)oxalamide (XB linker). (b) X-ray structure of the co-crystal of the monomer and XB linker. (c) X-ray structure of the co-crystal of the obtained polymer (poly(diiododiacetylene)) and XB linker. Colours are as follows. C: gray; H: white; O: red; N: blue; I: magenta. Adapted with permission from ref. 18. Copyright 2006, American Association for the Advancement of Science. | ||
Recently, solution-phase catalysis induced by XB has become an emerging area in organic synthesis.22–24 XB induces an elongation of the R–X bond or a polarization of B, thereby catalysing organic reactions and polymerizations in solution phases. In organic chemistry, XB was used to activate less reactive substrates to achieve addition and cyclization reactions, for example.25,26 On the other hand, only few examples on XB-catalyzed polymerization have been reported. Coulembier et al. reported an XB-catalyzed ring opening polymerization (ROP) of L-lactide (Scheme 1a) in 2010, in which iodine trichloride (catalyst) coordinates the monomer via XB and induces the polymerization.27 Takagi et al. reported an XB-mediated living cationic polymerization of isobutyl vinyl ether (Scheme 1b) in 2017, in which 2-iodoimidazolium derivatives (catalysts) coordinate the alkyl halide dormant species via XB to generate a propagating carbon-centred cation.28
 | ||
| Scheme 1 XB-catalyzed (a) ring opening polymerization and (b) living cationic polymerization. | ||
Living (or reversible-deactivation) radical polymerization (LRP) is a powerful technique for synthesizing well-defined polymers with narrow dispersities. Examples of the LRP systems are nitroxide-mediated polymerization (NMP),29 atom transfer radical polymerization (ATRP),30–32 reversible addition–fragmentation chain transfer (RAFT) polymerization,33,34 iodine-transfer polymerization (ITP),35 organotellurium-mediated radical polymerization (TERP),36 and cobalt-mediated polymerization.37 The basic concept of LRP is the reversible activation of a dormant species (polymer–X) to a propagating radical (polymer˙) (Scheme 2a).38
 | ||
| Scheme 2 Reversible activation: (a) general scheme in LRP, (b) RTCP, and (c) RCMP. | ||
Our research group developed organocatalyzed LRP systems using alkyl iodides as initiating dormant species and organic molecules as catalysts, which are reversible chain transfer catalyzed polymerization (RTCP) developed in 200639 and reversible complexation mediated polymerization (RCMP) developed in 2011.40 RTCP and RCMP are mechanistically different, as described below, and RCMP uses XB catalysis. RCMP would be a good example highlighting the power of XB in polymer chemistry. In this mini-review, we summarize the recent development and applications of RTCP and RCMP with a particular focus on RCMP via XB catalysis. This mini-review refers particularly to our research for dedicating to the 2021 Polymer Chemistry Pioneering Investigators issue.
2. Polymerizations and applications
2.1. Mechanisms, catalysts, and typical polymerization behaviours of methyl methacrylate (MMA)
Both RTCP and RCMP use an alkyl iodide (R–I) as an initiating dormant species and an organic molecule as a catalyst. Mechanistically, RTCP uses a chain transfer between the dormant species and catalyst (Scheme 2b). RCMP uses an XB catalysis between the dormant species and catalyst (Scheme 2c).An RTCP system consists of an R–I dormant species, a deactivator catalyst (A–I), and a conventional radical initiator such as an azo compound (Scheme 2b).39 In RTCP, polymer˙, which is originally supplied by the conventional radical initiator, reacts with A–I, generating an activator radical (A˙) as well as polymer–iodide (polymer–I). Subsequently, A˙ reversibly reacts with polymer–I to generate polymer˙ and A–I. This reversible chain transfer between the polymer and catalyst results in a reversible activation of polymer–I. A˙ radicals are germanium, phosphorus, nitrogen, oxygen, and carbon centred radicals, and examples of A–I are GeI4, PI3, and N-iodosuccinimide.39,41–44
An RCMP system consists of an R–I dormant species and an activator catalyst (Scheme 2c).40 Polymer–I and the catalyst form an XB complex (polymer–I⋯catalyst), and the complex subsequently reversibly generates polymer˙ and an I˙⋯catalyst complex. This reversible complexation mediates the polymerization, and hence the polymerization is termed RCMP. In organic chemistry, the reactions of alkyl halides (R–X) with amine bases (B) to generate the alkyl radicals via XB catalysis (R–X⋯B) have been well studied,45–48 but the reactions were irreversible in all cases. Our research group found the reversible reactions using alkyl iodides, enabling the use of XB catalysis to LRP. Since our first report in 2011, we have developed several types of RCMP catalysts, including amines,40,49 iodide anion (I−),50–54 organic superbases,55 pseudohalide anions,56 pyridine N-oxides,57 and oxyanions58 (Fig. 3). The reactivity of RCMP catalysts depends on their XB-forming ability, i.e., basicity or nucleophilicity, in principle. A representative catalyst is I−. I− is used in the form of salts such as tetrabutylammonium iodide (Bu4N+I−) (BNI).50
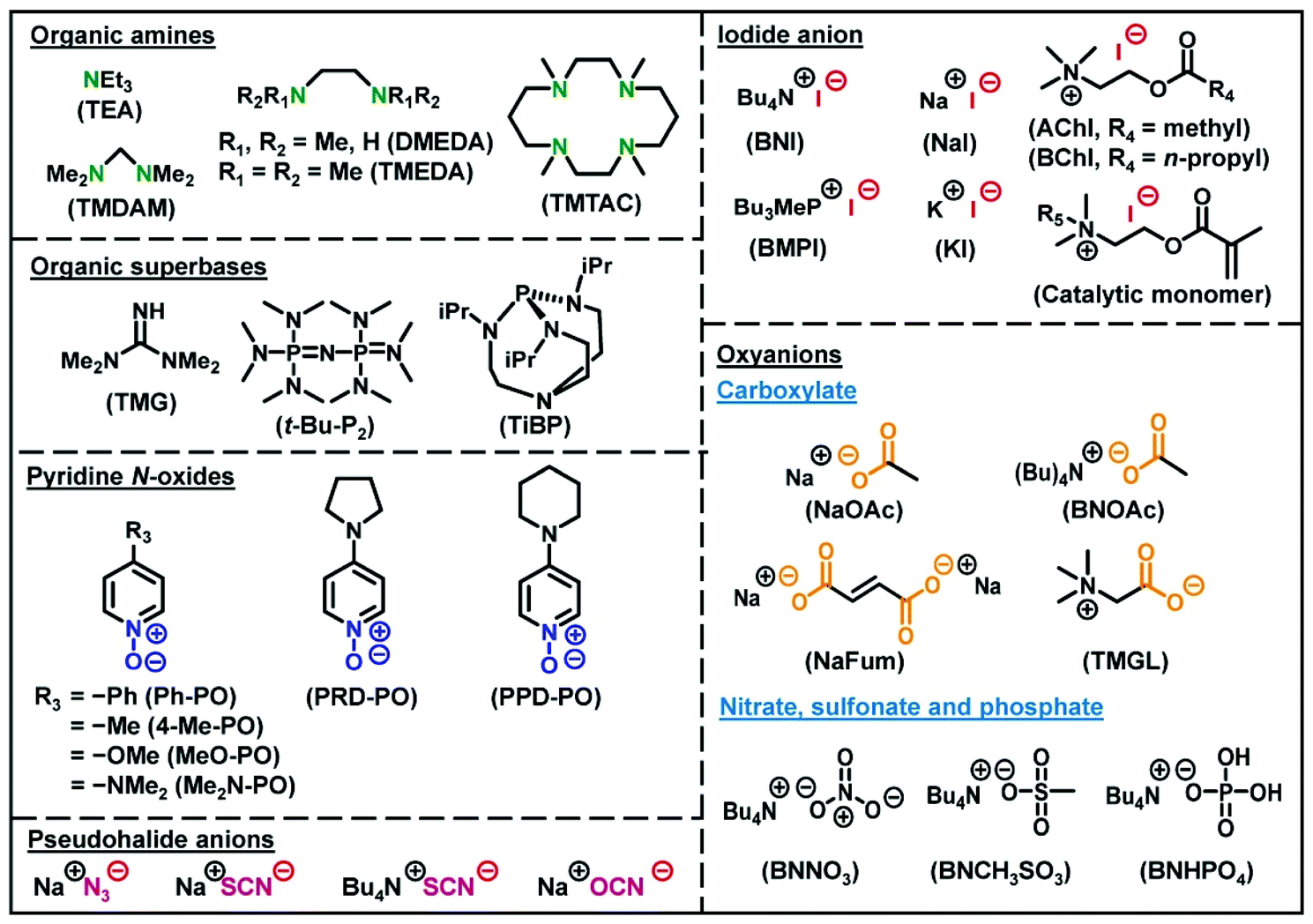 | ||
| Fig. 3 Structures and abbreviations of RCMP catalysts. | ||
Fig. 4 (circles) show an example of RCMP of methyl methacrylate (MMA) (100 eq.) using iodo-2-methylpropionitrile (CP–I (Fig. 5a)) (1 eq.) as an initiating R–I and BNI (1 eq.) as an organic catalyst at 70 °C.50 The number-average molecular weight (Mn) agreed with the theoretical value, and the dispersity (Đ) (= Mw/Mn) remained 1.1–1.2 throughout the polymerization, where Mw is the weight-average molecular weight. The Mn value was able to increase to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (Fig. 4 (triangles)) and up to 140000 g mol−1 (ref. 50) with relatively small Đ values (<1.5) for MMA.
