
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Epoxy-functionalised 4-vinylguaiacol for the synthesis of bio-based, degradable star polymers via a RAFT/ROCOP strategy†
Thomas M.
McGuire
a,
Masato
Miyajima
b,
Mineto
Uchiyama
 b,
Antoine
Buchard
*a and
Masami
Kamigaito
*b
b,
Antoine
Buchard
*a and
Masami
Kamigaito
*b
aCentre for Sustainable and Circular Technologies, Department of Chemistry, University of Bath, Claverton Down BA2 7AY, Bath, UK. E-mail: a.buchard@bath.ac.uk
bDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: kamigait@chembio.nagoya-u.ac.jp
First published on 19th August 2020
Abstract
An epoxy derivative of a naturally occuring vinylphenolic compound, 4-vinylguaiacol (4VGEP), was used for the synthesis of a well-defined (Mw/Mn = 1.08–1.14), bio-based styrene-type polymer. Block copolymers of 4VGEP with styrene and diethylacrylamide were also prepared and used as macro-monomers in metal, and metal-free, ring-opening copolymerisation (ROCOP) with cis-4-cyclohexene-1,2-dicarboxylic anhydride to form ester cross-linked star polymers in high yields (82–90%), with narrow dispersity (Mw/Mn = 1.27–1.40). Finally, the selective degradation of the ester core of the styrene-based star was achieved under acidic conditions.
Introduction
The valorisation of renewable resources for the preparation of degradable polymers is a rapidly developing field in macromolecular chemistry in view of sustainability.1–4 In addition to renewability, the characteristic chemical structures of natural products can enhance properties and functionalities of synthetic polymers. More specifically, the oxygen-rich nature of renewable feedstocks can act as a useful chemical handle for stimuli-responsive degradability.5Star polymers are composed of a core attached to three or more linear polymers or arms.6 Advances in the controlled synthesis of star polymers have led to their applications being posited in many areas of chemistry, including in drug delivery,7,8 drug encapsulation,9 gene delivery,10 imaging,11–13 and catalysis.14,15 Reversible-addition–fragmentation chain transfer (RAFT) polymerisation is a highly versatile method for the preparation of functional polymers, including star polymers.16,17 Broadly, there are two strategies for the synthesis of star polymers using RAFT polymerisation: arm-first or core-first. In a core-first approach, linear arms can be polymerised from a multi-functional initiating core. Alternatively, when using an arm-first approach, linear polymers are synthesised initially and a core is grafted from the arms through careful choice of polymerisation conditions. Typically, star polymers synthesised using RAFT in an arm-first approach, are formed by cross-linking vinyl-functionalised polymers with multi-vinyl compounds.6,18–21 This creates a core linked by C–C bonds, the controlled degradation of which is challenging.
Some of us have recently reported the polymerisation of 4-vinylguaiacol (4VG), derived by decarboxylation of naturally occurring ferulic acid.22–25 The presence of a hydroxyl group in 4VG allows for easy functionalisation of 4VG-derived monomers. One potential modification could be through the incorporation of an epoxide moiety, which are versatile functionalities for a number of chemical transformations, including ring opening copolymerisation (ROCOP) with cyclic anhydrides to form polyesters.26 Practically, ROCOP can offer advantages over ring opening polymerisation (ROP) of lactones as the properties of the resultant polymer can be tuned through selection of the appropriate co-monomers, thus avoiding lengthy lactone syntheses.
The reactivity of epoxy and vinyl functional groups is generally orthogonal, and has been exploited in one-pot sequential,27,28 simultaneous29 or switchable30–33 block-copolymer syntheses through design of bespoke catalyst/initiator systems.34 However, the number of epoxide-functionalised, styrene-type polymers remain relatively small,35–37 and we envisaged that epoxy-functionalised 4VG (4VGEP) could act as a bifunctional monomer, which may be suitable for both RAFT and ROCOP, and useful for sequential star-polymer synthesis. Moreover, as ester linkages are hydrolytically sensitive, this could enable selective degradation. Such a strategy has been employed by Wu and coworkers with poly(propylene carbonate)-block-poly(4-vinylcatechol acetonide) in which the polycarbonate component was selectively degraded under mildly alkaline conditions to form membranes with monodisperse nanopores.28 Tsarevsky and coworkers have also reported on the atom-transfer-radical-polymerisation (ATRP) and RAFT-polymerisation of 4-vinylphenyl glycidyl ether derivatives to give linear and star-shaped epoxide-containing, but non-degradable, polyolefins.36,38 Using a dual-functional initiator strategy, Qiao and coworkers have combined ATRP of vinyl monomers with ROP of lactones for the synthesis of mixed poly(olefin/ester) star polymers with various architectures.39,40 The polyester component of these star polymers could be selectively hydrolysed, although scope for core or arm functionality would here be limited by the availability and polymerisability of (di)lactones used.
Presented here is the polymerisation of 4VGEP, a fully-bio derived, epoxide-functionalised, styrene-type monomer which is readily synthesised.41 Block copolymers with styrene diethylacrylamide (DEAA) have been used to synthesise star polymers, by an arm first approach, using a sequential, tandem living RAFT/ROCOP-based strategy. Controlled degradation of the styrene-based star polymer's core to linear polymers was achieved under acidic conditions.
Results and discussion
Decarboxylation of ferulic acid in the presence of triethylamine (NEt3) afforded 4VG as previously reported (Scheme 1).22 4VGEP was synthesised in good yield by reaction of 4VG with epichlorohydrin (which may be derived from glycerol42) in the presence of the phase transfer catalyst, BnEt3NCl.41 | ||
| Scheme 1 Synthesis of 4VGEP from ferulic acid and epichlorohydrin. | ||
Recrystallisation from hexane afforded the monomer in sufficient purity for RAFT-polymerisation (Fig. 1a).
