
Synthesis of poly(stearyl methacrylate)-poly(2-hydroxypropyl methacrylate) diblock copolymer nanoparticles via RAFT dispersion polymerization of 2-hydroxypropyl methacrylate in mineral oil†

Abstract
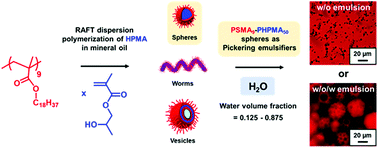
Poly(stearyl methacrylate)-poly(2-hydroxypropyl methacrylate) (PSMA-PHPMA) diblock copolymer nanoparticles are synthesized via reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) in mineral oil at 90 °C. The relatively short PSMA precursor (mean degree of polymerization = 9) remains soluble in mineral oil, whereas the growing PHPMA block quickly becomes insoluble, resulting in polymerization-induced self-assembly (PISA). Relatively high HPMA monomer conversions (≥98%) were achieved within 70 min as confirmed by in situ1H NMR spectroscopy studies, while gel permeation chromatography (GPC) analyses indicated high blocking efficiencies and relatively narrow molecular weight distributions (Mw/Mn ≤ 1.37) for all PISA syntheses. Depending on the precise synthesis conditions, this PISA formulation can produce diblock copolymer spheres, worms or vesicles; a pseudo-phase diagram has been constructed to enable reproducible targeting of each pure phase. Thus this is a rare example of the use of a commercially available polar monomer for PISA syntheses in non-polar media that offers access to the full range of copolymer morphologies. The resulting nanoparticles were characterized using dynamic light scattering (DLS), transmission electron microscopy (TEM), oscillatory rheology and small-angle X-ray scattering (SAXS). Interestingly, PSMA9-PHPMA70 worms undergo an unusual (partial) worm-to-vesicle transition at elevated temperature. Finally, PSMA9-PHPMA50 spheres were evaluated as putative Pickering emulsifiers. Using lower water volume fractions produced water-in-oil (w/o) emulsions after high shear homogenization, as expected. However, using higher water volume fractions, shear rates or copolymer concentrations favored the formation of w/o/w Pickering double emulsions.
- This article is part of the themed collections: Polymerization-Induced Self-Assembly (PISA) and Celebrating our 2020 Prize and Award winners
 Please wait while we load your content...
Please wait while we load your content...