000 g mol−1 (Fig. 4 (triangles)) and up to 140000 g mol−1 (ref. 50) with relatively small Đ values (<1.5) for MMA.
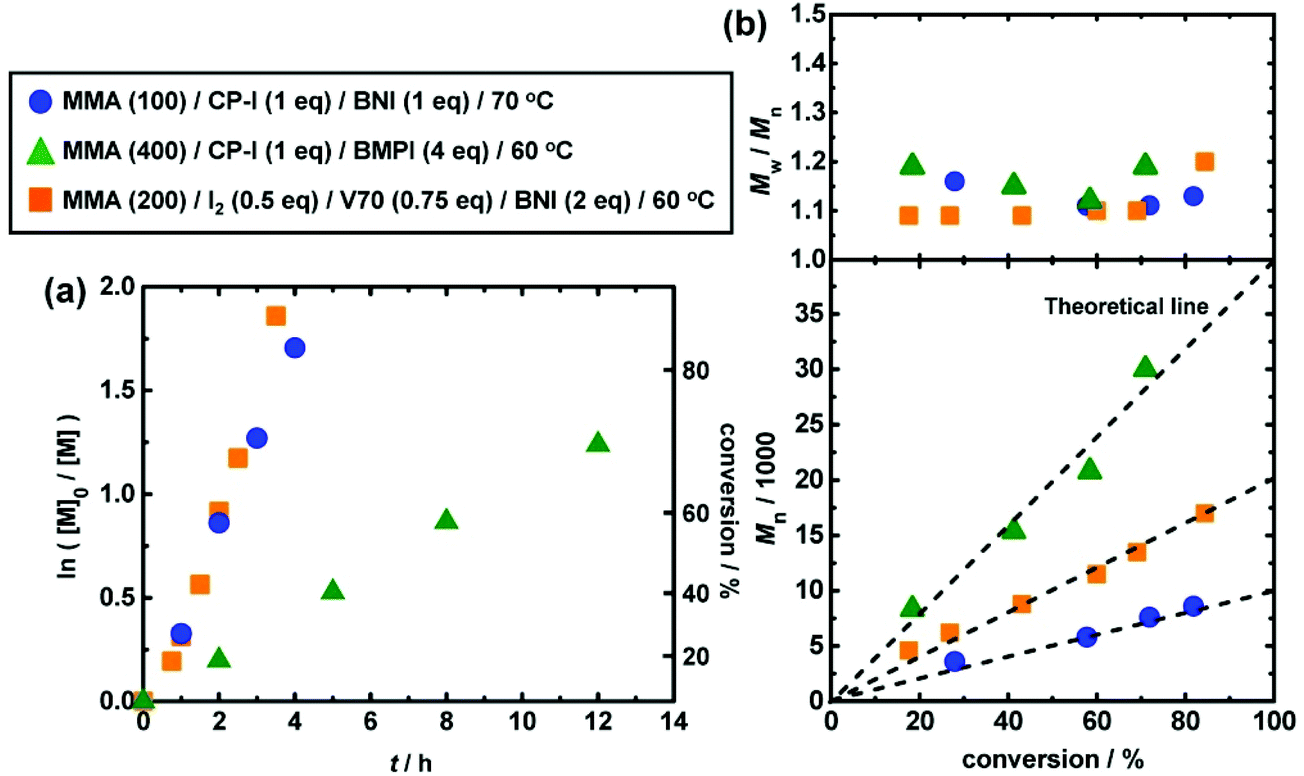 | ||
| Fig. 4 Plots of (a) ln([M]0/[M]) vs. t and (b) Mn and Mw/Mnvs. conversion for RCMP of MMA. [M] is the concentration of monomer. The symbol and reaction conditions are indicated in the figure. V70 is 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile). Adapted with permission from ref. 50. Copyright 2013, American Chemical Society. | ||
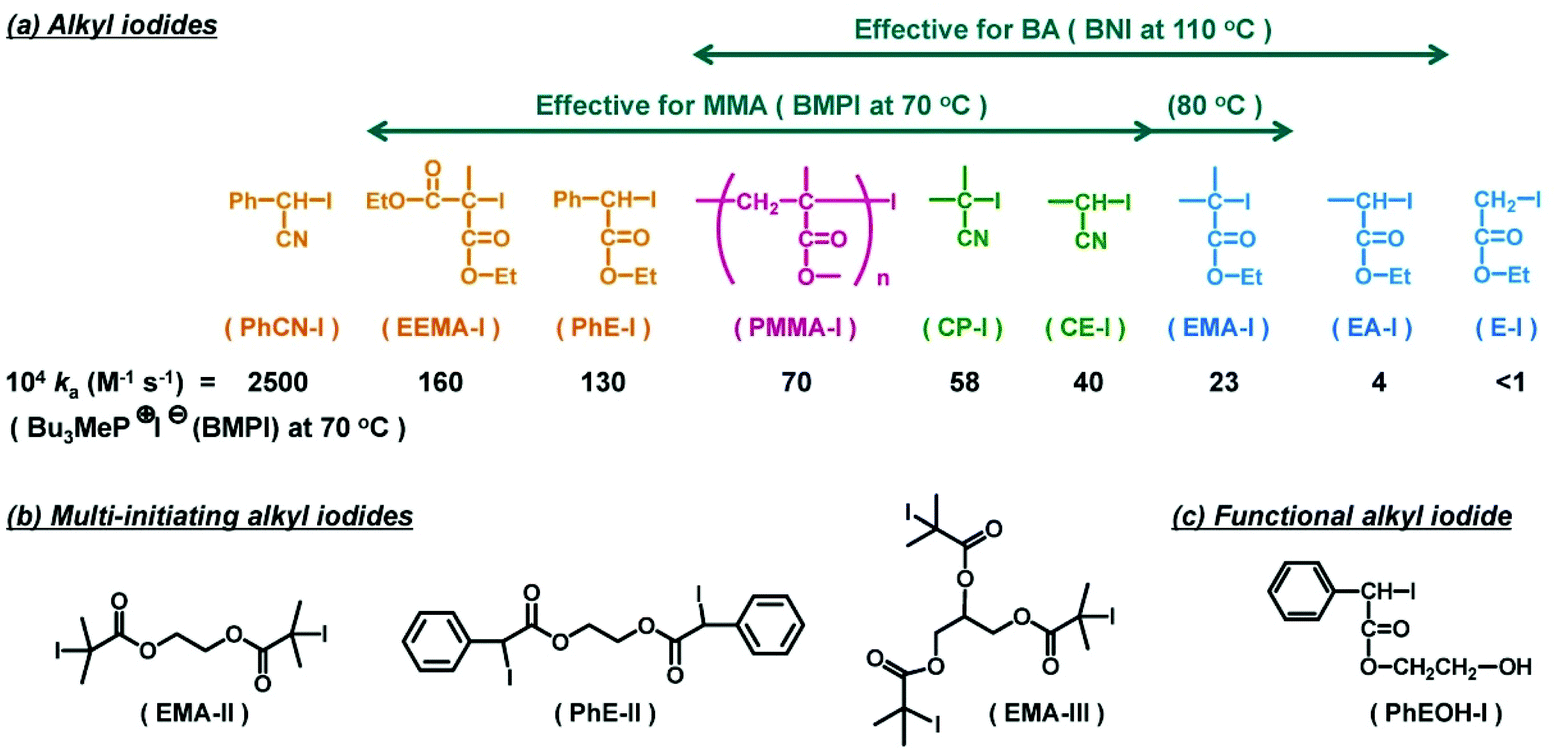 | ||
| Fig. 5 Examples of R–I used in RCMP. (a) Mono-initiating alkyl iodides and the ka values with tributylmethylphosphonium iodide (BMPI) in the toluene-d8/acetonitrile-d3 (90/10) mixture at 70 °C. The arrows indicate the ranges of effective alkyl iodides in the polymerizations of MMA and BA. (b) Multi-initiating alkyl iodides. (c) Functional alkyl iodide. Adapted with permission from ref. 60. Copyright 2014, American Chemical Society. | ||
The activation rate constant (ka (Scheme 2c)) of PMMA–I with BNI was experimentally determined to be 0.050 M−1 s−1 in bulk MMA at 70 °C, where PMMA–I is poly(methyl methacrylate)–iodide.50 This values mean that, with 80 mM of BNI (a typical catalyst concentration), PMMA–I is activated every 4 min, quantitatively confirming the high frequency of the catalytic activation and explaining why this RCMP system can provide low-dispersity polymers from an early stage of polymerization. In the I− catalyst system, polymer–I is activated by I− (activator) to generate polymer˙ and I2˙− (deactivator). I2˙− is not a stable radical and recombines with another I2˙− to generate I− (activator) and I3− (working as a deactivator).50
The free radical nature of the propagating species was supported by a radical trap experiment in low-mass model systems.49–52,55–58 For example (Fig. 6a), an alkyl iodide CP–I (10 mM), an I− catalyst (80 mM), and a radical trap 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) (20 mM) were heated in a solvent at 60 °C.50 If CP–I reacts with the catalyst to generate an alkyl radical CP˙, CP˙ is trapped by TEMPO, thereby yielding CP–TEMPO. Fig. 6a shows the 1H NMR spectra before and after the heating. The signals (peaks a′, b′ and c′) of CP–TEMPO was observed after the heating, proving the radical generation from CP–I.
