DOI:
10.1039/D0PY00799D
(Paper)
Polym. Chem., 2020,
11, 5279-5285
Supramolecular behaviour and fluorescence of rhodamine-functionalised ROMP polymers†
Received
2nd June 2020
, Accepted 14th July 2020
First published on 16th July 2020
Abstract
Inherently fluorescent polymers are of interest in materials and medicine. We report a ring-opening metathesis polymerisation (ROMP) platform for creation of amphiphilic block copolymers in which one block is formed from rhodamine B-containing monomers. The polymers self-assemble into well-defined micelles which are able to sequester molecular dyes and further interact with them by energy transfer. Despite incorporating a cationic dye known to bind DNA, the polymer micelles do not interact with DNA, indicating that they are potentially safe for use in bioanalytical applications.
Introduction
Polymer-dye conjugates have uses including optical imaging, photochromic materials, light harvesting, and as fluorescent tools for polymer chain association and conformational studies, as well as in biological diagnostic applications.1 The majority of polymer-dye conjugates are prepared using free radical polymerisation, which is not compatible with all dyes as some groups can act as radical scavengers.2 In parallel, synthetic polymer therapeutics have become an established component of modern medicine,3,4 including water-soluble polymers, polymeric drugs, and polymer–drug conjugates as systems developed with applications in bionanotechnology.5–7 A range of polymerisation methods have been used to synthesise the conjugates, amongst which ring-opening metathesis polymerisation (ROMP) has gained increasing prominence.8–11 The living nature of the ROMP reaction allows for the preparation of a vast array of copolymers with excellent control over chain length, functional density, and monomer variety, and has led to the preparation of a wide range of bio-related and therapeutic ROMP polymers.12–22 In a wider context, ROMP also allows for the incorporation of dyes that are not suitable for free radical polymerisation processes, and the control ROMP affords can deliver polymer conjugates below 45 kDa, as required to allow renal clearance in eventual in vivo applications.8
We here report the synthesis and analysis of a new fluorophore-rich block copolymer prepared by ROMP. Previous work in our laboratories has focused on polymer–drug conjugates23,24 and we wished to explore the incorporation of fluorophores25 as these have a range of important medical applications, enabling visualisation of cellular uptake, and are complementary to our luminescence investigations of uptake of potential therapeutics in resistant cancer lines.26 We were particularly interested in self-assembled systems which possess “stealth” properties and to this end we used polyethylene glycol (PEG) as a clinically established copolymerisation adduct; PEG interacts minimally with biological fluid components, is hydrophilic, biologically inert, and can be used to control self-assembly outcomes, which in turn affects blood circulation and elimination.5 Rhodamine B (RhB) was chosen as the fluorophore. The rhodamine group of dyes are second only to the xanthene fluorescein group with respect to polymer dye conjugates used for labelling2 and yet there are few ROMP polymer conjugate examples in the literature.20,25,27–32 Our ROMP polymer shows robust self-assembly into well-defined nanoparticles, which can interact both supramolecularly and photophysically with molecular dyes, while showing no interactions with DNA which could lead to toxicity. Such nanoparticles which display high fluorescence and minimal interactions with biomolecules could be useful in monitoring drug-delivery processes at the cellular level.
