
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh molar mass cyclic poly(L-lactide) obtained by means of neat tin(II) 2-ethylhexanoate†
Hans R.
Kricheldorf
*a and
Steffen M.
Weidner
 b
b
aInstitut für Technische und Makromolekulare Chemie, Bundesstrasse 45, D-20246 Hamburg, Germany. E-mail: hrkricheldorf@aol.de
bBAM, Federal Institute for Material Research and Testing, Richard-Willstätter-Strasse 11, D-12489 Berlin, Germany
First published on 16th July 2020
Abstract
L-Lactide was polymerized in bulk at 120, 140, 160 and 180 °C with neat tin(II) 2-ethylhexanoate (SnOct2) as the catalyst. At 180 °C the Lac/Cat ratio was varied from 25/1 up to 8000/1 and at 160 °C from 25/1 up to 6000/1. The vast majority of the resulting polylactides consist of cycles in combination with a small fraction of linear chains having one octanoate and one COOH end group. The linear chains almost vanished at high Lac/Cat ratios, as evidenced by MALDI-TOF mass spectrometry and measurements of intrinsic viscosities and dn/dc values. At Lac/Cat ratios <1000/1 the number average molar masses (Mn) are far higher than expected for stoichiometic initiation, and above 400/1 the molar masses vary relatively little with the Lac/Cat ratio. At 180 °C slight discoloration even at short times and degradation of the molar masses were observed, but at 160 °C or below colorless products with weight average molar masses (Mw) up to 310![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were obtained. The formation of high molar mass cyclic polylactides is explained by a ROPPOC (Ring-Opening Polymerizatiom with simultaneous Polycondensation) mechanism with intermediate formation of linear chains having one Sn–O–CH end group and one mixed anhydride end group. Additional experiments with tin(II)acetate as the catalyst confirm this interpretation. These findings together with the detection of several transesterification mechanisms confirm the previous critique of the Jacobson–Stockmayer theory.
000 g mol−1 were obtained. The formation of high molar mass cyclic polylactides is explained by a ROPPOC (Ring-Opening Polymerizatiom with simultaneous Polycondensation) mechanism with intermediate formation of linear chains having one Sn–O–CH end group and one mixed anhydride end group. Additional experiments with tin(II)acetate as the catalyst confirm this interpretation. These findings together with the detection of several transesterification mechanisms confirm the previous critique of the Jacobson–Stockmayer theory.
Introduction
In addition to certain derivatives of starch or cellulose, polylactides are the most widely used and produced biosourced polymer.1–5 Polylactides have the additional advantage of being hydrolytically degradable without needing the presence of microorganisms, as is typical of composting. Since the pollution of nature with “immortal” plastics rapidly increases, their substitution by biosourced, hydrolytically degradable polymers is an important contribution to the stabilization of climate and reduction of environmental pollution, even when this substitution cannot reach the level of 100%. In addition to the substitution of immortal plastics homo- and copolymers of lactic acid have found numerous applications in medicine and pharmacy, beginning with their application as resorbable medical sutures.6–9 At least seven chemical companies have meanwhile established a technical production of poly(L-lactide) with a production of more than 500000 t a−1, and their number is increasing.
Almost all homo- and copolyesters based on lactide are technically produced by ring-opening polymerization (ROP) of lactide using tin(II) 2-ethylhexanoate (SnOct2) as the catalyst. The characteristics of this catalyst are high effectiveness, low costs and stability on storage. However, it is cytotoxic like all soluble tin compounds, which means that it is poisonous for almost all living organisms, for fungi, microbes and higher animals, but its toxicity for humans is rather low when compared to that of organotin compounds. Therefore, it has been admitted as a food preservative by the American FDA. Hence, a large number of publications dealing with SnOct2 catalyzed ROPs of lactide appeared after 1972, and ref. 10–39 do not claim to be a complete list of such studies. It is known for almost four decades that the catalytic activity of SnOct2 in the ROPs of lactides and lactones is slightly enhanced by the addition of alcohols.39 Because of its low toxicity, isopropanol is the most widely used cocatalyst/initiator for the technical production of polylactides. Addition of an alcohol, which is incorporated as the ester end group in the lactide chain, has the additional benefit that it allows for the control of the average molar mass via the lactide/alcohol ratio. At low temperatures such ROPs have the character of living polymerizations, whereas under conditions of technical production the formation of cycles by backbiting and other transesterification reactions complicate the course of the polymerization.30,31,37
This situation has the consequence that relatively little is known about ROPs of lactide catalyzed by neat SnOct2. The first study in this direction was published by Sinclair and Gynn in 1972.9 That publication and most later papers reporting on ROPs of lactides catalyzed by neat SnOct2 served preparative purposes and did not provide any information on end groups, topology and polymerization mechanisms. The first hypothesis about the initiation mechanism was published by Pennings and co-workers in 1987 and 1992.11,14 In the first paper the formation of a Sn–O–Sn bond by reaction of SnOct2 with traces of water was assumed to be the true catalytic species, but neither an initiation nor a propagation step was outlined. The initiation step presented in Scheme 1 was formulated in the second paper, but once again neither propagation steps were formulated nor the formation of cyclics was discussed. Zhang et al. also postulated that the Sn–OH or Sn–OR groups resulting from the reactions of SnOct2 with traces of water or alcohols are the true initiators, but they did not formulate a polymerization mechanism.18 Dahlmann and Rafler postulated again that the true initiator is a Sn–OH or Sn–OR group resulting from the reaction of SnOct2 with moisture or alcohols,10,12,13 but they also outlined insertion and propagation steps according to the coordination/insertion mechanism published before by Kricheldorf et al. (Scheme 2).40
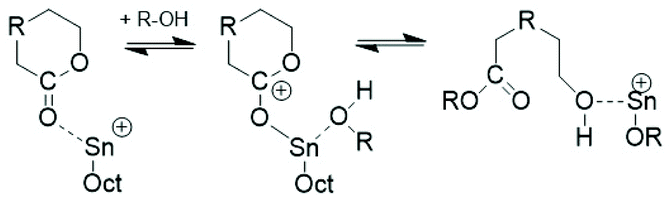 | ||
| Scheme 1 Hypothetical initiation mechanism proposed by Pennings and coworkers.14 | ||
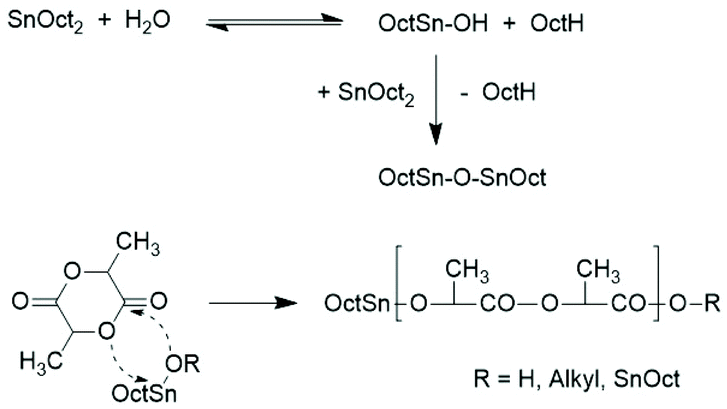 | ||
| Scheme 2 ROP mechanism proposed by Dahlmann and Rafler.10,12,13 | ||
Two mechanistic studies were contributed by Kricheldorf et al. in 1995 and 2000 with variation of the Lac/Cat ratio between 5/1 and 200/1.19,28 Polymerizations with and without addition of benzyl alcohol were compared at 120 °C. It was found by 1H NMR and 13C NMR spectroscopy for the lowest Lac/Cat ratios that approximately 10% of the octanoate groups added via the catalyst were incorporated into the polylactide chains as ester end groups. The reaction mechanism outlined in Scheme 3 was formulated as the hypothetical explanation of this result.
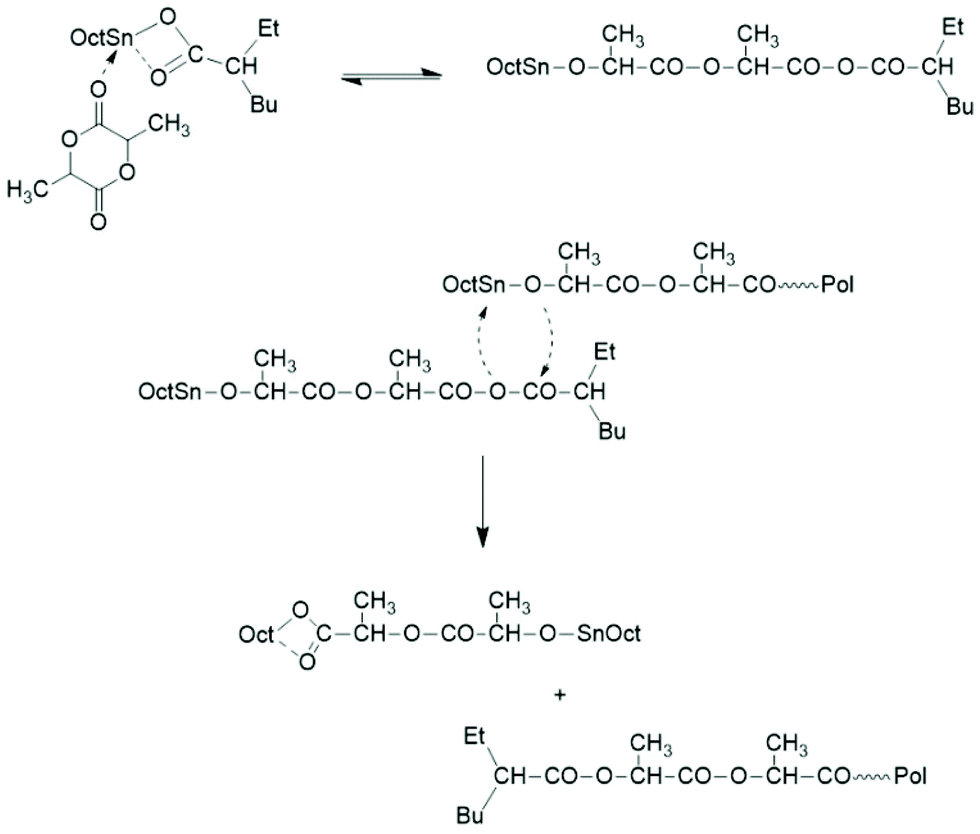 | ||
| Scheme 3 Initiation mechanism yielding a mixed anhydride group proposed by Kricheldorf et al.19,28 | ||
Intramolecular reactions of the mixed anhydride group formed by the initiation step were not considered and the formation of cyclic polylactides was not discussed. A further mechanistic study was contributed by Schwach et al. in 1997.21 The reaction product obtained at 140 °C from extremely low Lac/Cat ratios (1.1, 2.5 and 13.5) was characterized and oligomers having one octanoyl and one carboxylic acid end group were identified by 1H NMR spectroscopy. A cationic reaction mechanism was postulated (Scheme 4).