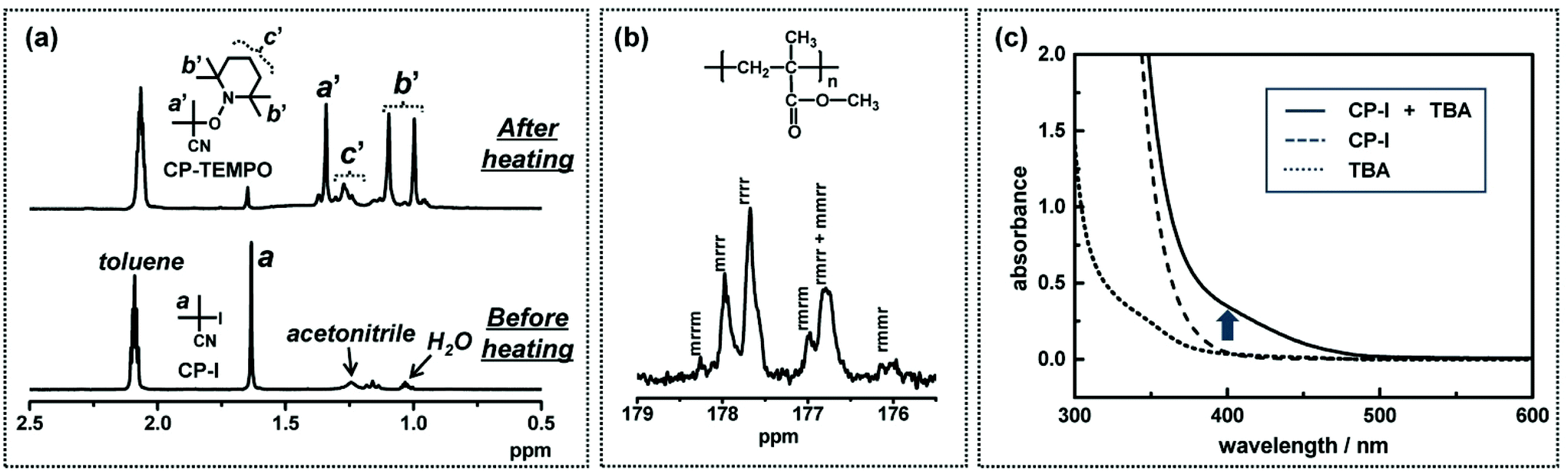 | ||
| Fig. 6 (a) 1H NMR spectra (in the range of 0.5–2.5 ppm) of the solution of CP–I (10 mM), ethylmethyl imidazolium iodide (80 mM), and TEMPO (20 mM) heated at 60 °C for 0 and 15 h. The solvent was the mixture of toluene-d8 and acetnitrile-d3 (9:1). The mixture of toluene-d8 (dielectric constant ε = 2.4) and acetonitrile-d3 (ε = 37.5) is a model of MMA medium (ε = 7.9). (b) 13C NMR spectrum (in the range of 175.5–179 ppm) of the polymer produced in the polymerization of MMA (100 eq.) with CP–I (1 eq.), triethylamine (0.5 eq.), and I2 (1 eq.) at 90 °C for 3 h (monomer conversion = 76%, Mn = 7300, and Đ = 1.24). (c) UV-Vis spectra of TBA (80 mM) only (dotted line), CP–I (80 mM) only (dashed line), and a mixture of CP–I (80 mM) and TBA (80 mM) (solid line) in MMA (ambient temperature). Adapted with permission from ref. 40, 49, and 50. Copyright 2011 and 2013, American Chemical Society. | ||
The free radical nature of the propagating species was also confirmed from the tacticity of the polymer obtained in the polymerization of MMA with triethylamine as a catalyst.40 The 13C NMR spectrum (Fig. 6b)40 showed that the pentad distribution (for the carbonyl carbon) is virtually the same as that for a conventional free radical polymerization. The polymerization was also completely inhibited in the presence of a radical trap TEMPO, supporting the radical mechanism during the polymerization.40
The XB complex (R–I⋯catalyst) formation was supported by a UV-Vis measurement in a low-mass model system containing CP–I as an alkyl iodide and tributylamine (TBA) as a catalyst.49Fig. 6c shows the absorption spectra of TBA (80 mM) only, CP–I (80 mM) only, and a mixture of CP–I (80 mM) and TBA (80 mM) in bulk MMA. A new shoulder peak appeared for the mixture of CP–I and TBA at approximately 400 nm and ranged from 350 nm to 500 nm (solid line). The new peak would correspond to an XB complex of CP–I and TBA. Such red-shifted absorption was reported for complexes of alkyl chlorides/bromides with amines.59
Various alkyl iodides were used as initiating R–I dormant species (Fig. 5). Fig. 5a shows the ka values of R–I with an I− catalyst systematically determined in a polar solvent at 70 °C.60 The ka value tended to increase in the order of primary (E–I) < secondary (EA–I) < tertiary (EMA–I) alkyl groups, increase in the order of phenyl ≈ ester (EMA–I) < cyano (CP–I) stabilizing substituents, and increase in the order of one (EMA–I) < two (PhE–I) stabilizing substituents. Fig. 5a also depicts the range of effective R–I for the MMA and butyl acrylate (BA) polymerizations at different temperatures. In principle, an efficient R–I should have a sufficiently large ka value, and also the released alkyl radical should be reactive enough for monomer addition. PhCN–I (Fig. 5a) releases a too stable alkyl radical and is not suitable for an initiating dormant species.
A possible drawback of RCMP is the use of R–I. Some alkyl iodides are not very stable for long-term storage. To address this issue, instead of an isolated R–I, an R–I generated in situ in the polymerization was also used. Molecular iodine (I2) and an azo compound (R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R) were used as starting compounds, and for the polymerization, the R–I in situ formed was used. The I2/azo method was originally developed by Lacroix-Desmazes et al. for ITP (reverse ITP (RITP))35,61 and subsequently used in RCMP.50,51Fig. 4 (squares) shows the polymerization of MMA (200 eq.) with I2 (0.5 eq.), an azo compound (2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V70)) (0.75 eq.), and BNI (2 eq.) at 70 °C.50 Virtually no polymerization occurred at 0.5 h, during which R–I was generated from I2 and V70. After this period, the polymerization proceeded in a controlled manner. Another useful method to in situ generate R–I was a halogen exchange from the bromide precursors (R–Br) during the polymerization.62–66
N–R) were used as starting compounds, and for the polymerization, the R–I in situ formed was used. The I2/azo method was originally developed by Lacroix-Desmazes et al. for ITP (reverse ITP (RITP))35,61 and subsequently used in RCMP.50,51Fig. 4 (squares) shows the polymerization of MMA (200 eq.) with I2 (0.5 eq.), an azo compound (2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V70)) (0.75 eq.), and BNI (2 eq.) at 70 °C.50 Virtually no polymerization occurred at 0.5 h, during which R–I was generated from I2 and V70. After this period, the polymerization proceeded in a controlled manner. Another useful method to in situ generate R–I was a halogen exchange from the bromide precursors (R–Br) during the polymerization.62–66
2.2. Monomers and dispersity control
RCMP is amenable to a range of hydrophobic and hydrophilic functional methacrylates and also acrylates, styrene, and acrylonitrile (Fig. 7). The Mn value ranged from 103 to 105 g mol−1 and up to 290000 g mol−1 with relatively small Đ values (<1.5).50,67 Recently, RCMP was successfully used for biomass-derived itaconate derivatives (Fig. 7)68 and biocompatible monomers (Fig. 7) such as 2-hydroxyethyl methacrylate (HEMA), poly(ethylene glycol) methyl ether methacrylate (PEGMA), 2-methoxyethyl acrylate (MEA), poly(ethylene glycol) methyl ether acrylate (PEGA), 2-methacryloyloxyethyl phosphoryl choline (MPC betaine monomer), and [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (SBMA betaine monomer).52 Green RCMP systems of such biocompatible monomers using biocompatible catalysts such as choline iodide analogues (AChI and BChI (Fig. 3)) and carboxylates (NaFum and TMGL (Fig. 3)) and non-toxic solvents such as ethyl lactate, ethanol, and water were also developed, being attractive for sustainable society.52,58
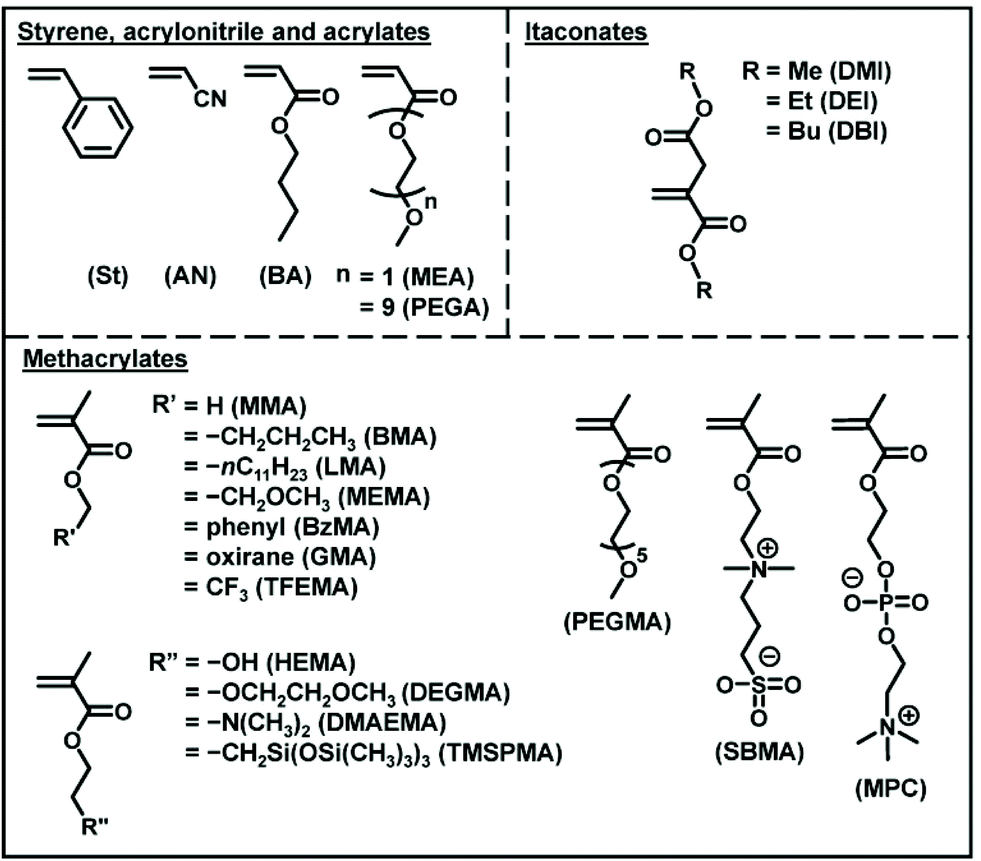 | ||
| Fig. 7 Examples of the monomers applicable in RCMP. | ||
Hunter et al. reported that the stability of XB is relatively insensitive to the solvent polarity.69 The association constant of an I2/urea XB complex ([complex]/([I2][urea])) decreased by only one order of magnitude by changing the solvent from alkanes (n-octane) to alcohols (methanol), for example. The solvent-insensitivity of XB is in sharp contrast to the significant solvent-sensitivity of hydrogen bonding. The stability (association constant) of a phenol/urea hydrogen bonding complex decreased by as much as three orders of magnitude by changing the solvent from n-octane to methanol. This weak sensitivity of XB to reaction media would rationalize the amenability of RCMP to a wide range of hydrophobic and hydrophilic monomers and solvents.