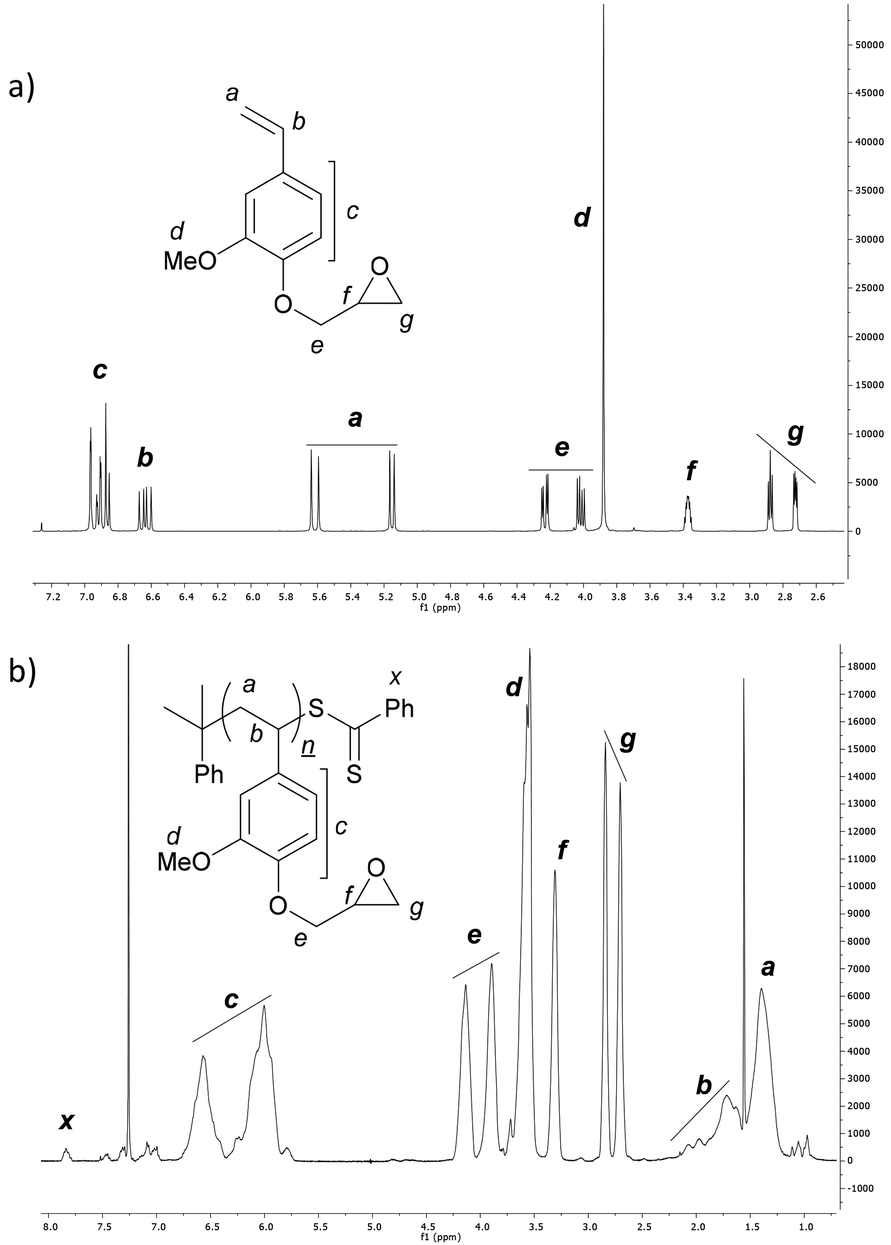 | ||
| Fig. 1 1H NMR spectrum of (a) 4VGEP (CDCl3, 25 °C) and (b) P4VGEP (CDCl3, 60 °C). | ||
The homopolymerisation of 4VGEP was initiated by azobisisobutyronitrile (AIBN) in the presence of cumyl dithiobenzoate (CDB) at 60 °C (Fig. 2). The polymerisation demonstrated typical living characteristics, with good agreement between theoretical and observed molecular weights (Mn,SEC) and narrow molecular weight distributions (ĐM = 1.08–1.14) as determined by size exclusion chromatography (SEC) (Fig. 2b and c). As expected, 1H NMR spectroscopy showed that the epoxide functionality remained intact post-RAFT polymerisation (Fig. 1b).