Results and discussion
Monomer synthesis
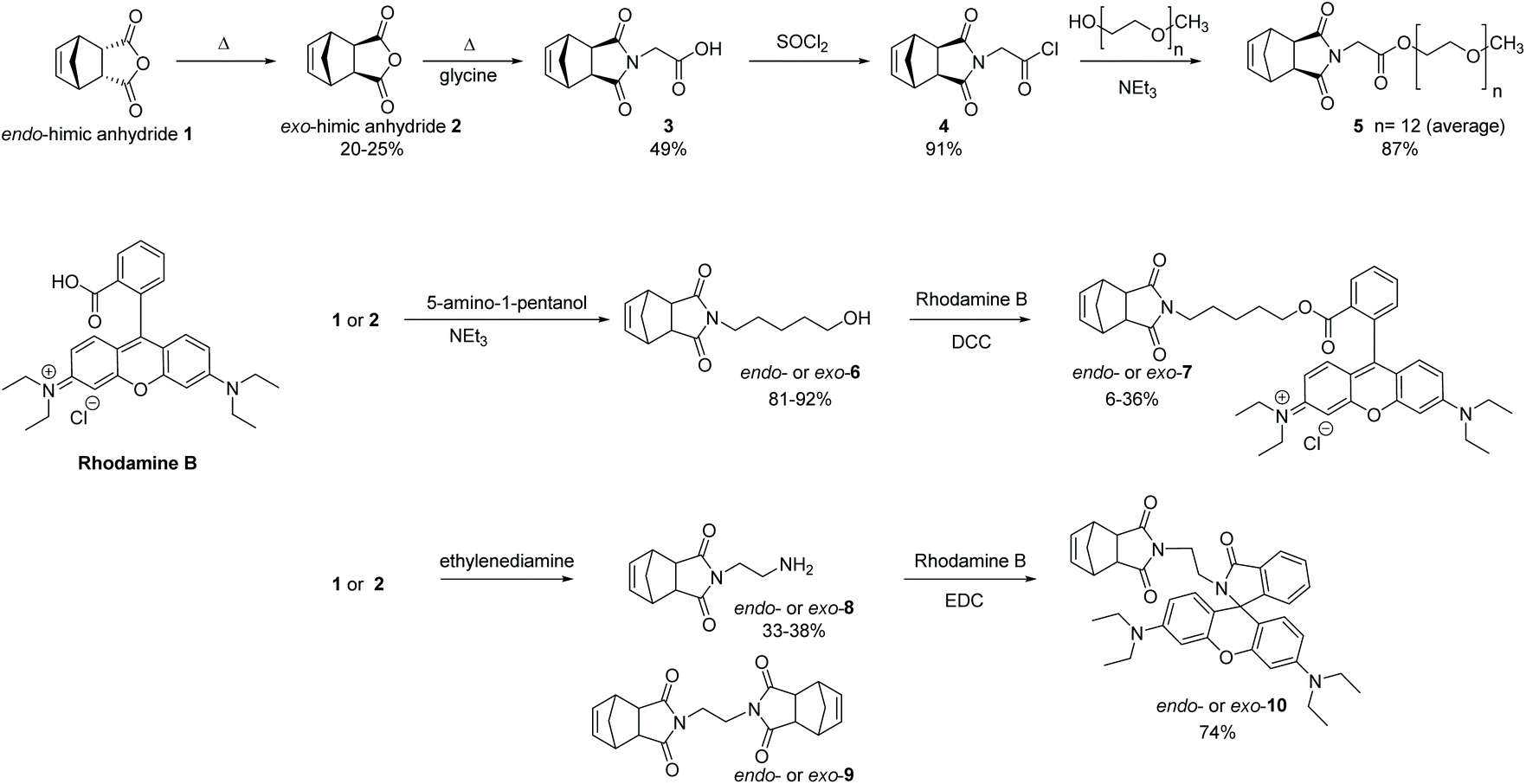
The incorporation of PEG units onto ROMP polymers is well-established with some early examples reported by the Grubbs and Nguyen groups amongst others.33–36 For our studies, we chose to synthesise the norbornene PEG-derivative, 5, as it possesses a symmetrical structure which minimizes head-to-tail effects, and its preparation and polymerisation characteristics have previously been reported by our group.37 The synthesis is a three step procedure from exo-himic anhydride 2 (Scheme 1), itself prepared from the commercially available endo-himic anhydride 1 according to a well-established procedure.38 The glycine derivative, 3, is a known solid and here we report single crystal diffraction data and structure for the first time (see ESI†). RhB possesses a carboxylic acid group, and we originally envisaged synthesizing the required monomer derivative, exo-7, via a Steglich esterification39 with N-(hydroxypentanyl)-cis-5-norbornene-endo-2,3-dicarboximide, 6, the latter available following a literature procedure.40 The reaction sequence was first tested with the more readily available endo-himic anhydride to establish suitable reaction conditions. The desired ester derivatives, endo- and exo-7, were obtained but repeated purification steps were required to remove urea by-products, and traces of RhB persisted, suggesting that hydrolysis occurred during the purification process, giving low overall yields. We therefore switched our attention to an amide derivative of RhB as these are less prone to hydrolysis and the norbornene-amino derivative, 8, was identified as a suitable adduct to couple with the RhB. Upon searching the literature, Davies et al.41 reported that the reaction with endo carbic anhydride 1 was possible using an excess of ethylenediamine but stated that attempts with the exo isomer 2 had failed, an unexpected observation given the similarity of other reactions such as shown in Scheme 1. The Kruger group42 stated that the product, endo-8, was obtained but not fully purified, the authors arguing that its purity would not affect subsequent steps. Successful reactions of ethylenediamine with 2, have been reported by other groups but the purification procedures varied from column chromatography43–45 to no work-up at all.46 For the purposes of this project, it was necessary to develop a work-up procedure to isolate and characterise the pure product. In our hands, the required endo- or exo-8 derivatives are formed using an excess of ethylenediamine, followed by a work-up using toluene which selectively removes unwanted disubstituted product, 9. Subsequently the desired norbornene-RhB derivative, 10, was prepared as outlined in Scheme 1. For both endo- and exo-10 derivatives, the final products were crystalline solids which were fully characterized by NMR analysis and the structures confirmed by single crystal X-ray diffraction. The crystal structures show that the products in each case had equilibrated to the neutral, lactam form (Fig. 1).