Catalytic monomers containing a polymerizable methacrylate or acrylate moiety and a catalytic quaternary ammonium iodide (QAI) moiety were developed as a new type of monomers.53 The catalytic function incorporated in the monomer enabled a self-catalysed RCMP. Also interestingly, the obtained polymers bore QAI moieties at the side chains and may be used for antibacterial applications.
The dispersity (Đ) is an important parameter determining polymer properties such mechanical strength, miscibility, stimuli-responsive transition, and self-assemble behavior.70,71 Thus, the modulation of the Đ value is an emerging interest in polymer chemistry.71–76 RCMP was also used to modulate the Đ value exploiting a unique temperature-selective activation of polymer–I (Fig. 8a).77 The RCMP of a methacrylate was conducted in the presence of a small amount of an acrylate (4–10%) at a mild temperature of 60 °C. At this mild temperature, the RCMP of the methacrylate smoothly proceeded, but once the dormant polymer chain contained an acrylate at the terminal unit, the chain was hardly activated because of its strong carbon–iodide bond. Thus, the acrylate-terminal chain accumulated over the polymerization, thus increasing the Đ value (Fig. 8a). The Đ value was finely tuned by varying the amount of the acrylate. Importantly, such an acrylate-terminal dormant chain was not a dead polymer but was able to be re-activated at an elevated temperature of 110 °C (Fig. 8b), enabling the synthesis of di-block, multi-block, star-shaped, and brush-shaped block copolymers with tailored Đ values.
 | ||
| Fig. 8 (a) One-pot synthesis of polymer with high dispersity and high iodide chain-end fidelity. (b) Applications to various types of block copolymers. Adapted with permission from ref. 77. Copyright 2019, John Wiley and Sons. | ||
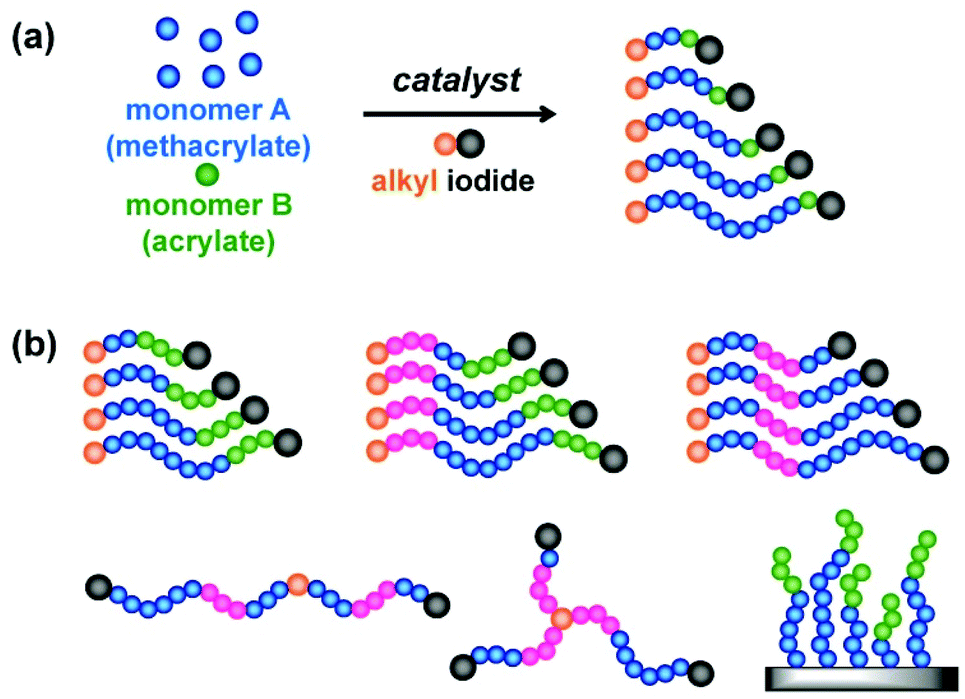
2.3. Polymers with complex architectures
RCMP can yield polymer–I with high degrees of iodide-chain-end fidelity (livingness) (92–99% with ±5% experimental error) up to relatively high monomer conversions (e.g., a 78% monomer conversion).50 Such high iodide-chain-end fidelity allows efficient block copolymerizations and chain-end modifications. RCMP was used to synthesize a range of di-block, tri-block, and multi-block copolymers78–80 and polymers with complex architectures such as star polymers51,78,81 and hyper-branched copolymers.82,83 Tri-block copolymers were synthesized from alkyl di-iodides with two initiating moieties (Fig. 5b).78 For example, PMMA-b-PBA-b-PMMA and PBA-b-PMMA-b-BA with different (ABA and BAB) sequences were obtained, where PBA is poly(butyl acrylate). 3-Arm star polymers were synthesized from an alkyl tri-iodide (Fig. 5b).78 Hyper-branched polymers were synthesized using an inimer containing a polymerizable vinyl group and an alkyl iodide moiety.83A temperature-selective radical generation from a designed alkyl di-iodide (I–R2–R1–I) initiator, 2-iodo-2-(4′-(2′′-iodopropionyloxy)phenyl-acetate) (I–MEPE–I) (Fig. 9), opened up the synthesis of asymmetric CABC tetra-block copolymers.79 The reactivities of I–R2 and R1–I were largely different; R1–I initiated at a mild temperature (60 °C) and I–R2 initiated only at an elevated temperature (110 °C). Methacrylates A and B were polymerized from the R1–I (PE–I) site at 60 °C, at which temperature the I–R2 (I–ME) site remained unreacted (Fig. 9). Subsequently, an acrylate C was polymerized at 110 °C, at which temperature the polymerization occurred from both chain ends, yielding asymmetric CABC tetra-block copolymers (Fig. 9). The temperature-selective radical generation was further exploited for the synthesis of ABC-type miktoarm star copolymers.81
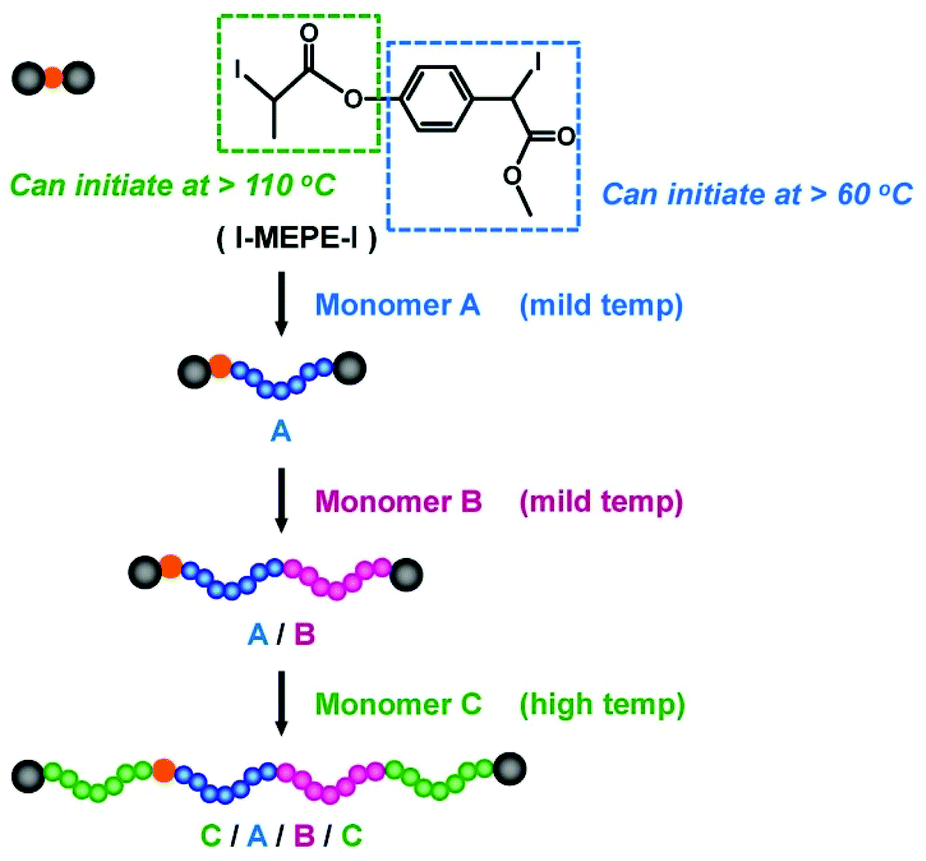 | ||
| Fig. 9 Temperature-selective initiation from I–MEPE–I for the synthesis of CABC tetra-block copolymer. Adapted with permission from ref. 79. Copyright 2018, John Wiley and Sons. | ||
Polymer–I was used as a macroinitiator to synthesize di-block copolymers. When polymer–I is a polymethacrylate–iodide, owing to the weak carbon–iodide bond, polymer–I is sometimes not very stable upon long-term storage. To address this issue, a PMMA containing an unsaturated chain-end (PMMA–Y) was used as a macroinitiator to synthesize block copolymers, where Y is CH2C(CH2)COOCH3.84,85 PMMA–Y was converted to PMMA˙ via an addition fragmentation chain transfer (AFCT), and the generated PMMA˙ was used for the block polymerization via RCMP, yielding block copolymers. The use of PMMA–Y as a macroinitiator is a convenient method to obtain block copolymers.