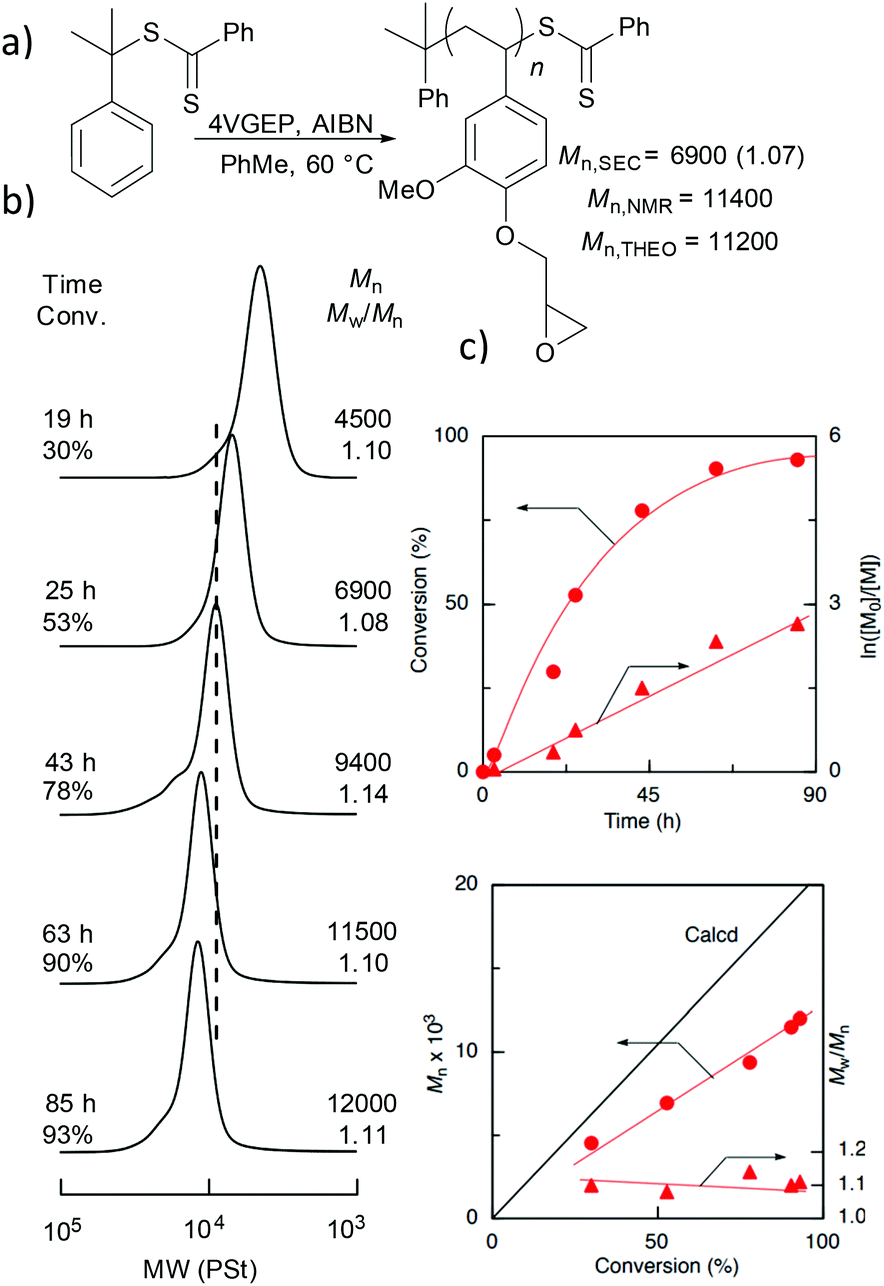 | ||
Fig. 2 (a) Homopolymerisation of 4VGEP at 60 °C at [4VGEP]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CDB]0:[AIBN]0 loadings of 100:1:0.25 in toluene. [4VGEP]0 = 3.0 mol L−1 (b) associated SEC chromatograms (c) time vs. conversion and conversion vs. Mn and vs. ĐM plots. :[CDB]0:[AIBN]0 loadings of 100:1:0.25 in toluene. [4VGEP]0 = 3.0 mol L−1 (b) associated SEC chromatograms (c) time vs. conversion and conversion vs. Mn and vs. ĐM plots. | ||
Next, block copolymers of 4VGEP and styrene (St-b-4VGEP), and 4VGEP and diethylacrylamide (DEAA, DEAA-b-4VGEP) were synthesised (Fig. 3). Styrene and DEAA were chosen as comonomers as they are known to form hydrophobic and hydrophilic polymers, respectively. First, polystyrene (PSt) and polyDEAA (PDEAA) were prepared and isolated as macro-RAFT agents, targeting a degree of polymerisation (DP) between 100–200. The polymerisations were performed at [monomer]0:[CDB]0 ratios of 200:1. For styrene, the reaction was performed in bulk in the absence of initiator, whereas for DEAA, the polymerisation was carried out at 60 °C using AIBN ([DEAA]0:[AIBN]0:[CDB]0 = 200:0.2:1). The reactions were terminated once monomer conversion reached over 50%.
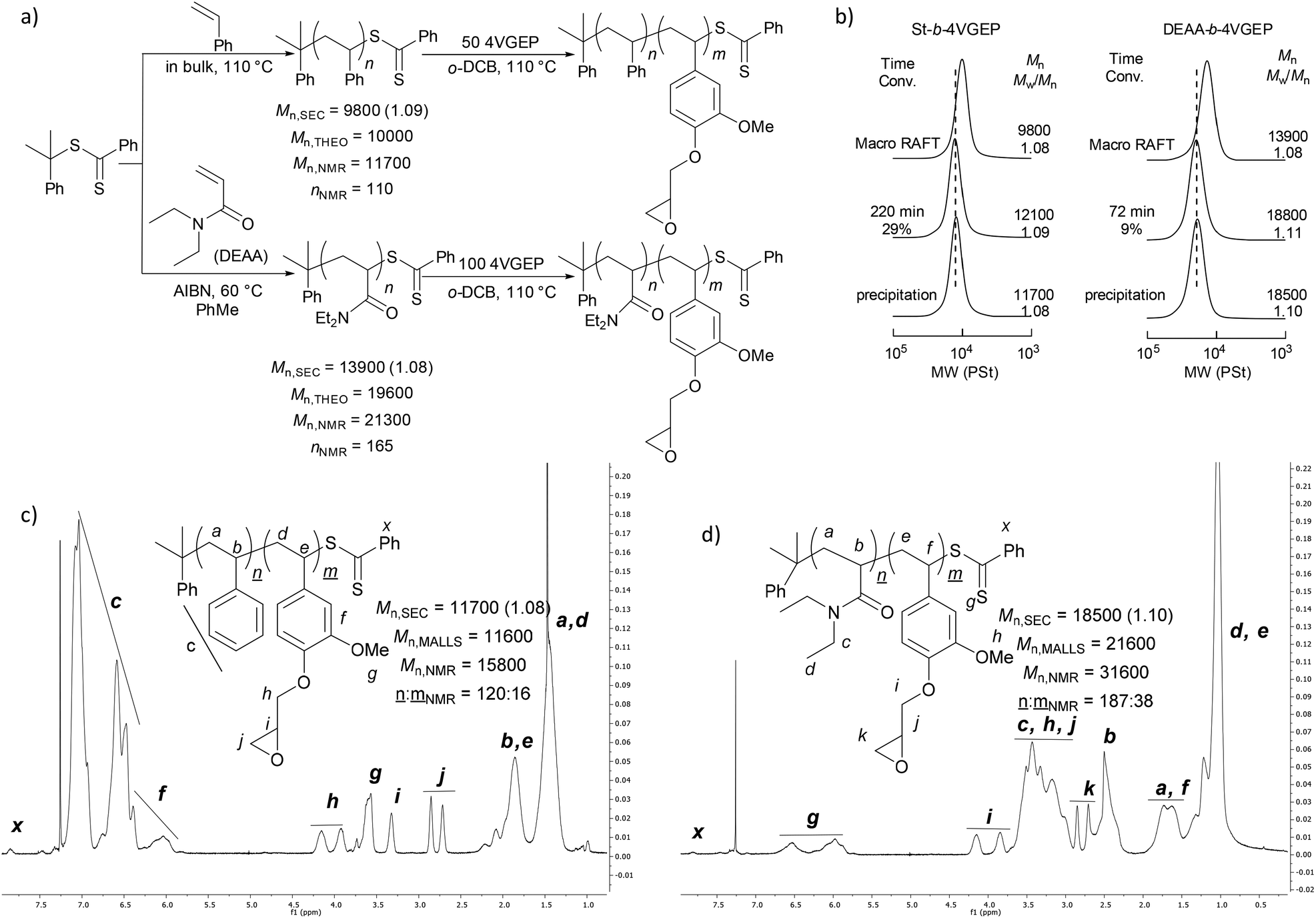 | ||
| Fig. 3 (a) Synthesis of St-b-4VGEP and DEAA-b-4VEP. (b) SEC chromatograms for the block copolymerisation of PSt and 4VGEP at [PSt]0:[4VGEP]0 50:1 and PDEAA and 4VGEP at [PDEAA]0:[4VGEP]0 of 100:1 at 110 °C in o-DCB. [4VGEP]0 = 2.0 mol L−1. (c) 1H NMR spectrum of St-b-4VGEP (CDCl3, 60 °C) and (d) DEAA-b-4VEP (CDCl3, 60 °C). | ||
With these macro-RAFT agents in hand, copolymerisations with 4VGEP were attempted, targeting the formation of block copolymers with a short 4VGEP segment at a styrene:4VGEP and DEAA:4VGEP ratio of approximately 10:1 (Fig. 3a). The initial reaction was carried out with the macro-RAFT PSt at loadings of [4VGEP]0:[AIBN]0:[PSt]0 of 20:0.2:1 at 60 °C in toluene. The large amount of toluene necessary to completely solubilise both 4VGEP and