 |
| | Scheme 1 Monomer synthesis. | |
 |
| | Fig. 1 Crystal structures of (a) endo-10 and (b) exo-10 in their ring-closed lactam forms. Hydrogen atoms, solvent molecules, and disorder omitted for clarity. See Fig. S16 and S19 (ESI†) for thermal ellipsoid plots. | |
Synthesis of polymers
The polymerisation characteristics of the exo-norbornene-PEG monomer 5 have been determined previously24,31 and so we focused on investigating the homopolymerisation of exo-norbornene-RhB monomer exo-10. The Grubbs G3 initiator was chosen as this has been shown to possess excellent activity even with complex pendant groups.13,47 A kinetic study of the polymerisation in deuterated-chloroform showed that the polymerisation commenced rapidly and the majority of the monomer was consumed after 30 min, although approximately 80 min were required for complete conversion as evidenced by the disappearance of the monomer peak at 2.5 ppm (Fig. 2).
 |
| | Fig. 2 Kinetics of ROMP of monomer exo-10 measured by 1H NMR in CDCl3. | |
The block copolymerization was performed starting with 5 which is known to polymerise within 10 min,24,37 followed by addition of exo-10. The homopolymer of 5 was formed at a [M]0/[C]0 of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at an initial concentration of 0.045 M in CH2Cl2 and GPC analysis of a sample showed a polydispersity index of 1.36 and an average molecular weight of 11 kDa; this was followed by addition of an equimolar amount of exo-10 which was allowed to react for 100 min and the reaction then terminated with ethyl vinyl ether, to give poly 5-b-exo-10 as a final polymer product with an average molecular weight of 21.8 kDa and a Đ of 1.29 (Fig. S22, ESI†). NMR analysis of poly 5-b-exo-10 was consistent with that expected from a combination of the homopolymer spectra.
:1 at an initial concentration of 0.045 M in CH2Cl2 and GPC analysis of a sample showed a polydispersity index of 1.36 and an average molecular weight of 11 kDa; this was followed by addition of an equimolar amount of exo-10 which was allowed to react for 100 min and the reaction then terminated with ethyl vinyl ether, to give poly 5-b-exo-10 as a final polymer product with an average molecular weight of 21.8 kDa and a Đ of 1.29 (Fig. S22, ESI†). NMR analysis of poly 5-b-exo-10 was consistent with that expected from a combination of the homopolymer spectra.
Self-assembly of block copolymer
Poly 5-b-exo-10 was tested under multiple buffer conditions to assess its interaction with both single-stranded and double-stranded DNA. The polymer (1 mg) was dissolved in acetone (100 μL), and 900 μL of the aqueous medium of choice (unbuffered water; tris-borate-EDTA (TBE, pH 8) buffer; tris-acetate-magnesium (TAMg, pH 8) buffer; and acetate (pH 5) buffer) was added in portions (15 × 10 μL, 10 × 20 μL, 5 × 50 μL, 3 × 100 μL) whilst stirring.
DLS analysis of the micellar solutions (Fig. 3a, S23–S25, ESI†) gave particle diameters of 27 nm (water), 38 nm (TBE), and 37 nm (TAMg). The size distributions of the pH buffered particles were very low (dispersity indices of 0.04 and 0.05 for TBE and TAMg respectively), while the distribution in unbuffered water was noticeably broader (dispersity index of 0.24). Examination of the size distribution graphs (Fig. S23–S25, ESI†) makes it clear that taking into account the broader distribution in water, the differences in average size are not significant. The size of the micelles was not concentration dependent. The emission intensity of the RhB unit was also measured (Fig. S28–S30, ESI†) and again the buffered systems were distinctly different from the unbuffered solution, which was noticeably less intense – this suggests that the interior arrangement of chains in water promotes self-quenching to a greater degree. The micelles in water were also analysed by TEM (Fig. 3a, b and S26, S27, ESI†), giving size distributions slightly smaller than those in solution using DLS (22 ± 2 nm). This is as expected, since TEM does not record a hydration sphere and uses a vacuum system which frequently contracts the diameter of the self-assembled systems, whereas DLS observes particles including their solvation sphere. The size distribution was remarkably uniform, showing that the low dispersity polymers also have minimal variation in aggregation number as well.
 |
| | Fig. 3 Characterisation of poly 5-b-exo-10 in water by (a) DLS and (b) TEM imaging (stained with uranyl acetate) and (c) counting of TEM particle sizes. Data for TBE and TAMg can be found in the ESI.† | |
Fluorescence and energy transfer
Typically in fluorophore-functionalised ROMP polymers, fluorescent monomers are copolymerised with monomers that provide different imaging functionalities such as MRI contrast.48,49 When copolymerised, the fluorescent monomers tend to undergo quenching to varying degrees and this can be caused by several phenomena including aggregation-caused quenching (ACQ)50 and chemical interaction with other functional groups.51–53 In the system reported here, since the hydrophobic block is formed from just one monomer, it is possible for quenching or other photophysical interactions to occur both at intra- and interchain levels. Intrachain effects will be independent of the local concentration of the polymer, whereas interchain effects will depend on self-assembly and change of local environment. Fluorescence measurements at concentrations between 0.0001 to 1 mg mL−1 show that emission intensity increases with concentration above that expected for simple concentration effects, indicating increased efficiency of emission likely due to a more hydrophobic environment,54 as a result of micellization. However, no further increase is observed with concentration, and from 0.01 to 1 mg mL−1 emission intensity decreases, consistent with significant interchain quenching (Fig. 4a).