2.4. Chain-end functionalization
Chain-end functionalized polymers are of great interest for their use as building blocks to create architecturally designed polymers or to graft on solid surfaces for surface modification.86,87 The use of functional R–I initiators (Fig. 5c) in RCMP provided polymers with functional groups at the initiating chain end.51,61 Another approach was the post iodide-chain-end transformation of polymer–I, providing polymers with functional groups at the growing chain end. The weak carbon–iodide bond facilitated the chain-end transformation.For polyacrylates, polymer–I was reacted with primary amines (NH2R), successfully generating polymers (polymer–NHR) with, e.g., phenyl, alkyl, OH, NH2, SH, and Si(OEt)3 functionalities at the R moiety.88 A photo-selective chain-end transformation of polymer–I was conducted using bifunctional cysteamine (NH2CH2CH2SH) (Fig. 10a).89 Without photo irradiation, the chain-end iodide reacted with the amino group of cysteamine to generate a thiol-terminated polymer (polymer–SH) (Fig. 10c). With UV irradiation, the carbon–iodide bond was homolytically cleaved to give a carbon-centred radical (polymer˙), which subsequently underwent a radical chain transfer with the thiol group of cysteamine to generate a hydrogen-terminated polymer (polymer–H) (Fig. 10c). Thus, by switching the UV irradiation on and off, polymer–H and polymer–SH were selectively obtained from the same reactants (polymer–I and cysteamine). Another photo-selective chain-end transformation of polymer–I was conducted co-using NH2R and formic acid (Fig. 10b), which widened the scope of the chain-end functionality.90 Without photo irradiation, polymer–I reacted with NH2R to generate polymer–NHR with, e.g., phenyl, alkyl, OH, NH2, SH, and Si(OEt)3 functionalities at the R moiety, while with UV irradiation, polymer–I was converted to polymer–H via a photochemical reaction with formic acid (Fig. 10c).
 | ||
| Fig. 10 Photo-selective chain-end functionalization with (a) cysteamine and (b) primary amines and formic acid and (c) the mechanisms. Adapted with permission from ref. 89 and 90. Copyright 2018 and 2019, Royal Society of Chemistry. | ||
Pseudo-halide anions such azide (N3−), thiocyanate (SCN−), and cyanate (OCN−) anions form XB with polymer–I and are good catalysts of RCMP. Interestingly, the reactions of polymer–I with the pseudo-halide anions were solvent-selective.56 In non-polar solvents, N3− served as a catalyst for polymer–I to reversibly generate polymer˙. In polar solvents, N3− worked as a nucleophile for polymer–I to undergo a substitution reaction generating an azide chain-end functionalized polymer–N3. Exploiting this solvent selectivity, one-pot synthesis of azide chain-end PMMA (PMMA–N3) was conducted; namely, the RCMP of MMA with sodium azide (NaN3) was conducted in a non-polar solvent to generate PMMA–I, and subsequently to the same reaction solution, a polar solvent was added to convert PMMA–I to PMMA–N3 in one-pot.
Tetrabutylammonium azide (BNN3) was also used instead of NaN3.91 Different from NaN3, BNN3 led to the substitution even in non-polar solvents. This feature enabled the post-azidation of fluorinated (super-hydrophobic) polymer–I in non-polar solvents, which are otherwise insoluble in polar solvents. Azide chain-end polymers are highly useful to construct complex macromolecules through a click reaction.92
2.5. Photo-polymerization
Photo-controlled LRPs have widely been explored.93–96 Photo-controlled ATRP and RAFT polymerization using organic catalysts are emerging techniques attracting great attention.97–102 Photo-controlled RCMP (photo-RCMP) uses an initiating R–I and a light-absorbing organic catalyst.49,103 A possible mechanism (Fig. 11) is that the catalyst coordinates polymer–I to form the polymer–I⋯catalyst complex and that the catalyst serves as an antenna to absorb the light. The energy is subsequently transferred from the catalyst to the C–I bond, giving rise to the C–I cleavage. Notably, several organic molecules with different absorption wavelengths were feasible, covering the entire visible light region (350–750 nm) (Fig. 11).103 The polymerization was also an ideal on–off switchable system with and without the light irradiation. An application was a one-pot selective regulation of RCMP and another type of polymerization (ring opening polymerization (ROP)) (Scheme 3). Using different irradiation wavelengths (550–750 nm for RCMP and 350–380 nm for ROP), the polymerizations of MMA and δ-valerolactone (VL) were initiated and controlled from a dual initiator of RCMP and ROP in one-pot, generating a block copolymer of MMA and VL.103 | ||
| Fig. 11 Possible mechanism for photo-RCMP and photo-catalysts. Adapted with permission from ref. 103. Copyright 2015, American Chemical Society. | ||
 | ||
| Scheme 3 Synthesis of poly(methyl methacrylate)-b-poly(δ-valerolactone) block copolymer via photo-polymerization. Adapted with permission from ref. 103. Copyright 2015, American Chemical Society. | ||
Zhang, Cheng, and coworkers developed visible-light-controlled RCMP systems. One system used polar solvents such as dimethyl sulfoxide as catalysts to form the polymer–I⋯solvent complex.104 Another system used amine-functional monomers as catalytic monomers to form the polymer–I⋯monomer complex.105 Both systems were driven via visible-light irradiation using LEDs or natural sunlight. Near-infrared (NIR)-controlled RCMP was achieved using the polymer–I⋯(carbonyl compound) complex.106 The high-penetration of the NIR light enabled the successful polymerizations even through thick barriers such as pork skin and A4 paper between the light source and the polymerization solution. Water was also used as a catalyst to form the polymer–I⋯water complex under blue-LED irradiation.107 Li et al. demonstrated the in situ bromide–iodide transformation of the initiator and the subsequent photo-RCMP, both of which were controlled by white-LED irradiation.108 Matyjaszewski et al. showed the successful iodine-mediated photo ATRP in aqueous media with oxygen tolerance under visible light irradiation, offering a green and highly efficient photo polymerization.66
2.6. Heterogeneous polymerization and self-assembly
RTCP and RCMP were not only conducted in homogeneous (bulk and solution) systems but also in heterogeneous systems. The first report was an aqueous microsuspension RTCP of MMA using CP–I (R–I initiator), N-iodosuccinimide (NIS) (catalyst), and n-tetradecyltrimethyl ammonium bromide (surfactant), in which a high monomer conversion (93%) was attained within 2 h.109 Instead of water, supercritical carbon dioxide was also used in a dispersion RTCP of styrene, giving relatively small Đ values (1.3–1.5).110Amphiphilic block copolymers can self-assemble in selective solvents. The obtained micelles, worms, vesicles, and other structures are widely utilized as delivery containers, nano-reactors, and imaging materials, for example.111 Such self-assemblies can be obtained using pre-synthesized block copolymers or obtained during block polymerizations, the latter of which is known as polymerization induced self-assembly (PISA).112–116 A significant advantage of PISA is the high polymer concentration (solid content) attainable in the reaction mixture. In PISA, a macroinitiator, which is soluble in the solvent, undergoes chain-extension with another monomer to generate an amphiphilic block copolymer. During the polymerization, as the second block (solvent-insoluble) segment grows, the block copolymer becomes insoluble in the solvent, in situ generating self-assemblies.
The combination of PISA with RCMP provided micelles, worms, and vesicles.64,117,118Fig. 12 shows the morphology diagram in a PISA using PMAA and PMMA as hydrophilic and hydrophobic segments, respectively, where PMAA is poly(methacrylic acid).117 The assembly structure shifted from micelles to worms and vesicles with an increase in the fraction of the hydrophobic PMMA segment in the block copolymer. A biocompatible poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA) was also used as a hydrophilic segment, forming micelles and vesicles with biocompatible surfaces.118 The encapsulation of an external molecule was also tested.118 Instead of thermal RCMP, Zhu et al. utilized photo-RCMP in a PISA of benzyl methacrylate under blue LED light, successfully obtaining micelles, worms, and vesicles.64 RCMP is free from sulfur and transition metals. The obtained nano-particles and nano-capsules may be useful for biomedical, cosmetic, and agrochemical applications.