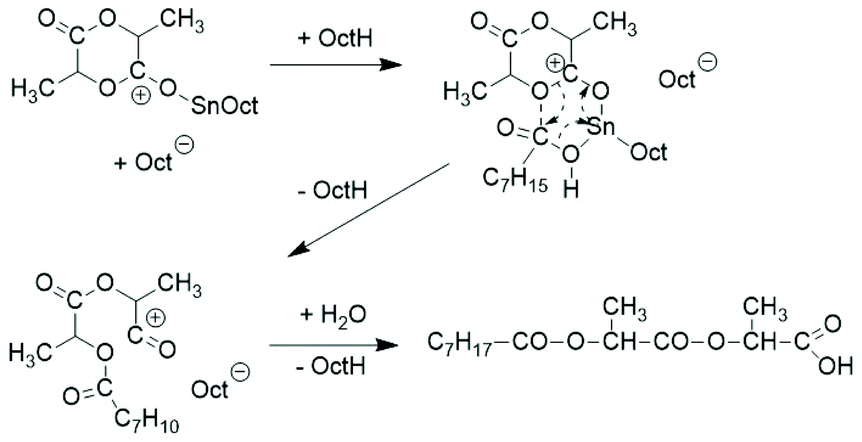 | ||
| Scheme 4 Cationic initiation mechanism proposed by Schwach et al.21 | ||
Two more papers dealing with neat SnOct2 were then published by Witzke et al. and by Shinno et al. in 1997.22,24 These studies were focused on kinetic aspects. The exclusive formation of linear chains was assumed, but not proven, and mechanistic aspects were not discussed. In two more recent papers focused on the preparative aspects of ROPs catalysed by neat SnOct2 or its combination with triphenylphosphine no information on the polymerization mechanism or topology is disclosed.32,33
In this context, the present study served three purposes. First, it should be revealed to what extent the molar mass depends on the Lac/Cat ratio and on the temperature. Second, a more reliable identification of end groups should be contributed. Third, it should be elucidated to what extent cycles are formed. Fourth, a polymerization mechanism explaining the experimental results should be formulated. Since SnOct2 is the most widely used catalyst for research purposes and technical production of polylactides, it seems to be worthwhile to shed more light on the catalytic properties of neat SnOct2.
Experimental
Materials
L-Lactide, a product of Corbion Purac, was kindly supplied by Thyssen-Uhde AG (Berlin). It was once recrystallized from toluene (99.89%, extra dry, ACROS Chemicals, Geel, Belgium). SnOct2 (purity >96%) was purchased from Alfa Aesar (Karlsruhe, Germany), because to the best knowledge of the authors, this product had the highest purity of all commercial SnOct2 products. When a 50 g sample of SnOct2 was subject to azeotropic drying with toluene, no water was observed. Hence, a solution of commercial SnOct2 in toluene was used for all experiments without additional purification. 0.5 M, 0.2 M solutions of SnOct2 in pure and dry toluene were prepared and used for an exact dosage of the catalyst at high Lac/Cat ratios. Tin(II) acetate (SnAc2) was also purchased from Alfa Aesar and dried in a vacuum at 40 °C in the presence of solid KOH to remove traces of acetic acid. Purapol L105, a linear poly(L-lactide) initiated with isopropanol and containing approx. 2 weight% of cyclic oligomers, was purchased from Corbion Purac, whereas the linear poly(L-lactide) NW3251D was purchased from Nature Works. Ligroin, kindly supplied by BASF, was used as received.Polymerizations with SnOct2 as the catalyst (Tables 1–3)
| Exp. no. | Lac/Cat | Time (h) | M n (theor.)a | M n (found)b | M w (found)b | Đ |
|---|---|---|---|---|---|---|
| a Calculated for 100% conversion by assuming that all octanoate residues added to the reaction mixture in the form of SnOct2 have formed an ester end group. b Determined by PS calibrated SEC measurements of the virgin samples. | ||||||
| 1 | 25/1 | 0.5 | 1900 | 11000 |
21000 |
1.9 |
| 1 | 50/1 | 0.5 | 3750 | 17500 |
45000 |
2.6 |
| 2 | 100/1 | 0.5 | 7500 | 30000 |
77000 |
2.6 |
| 3 | 200/1 | 0.5 | 15000 |
39000 |
108000 |
2.8 |
| 4 | 400/1 | 0.5 | 29000 |
79000 |
197000 |
2.5 |
| 5 | 600/1 | 0.5 | 43500 |
99000 |
220000 |
2.2 |
| 6 | 1000/1 | 1.0 | 72200 |
53000 |
157000 |
3.0 |
| 7 | 2000/1 | 1.0 | 144300 |
55000 |
160000 |
2.9 |
| 8 | 4000/1 | 2.0 | 288500 |
47000 |
136000 |
2.9 |
| 9 | 6000/1 | 2.0 | — | 49000 |
126000 |
2.6 |
| 10 | 8000/1 | 2.0 | — | 52000 |
132000 |
2.5 |
| Exp. no. | Lac/Cat | Time (h) | M n (theor.)a | M n | M w | Đ |
|---|---|---|---|---|---|---|
| a Calculated for 100% conversion assuming that all octanoate residues added to the reaction mixtures in the form of SnOct2 have formed ester end groups. | ||||||
| 1 | 25/1 | 0.5 | 1900 | 13000 |
25000 |
1.9 |
| 2 | 50/1 | 0.5 | 3600 | 34500 |
63000 |
1.8 |
| 3 | 100/1 | 0.5 | 7500 | 54000 |
106000 |
2.0 |
| 4 | 200/1 | 0.5 | 15000 |
94000 |
170000 |
1.8 |
| 5A | 400/1 | 1.0 | 29000 |
137000 |
275000 |
2.0 |
| 5B | 400/1 | 8.0 | — | 54000 |
133000 |
2.5 |
| 6A | 600/1 | 1.0 | 43500 |
135000 |
310000 |
2.3 |
| 6B | 600/1 | 8.0 | — | 71000 |
155000 |
2.2 |
| 7A | 1000/1 | 1.0 | 72200 |
126000 |
297000 |
2.4 |
| 7B | 1000/1 | 8.0 | — | 80000 |
171000 |
2.1 |
| 8 | 2000/1 | 1.0 | 144300 |
123000 |
310000 |
2.5 |
| 9 | 4000/1 | 1.0 | 288000 |
165000 |
315000 |
1.9 |
| 10 | 6000/1 | 2.0 | 432000 |
115000 |
250000 |
2.2 |
| Exp. no. | Lac/Cat | Temp. (°C) | Time (h) | M n | M w | Đ |
|---|---|---|---|---|---|---|
| 1 | 1000/1 | 140 | 0.5 | 107000 |
195000 |
1.8 |
| 2 | 1000/1 | 140 | 1 | 105000 |
212000 |
2.0 |
| 3 | 1000/1 | 140 | 2 | 103000 |
235000 |
2.3 |
| 4 | 1000/1 | 140 | 4 | 90000 |
217000 |
2.4 |
| 5 | 1000/1 | 140 | 9 | 84000 |
205000 |
2.4 |
| 6 | 1000/1 | 140 | 24 | 79000 |
190000 |
2.4 |
| 7 | 1000/1 | 120 | 4 | 103000 |
233000 |
2.3 |
| 8 | 1000/1 | 120 | 9 | 54000 |
152000 |
2.8 |
Polymerizations catalyzed with Sn(II) acetate (no. 1, Table 4)
Sn(II)acetate (2.0 mmol) was weighed into a flame-dried 50 mL Erlenmeyer flask; L-lactide (50 mmol) and a magnetic bar were added under a blanket of argon. The reaction vessel was immersed in an oil bath thermostated at 160 °C. After 30 min part of the polylactide was removed from the hot melt with a spatula and pincer. Analogous experiments were performed with smaller amounts of SnAc2. Conversions around 97% corresponding to the thermodynamic equilibrium were found in all experiments.| Exp. no. | Reaction conditions | M n | M w | Cycles |
|---|---|---|---|---|
| a Due to a Lac/initiator ratio of 100/1 and conversion around 99%, the degree of polymerization was ∼100. | ||||
| 1 | ROP initiated with ethyl lactate at 120 °C/4 h followed by acetylation (Lc) | 22000 |
49000 |
− |
| 2 | Lc treated with SnOct2 (50/1) at 180 °C/0.5 h in bulk | 7500 | 17000 |
+ |
| 3 | Lc treated with SnOct2 (50/1) at 160 °C/1 h in 1,2-dichlorobenzene | 8500 | 20000 |
+ |
| 4 | Lc treated with SnOct2 (50/1) at 140 °C/1 h in 1,2-dichlorobenzene | 10500 |
23000 |
+ |
Syntheses and reactions of linear poly(L-lactide)s (Table 5)
000; Mw = 43000 g mol−1.
| Exp. no. | Lac/Cat | Time (h) | M n | M w | Đ |
|---|---|---|---|---|---|
| 1 | 25/1 | 0.5 | 7500 | 17700 |
2.2 |
| 2 | 50/1 | 0.5 | 10000 |
30000 |
3.3 |
| 3 | 100/1 | 0.5 | 24500 |
64500 |
2.6 |
| 4 | 200/1 | 1.0 | 55500 |
133000 |
2.4 |
| 5 | 400/1 | 1.0 | 82000 |
200000 |
2.4 |
| 6 | 600/1 | 1.0 | 108000 |
247000 |
2.3 |
| 7 | 1000/1 | 1.0 | 165000 |
315000 |
1.9 |
| 8 | 2000/1 | 1.0 | 140000 |
250000 |
1.8 |
000, Mw = 49000 g mol−1. The measured Mn values of 21000/22000 g mol−1 agree well with the theoretical Mn of 14300 g mol−1, because calibration of SEC measurements with polystyrene standards results in an overestimation of linear polylactides by 50–55% corresponding to a correction factor of 0.67 (±0.01).41
Methods
400 MHz 1H NMR spectra were recorded on a Bruker Avance 400 FT spectrometer in 5 mm sample tubes. 150 MHz 13NMR spectra were recorded with a Bruker Avance III 600 FT spectrometer in 5 mm sample tubes. For both kinds of NMR measurements CDCl3 containing TMS served as the solvent.An AutoflexMax MALDI mass spectrometer (Bruker Daltonik, Bremen, Germany) equipped with a Nd-YAG laser (355 nm) was used for mass spectrometric analysis. The laser repetition rate was 2000 Hz. Spectra were always recorded in the linear instrument mode. Premixed solutions of 50 μL matrix (DCTB – trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile, 10 mg mL−1, chloroform), 20 μL of dissolved samples (5 mg mL−1, chloroform) and 5 μL of potassium trifluoroacetate (KTFA 2 mg ml−1) were prepared and finally, 1 μL of the mixed solution was deposited on a stainless-steel target. The manufactures software (FlexControl and FlexAnalysis) was used for data recording and data evaluation. Typically, 10000 single spectra recorded on 5 different positions were accumulated for one spectrum.