 |
| | Fig. 4 (a) Effect of dilution upon fluorescence intensity in water (note log scales on both axes). The samples were excited at 550 nm, and emission collected over 560–760 nm, with λmax = 580 nm. (b) Emission spectra for FRET experiments. λex (proflavine) = 460 nm, λex (6-FAM) = 490 nm, λex (RhB) = 550 nm. | |
Förster resonance energy transfer (FRET),55 which is dependent upon the distance between a pair of donor–acceptor fluorophores, was used to further probe the polymer micelles using a supramolecular approach through encapsulation of hydrophobic dyes within the polymer micelles. For efficient FRET, there must be both a short distance between the two dyes (<10 nm), and good spectral overlap between the emission peak of the directly excited (donating) dye and the excitation peak of the accepting dye. The overall effect of FRET is a reduction in the emission intensity of the donating dye and an increase in that of the accepting dye. 6-Carboxyfluorescein (6-FAM) was selected for these studies as its emission maximum 520 nm (excitation maximum 490 nm) is broad enough to overlap with RhB excitation (λmax = 550 nm). At constant 6-FAM concentration, the presence of poly 5-b-exo-10 micelles in water caused the 6-FAM emission at 520 nm to drop by 11%, while a shoulder appeared at 580 nm on the emission spectra, corresponding exactly to RhB emission (Fig. 4b). Changing 6-FAM for proflavine (emission λmax = 510 nm) the same effect was seen, with the proflavine peak decreasing coincidentally by the same value (11%) while RhB emission appeared at 580 nm. In both cases, the integrated emission intensity decreased by 14%, indicating very little non-radiative energy loss, given that under optimal conditions, the quantum yield for direct excitation of RhB is no higher than 0.8, and typically closer to 0.6.56 These results indicate that the micelles are both capable of noncovalently binding organic molecules and participating in FRET processes with such guests. This is potentially a useful tool for study of polymer-mediated drug delivery: if an encapsulated drug (e.g. doxorubicin) changes the photophysics of the polymer (either by FRET or some other measurable effect), it becomes possible to distinguish between the bound and free drug using fluorescence microscopy without making assumptions about colocalisation.
Interaction with DNA
Due to its cationic and aromatic nature, RhB can bind to DNA and has been found to interact with the minor groove of the B-DNA double helix, leading to a reduction in fluorescence.57 For our system to be useful as a tool for examining drug delivery, binding of DNA should be minimised. We examined the interaction of poly 5-b-exo-10 with both single-stranded and double stranded DNA (ssDNA and dsDNA) oligomers (20 mers). We observed no meaningful changes in dimension (DLS Fig. S31–S33, TEM Fig. S35–S40, ESI†) or fluorescence (Fig. S34, ESI†) upon mixing at a 1:1 ratio of DNA bases to RhB monomer, in either water, or TBE, or TAMg buffer. The reason for this lack of interaction became clear when we performed analysis by agarose gel electrophoresis (Fig. 5, S43, ESI†): the micelle migrated in the same direction as DNA, towards the anode, forming a very well-defined band, which again demonstrates their uniformity. The direction of mobility remained the same despite adjusting the pH to 5 (acetate buffer) to be entirely sure that the RhB units are in their cationic form, and even after annealing the samples from 95 °C to 4 °C over an hour to overcome kinetic effects. Electrophoretic mobility is determined by the surface charge, and in this case, there is PEG on the surface, not cationic RhB, regardless of pH effects. The surface charge includes associated ions, and this is usually modelled as the electrostatic double layer – the chemistry on the exterior binds either anions or cations as a first layer, and a second layer forms of the opposite ions. Here, we believe that the PEG binds cations through the oxygen atoms, resulting in an anionic second sphere which leads to the observed electrophoresis result. The negative surface charge would therefore repel the similarly-charged DNA and prevent complexation.
 |
| | Fig. 5 Assessing interaction of poly 5-b-exo-10 with DNA by agarose gel electrophoresis (2.5% agarose, TBE buffer, 1:1 ratio of RhB:nucleobase, stained with GelRed, contrast enhanced). | |
Conclusion
We have shown that ROMP can be used to create RhB-rich block copolymers which self-assemble into well-defined micelles whose fluorescence can be modulated through non-covalent inclusion of molecular dyes. Despite the propensity of RhB to interact with DNA, the micellization results in safe confinement of the RhB within the micelle core, while the PEG shell is expected to provide biological “stealthing”. The true innocence of fluorescent dyes used to image processes such as cellular uptake of nanostructures can be dubious;58 we believe that this system provides a robust route to high emission/low interaction nanostructures which could be used to enhance analytical studies in bionanotechnology.
Conflicts of interest
The authors declare no competing financial interests. All authors have given approval to the final version of the manuscript.
Acknowledgements
This work was supported by the School of Physical Sciences, University of Kent Graduate Training Scheme, and by the EPSRC Doctoral Training Programme. We gratefully acknowledge Dr Andrew Morrell for advice and help with and LC and GC analyses, and Dr Ian Brown for TEM analyses.
References
- M. Beija, M. T. Charreyre and J. M. G. Martinho, Prog. Polym. Sci., 2011, 36, 568–602 CrossRef CAS.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2013, 65, 60–70 CrossRef CAS PubMed.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- D. Smith, E. B. Pentzer and S. T. Nguyen, Polym. Rev., 2007, 47, 419–459 CrossRef CAS.
- B. M. deRonde and G. N. Tew, Biopolymers, 2015, 104, 265–280 CrossRef CAS PubMed.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- Y. Chen, M. M. Abdellatif and K. Nomura, Tetrahedron, 2018, 74, 619–643 CrossRef CAS.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- J. M. Fishman, D. B. Zwick, A. G. Kruger and L. L. Kiessling, Biomacromolecules, 2019, 20, 1018–1027 CrossRef CAS PubMed.
- S. C. G. Biagini, S. M. Bush, V. C. Gibson, L. Mazzariol, M. North, W. G. Teasdale, C. M. Williams, G. Zagotto and D. Zamuner, Tetrahedron, 1995, 51, 7247–7262 CrossRef CAS.
- S. C. G. Biagini, R. G. Davies, V. C. Gibson, M. R. Giles, E. L. Marshall, M. North and D. A. Robson, Chem. Commun., 1999, 235–236 RSC.
- A. L. Parry, P. H. H. Bomans, S. J. Holder, N. A. J. M. Sommerdijk and S. C. G. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS PubMed.
- M. Neqal, J. Fernandez, V. Coma, M. Gauthier and V. Héroguez, J. Colloid Interface Sci., 2018, 526, 135–144 CrossRef CAS PubMed.
- Y. Zhang, Q. Yin, H. Lu, H. Xia, Y. Lin and J. Cheng, ACS Macro Lett., 2013, 2, 809–813 CrossRef CAS PubMed.
- P. Bertrand, C. Blanquart and V. Héroguez, Biomolecules, 2019, 9, 60 CrossRef PubMed.
- C. E. Callmann, C. V. Barback, M. P. Thompson, D. J. Hall, R. F. Mattrey and N. C. Gianneschi, Adv. Mater., 2015, 27, 4611–4615 CrossRef CAS PubMed.
- S. C. G. Biagini, V. C. Gibson, M. R. Giles, E. L. Marshall and M. North, Chem. Commun., 1997, 1097–1098 RSC.
- S. Akkad and C. J. Serpell, Macromol. Rapid Commun., 2018, 39, 1–5 CrossRef PubMed.
- S. Shehata, C. J. Serpell and S. C. G. Biagini, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9884114.v1.
- E. K. Riga, D. Boschert, M. Vöhringer, V. T. Widyaya, M. Kurowska, W. Hartleb and K. Lienkamp, Macromol. Chem. Phys., 2017, 218, 1700273 CrossRef.
- S. J. Thomas, B. Balónová, J. Cinatl, M. N. Wass, C. J. Serpell, B. A. Blight and M. Michaelis, ChemMedChem, 2020, 15, 349–353 CrossRef CAS PubMed.
- K. Miki, K. Oride, S. Inoue, Y. Kuramochi, R. R. Nayak, H. Matsuoka, H. Harada, M. Hiraoka and K. Ohe, Biomaterials, 2010, 31, 934–942 CrossRef CAS PubMed.
- K. Miki, A. Kimura, K. Oride, Y. Kuramochi, H. Matsuoka, H. Harada, M. Hiraoka and K. Ohe, Angew. Chem., Int. Ed., 2011, 50, 6567–6570 CrossRef CAS PubMed.
- E. M. Kolonko and L. L. Kiessling, J. Am. Chem. Soc., 2008, 130, 5626–5627 CrossRef CAS PubMed.
- S. L. Mangold, R. T. Carpenter and L. L. Kiessling, Org. Lett., 2008, 10, 2997–3000 CrossRef CAS PubMed.
- N. Xie, K. Feng, J. Shao, B. Chen, C. H. Tung and L. Z. Wu, Biomacromolecules, 2018, 19, 2750–2758 CrossRef CAS PubMed.
- F. Gueugnon, I. Denis, D. Pouliquen, F. Collette, R. Delatouche, V. Héroguez, M. Grégoire, P. Bertrand and C. Blanquart, Biomacromolecules, 2013, 14, 2396–2402 CrossRef CAS PubMed.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, Macromolecules, 2000, 33, 6239–6248 CrossRef CAS.
- K. J. Watson, D. R. Anderson and S. B. T. Nguyen, Macromolecules, 2001, 34, 3507–3509 CrossRef CAS.
- A. Carrillo, K. V. Gujraty, P. R. Rai and R. S. Kane, Nanotechnology, 2005, 16, S416–S421 CrossRef PubMed.
- J. J. Murphy, T. Kawasaki, M. Fujiki and K. Nomura, Macromolecules, 2005, 38, 1075–1083 CrossRef CAS.
- S. C. G. Biagini and A. L. Parry, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3178–3190 CrossRef CAS.
- D. Craig, J. Am. Chem. Soc., 1951, 73, 4889–4892 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- Y. C. Teo and Y. Xia, Macromolecules, 2015, 48, 5656–5662 CrossRef CAS.
- R. G. Davies, V. C. Gibson, M. B. Hursthouse, M. E. Light, E. L. Marshall, M. North, D. A. Robson, I. Thompson, A. J. P. White, D. J. Williams and P. J. Williams, J. Chem. Soc., Perkin Trans. 1, 2001, 3365–3381 Search PubMed.
- F. M. Pfeffer, T. Gunnlaugsson, P. Jensen and P. E. Kruger, Org. Lett., 2005, 7, 5357–5360 CrossRef CAS PubMed.
- M. Schaefer, N. Hanik and A. F. M. Kilbinger, Macromolecules, 2012, 45, 6807–6818 CrossRef CAS.
- M. Alizadeh and A. F. M. Kilbinger, Macromolecules, 2018, 51, 4363–4369 CrossRef CAS.
- X. Liu, G. Qiu, L. Zhang, F. Liu, S. Mu, Y. Long, Q. Zhao, Y. Liu and H. Gu, Macromol. Chem. Phys., 2018, 219, 1800273 CrossRef.
- M. Zhang, P. Vedantham, D. L. Flynn and P. R. Hanson, J. Org. Chem., 2004, 69, 8340–8344 CrossRef CAS PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, Organometallics, 2001, 20, 5314–5318 CrossRef CAS.
- M. A. Sowers, J. R. Mccombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- S. Mukherjee, H. Dinda, L. Shashank, I. Chakraborty, R. Bhattacharyya, J. Das Sarma and R. Shunmugam, Macromolecules, 2015, 48, 6791–6800 CrossRef CAS.
- J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426–429 RSC.
- A. Rostami and M. S. Taylor, Macromol. Rapid Commun., 2012, 33, 21–34 CrossRef CAS PubMed.
- H. Kim, Y. Kim and J. Y. Chang, Macromol. Chem. Phys., 2014, 215, 1274–1285 CrossRef CAS.
- Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS PubMed.
- K. G. Casey and E. L. Quitevis, J. Phys. Chem., 1988, 92, 6590–6594 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- M. M. Islam, M. Chakraborty, P. Pandya, A. Al Masum, N. Gupta and S. Mukhopadhyay, Dyes Pigm., 2013, 99, 412–422 CrossRef CAS.
- A. Lacroix, E. Vengut-Climent, D. De Rochambeau and H. F. Sleiman, ACS Cent. Sci., 2019, 5, 882–891 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2005040–2005042. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py00799d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sara
Shehata
,
Sara
Shehata