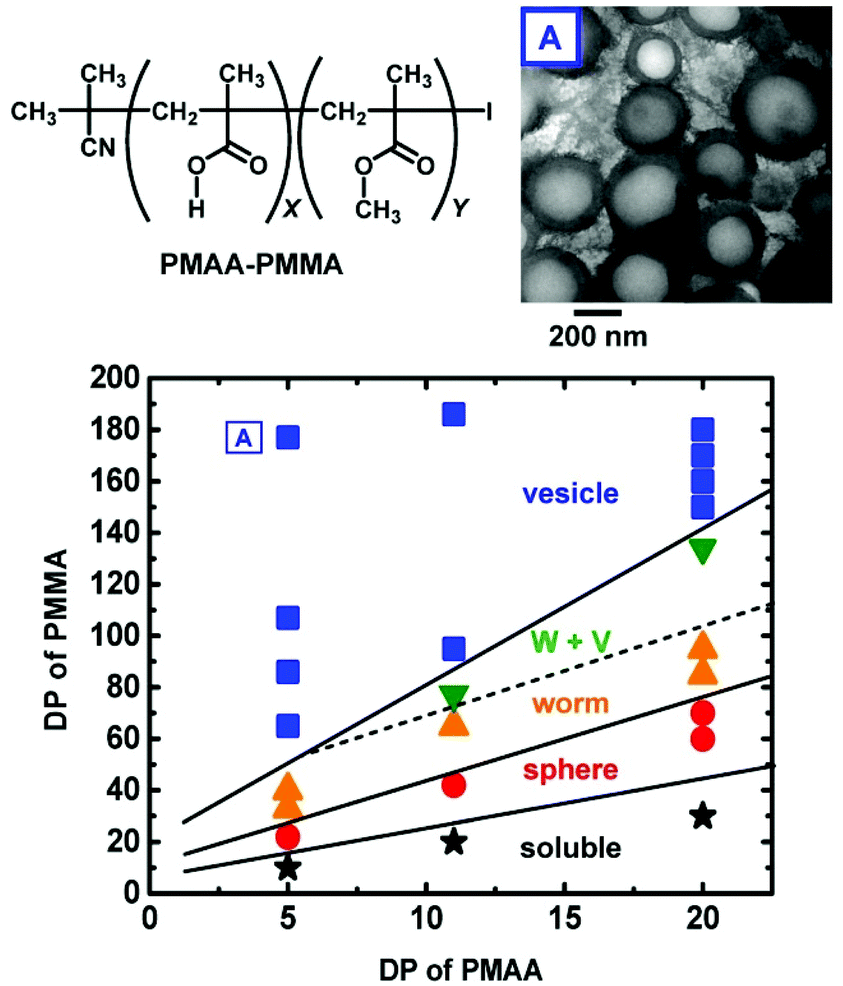 | ||
| Fig. 12 Morphology diagram of the self-assemblies generated in the PISA process for PMAA–PMMA in ethanol, structure of PMAA–PMMA, and TEM image of vesicle. The solid content was 5–9 wt%. W + V denotes a mixture of worms and vesicles. DP is the degree of polymerization. Adapted with permission from ref. 117. Copyright 2018, Royal Society of Chemistry. | ||
Pre-synthesized block copolymers via RCMP were also used to obtain self-assemblies. Amphiphilic CABC tetra-block copolymers (section 2.3) gave unique particles such as Janus-type star particles and flower-like particles, depending on the A, B, and C segments (Fig. 13).79,80,119 Interestingly, the Janus-type star particle contains dual cores and may serve as a dual container of two different guest molecules in a co-delivery system (Fig. 13). The use of a thermo-responsive polymer in the C segment enabled reversible transformation between the star and flower (Fig. 13), which was used in encapsulation and release of guest molecules and temperature-dependent shielded and exposed functional surfaces.80 Not only the flower but also discs, toroids, and a porous structure were also obtained.119 Besides CABC tetra-block copolymers, an amphiphilic star polymer with three different arms formed a large compound micelle,81 and an amphiphilic rod-coil di-block copolymer yielded micelles and vesicles.68
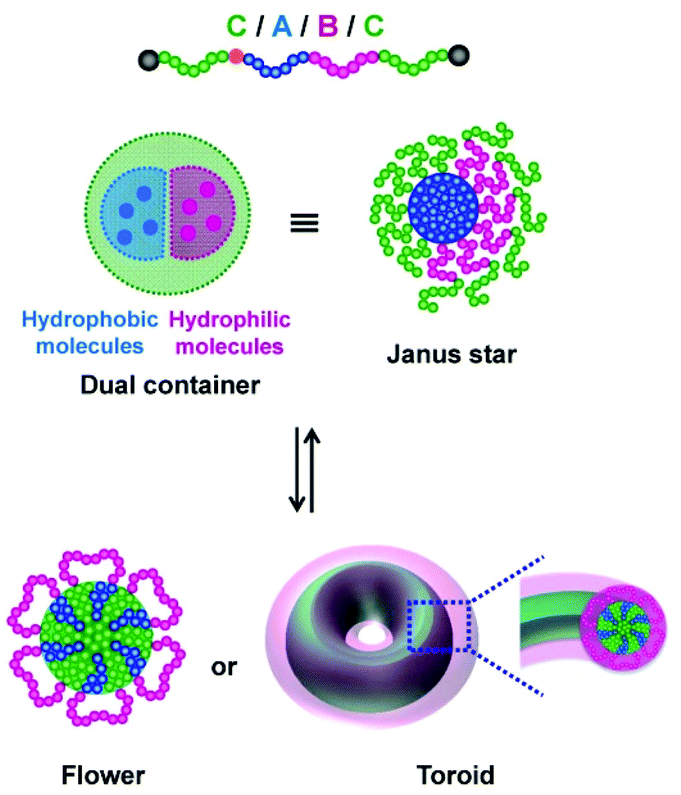 | ||
| Fig. 13 (a) Reversible morphological transformation between Janus star micelle and flower micelle or toroid using CABC tetra-block copolymer. Adapted with permission from ref. 75. Copyright 2020, John Wiley and Sons. Adapted with permission from ref. 119. Copyright 2020, Royal Society of Chemistry. | ||
2.7. Polymer brushes
Surface-initiated LRP (LRP from surface-bound initiators) is an enabling technique for fabricating concentrated polymer brushes on solid surfaces.120–122 Patterned polymer brushes are of interest because polymer brushes with different physical properties are spatially arranged.123,124 Patterned polymer brushes find applications to bio-microarray, molecular recognition, and microelectronic devices, for example.124Polymer brushes were obtained via thermal surface-initiated RTCP78 and RCMP.125 Patterned polymer brushes were obtained via photo-controlled surface-initiated RCMP (photo-SI-RCMP).125 Positive-type patterned brushes were prepared via photo-SI-RCMP under visible light (500–600 nm) using photomasks (Fig. 14a). Negative-type patterned brushes were also prepared (Fig. 14b). The R–I initiator carries a weak carbon–iodide bond and could be degraded very rapidly (within 1 min) under UV light (250–385 nm). The degradation of the R–I initiator was conducted under UV light using photomasks, and the subsequent photo-SI-RCMP under visible light generated negative-type patterned brushes (Fig. 14b). Sequential combination of the positive and negative patterning afforded complex patterned brushes such as patterned binary brushes (Fig. 14c) and patterned block copolymer brushes.125 The graft density of polymer brush was also tailored by adjusting UV-irradiation energy in the initiator degradation step, by which three-dimensional (3D) patterned brushes with high-density (concentrated), moderate-density (semi-diluted), and low-density (diluted) brush areas were prepared (Fig. 14d).67 The size exclusion of external molecules from the polymer brush depends on the graft density.122 Thus, the patterned brushes with different graft densities may serve as molecular recognition interfaces. Patterned cross-linked polymer brushes were also prepared.126 Crosslinking and decrosslinking were reversibly triggered by thermal, chemical, and photo stimuli, which was used to modulate the surface wettability and create and delete the brush patterns.