For SEC measurements a modular system consisting of an isocratic pump (Chromophor C 1130) (1 mL min−1), a column oven (40 °C) and a manual injection valve (Rheodyne) was used. The injected sample volume was 100 μL. The sample concentration was typically 2–4 mg mL−1. A triple detector combination consisting of a refractive index detector RI-501 (Shodex), a multi-angle laser light scattering detector DAWN and a viscometer Viscostar (both Wyatt Germany, Dernbach) was used. Clarity software (GPC extension, DataApex) was used for instrument control and molar mass calculation, whereas ASTRA 6 (Wyatt) was used to perform the MHS calculations. The calibration of SEC was performed using commercially available polystyrene standard sets (Polymer Standards Service – PSS, Mainz, Germany). Due to a “spike” in the baseline around 600–800 g mol−1, the number average (Mn) and weight average (Mw) molar masses and the dispersities Đ were calculated from 1100 g mol−1 onwards, and not as in our previous publications usually from 300 g mol−1 onwards. Therefore, not all oligomers were included in the calculation. This difference has only little influence on Mw, but the Mn values may be 30–60% too high, and the dispersities correspondingly underestimated.
Results and discussion
Polymerizations catalyzed by SnOct2
The SnOct2 used throughout this work was purchased from Alfa Aesar, because it had the highest purity (>96%) among the commercial products the authors have found. It was dried by azeotropic distillation with toluene, but water was not detectable. It certainly contained a small amount of free 2-ethylhexanoic acid. Yet, when the experiments no. 6 of Table 1 and no. 7A of Table 2 were repeated with a distilled SnOct2 no significant difference was detectable. Therefore, all experiments of this work were conducted with the commercial SnOct2 as received.A first series of polymerizations was performed at 180 °C, a temperature immediately above the normal “melting point” and the highest temperature used for the technical production. The Lac/Cat ratio was varied to reveal its influence on the molar mass and on the topology of the resulting polylactides. With Lac/Cat ratios <1000/1 the viscosity increased rapidly within 5 min and the polymerization was stopped after 0.5 h. With the highest Lac/Cat ratios of 6000/1 and 8000/1 a high viscosity was achieved after approx. 15 min and these polymerizations were worked up after 2 h, but a duration of 1 h might suffice for maximum molar masses. In all cases the virgin reaction products were characterized by MALDI-TOF mass spectrometry and by SEC measurements including Mark–Houwing–Sakurada (MHS) measurements. These measurements were performed with triple detection and allowed for comparison of the intrinsic viscosities of the polymerization products with a commercial linear standard sample (Purapol L105).
The SEC data revealed that the molar masses increased slowly with the Lac/Cat ratio up to 600/1, in a series of polymerizations that had an identical reaction time of 0.5 h. The molar masses were lower for higher Lac/Cat ratios, but the reaction times were longer, suggesting that these results were influenced by thermal degradation and not by the Lac/Cat ratio. This conclusion was supported by the following findings. First, the molar masses were significantly higher when the temperature was lowered to 160 °C (A-experiments in Table 2). Second, the polylactides prepared at 160 °C were colourless, whereas the polylactides polymerized at 180 °C had a yellowish colour at short reaction times (0.5 h) and an orange colour at longer times. Third, the MALDI-TOF spectra of the 180 °C products displayed weak signals at masses ±16 Da relative to the masses of cycles (see discussion in the next section), whereas these peaks were barely detectable in most samples, when the reaction temperature was lowered to 160 °C, 140 °C or below.
The MHS measurements of the samples prepared with Lac/Cat ratios of 400/1 or higher evidenced that the vast majority of the reaction products had a cyclic topology in the mass range from 50000 to 500000 Da regardless of the temperature (Fig. 1 and Fig. S1 in the ESI†). These measurements are not highly accurate and do not allow for a distinction between 95 and 99% cyclization. The intrinsic viscosities showed values around 65–70% of the intrinsic viscosity measured for a commercial linear polylactide (Purapol L105) under identical conditions. These lower intrinsic viscosities are a consequence of the lower hydrodynamic volume and viscosity ratios of 0.65–0.70/1 have also been reported by other research groups for cyclic polylactides42 and various other cyclic polymers.43,44 All these MHS measurements indicated that the poly(L-lactide)s prepared at 160 °C with Lac/Cat ratios of 400/1 and above mainly consist of cyclic polylactides. In order to confirm these unexpected results by an independent method, dn/dc ratios were determined for three samples (no. 7A, 8 and 10, Table 2). The higher coil density of cyclic polymers has the consequence that the change of the refractive increment with concentration is smaller than that in the case of linear polymers having similar molar masses. This method is again not accurate, but reliable and independent of any software. For the three samples identical values of 0.0216/0.0217 were obtained, and these values agreed with the dn/dc of polylactides prepared by REP with cyclic tin catalysts. In contrast, a dn/dc of 0.0259/0.0260 was determined for the linear commercial polylactides Purapol L105 and NW 3251D.
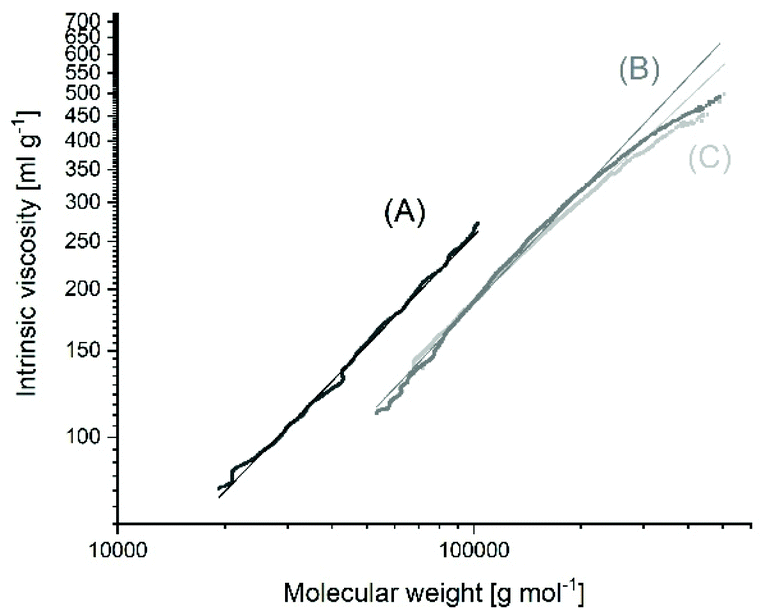 | ||
| Fig. 1 Intrinsic viscosity (MHS) measurements of SnOct2-catalyzed poly(L-lactide)s: (A) commercial, linear Purapol L105 initiated with isopropanol, (B) prepared at 160 °C with Lac/Cat = 2000/1 (no. 8, Table 2), (C) prepared at 160 °C with Lac/Cat = 6000/1 (no. 10, Table 2). | ||
One more interesting aspect of the A – experiments at 160 °C concerns the SEC measurements which demonstrate that the molar masses depend were little on the Lac/Cat ratio (factor 1.2), when the Lac/Cat ratio was varied from 400/1 to 4000/1 (factor 10). Such a result is characteristic when the catalyst is a true catalyst over the whole course of the polymerization and does not act as an initiator (although the first reaction step is of course an initiation step). This finding supports the polymerization mechanism discussed below in the section “mechanistic aspects”. This result also supports the afore-mentioned conclusion that the molar masses measured for the “180 °C samples” are affected by thermal degradation.
Polymerizations were also performed at 140 and 120 °C to study the influence of temperature (and time) on molar masses and topology (Table 3). The experiments listed in Table 3 gave the following results. At both temperatures Mw values above 220000 g mol−1 were obtained. The maximum values that can be achieved depend above all on the optimization of the reaction time, since the data compiled in Table 3 demonstrate that even at these relatively low temperatures the rapid chain growth process is followed by steady degradation. Therefore, in technical production a catalyst poison is added as soon as the desired molar mass has been achieved. Furthermore, sample no. 4A (Table 3) was subject to dn/dc (0.0216) and MHS measurements (Fig. S2†). The results confirmed that this sample almost completely consisted of cycles also in the high molar mass fraction. Hence, a perfect agreement with the corresponding data found for the 160 °C samples was obtained.