PSt at 60 °C resulted in [4VGEP]0 of 0.75 mol L−1, which is relatively low for RAFT polymerization to be effective. Consequently, while the addition of short blocks of 4VGEP to the macro-RAFT PSt chains was observed by 1H NMR spectroscopy at conversions of 19% and over, only slight increases in molecular weight and small shifts in the SEC curves were detected, with long reaction times resulting in small shoulders at higher molecular weight in the associated SEC chromatograms. These observations suggested a non-negligible occurrence of radical–radical termination reactions along with a slow polymerisation due to the low concentration of 4VGEP, and indicated that the initial [4VGEP]0 and temperature were insufficient for the block copolymerisation to proceed as intended.
A second reaction was carried out at 110 °C in ortho-dichlorobenzene (o-DCB) in the absence of initiator at [4VGEP]0:[PSt]0 of 50:1, giving a [4VGEP]0 = 2.0 mol L−1 (Fig. 3b). No initiator is necessary for RAFT polymerization of styrene and its derivatives at high temperature because thermal initiation generally occurs.22 Satisfyingly, the polymerisation proceeded well, with a significant increase in the copolymer Mn,SEC and no shoulders detected in the SEC chromatograms. At 29% conversion, the reaction was quenched to get a short block of 4VGEP for the subsequent cross-linking reaction.
The conditions were then applied for the 4VGEP copolymerisation with PDEAA macro-RAFT agent, at loadings of [4VGEP]0:[PDEAA]0 of 100:1, to maintain [4VGEP]0 of 2.0 mol L−1. As expected, the polymerisation proceeded well as indicated by 1H NMR spectroscopy and SEC analysis (Fig. 3b). The reaction was quenched at 9% conversion. For both macro-RAFT agents, the epoxide remained intact following polymerisation (Fig. 3c and d).
Star polymer synthesis was then attempted using epoxide/anhydride ring opening copolymerisation (ROCOP) (Fig. 4). A ROCOP strategy was pursued as it was postulated that the resulting polyester core would allow for the selective degradation of the star polymers.
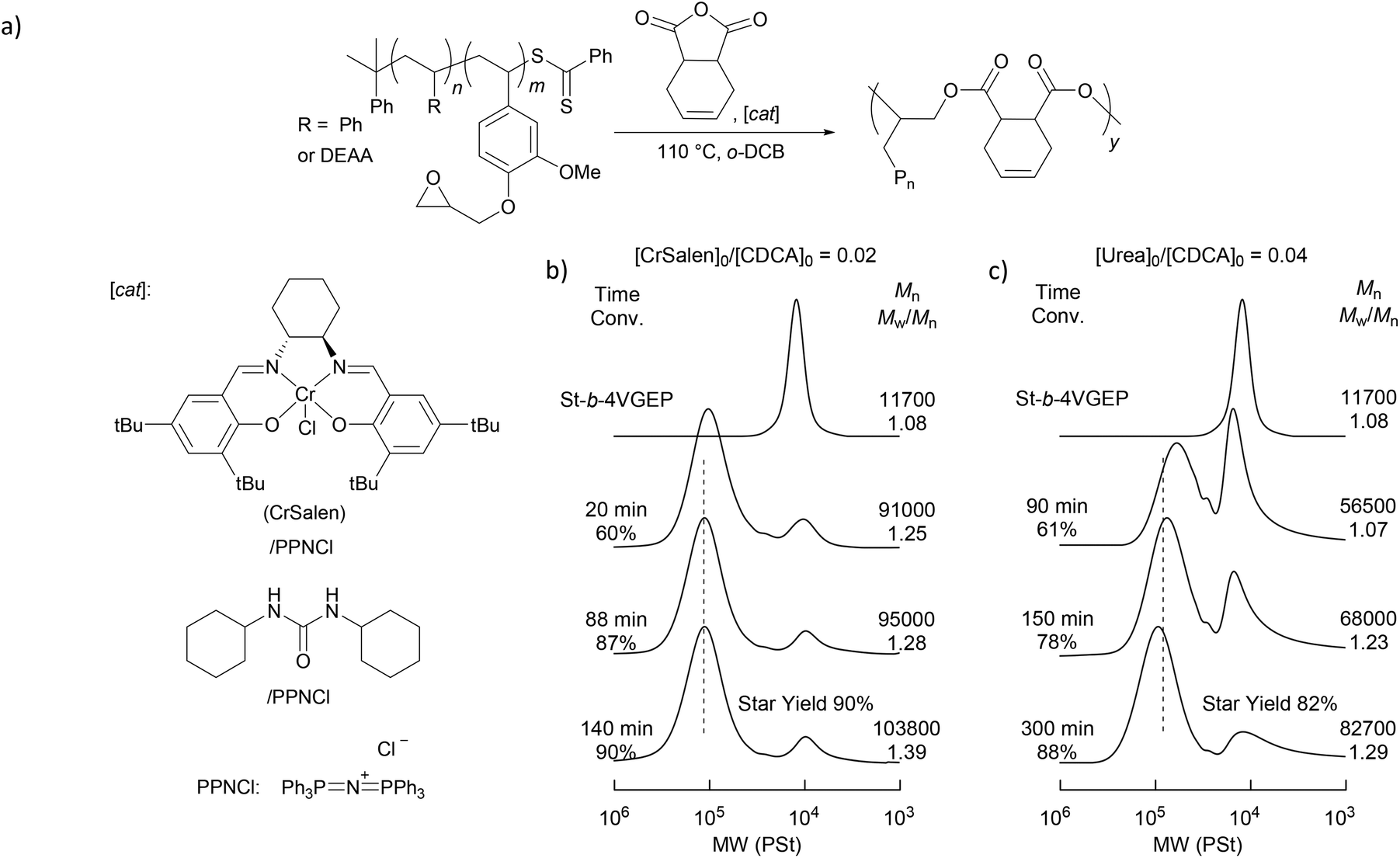 | ||
| Fig. 4 (a) Synthesis of star polymers from St-b-4VGEP and CDCA. SEC chromatograms for the (b) CrSalen (2 mol%) and (c) urea (4 mol%) catalysed ROCOP of St-b-4VGEP and CDCA. mol% given with respect to anhydride loading. [St-b-4VGEP] = 0.01 mol L−1. | ||
cis-4-Cyclohexene-1,2-dicarboxylic anhydride (CDCA) was chosen as the co-monomer as it is well studied in ROCOP and the presence of a double bond could allow for further functionalisation of the star polymer if desired. In addition, the parent diacid of CDCA may be sustainably-derived from 1,4-cyclohexadiene43 or the anhydride may be synthesised directly via a Diels–Alder reaction of renewably-sourced 1,3-butadiene44 and maleic anhydride.45
Reactions were initially carried out with the block copolymer St-b-4VGEP using a binary catalyst comprising a chromium salen chloride complex (CrSalen) and bis(triphenylphosphoranylidene)ammonium chloride salt (PPNCl). CrSalen was selected as it is a commercial and versatile ROCOP catalyst.46,47 A [St-b-4VGEP]0:[CDCA]0 ratio of 1:14 was chosen based on the copolymerisation NMR data, in order to achieve equal anhydride and epoxide concentrations. ROCOP was found to proceed readily at loadings of [St-b-4VGEP]0:[CDCA]0:[CrSalen]0:[PPNCl]0 of 10:140:2.8:2.8 and 10:140:1.4:1.4, with [St-b-4VGEP]0 = 0.01 mol L−1 (Table 1, entries 1 and 2; Fig. 5). Good control was exhibited in the ROCOP, with narrow ĐM (1.32–1.35) obtained, and absolute molecular weight approximately doubling upon decreasing the CrSalen loading from 2 mol% (Mn,MALLS = 189700 g mol−1) to 1 mol% (Mn,MALLS = 380800 g mol−1). Under otherwise analogous conditions, with [St-b-4VGEP]0 of 0.38 mol L−1, insoluble residues were formed, likely as a result of star-star coupling.