 | ||
| Fig. 14 (a) Positive-type patterned brush. (b) Negative-type patterned brush. (c) Patterned binary brush. (d) 3D-patterning with different graft density. Adapted with permission from ref. 125. Copyright 2018, John Wiley and Sons. Adapted with permission from ref. 67. Copyright 2019, American Chemical Society. | ||
Polymer brushes with patterned chain-end functionalities were also prepared. The brush chain-end iodide was converted to thiol, alkyne, and hydrogen in patterned manners using photo-selective reactions (section 2.4) with photomasks.89,90 The thiol chain-end was further labelled with a fluorescent maleimide via a thiol–maleimide click reaction, giving patterned fluorescent brushes,89 and the alkyne chain-end was copolymerized with a fluorinated vinyl monomer, giving refractive-index patterned brushes.90
2.8. Application in industry
Dainichiseika Color & Chemicals Mfg. Co., Ltd has commercially used RTCP/RCMP for manufacturing block copolymers at industrial scales (Fig. 15). They use the block copolymers as dispersants of pigments for printer inks and other applications. RTCP and RCMP use relatively inexpensive initiators and catalysts and are free from metals and odour, serving as a cost-effective manufacturing process. Various R–I initiators are available from Godo Shigen Co., Ltd. Some R–I initiators are also available from Tokyo Chemical Industry (TCI) Co., Ltd. | ||
| Fig. 15 Dispersion of pigment nano-particles using block copolymers and pilot reactor for industrial manufacturing in Dainichiseika Color & Chemicals Mfg. Co., Ltd. | ||
2.9. Solid-phase polymerization
Solid-phase polymerization (SPP) of solid monomer (co)crystals is widely used in polymer synthesis.127–130 As mentioned, the use of XB-based monomer (co)crystals in topochemical SPP enabled the synthesis of structurally controlled polymers.20,21 Recently, XB-based monomer co-crystals were for the first time used in free radical SPP.131 Because hydrogen bonding had almost predominantly been used to obtain monomer (co)crystals for free radical SPP, the use of XB widened the monomer scope for free radical SPP (encompassing monomers without hydrogen bonding moieties). The XB-based monomer co-crystals were prepared using electron-donating monomers such as vinyl pyridine and an electron-accepting linker (1,4-diiodotetrafluorobenzene) (Fig. 16). The monomers were regularly aligned in the crystals, and the adjacent vinyl groups were close enough to undergo chain propagation. Free radical SPP was conducted thermally and photochemically, yielding polymers with large Mn values and small Đ values because of the monomer alignment in the crystals. Interestingly, liquid monomers (e.g., vinyl pyridine) were converted to solid monomer co-crystals before the polymerization (Fig. 16). This feature enabled the construction of pre-shaped desired architectures such as sheets before the polymerization, and the architectures were fixed via SPP (Fig. 16). Complex architectures such as a 3D-shaped architecture and a layered sheet actuator were also obtained. This approach may serve as a useful polymer engineering tool for using liquid monomers to constitute pre-determined polymer material architectures.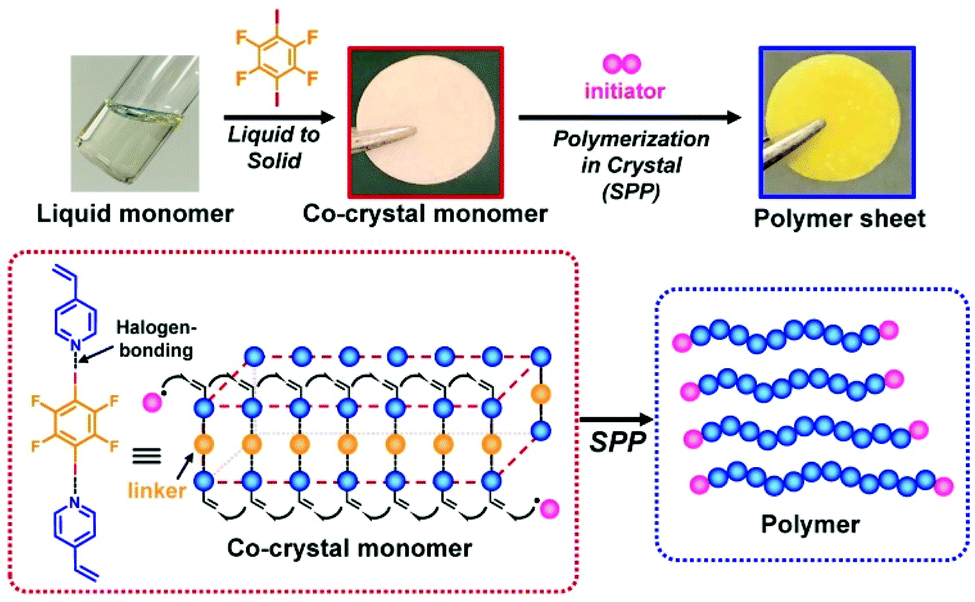 | ||
| Fig. 16 XB-based monomer co-crystal and SSP. Adapted with permission from ref. 131. Copyright 2020, John Wiley and Sons. | ||
3. Conclusions and outlook
Because of the abundance of XB synthons, high complementarity, and compatibility in polar and nonpolar environments, XB offers useful catalytic reactions. RCMP would be one of the most promising examples of XB-catalysis. RCMP is attractive in its broad monomer scope, accessibility to a range of polymer structures, mild polymerization conditions, and absence of metals and odour. The catalysts encompassed amines, iodide anions, pseudo-halide anions, and oxygen-centred anions. Various block copolymers and branched polymers were obtained using designed R–I initiators. The post-transformation of the chain-end iodide offered various chain-end functionalized polymers. Polymer nanoparticles and (patterned) polymer brushes were accessed via heterogeneous and photo-controlled polymerizations. Besides RCMP, free-radical SPP of XB-based monomer co-crystals provided pre-shaped polymer material architectures using liquid monomers.Like other XB catalysis, RCMP can use organic compounds as catalysts. The use of non-metal, less toxic, and colorless catalysts would be attractive in electronic, biomedical, and optical applications. I−, pseudo-halide anion, and oxyanion catalysts are used in the form of organic salts such as Bu4N+I− or alkali metal salts such as Na+I−. Na+ is not a heavy metal and is still biologically safe. A possible drawback of RCMP is the use of R–I initiators, some of which gradually degrade upon long-term storage. The use of the R–I in situ generated via, e.g., the I2/azo method and halogen exchange would be a possible solution. As a future direction, further development of efficient catalysts and use of environmental–friendly conditions are important for widened applications and sustainable society. Such efficient catalysts are desired to expand the monomer scope, molecular weight range, and accessible polymer structures. The use of environmentally–friendly catalysts and solvents and also the development of robust processes for the removal of catalysts and chain-end iodide from the product polymers would further increase the feasibility of RCMP for industrial use. This mini-review summarised the state-of-the-art of RCMP. We hope that the mini-review will be helpful for further exploration of XB catalysis and further development of RCMP.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation (NRF), Singapore, under its National Research Foundation Investigatorship (NRF-NRFI05-2019-0001) and Ministry of Education, Singapore, under its Academic Research Fund (AcRF) Tier 2 (MOE2017-T2-1-018).References
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- J.-C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC.
- B. Li, S.-Q. Zang, L.-Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS.
- J.-C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281–9290 CrossRef CAS PubMed.
- G. Berger, P. Frangville and F. Meyer, Chem. Commun., 2020, 56, 4970–4981 RSC.
- G. Berger, J. Soubhye and F. Meyer, Polym. Chem., 2015, 6, 3559–3580 RSC.
- R. Bertani, P. Metrangolo, A. Moiana, E. Perez, T. Pilati, G. Resnati, I. Rico-Lattes and A. Sassi, Adv. Mater., 2002, 14, 1197–1201 CrossRef CAS.
- J. Xu, X. Liu, T. Lin, J. Huang and C. He, Macromolecules, 2005, 38, 3554–3557 CrossRef CAS.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030–1034 CrossRef CAS PubMed.
- C. Wilhelm, S. A. Boyd, S. Chawda, F. W. Fowler, N. S. Goroff, G. P. Halada, C. P. Grey, J. W. Lauher, L. Luo, C. D. Martin, J. B. Parise, C. Tarabrella and J. A. Webb, J. Am. Chem. Soc., 2008, 130, 4415–4420 CrossRef CAS PubMed.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef CAS.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- J. Bamberger, F. Ostler and O. G. Mancheño, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- J.-P. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052–12055 RSC.
- A. Dreger, P. Wonner, E. Engelage, S. M. Walter, R. Stoll and S. M. Huber, Chem. Commun., 2019, 55, 8262–8265 RSC.
- O. Coulembier, F. Meyer and P. Dubois, Polym. Chem., 2010, 1, 434–437 RSC.
- K. Takagi, K. Yamauchi and H. Murakata, Chem. – Eur. J., 2017, 23, 9495–9500 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 2603–2614 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS PubMed.
- S. Yamago, Chem. Rev., 2009, 109, 5051–5068 CrossRef CAS PubMed.
- A. Debuigne, R. Poli, C. Jerome, R. Jerome and C. Detrembleur, Prog. Polym. Sci., 2009, 34, 211–239 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- A. Goto, H. Zushi, N. Hirai, T. Wakada, Y. Tsujii and T. Fukuda, J. Am. Chem. Soc., 2007, 129, 13347–13354 CrossRef CAS PubMed.
- A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsujii and H. Kaji, Macromolecules, 2011, 44, 8709–8715 CrossRef CAS.
- A. Goto, Y. Tsujii and T. Fukuda, Polymer, 2008, 49, 5177–5185 CrossRef CAS.
- A. Goto, N. Hirai, T. Wakada, K. Nagasawa, Y. Tsujii and T. Fukuda, Macromolecules, 2008, 41, 6261–6264 CrossRef CAS.
- A. Goto, N. Hirai, K. Nagasawa, Y. Tsujii, T. Fukuda and H. Kaji, Macromolecules, 2010, 43, 7971–7978 CrossRef CAS.
- P. Vana and A. Goto, Macromol. Theory Simul., 2010, 19, 24–35 CrossRef CAS.
- D. P. Stevenson and G. M. Coppinger, J. Am. Chem. Soc., 1962, 84, 149–152 CrossRef CAS.
- H. Ishibashi, S. Haruki, M. Uchiyama, O. Tamura and J. Matsuo, Tetrahedron Lett., 2006, 47, 6263–6266 CrossRef CAS.
- F. Sladojevich, E. McNeill, J. Bcrgel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712–3716 CrossRef CAS PubMed.
- Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442–1445 CrossRef CAS.