MALDI TOF mass spectrometry and NMR spectroscopy
The MALDI TOF mass spectra of all samples listed in Tables 1–3 and prepared with Lac/Cat ratios >100/1 looked nearly identical displaying peaks of odd and even cycles, as exemplarily illustrated in Fig. 2B and 3A. However, two less intense peak series with masses of 16–17 Da lower and higher than the mass of the cycles were detectable in the spectra of samples prepared with low Lac/Cat ratios (50/1, no. 2, Table 1 and Fig. 2A). These peaks almost vanished at higher Lac/Cat ratios (Fig. 2B and 3A). Yet these peaks became stronger at longer reaction times (Fig. 3B). These phenomena were observable at 180 and 160 °C. Therefore, it may be concluded that this peak indicates the beginning of thermal degradation favoured by a high concentration of the catalyst and longer reaction times.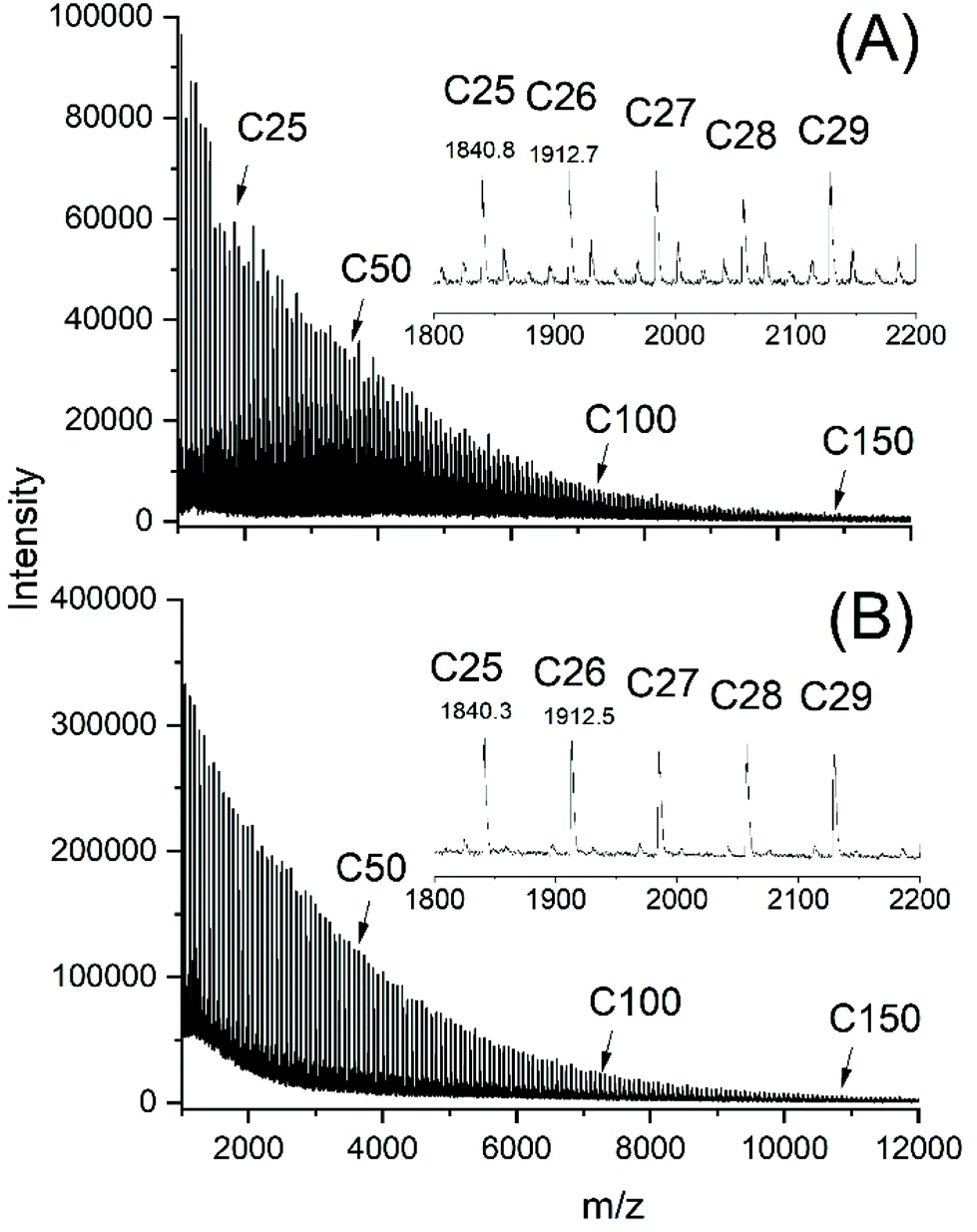 | ||
| Fig. 2 MALDI-TOF mass spectra of poly(L-lactide)s polymerized with SnOct2 at 180 °C: (A) Lac/Cat 50/1 (no. 2, Table 1), (B) Lac/Cat = 400/1 (no. 4, Table 1). | ||
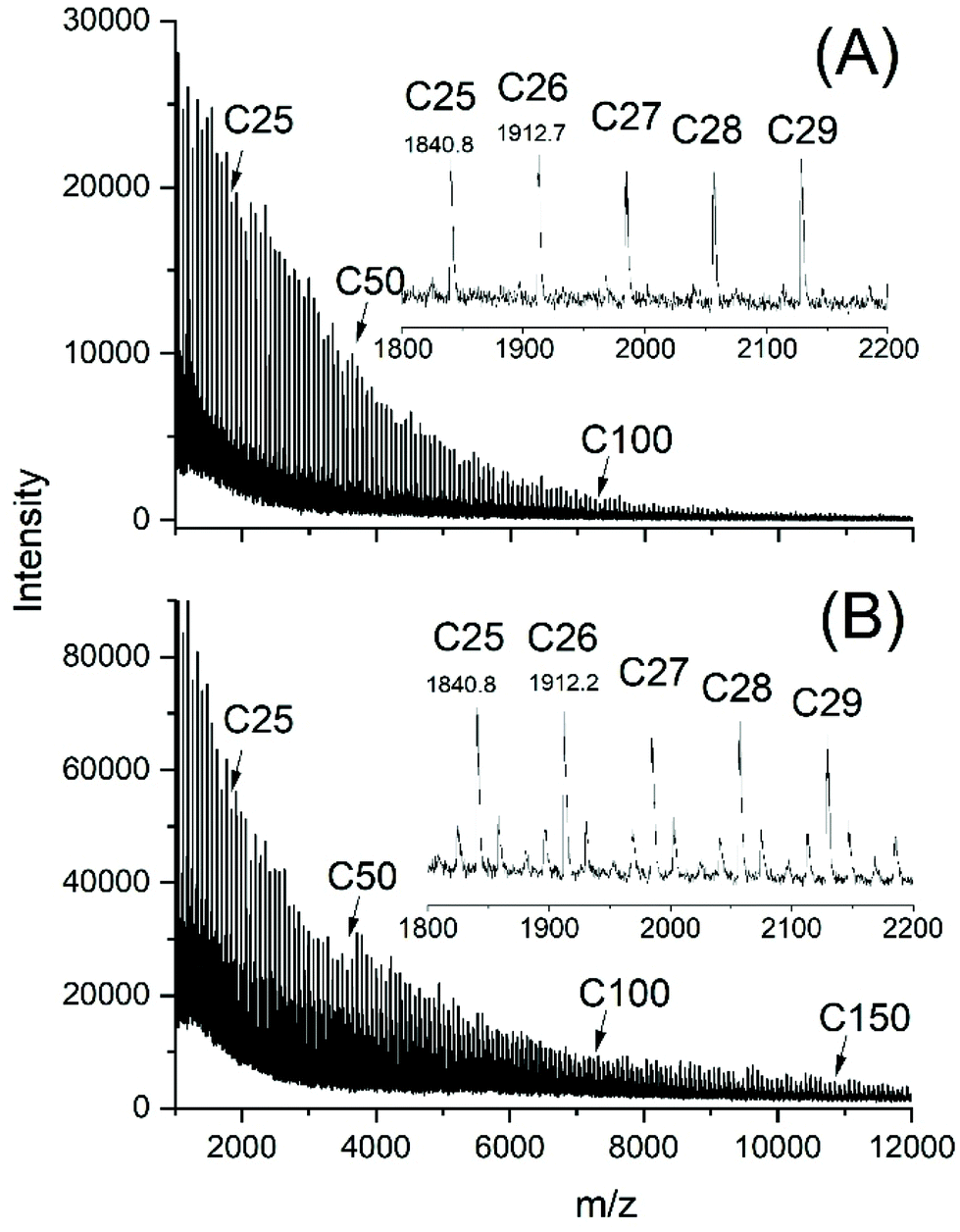 | ||
| Fig. 3 MALDI-TOF mass spectra of poly(L-lactide)s polymerized with SnOct2 at 160 °C: (A) Lac/Cat = 6000/1, 2 h (no. 10, Table 2), (B) Lac/Cat = 1000/1, 8 h (no. 6B, Table 2). | ||
Unfortunately, the interpretation of the MALDI-TOF mass spectra of polylactides prepared with SnOct2 as the catalyst is complicated by the fact that the mass of 2-ethyl hexanoic acid is identical to that of lactide. Therefore, the mass peaks representing the rings may also include the linear chains of the structure La (Scheme 5). However, when COOH end groups are present in significant quantities, they will form potassium (or sodium) salts when the MALDI sample targets were prepared with large amounts of dopants such as potassium trifluoroacetate or sodium iodide (La_K Scheme 5).
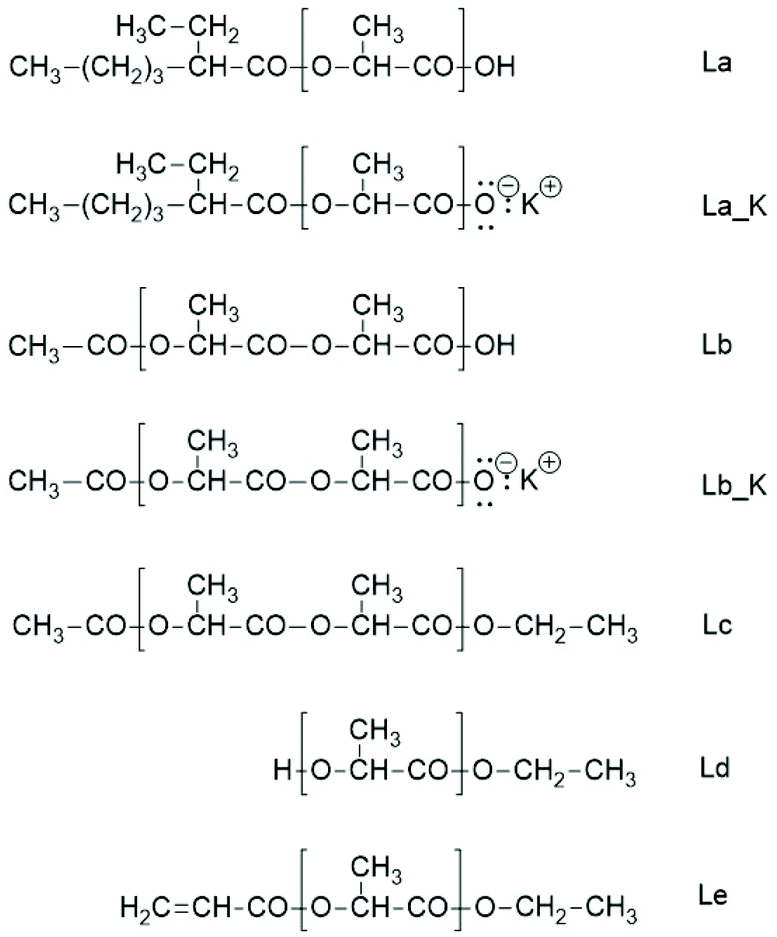 | ||
| Scheme 5 Structures and labels of linear polylactide chains discussed in this work. | ||
Hence, the MALDI TOF mass spectra show an additional peak with +38 Da relative to the mass peaks of the cycles. Yet, such peaks were absent, regardless how much dopant was added to the samples no. 1 or 2, Table 1. Therefore, it may be concluded that the molar fraction of La chains is so low (<10 mol%) that it was not detectable in the mass spectra.
This interpretation was confirmed using 13C NMR spectra. The 150 MHz spectrum presented in Fig. 4 revealed three interesting features. First, the symmetry of the CO signal (and of the CH signal) indicates the absence of racemization, because this signal is sensitive to the presence of L/D stereosequences. Secondly, it demonstrates the presence of six CH2 and CH3 signals of 2-ethyl hexanoate units, whereas CO signals of the COOH and ester end group in La chains are absent. Both signals are expected several ppm downfield of the CO backbone signal at 169.6 ppm. Furthermore, the CH-signal is absent. Considering that the magnetic coupling with 118Sn extremely broadens the CO2 and CH signal of SnOct2 (see Fig. S3†) in contrast to the spectrum of the 2-ethylhexanois acid (Fig. S4†), the 13C NMR spectrum presented in Fig. 4 looks like that of a mixture of cycles and unreacted SnOct2.
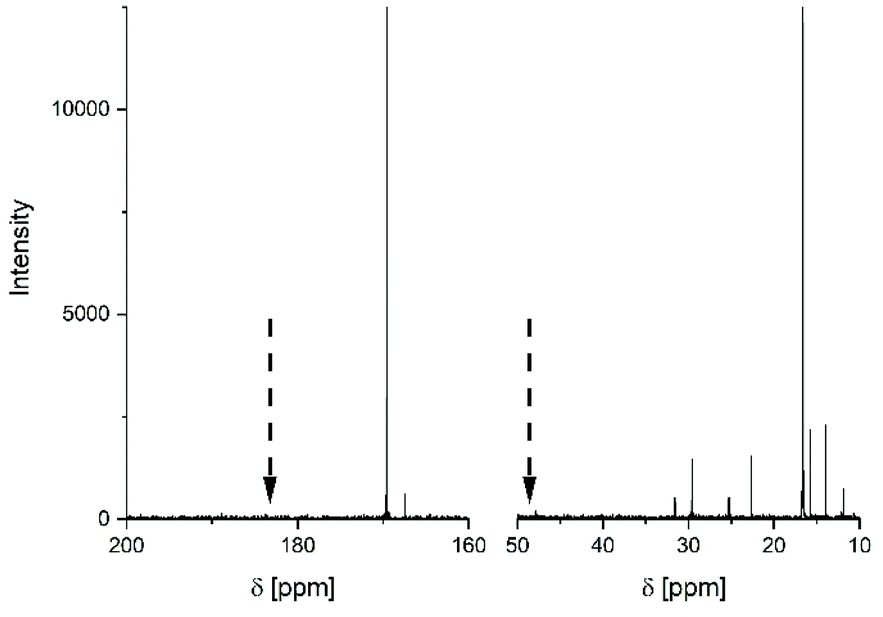 | ||
| Fig. 4 150 MHz 13C NMR spectrum of the poly(L-lactide) prepared at 160 °C with Lac/Cat = 25/1 (no. 1, Table 2). The arrows indicate the positions where CO and CH signals of 2-ethylhexanoate end groups are expected. | ||
This interpretation is supported by the 13C NMR spectrum obtained after precipitation of the polylactides no. 1 and 2, Table 1, into ligroin. After precipitation most of the SnOct2 should be removed in contrast to the 2-ethylhexanoate end groups. Since no octanoate signals were observable in the 13C NMR spectra of the precipitated samples the signals of the virgin samples must have originated from SnOct2 and not from end groups. Further confirmation of this conclusion comes from the work of Dahlmann and Rafler,10 who studied the reaction of equimolar amounts of D,L-lactide and SnOct2 at 120 °C. They obtained a polylactide having a Mn of 9900 g mol−1 and extracted 90% of unreacted SnOct2.
Weak signals of octanoate groups were detectable in the more sensitive 1H NMR spectra after two precipitations suggesting that a low percentage of La chains was present. This observation agrees well with the previous study of the first author. Nonetheless, both mass spectra and 13C NMR spectra agree in that the vast majority of the polymerization products have a cyclic topology, and this result is complementary to the information derived from MHS and dn/dc measurements.
With higher Lac/Cat ratios the percentage of La chains will further decrease, so that the “contamination” of the cycles with La chains will fall below a level of 1 mol or 1 weight%. This conclusion is confirmed by the MHS measurements of the “160 °C samples” (Table 2) mentioned above and illustrated by Fig. 1 and by Fig. S5–7 in the ESI.† In summary it may be said that polymerization of L-lactide catalyzed with neat SnOct2 at temperatures around 160 or 140 °C is a convenient and efficient method for the preparation of high molar mass cyclic poly(L-lactide)s free of racemization and without discoloration.
Mechanistic aspects
The basis for the formulation of the polymerization mechanism underlying the afore-mentioned results are experiments with the catalysts summarized in Scheme 645–47 plus analogous experiments with commercial tributyltin chloride and diphenyltin dichloride.48 These polymerizations are primarily ROPs proceeding via the coordination–insertion mechanism, which is characteristic of all covalent metal compounds.40 However, it turned out that increasing reactivity (electrophilicity) of the ester end groups formed in the initiation step ((1) in Scheme 7) favors end-to-end cyclization, so that the most reactive end groups yield high molar mass polylactides almost exclusively consisting of cycles. Cyclic oligomers are, of course, also formed by “back-biting” ((3) in Scheme 7). This course of these ROPs is confirmed by the detection of even-numbered linear chains having an activated ester end group. Hence, this class of polymerizations combines ring-opening polymerization with simultaneous polycondensation and end-to-end cyclization and has been labeled ROPPOC.
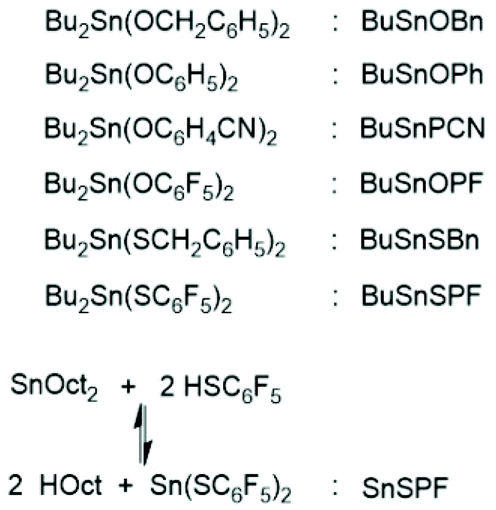 | ||
| Scheme 6 Structures and acronyms of catalysts used for ROPPOC of L-lactide.45–47 | ||
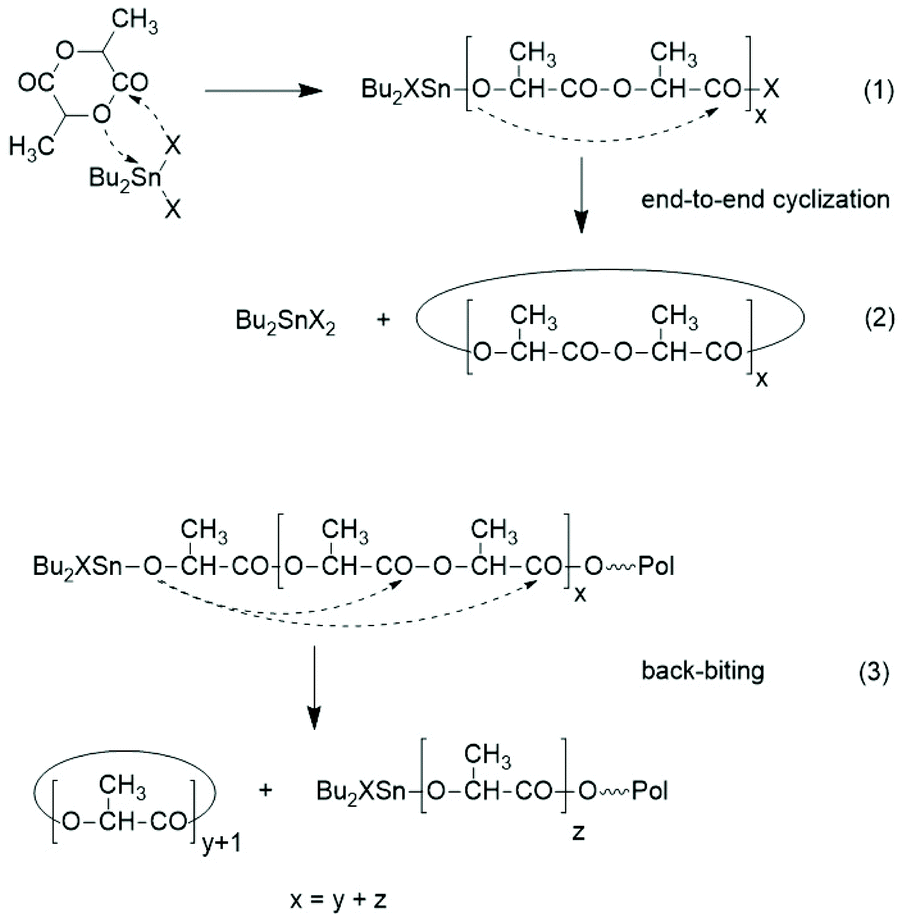 | ||
| Scheme 7 ROPPOC mechanism as explored and discussed in ref. 45–47. | ||
Considering these results, an analogous ROPPOC mechanism is the most likely explanation of the results obtained in this work for polymerizations catalyzed with neat SnOct2 (Scheme 8). The initiation step proceeds according to the coordination–insertion mechanism and yields a tin alkoxide end group. This group is several orders of magnitude more reactive (nucleophilic) than the Sn-carboxylate group, so that propagation is much faster than initiation and long chains are formed after a few minutes. Kinetic measurements by Witzke et al. support this hypothesis.22 A consequence of this scenario (kp ≫ ki) is that not all SnOct2 reacts at the same time. Hence, the molar concentration of the activated polylactide chains is very low and this fact favors intramolecular reactions according to the Ruggli-Ziegler dilution principle.
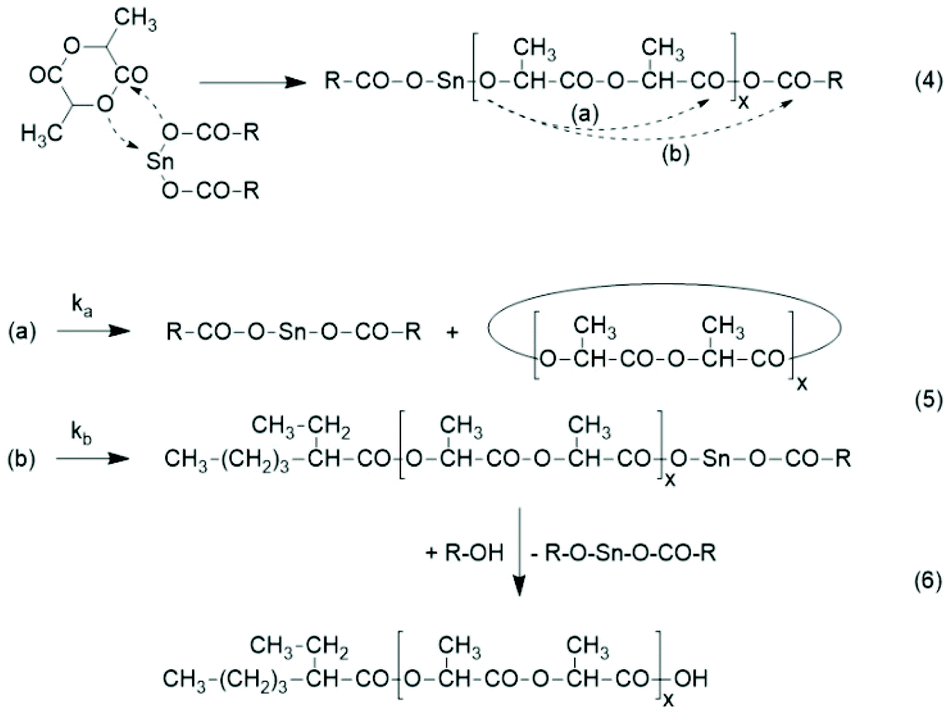 | ||
| Scheme 8 ROPPOC mechanism explaining the results obtained with neat SnOct2. | ||
The reaction of the nucleophilic end group with the mixed anhydride end group may proceed via route (a) and via (b) (Scheme 6). Route (a) is highly favored, because the electrophilicity of the lactyl CO-group is at least by a factor of 102 higher than that of the 2-ethylhexanoyl group. This conclusion is based on two facts. First, it is well known that polylactide is considerably more sensitive to hydrolysis than poly(ε-caprolactone). Second, an ester group of 2-ethyhexanoic acid is less sensitive to hydrolysis or any other nucleophilic attack than an ester group of ε-hydroxycaproic acid due to the branching in the α-position, which has a positive inductive effect and also causes steric hindrance for a nucleophilic attack. For the purpose of this work it does not matter, if the rate constant ka is 102, 103 or 104 higher than kb, because the entire polymerization process is not perfectly under kinetic control. The kinetic control certainly predominates during the first minutes but with increasing time equilibration reactions modify the structure and molar ratio of the reaction products. Nonetheless, ka ≫ kb is one reason, why the vast majority of the reaction products are cycles and only a small fraction of the 2-ethylhexanoyl residues is incorporated as ester end groups. The second reason is elimination of the catalyst by end-to-end cyclization ((a) in Scheme 4).
Concerning the equilibration reactions which influence the course of these ROPPCs, three mechanisms can be identified. At first, the formation of cyclic oligomers by back-biting should be mentioned as illustrated by eqn (3) in Scheme 7. This reaction, which establishes a ring-chain equilibration, is known from alcohol-initiated polymerizations at temperatures above 140 °C (ref. 37) and also from the previous ROPPOC experiments.45–48 The second type of equilibration process is an intermolecular transesterification with random chain scission (Scheme 9). This transesterification has recently been shown to proceed around and above 120 °C, and thus, is faster than backbiting.37
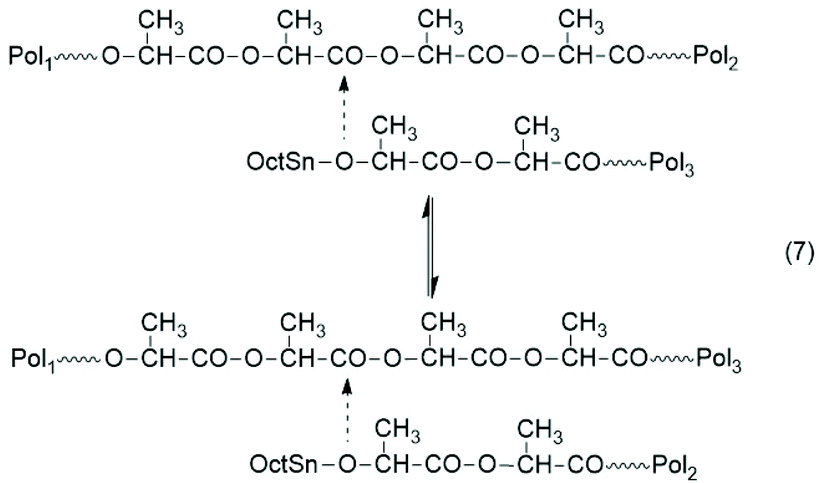 | ||
| Scheme 9 Reversible transesterification between linear chains. | ||
The third kind of transesterification is an attack of SnOct2 onto a polylactide chain (Scheme 10).
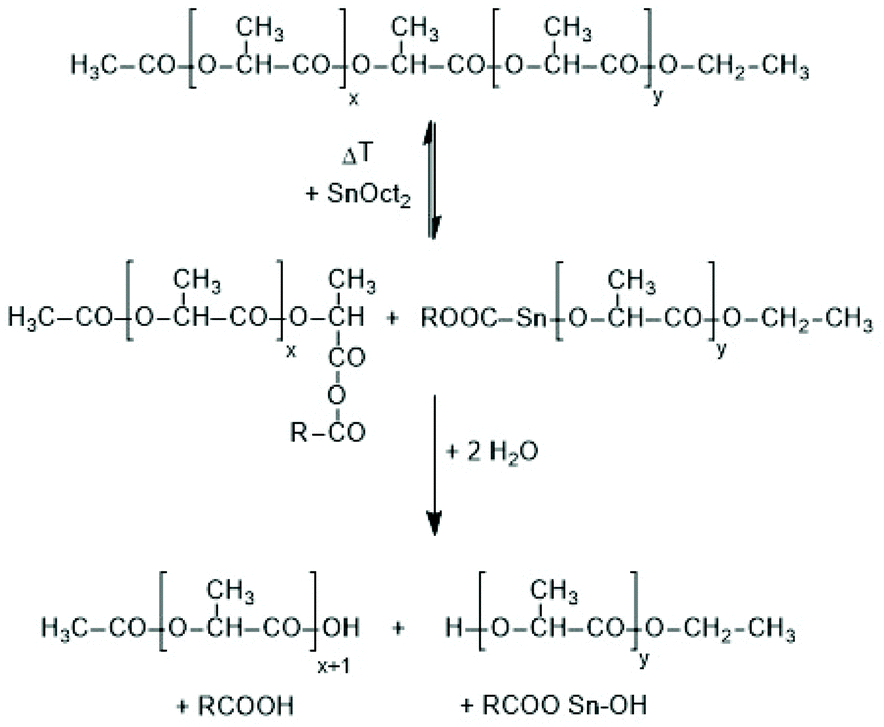 | ||
| Scheme 10 Model reaction of a reversible chain cleavage of polylactides by SnOct2 followed by work up in the presence of moisture. | ||
Experimental evidence for the existence of this reaction at temperatures around or above 140 °C has been achieved in this work in the following way. At first, a ROP of L-lactide was initiated at 120 °C with ethyl L-lactate and the resulting HO–CH-terminated chain was acetylated with acetic anhydride to yield the linear Ac-PLA-Et chains (Lc in Scheme 4). These chains were mixed with SnOct2 in CH2Cl2 solution and after evaporation of the solvent the remaining reaction mixture was kept in the molten state at 180 °C for 0.5 h. The virgin reaction mixture was then characterized by MALDI-TOF mass spectrometry. Analogous experiments were performed at 160 °C/1 h and 140 °C/1 h with a 50% w/v solution of the acetylated polylactide in dichlorobenzene doped with SnOct2 (Lac/Cat = 100/1).
The MALDI-TOF mass spectra (exemplarily shown in Fig. 5 and S5†) carry all the same message, namely chain scission followed by total equilibration of all components of the reaction mixture. These two figures demonstrate that the reaction products one may expect from this kind of transesterification are formed (Lb and Lb chains). Equal amounts of odd and even species proved that complete equilibration had occurred. Furthermore, the mass peaks of cyclics were clearly detectable (labeled C in Fig. 5). When the catalyst attacks cyclic polylactide linear chains of structure La are formed (after hydrolysis of the Sn–O bond), yet, this ring cleavage is reversible and thus, establishes a second type of ring-chain equilibration. When the cycles are large, so that their population of conformations is almost identical to that of the linear counterpart, the ring closing reaction is thermodynamically favored by a gain in entropy. This is presumably the reason why the polymerization of lactide with neat SnOct2 favors the formation of large cycles in combination with (seemingly) unreacted SnOct2.
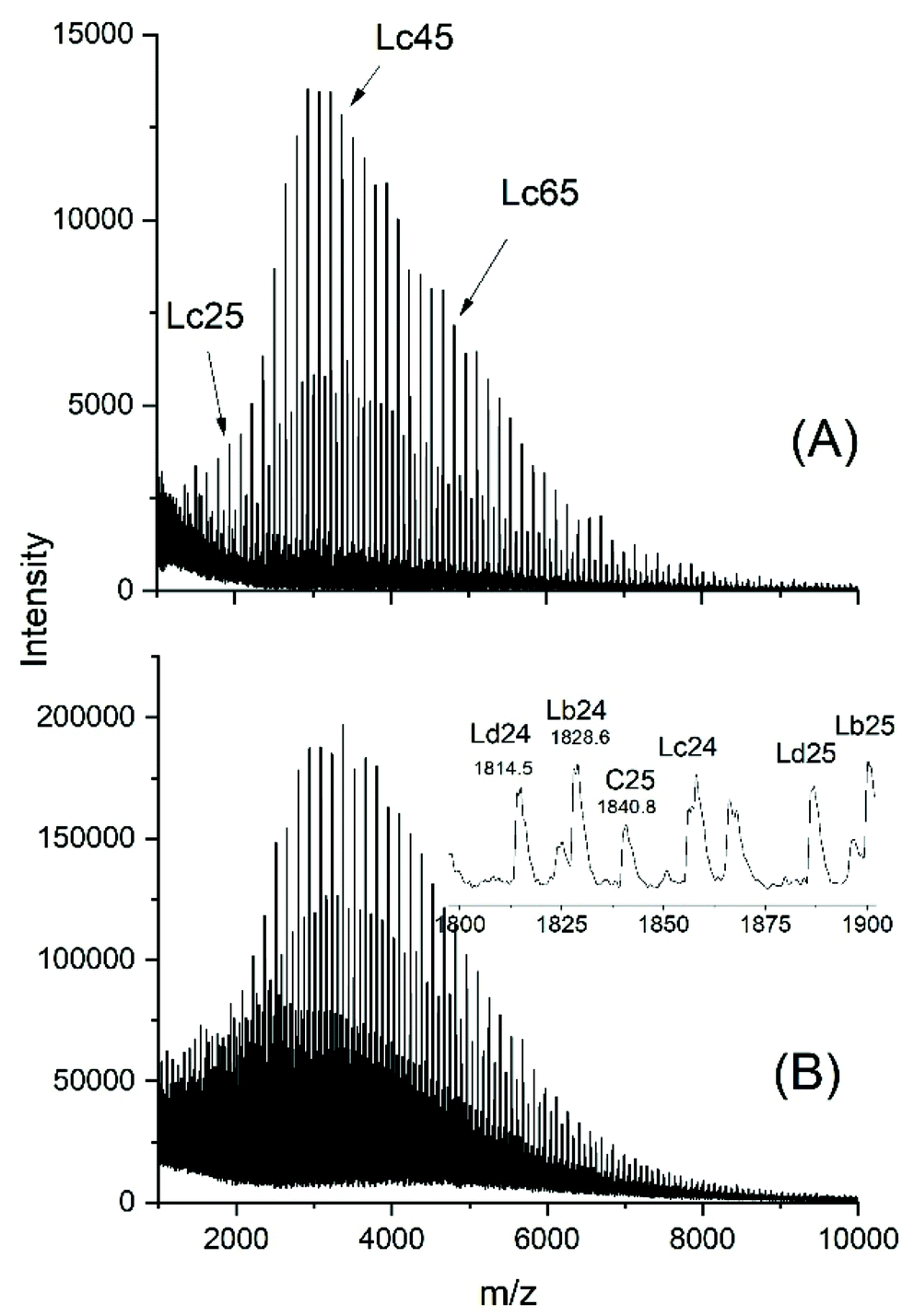 | ||
| Fig. 5 MALDI-TOF mass spectra of (A) the linear Ac-PLA-Et (no. 1, Table 5), (B) Ac-PLA-Et after heating with SnOct2 (Lac/Cat = 50/1) at 140 °C/1 h (no. 4, Table 5). | ||
All these transesterification reactions contribute to a broadening of the molar mass distribution (relative to a ROP solely under kinetic control) and result in a rapid decrease of the average molar masses, when the polymerizations were performed at 180 °C.
Finally, two experiments were conducted to elucidate the role of chain scission. From the studies of several research groups it is known that bond dissociation into radicals requires temperatures above 300 °C.49–52 The only kind of chain scission that may occur below 250 °C is the s.c. β-cis elimination, which yields acrylate and COOH end groups (Scheme 11). However, the authors have not found any information about the occurrence of this reaction at 180 °C or below. Therefore, Ac-PLA-Et was heated to 180 °C either for 1 h or for 4 h, and the reaction product was characterized by MALDI-TOF mass spectrometry. However, mass peaks that could be attributed to Lb and Lf chains were not detected. Since the signal-to-noise ratio of these mass spectra was not high, up to 5 mol% of these degradation products might have been present. Nonetheless, these experiments suggest that the formation of low molar cycles is mainly responsible for the degradation of the molar masses at reaction times <8 h.
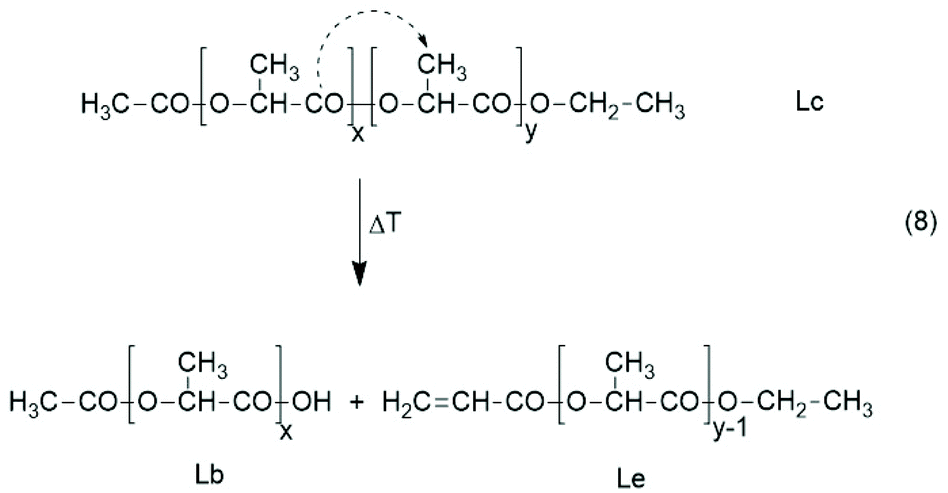 | ||
| Scheme 11 Chain scission by thermal β-elimination. | ||
All these results together indicate that the initially rapid and kinetically controlled polymerizations are more and more influenced by thermodynamically controlled equilibration reactions, which are the main reason for the decrease of the average molar masses. This interpretation is supported by the SEC elution curves as illustrated in Fig. 6 and Fig. S6† for the polymerizations performed at 160 °C. At longer polymerization times (8 h) the SEC curves display a weak shoulder between 20 and 26 min, suggesting an increase of low molar mass cycles.
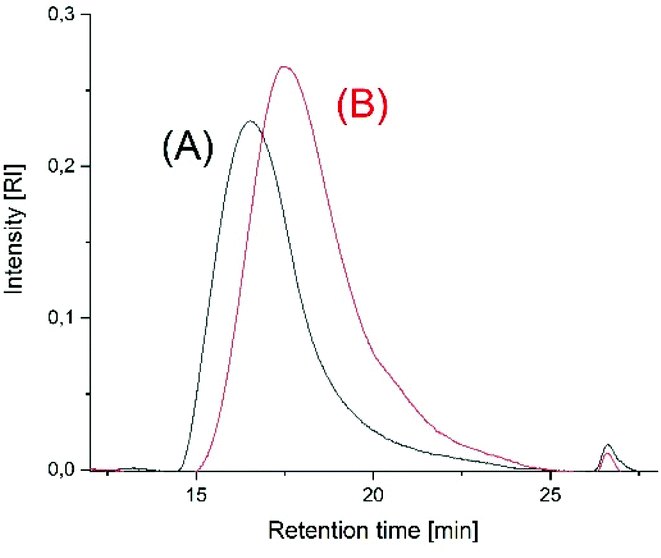 | ||
| Fig. 6 SEC elution curves of poly(L-lactide)s prepared at 160 °C with Lac/Cat = 400/1 (A) time 1 h (no. 4A, Table 2), (B) time 8 h, (no. 4B, Table 2). | ||
Polymerizations catalyzed by Sn(II)acetate
Sn(II)acetate is a commercial chemical, which was used as a catalyst for four polymerization experiments with low Lac/Cat ratios (Table 4). The reason for these experiments is an analytical advantage for the interpretation of the MALDI TOF mass spectra. The linear chains of structure Lb or Lb_K (Scheme 5) don't have the same mass as the cyclic polylactides and thus, allow for an easy detection when present. Furthermore, the authors have not found any detailed study dealing with ROP of lactides catalysed by neat SnAc2.The MALDI-TOF mass spectrum of the sample with the lowest Lac/Cat ratio (25/1, no. 1, Table 4) is presented in Fig. 7. It reveals that the vast majority of the reaction product are acetate terminated linear chains having a free CO2H end group (Lb in Scheme 5, s. Fig. 7A). When more potassium trifluoroacetate is added, the small peaks of the Lb potassium salt observable in Fig. 7A increases in intensity, whereas the peaks of the Lb chains having free CO2H groups decrease (Fig. 7B). Since this effect is not detectable when SnOct2 is used as a catalyst (no. 1, Table 2), these findings support the above conclusion that with SnOct2 mainly cycles are formed.
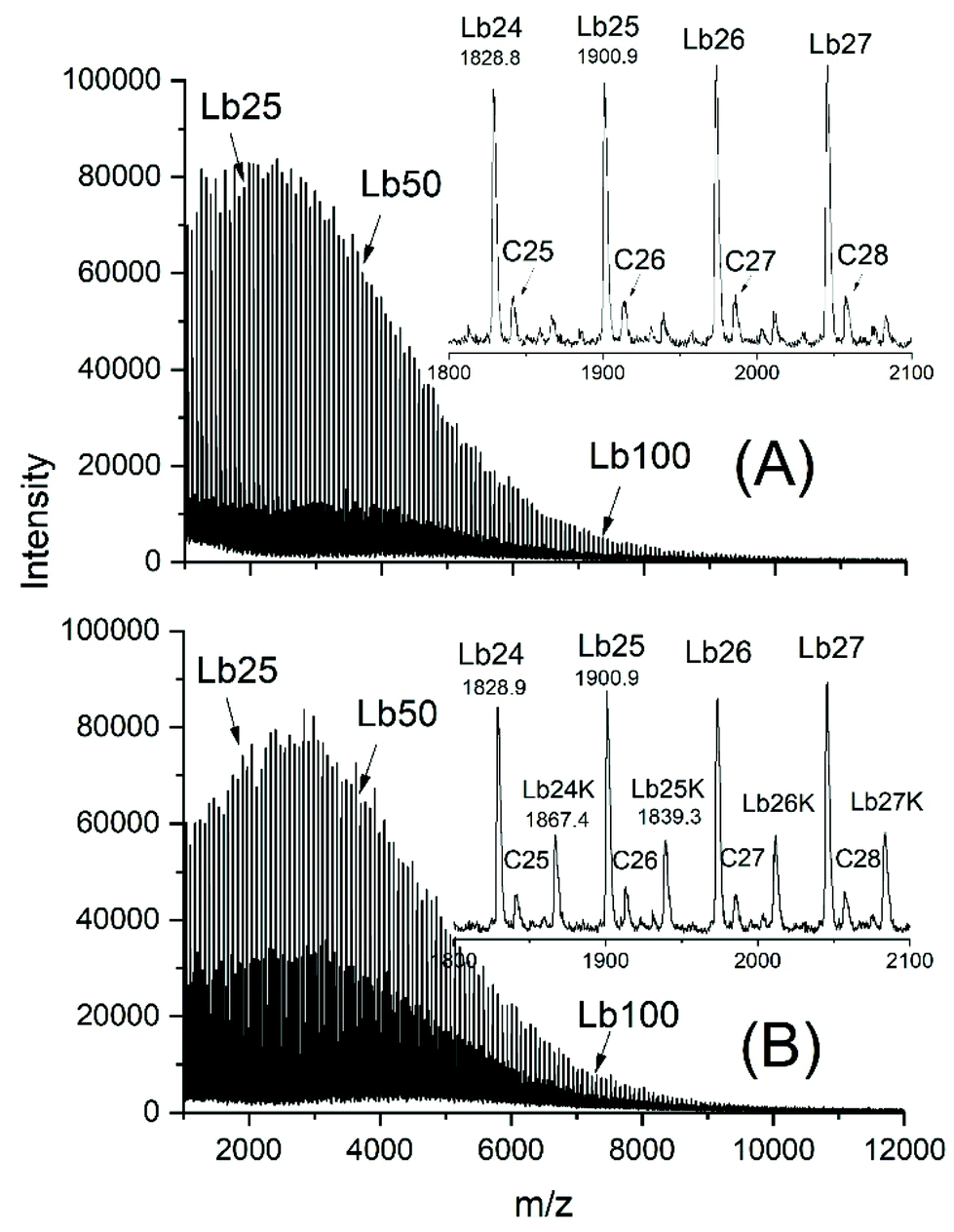 | ||
| Fig. 7 MALDI-TOF mass spectrum of poly(L-lactide)s polymerized with Sn(II)acetate at 160 °C: (A) Lac/Cat = 25/1 (no. 1, Table 4), normal doping, (B) the same sample after excessive doping with potassium trifluoroacetate. | ||
The higher fraction of linear chains (Lb) at low Lac/Cat ratios of SnAc2 may be understood on the basis of the ROPPOC mechanism discussed above. The acetyl group is significantly more electrophilic than the 2-ethylhexanoyl residue because the positive inductive effect and the steric hindrance inferred by branching are lacking. Therefore, the ka/kb ratio is lower and favours a higher fraction of linear chains. Furthermore, the transesterification reaction formulated in Scheme 10, which also yields Lb chains when the catalyst attacks cycles, may be somewhat more favourable for linear chains, when SnAc2 is used instead of SnOct2.
The mass spectra also indicated that the molar fraction of the cycles strongly increases at the expense of the linear (Lb) chains (Fig. S7A†), the peaks of which almost disappear at Lac/Cat ratios of 400/1 and higher (Fig. S7B†).
The dn/dc measurement of sample no. 7, Table 5, gave a value of 2.017 corresponding to a cyclic topology and the MHS measurements illustrated in Fig. S8† confirm the conclusion that with increasing Lac/Cat ratio the cyclic polylactides become the predominant species.
The 13C NMR spectrum of the polylactide polymerized with a Lac/Cat ratio of 25/1 (no. 1, Table 5, see Fig. S9†) displays the signals of the acetate and of the COOH end groups in contrast to the 13C NMR spectrum of the analogous SnOct2-catalyzed sample (no. 1, Table 2). Therefore, it confirms the interpretation of the MALDI-TOF and 13C NMR spectra given above.
The SEC measurements revealed the following trends. The Mn's increased with higher Lac/Cat ratio up to 1000/1 and a maximum Mw above 300000 g mol−1 was achieved in agreement with analogous polymerizations catalysed by SnOct2 (Table 2). All these findings are in agreement with the ROPPOC mechanism and the results discussed for SnOct2-catalyzed polymerizations.
SEC measurements
As already mentioned above, calibration with polystyrene overestimates the real molar masses of linear polylactides (and other aliphatic polyesters), because the hydrodynamic volume of polystyrene is about 1/3 smaller than that of polylactide. Therefore, a correction factor around 0.68 is needed to transform the measured values into realistic ones. However, the hydrodynamic volume of cyclic polylactide is also about 1/3 lower than that of the linear counterparts, and thus a correction is not needed. Therefore, the uncorrected Mn and Mw data are listed in Tables 1–3 and 5 and in previous publications dealing with cyclic poly(L-lactide)s.45–48 Another aspect which merits discussion are the dispersities, most of which fall into the range of 1.9–2.9 (Tables 1–3 and 5). These values are somewhat lower than the real dispersities, because for technical reasons (see Experimental) not all oligomers were included in the calculation of Mn (and Mw). This deficit does not affect Mw but results in higher Mn values and dispersities <2.0, which are impossible for equilibrated polymers. Niehaus and Jackson have studied nylons and polyethylene terephthalate prepared by reversible polycondensations with conversions around 99%.53 As expected from the Florys theory,54 the dispersities of the linear chains alone were around 2.0, whereas the dispersities of cyclic oligomers + linear chains are >4. A theoretical treatment of equilibrated cyclic polymers does not exist to the best knowledge of the authors, but dispersities between 2 and 4 are most likely in good agreement with the data obtained in this work considering a slight overestimation of Mn.For cyclic polylactides prepared by zwitterionic polymerization in solution several research groups have reported dispersities <1.5, and for linear polylactides numerous researchers have found dispersities as low as 1.1.55–57 Such low dispersities were considered to be a particularly important achievement of their synthetic methods. However, the authors have not been aware of any commercial polylactide the application of which requires such low dispersities (even after consulting several experts in the field of drug-delivery systems). For almost all applications requiring thermal processing (e.g. extrusion injection moulding, bottle blowing) low dispersities are not only useless, they are even a serious disadvantage, because polymers having broader dispersities show a better balance of moderate melt viscosity and good mechanical properties. When low and high dispersity polymers are compared at identical Mn, the broadly dispersed polymer reveals better mechanical properties due to a larger fraction of high molar mass species. When compared at identical Mw, the broadly dispersed polymers show a lower melt viscosity due to a larger fraction of oligomers which act as the “solvent”. Therefore, the high dispersities of the cyclic polylactides obtained in this work are an useful property for all applications requiring processing from the melt.
Conclusions
Four important conclusions may be drawn from the present study. First, neat SnOct2 yields cyclic polylactides and not linear chains as assumed in previous decades. From the preparative point of view polymerizations in bulk at a temperature of 160 °C with reaction times around 0.5–2.0 h (depending on the Lac/Cat ratio) seem to be an optimum. Cyclic polylactides having Mw's in the range of 250000–320000 g mol−1 can be obtained, without racemization and without discoloration. Since SnOct2 has been accepted by the U.S. FDA as a food stabilizer and polymerization catalyst, the polymerization conditions studied in this work are suited for upscaling and technical production.
Second, these results are best explained by a ROPPOC mechanism based on initiation and propagation steps via the coordination–insertion mechanism followed by ring closure via the temporarily formed mixed anhydride end group. Third, comparison of SnOct2 and SnAc2 catalysis indicates that the nature of carboxylic acid has a significant influence on the course of the polymerization and thus, on the structure of the resulting polylactides. This aspect certainly deserves further exploration, and part of our current research activities is focused on this point. Fourth, it is demonstrated that ROPPOC catalyzed by neat SnOct2 involves at least three different transesterification mechanisms (in addition to propagation), and thus certainly belongs to the group of reversible polycondensations. Together with the detection of a large fraction of high molar mass cycles the formation of which requires end-to-end cyclization, these results confirm the results and conclusions recently published in a critique of the Jacobson–Stockmayer theory, which ignored the existence of end-to-end cyclization in reversible polycondensations.58
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Dr U. Mühlbauer (Thyssen-Ude, Berlin) for kindly supplying L-lactide and Mrs A. Myxa (BAM Berlin) for the SEC measurements.Notes and references
- G. Kharas, F. Sanchez-Riera and D. Severson, Polymers of Lactic Acid, in Plastics from Microbes, ed. D. P. Mobley, Hanser Gardner Publications, Inc., 1994 Search PubMed.
- L.-T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, John Wiley & Sons, 2011 Search PubMed.
- M. L. Di Lorenzo and R. Androsch, Synthesis, Structure and Properties of Poly (lactic acid), Springer, 2018 Search PubMed.
- M. L. Di Lorenzo and R. Androsch, Industrial Applications of Poly (lactic acid), Springer, 2018 Search PubMed.
- H. K. Makadia and S. J. Siegel, Polymer, 2011, 3, 1377–1397 CAS.
- W. Hadasha and D. Bezuidenhout, in Industrial Applications of Poly (lactic acid), Springer, 2017, pp. 51–77 Search PubMed.
- J. Li, J. Ding, T. Liu, J. F. Liu, L. Yan and X. Chen, in Industrial Applications of Poly (lactic acid), Springer, 2017, pp. 109–138 Search PubMed.
- R. G. Sinclair and G. M. Gynn, Preparation and evaluation of glycolic and lactic acid-based polymers for implant devices used in management of maxillofacial trauma, Battelle Columbus Div OH, 1972 Search PubMed.
- J. Dahlmann and G. Rafler, Acta Polym., 1993, 44, 103–107 CrossRef CAS.
- A. Nijenhuis, D. Grijpma and A. Pennings, Macromolecules, 1992, 25, 6419–6424 CrossRef CAS.
- G. Rafler and J. Dahlmann, Acta Polym., 1990, 41, 611–617 CrossRef CAS.
- J. Dahlmann, G. Rafler, K. Fechner and B. Mehlis, Br. Polym. J., 1990, 23, 235–240 CAS.
- J. W. Leenslag and A. J. Pennings, Makromol. Chem., 1987, 188, 1809–1814 CrossRef CAS.
- F. Kohn, J. Van Ommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- A. K. Schneider, US Pat., US32397033/3463158, 1967 Search PubMed.
- H. R. Kricheldorf and J. Meier-Haack, Makromol. Chem., 1993, 194, 715–725 CrossRef CAS.
- X. Zhang, D. A. MacDonald, M. F. Goosen and K. B. McAuley, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2965–2970 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and C. Boettcher, Polymer, 1995, 36, 1253–1259 CrossRef CAS.
- Y. J. Du, P. J. Lemstra, A. J. Nijenhuis, H. A. Van Aert and C. Bastiaansen, Macromolecules, 1995, 28, 2124–2132 CrossRef CAS.
- G. Schwach, J. Coudane, R. Engel and M. Vert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3431–3440 CrossRef CAS.
- D. R. Witzke, R. Narayan and J. J. Kolstad, Macromolecules, 1997, 30, 7075–7085 CrossRef CAS.
- P. J. In't Veld, E. M. Velner, P. Van De Witte, J. Hamhuis, P. J. Dijkstra and J. Feijen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 219–226 CrossRef.
- K. Shinno, M. Miyamoto, Y. Kimura, Y. Hirai and H. Yoshitome, Macromolecules, 1997, 30, 6438–6444 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- S. Penczek, A. Duda, A. Kowalski, J. Libiszowski, K. Majerska and T. Biela, Macromol. Symp., 2000, 157, 61–70 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- M. Jalabert, C. Fraschini and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1944–1955 CrossRef CAS.
- Y. Yu, G. Storti and M. Morbidelli, Macromolecules, 2009, 42, 8187–8197 CrossRef CAS.
- Y. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS.
- P. Kaur, R. Mehta, D. Berek and S. N. Upadhyay, Macromol. Symp., 2012, 315, 106–111 CrossRef CAS.
- S. Zavrazhnov, V. Fomin, L. Beloded and T. Lobaeva, Russ. J. Appl. Chem., 2012, 85, 1264–1268 CrossRef CAS.
- V. Balasanthiran, T. L. Beilke and M. H. Chisholm, Dalton Trans., 2013, 42, 9274–9278 RSC.
- Y. Yu, E. J. Fischer, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2014, 53, 7333–7342 CrossRef CAS.
- S. Sosnowski and P. Lewinski, Polym. Chem., 2015, 6, 6292–6296 RSC.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2017, 218, 1600331 CrossRef.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2018, 219, 1800445 CrossRef.
- A. Schindler, Y. Hibionada and C. Pitt, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 319–326 CrossRef CAS.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS.
- A. Kowalski, J. Libiszowski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 1964–1971 CrossRef CAS.
- X. Y. Zhang and R. M. Waymouth, ACS Macro Lett., 2014, 3, 1024–1028 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- A. K. Acharya, Y. A. Chang, G. O. Jones, J. E. Rice, J. L. Hedrick, H. W. Horn and R. M. Waymouth, J. Phys. Chem. B, 2014, 118, 6553–6560 CrossRef CAS PubMed.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3767–3775 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1915–1925 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 952–960 CrossRef CAS.
- O. Wachsen, K. Platkowski and K.-H. Reichert, Polym. Degrad. Stab., 1997, 57, 87–94 CrossRef CAS.
- Y. Fan, H. Nishida, Y. Shirai and T. Endo, Polym. Degrad. Stab., 2004, 84, 143–149 CrossRef CAS.
- H. Nishida, T. Mori, S. Hoshihara, Y. Fan, Y. Shirai and T. Endo, Polym. Degrad. Stab., 2003, 81, 515–523 CrossRef CAS.
- Y. Wang, B. Steinhoff, C. Brinkmann and I. Alig, Polymer, 2008, 49, 1257–1265 CrossRef CAS.
- D. E. Niehaus and C. Jackson, Polymer, 2000, 41, 259–268 CrossRef CAS.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- W. Jeong, E. J. Shin, D. A. Culkin, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2009, 131, 4884–4891 CrossRef CAS PubMed.
- H. A. Brown, A. G. De Crisci, J. L. Hedrick and R. M. Waymouth, ACS Macro Lett., 2012, 1, 1113–1115 CrossRef CAS.
- G. Si, S. Zhang, W. Pang, F. Wang and C. Tan, Polymer, 2018, 154, 148–152 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2020, 11, 2595–2604 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00811g |
| This journal is © The Royal Society of Chemistry 2020 |