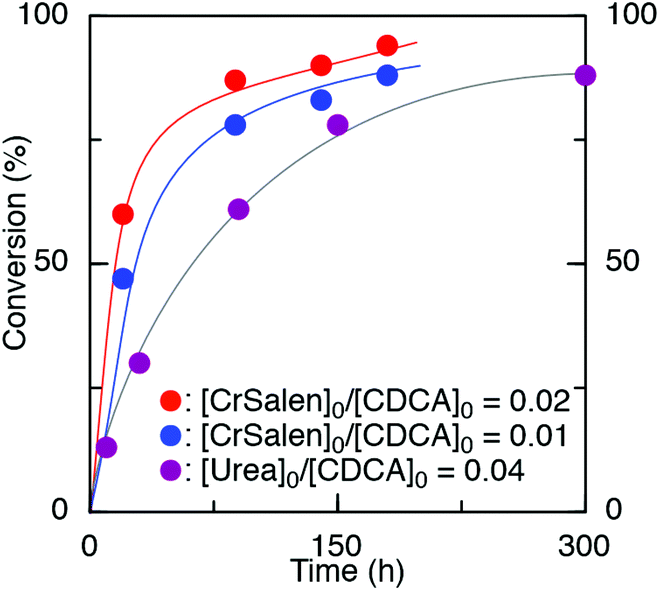 | ||
| Fig. 5 Time vs. % conversion for the CrSalen and urea catalysed ROCOP of St-b-4VGEP and CDCA at loadings of: (red) [St-b-4VGEP]0:[CDCA]0:[CrSalen]0:[PPNCl]0 = 10:140:2.8:2.8, (blue) [St-b-4VGEP]0:[CDCA]0:[CrSalen]0:[PPNCl]0 = 10:140:1.4:1.4 and (purple) [St-b-4VGEP]0:[CDCA]0:[urea]0:[PPNCl]0 = 10:140:5.6:5.6. mol% = [CrSalen]0/[CDCA]0. [St-b-4VGEP]0 = 0.01 mol L−1. | ||
| Entry | Block copolymer | Cata | CrSalen/CDCA | M n,SEC (ĐM)b | M n,MALLS (farm)c | Star yieldd |
|---|---|---|---|---|---|---|
| Reactions carried out at [block copolymer]0:[CDCA]0 of 1:14 at 110 °C in o-DCB. [block copolymer] = 0.01 mol L−1 unless otherwise stated.a Cat:PPNCl added in a 1:1 ratio.b Determined against a set of PSt standards, ĐM = Mw/Mn.c farm = (Mn,MALLS (Star) – 14 × conv × Mw (CDCA))/Mn,MALLS (St-b-4VGEP).d Taken from relative area of associated SEC chromatogram peak.e Reaction carried out at [DEAA-b-4VGEP]0:[CDCA]0 of 1:16. |
||||||
| 1 | St-b-4VGEP | CrSalen/PPNCl | 0.02 | 116800 (1.32) |
189700 (16.3) |
90% |
| 2 | St-b-4VGEP | CrSalen/PPNCl | 0.01 | 142400 (1.40) |
380800 (33.8) |
88% |
| 3 | St-b-4VGEP | Urea/PPNCl | 0.04 | 102800 (1.27) |
156100 (13.4) |
82% |
| 4e | DEAA-b-4VGEP | Urea/PPNCl | 0.04 | 91900 (1.33) |
468200 (21.6) |
89% |
Further study was carried out using a urea/PPNCl catalyst (Table 1, entry 3).48 Here, a ratio of [urea]0:[CDCA]0 of 1:25 was chosen as studies have shown that the urea catalyst is less active than the CrSalen.46 Again, the reaction was found to proceed readily at [St-b-4VGEP]0:[CDCA]0:[urea]0:[PPNCl]0 loadings of 10:140:5.6:5.6, albeit at a slower rate and with a slightly lower yield of star polymer obtained (Fig. 5). As expected from the higher catalyst loadings, absolute molecular weight was lower than for CrSalen (Mn,MALLS = 156100 g mol−1) while ĐM remained similar (1.27).
Given the low toxicity of dicyclohexyl urea and with potential biomedical applications in mind, the metal-free urea/PPNCl catalyst was chosen to carry out further study with the DEAA-b-4VGEP copolymer. The reaction proceeded as expected at [DEAA-b-4VGEP]0:[CDCA]0:[urea]0:[PPNCl]0 loadings of 10:170:6.8:6.8 (Table 1, entry 4, Fig. S5†). Good control was sustained in the ROCOP (ĐM 1.33) with high absolute molecular weights (Mn,MALLS 468200 g mol−1) obtained, demonstrating the versatility of the urea/PPNCL catalyst.
Selective degradation of the ester-linked core of the star polymer, star-St-b-4VGEP, was then attempted. We hypothesised that the ester-links would undergo hydrolysis whereas the vinyl polymers arms would remain intact, enabling controlled degradation of the core.39 Hydrolysis was attempted under acidic conditions using a 1:4 trifluoroacetic acid (TFA):H2O mixture. At room temperature, no reactivity was observed in a DCM solution of the star, up to 120 h, as confirmed by SEC analysis. However, satisfyingly, at 100 °C in toluene, near complete degradation of the core was possible after 48 h to form a polymer of Mn,SEC (ĐM) = 11200 (1.38) (Fig. 6 and S14†).
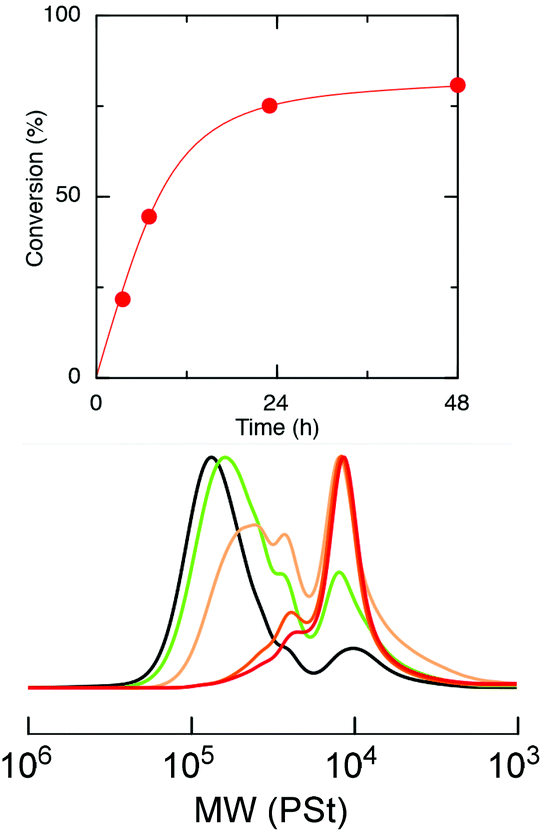 | ||
| Fig. 6 (Top) conversion of star-St-b-4VGEP to hydrolysis products vs. time. Conversions were calculated by the relative integration of the SEC traces of the star-St-b-4VGEP to linear polymer by RI detection. (Bottom) RI vs. Mn (PSt) for the acid hydrolysis of star-St-b-4VGEP at 100 °C (black = crude star polymer, green = 3.5 h aliquot, orange = 7 h aliquot, deep orange = 23 h aliquot and red = 48 h aliquot). | ||
Thermogravimetric analysis (TGA) showed that all novel-polymers had a temperature of 5% mass loss (Td5) of over 300 °C. The formation of the star polymers resulted in a slight decrease in Td5 which may be attributed to the incorporation of the thermally less-stable C–O bonds, formed as part of the ester linkages in the polymeric stars. Similarly, the homopolymer, P4VGEP, has a lower Td5 than both the block copolymers, St-b-4VGEP and DEAA-b-4VGEP, due to the greater density of epoxide-functionalised pendant arms (Fig. S13†).
Differential scanning calorimetry (DSC) indicated that all synthesised polymers were amorphous with Tg's ranging from 67–114 °C (Fig. 7). No microphase separation was observed between the styrene and 4VGEP or the DEAA and 4VGEP blocks copolymers. For both polymers, incorporation of the 4VGEP block resulted in a slight decrease in the polymers’ Tg (St-b-4VGEP −5 °C, DEAA-b-4VGEP −3 °C). Formation of the star polymers resulted in greater increase in Tg, consistent with the incorporation of a rigid cyclohexyl unit in the polymer (star St-b-4VGEP + 15 °C, star DEAA-b-4VGEP = +23 °C).
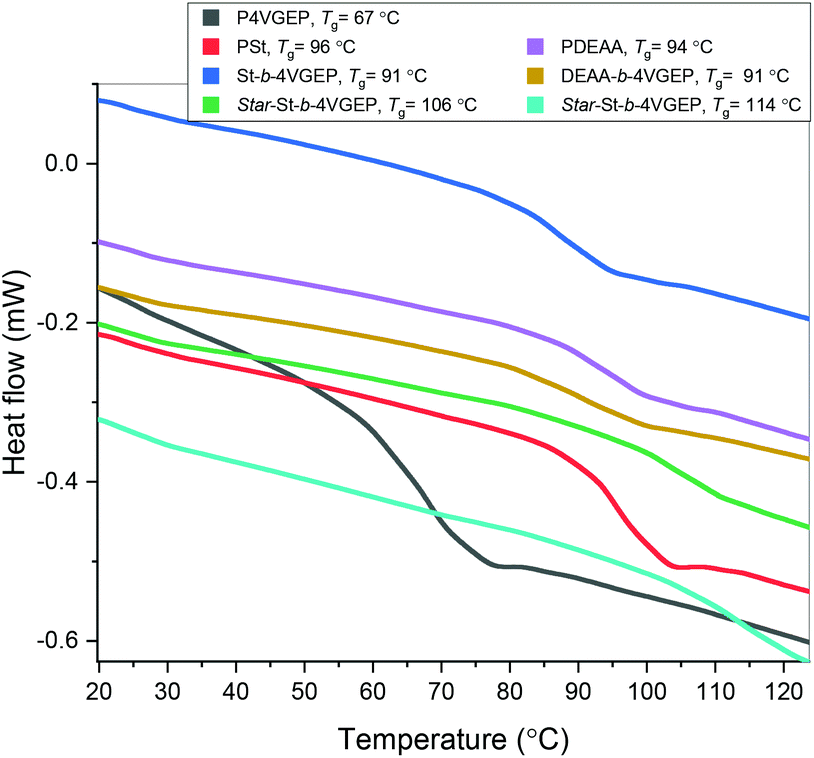 | ||
| Fig. 7 DSC thermograms of all polymers isolated in this study: P4VGEP (dark grey, Mn,SEC = 6900), PSt (red, Mn,SEC = 9800), St-b-4VGEP (blue, Mn,SEC = 11700), star-St-b-4VGEP (green, Mn,SEC = 116800), PDEAA (lilac, Mn,SEC = 13900), DEAA-b-4VGEP (gold, Mn,SEC = 18500) and star-St-b-4VGEP (cyan, Mn,sec = 91900). | ||
Conclusions
In conclusion, a novel epoxide functionalised styrene-type polymer has been synthesised from a ferulic acid and a glycerol derivative. Synthesis of PSt and PDEAA macro-RAFT agents followed by copolymerisation with 4VGEP formed the block copolymers, St-b-4VGEP and DEAA-b-4VGEP. Star polymer synthesis was then attempted by epoxide/anhydride ROCOP using a CrSalen/PPNCl and a urea/PPNCl catalyst. Initial study with St-b-4VGEP showed the ROCOP proceeded readily for both systems. The urea catalyst was then applied in the star polymer synthesis using the block copolymer, DEAA-b-4VEP, forming the star polymer, star DEAA-b-4VEGP in good yield. Finally, controlled degradation of the star polymer, star St-b4VGEP, was demonstrated under acidic conditions at 100 °C. All novel polymers were fully characterized by TGA and DSC. Future studies will focus on using 4VGEP to synthesise polymers with dynamic cross-links to enable the formation of materials with self-healing properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) Summer program fellowship (SP19105) granted to T. M. M. A. B. acknowledges the Royal Society (UF/160021 fellowship) for research funding.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 124684 CrossRef PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- G. L. Gregory, E. M. Lopez-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852–865 CrossRef CAS PubMed.
- C. Yang, S. Q. Liu, S. Venkataraman, S. J. Gao, X. Ke, X. T. Chia, J. L. Hedrick and Y. Y. Yang, J. Controlled Release, 2015, 208, 93–105 CrossRef CAS PubMed.
- Y. K. Chong, I. Zainol, C. H. Ng and I. H. Ooi, J. Polym. Res., 2019, 26, 79 CrossRef.
- T. K. Georgiou, Polym. Int., 2014, 63, 1130–1133 CrossRef CAS.
- Y. Wang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2012, 13, 2585–2593 CrossRef CAS PubMed.
- K.-I. Fukukawa, R. Rossin, A. Hagooly, E. D. Pressly, J. N. Hunt, B. W. Messmore, K. L. Wooley, M. J. Welch and C. J. Hawker, Biomacromolecules, 2008, 9, 1329–1339 CrossRef CAS PubMed.
- T. R. Bagby, S. Duan, S. Cai, Q. Yang, S. Thati, C. Berkland, D. J. Aires and M. Laird Forrest, Eur. J. Pharm. Sci., 2012, 47, 287–294 CrossRef CAS PubMed.
- V. Rodionov, H. Gao, S. Scroggins, D. A. Unruh, A.-J. Avestro and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 2570–2572 CrossRef CAS PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- Q. Qiu, G. Liu and Z. An, Chem. Commun., 2011, 47, 12685–12687 RSC.
- M. Uchiyama, K. Satoh, T. G. McKenzie, Q. Fu, G. G. Qiao and M. Kamigaito, Polym. Chem., 2017, 8, 5972–5981 RSC.
- N. Usuki, H. Okura, K. Satoh and M. Kamigaito, J. Polym. Sci., Polym. Chem., 2018, 56, 1123–1127 CrossRef CAS.
- H. Takeshima, K. Satoh and M. Kamigaito, Macromolecules, 2017, 50, 4206–4216 CrossRef CAS.
- H. Takeshima, K. Satoh and M. Kamigaito, Polym. Chem., 2019, 10, 1192–1201 RSC.
- H. Takeshima, K. Satoh and M. Kamigaito, Polymers, 2018, 10, 1404 CrossRef PubMed.
- W. S. J. Li, V. Ladmiral, H. Takeshima, K. Satoh, M. Kamigaito, M. Semsarilar, C. Negrell, P. Lacroix-Desmazes and S. Caillol, Polym. Chem., 2019, 10, 3116–3126 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- Y.-Y. Zhang, G.-W. Yang and G.-P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS.
- H.-J. Zhou, G.-W. Yang, Y.-Y. Zhang, Z.-K. Xu and G.-P. Wu, ACS Nano, 2018, 12, 11471–11480 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, X. Zhou, Z. Xue, X. Wang, X. Xie and R. Poli, Angew. Chem., Int. Ed., 2019, 58, 14311–14318 CrossRef CAS PubMed.
- A. Denk, S. Kernbichl, A. Schaffer, M. Kränzlein, T. Pehl and B. Rieger, ACS Macro Lett., 2020, 9, 571–575 CrossRef CAS.
- Y. Wang, Y. Zhao, S. Zhu, X. Zhou, J. Xu, X. Xie and R. Poli, Angew. Chem., 2020, 59, 5988–5994 CrossRef CAS PubMed.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef CAS.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef.
- D. C. McLeod and N. V. Tsarevsky, J. Polym. Sci., Polym. Chem., 2016, 54, 1132–1144 CrossRef CAS.
- D. C. McLeod and N. V. Tsarevsky, Polym. Int., 2014, 63, 868–875 CrossRef CAS.
- S. R. Woodruff, B. J. Davis and N. V. Tsarevsky, Macromol. Rapid Commun., 2014, 35, 186–192 CrossRef CAS PubMed.
- D. C. McLeod and N. V. Tsarevsky, Macromolecules, 2016, 49, 1135–1142 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 9018–9027 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, J. Polym. Sci., Polym. Chem., 2009, 47, 1485–1498 CrossRef CAS.
- E. Zago, E. Dubreucq, J. Lecomte, P. Villeneuve, F. Fine, H. Fulcrand and C. Aouf, New J. Chem., 2016, 40, 7701–7710 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- H. K. Hall, P. Nogues, J. W. Rhoades, R. C. Sentman and M. Detar, J. Org. Chem., 1982, 47, 1451–1455 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- T. Stosser, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 9 CrossRef PubMed.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical suppliers, purification of starting materials, instrumentation, synthetic procedures, characterisation of 4VGEP, 1H and 13C NMR spectra of 4VGEP and polymers, SEC traces, DSC thermograms and TGA thermograms. See DOI: 10.1039/d0py00878h |
| This journal is © The Royal Society of Chemistry 2020 |