- A. Ohtsuki, A. Goto and H. Kaji, Macromolecules, 2013, 46, 96–102 CrossRef CAS.
- A. Goto, A. Ohtsuki, H. Ohfuji, M. Tanishima and H. Kaji, J. Am. Chem. Soc., 2013, 135, 11131–11139 CrossRef CAS PubMed.
- J. Sarkar, L. Xiao and A. Goto, Macromolecules, 2016, 49, 5033–5042 CrossRef CAS.
- C.-G. Wang, F. Hanindita and A. Goto, ACS Macro Lett., 2018, 7, 263–268 CrossRef CAS.
- C.-G. Wang, X. Y. Oh, X. Liu and A. Goto, Macromolecules, 2019, 52, 2712–2718 CrossRef CAS.
- C.-G. Wang, J. J. Chang, E. Y. J. Foo, H. Niino, S. Chatani, S. Y. Hsu and A. Goto, Macromolecules, 2020, 53, 51–58 CrossRef CAS.
- L. Lei, M. Tanishima, A. Goto and H. Kaji, Polymers, 2014, 6, 860–872 CrossRef.
- C.-G. Wang and A. Goto, J. Am. Chem. Soc., 2017, 139, 10551–10560 CrossRef CAS PubMed.
- H. Xu, C.-G. Wang, Y. Lu and A. Goto, Macromolecules, 2019, 52, 2156–2163 CrossRef CAS.
- W. Mao, C.-G. Wang, Y. Lu, W. Faustinelie and A. Goto, Polym. Chem., 2020, 11, 53–60 RSC.
- W. J. Lautenberger, E. N. Jones and J. G. Miller, J. Am. Chem. Soc., 1968, 90, 1110–1115 CrossRef CAS.
- L. Lei, M. Tanishima, A. Goto, H. Kaji, Y. Yamaguchi, H. Komatsu, T. Jitsukawa and M. Miyamoto, Macromolecules, 2014, 47, 6610–6618 CrossRef CAS.
- P. Lacroix-Desmazes, R. Severac and B. Boutevin, Macromolecules, 2005, 38, 6299–6309 CrossRef CAS.
- L. Xiao, K. Sakakibara, Y. Tsujii and A. Goto, Macromolecules, 2017, 50, 1882–1891 CrossRef CAS.
- X. Liu, Q. Xu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2017, 8, 2538–2551 RSC.
- Q. Xu, C. Tian, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2019, 40, 1800327 CrossRef PubMed.
- B. Guo, L. Hou, Y. Li and L. Xiao, Polymer, 2020, 189, 122201 CrossRef CAS.
- S. Dadashi-Silab, G. Szczepaniak, S. Lathwal and K. Matyjaszewski, Polym. Chem., 2020, 11, 843–848 RSC.
- C.-G. Wang, H. W. Yong and A. Goto, ACS Appl. Mater. Interfaces, 2019, 11, 14478–14484 CrossRef CAS PubMed.
- K. Hu, J. Sarkar, J. Zheng, Y. H. M. Lim and A. Goto, Macromol. Rapid Commun., 2020, 41, 2000075 CrossRef CAS PubMed.
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- N. Corrigan, A. Almasri, W. Taillades, J. Xu and C. Boyer, Macromolecules, 2017, 50, 8438–8448 CrossRef CAS.
- D. T. Gentekos, J. Jia, E. S. Tirado, K. P. Barteau, D.-M. Smilgies, R. A. DiStasio and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 4639–4648 CrossRef CAS PubMed.
- M. Rubens and T. Junkers, Polym. Chem., 2019, 10, 6315–6323 RSC.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- X. Liu, C.-G. Wang and A. Goto, Angew. Chem., Int. Ed., 2019, 58, 5598–5603 CrossRef CAS PubMed.
- M. Tanishima, A. Goto, L. Lei, A. Ohtsuki, H. Kaji, A. Nomura, Y. Tsujii, Y. Yamaguchi, H. Komatsu and M. Miyamoto, Polymers, 2014, 6, 311–326 CrossRef.
- J. Zheng, C.-G. Wang, Y. Yamaguchi, M. Miyamoto and A. Goto, Angew. Chem., Int. Ed., 2018, 57, 1552–1556 CrossRef CAS PubMed.
- J. Zheng, C. Chen and A. Goto, Angew. Chem., Int. Ed., 2020, 59, 1941–1949 CrossRef CAS PubMed.
- Y. Ge, C. Chen, X. M. Sim, J. Zheng and A. Goto, Macromol. Rapid Commun., 2020, 41, 1900623 CrossRef CAS PubMed.
- H. Yang, Z. Wang, Y. Zheng, W. Huang, X. Xue and B. Jiang, Polym. Chem., 2017, 8, 2137–2144 RSC.
- H. Yang, Z. Wang, L. Cao, W. Huang, Q. Jiang, X. Xue, Y. Song and B. Jiang, Polym. Chem., 2017, 8, 6844–6852 RSC.
- J. J. Chang, L. Xiao, C.-G. Wang, H. Niino, S. Chatani and A. Goto, Polym. Chem., 2018, 9, 4848–4855 RSC.
- J. J. Chang, H. Niino, S. Chatani and A. Goto, Polym. Chem., 2019, 10, 5617–5625 RSC.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455–567 CrossRef CAS.
- A. Anastasaki, J. Willenbacher, C. Fleischmann, W. R. Gutekunsta and C. J. Hawker, Polym. Chem., 2017, 8, 689–697 RSC.
- C. Chen, L. Xiao and A. Goto, Macromolecules, 2016, 49, 9425–9440 CrossRef CAS.
- C. Chen, C. G. Wang, L. Xiao and A. Goto, Chem. Commun., 2018, 54, 13738–13741 RSC.
- C. Chen, C.-G. Wang, W. Guan and A. Goto, Polym. Chem., 2019, 10, 5913–5919 RSC.
- C.-G. Wang, A. M. L. Chong, Y. Lu, X. Liu and A. Goto, Chem. – Eur. J., 2019, 25, 13025–13029 CrossRef CAS PubMed.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junker, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- N. Corrigan, J. Yeow, P. Judzewitsch and C. Boyer, Angew. Chem., 2019, 58, 5170–5189 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- X. Pan, M. Lamson, J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS.
- A. Allushi, C. Kutahya, C. Aydogan, J. Kreutzer, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 1972–1977 RSC.
- A. Ohtsuki, L. Lei, M. Tanishima, A. Goto and H. Kaji, J. Am. Chem. Soc., 2015, 137, 5610–5617 CrossRef CAS PubMed.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 3576–3588 RSC.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Chem. Commun., 2016, 52, 10850–10853 RSC.
- C. Tian, P. Wang, Y. Ni, L. Zhang, Z. Cheng and X. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3910–3916 CrossRef CAS PubMed.
- Y. Ni, C. Tian, L. Zhang, Z. Cheng and X. Zhu, ACS Macro Lett., 2019, 8, 1419–1425 CrossRef CAS.
- F. Li, W. Yang, M. Li and L. Lei, Polym. Chem., 2019, 10, 3996–4005 RSC.
- M. Yorizane, T. Nagasuga, Y. Kitayama, A. Tanaka, H. Minami, A. Goto, T. Fukuda and M. Okubo, Macromolecules, 2010, 43, 8703–8705 CrossRef CAS.
- T. Kuroda, A. Tanaka, T. Taniyama, H. Minami, A. Goto, T. Fukuda and M. Okubo, Polymer, 2012, 53, 1212–1218 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. J. Monteiro and M. F. Cunningham, Macromolecules, 2012, 45, 4939–4957 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 2–27 CrossRef.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- J. Sarkar, L. Xiao, A. W. Jackson, A. M. van Herk and A. Goto, Polym. Chem., 2018, 9, 4900–4907 RSC.
- J. Sarkar, A. W. Jackson, A. M. van Herk and A. Goto, Polym. Chem., 2020, 11, 3904–3912 RSC.
- J. Zheng and A. Goto, Polym. Chem., 2020, 11, 3987–3993 RSC.
- J. Yan, M. R. Bockstaller and K. Matyjaszewski, Prog. Polym. Sci., 2020, 100, 101180 CrossRef CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS.
- Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Adv. Polym. Sci., 2006, 197, 1–45 CrossRef CAS.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2019, 1903850 Search PubMed.
- T. Chen, I. Amin and R. Jordan, Chem. Soc. Rev., 2012, 41, 3280–3296 RSC.
- C.-G. Wang, C. Chen, K. Sakakibara, Y. Tsujii and A. Goto, Angew. Chem., Int. Ed., 2018, 57, 13504–13508 CrossRef CAS.
- X. M. Sim, C.-G. Wang, X. Liu and A. Goto, ACS Appl. Mater. Interfaces, 2020, 12, 28711–28719 CrossRef CAS PubMed.
- S. N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Prog. Polym. Sci., 2005, 30, 10–37 CrossRef CAS.
- R. Mohanrao, K. Hema and K. M. Sureshan, Nat. Commun., 2020, 11, 865 CrossRef CAS PubMed.
- I. E. Claassens, L. J. Barbour and D. A. Haynes, J. Am. Chem. Soc., 2019, 141, 11425–11429 CrossRef CAS PubMed.
- R. Z. Lange, G. Hofer, T. Weber and A. D. Schlüter, J. Am. Chem. Soc., 2017, 139, 2053–2059 CrossRef CAS PubMed.
- H. T. Le, C.-G. Wang and A. Goto, Angew. Chem., Int. Ed., 2020, 59, 9360–9364 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |