
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pro-organic radical contrast agents (“pro-ORCAs”) for real-time MRI of pro-drug activation in biological systems†
Hung V.-T.
Nguyen‡
 abcd,
Alexandre
Detappe‡
bcde,
Peter
Harvey
f,
Nolan
Gallagher
a,
Clelia
Mathieu
cd,
Michael P.
Agius
cd,
Oksana
Zavidij
cd,
Wencong
Wang
a,
Yivan
Jiang
a,
Andrzej
Rajca
g,
Alan
Jasanoff
fhi,
Irene M.
Ghobrial
cd,
P. Peter
Ghoroghchian
*bcd and
Jeremiah A.
Johnson
*ab
abcd,
Alexandre
Detappe‡
bcde,
Peter
Harvey
f,
Nolan
Gallagher
a,
Clelia
Mathieu
cd,
Michael P.
Agius
cd,
Oksana
Zavidij
cd,
Wencong
Wang
a,
Yivan
Jiang
a,
Andrzej
Rajca
g,
Alan
Jasanoff
fhi,
Irene M.
Ghobrial
cd,
P. Peter
Ghoroghchian
*bcd and
Jeremiah A.
Johnson
*ab
aDepartment of Chemistry, Massachusetts Institute of Technology (MIT), 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
bDavid H. Koch Institute for Integrative Cancer Research, MIT, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: jaj2109@mit.edu; ppg@mit.edu
cDepartment of Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, Massachusetts 02215, USA
dHarvard Medical School, 25 Shattuck Street, Boston, Massachusetts 02115, USA
eCentre Paul Strauss, 3 Rue de la Porte de l'Hopital, 67000 Strasbourg, France
fDepartment of Biological Engineering, MIT, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
gDepartment of Chemistry, University of Nebraska, Lincoln, Nebraska 68588, USA
hDepartment of Brain and Cognitive Sciences, MIT, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
iDepartment of Nuclear Science and Engineering, MIT, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
First published on 26th June 2020
Abstract
Nitroxide-based organic-radical contrast agents (ORCAs) are promising as safe next-generation magnetic resonance imaging (MRI) tools. Nevertheless, stimuli-responsive ORCAs that enable MRI monitoring of prodrug activation have not been reported; such systems could open new avenues for prodrug validation and image-guided drug delivery. Here, we introduce a novel “pro-ORCA” concept that addresses this challenge. By covalent conjugation of nitroxides and drug molecules (doxorubicin, DOX) to the same brush-arm star polymer (BASP) through chemically identical cleavable linkers, we demonstrate that pro-ORCA and prodrug activation, i.e., ORCA and DOX release, leads to significant changes in MRI contrast that correlate with cytotoxicity. This approach is shown to be general for a range of commonly used linker cleavage mechanisms (e.g., photolysis and hydrolysis) and release rates. Pro-ORCAs could find applications as research tools or clinically viable “reporter theranostics” for in vitro and in vivo MRI-correlated prodrug activation.
Introduction
Theranostic agents—systems that simultaneously provide therapeutic and diagnostic modalities—are promising tools for personalized medicine.1–4 These agents can, in principle, afford molecular insights into disease states and non-invasively report in real-time on the accumulation, release, and efficacy of therapeutic agents, thereby enabling real-time therapeutic selection.3,5,6 Since their multifunctional requirements are difficult to achieve with small molecules, theranostic systems are often based on nanoscale materials, which themselves possess useful properties such as controllable size and shape, high loading/multiplexing capability, tunable circulation half-life, predictable biodistribution, and targeted tissue accumulation.7–9 Indeed, a plethora of theranostic agents based on nanoscale material scaffolds (e.g., liposomes, micelles, dendrimers, and polymers) carrying a wide range of therapeutic entities (e.g., immuno/chemotherapies and gene therapies) and high sensitivity (fluorescence, PET) or high-resolution (CT, MRI) imaging modalities have been reported.10–24“Smart” or “reporter” theranostics that leverage their imaging component to elucidate material-disease tissue interactions such as drug release kinetics, efficacy, or acquired therapeutic resistance are especially powerful.3,25–28 Elegant work using fluorescence- or FRET-based reporters of cellular apoptosis or drug release within the tumor microenvironment have been developed.29–36 Despite the highly innovative nature of these approaches and their immense utility as research tools, the translational potential of fluorescence imaging may be limited by light penetration depth and tissue autofluorescence. Alternatively, MRI offers high resolution, safety, and established clinical utility.24,25 “Reporter” theranostic materials that allow for correlation of drug release with MRI signal changes upon release of Gd-based contrast agents (CAs) or changes to the delivery vehicle itself have been reported.37–40 Though metal-based MRI CAs have found widespread clinical utility, they continue to face safety concerns41,42 that have led to discontinuation or withdrawal of several products.43–47 Metal-free organic radical contrast agents (ORCAs) have been intensely studied in recent years to overcome the safety issues of metal-based CAs;48–58 however, to our knowledge, there are no reports of theranostic ORCAs for MRI-correlated drug release.
We have extensively investigated bottlebrush and related brush-arm star polymer (BASP) polymer architectures for applications in self-assembly, energy storage, drug delivery and imaging, including as ORCAs for MRI.59–71 BASPs can be readily covalently conjugated to biocompatible components such as polyethylene glycol (PEG) as well as single or multiple prodrug payloads or imaging agents through the use of branched macromonomers (MMs).59–67,71 PEG-based BASPs have displayed excellent safety profiles in mice and higher species, their synthesis is highly scalable and reproducible, and through MM design, the BASP prodrug activation mechanism and rate can be precisely defined.59–67 BASPs carrying a high concentration of spirocyclohexyl nitroxides (chex72) currently represent promising ORCAs for MRI, proving useful for T2-weighted MRI of tumors in vivo.64–66,71
A significant challenge in the field of polymer-based prodrugs, including BASP prodrugs, lies in the real-time monitoring of prodrug activation, i.e., conversion of polymer-bound prodrug into free drug, in biological systems. Prodrug activation kinetics can significantly impact therapeutic outcomes: activation that occurs too quickly or too slowly can be detrimental.62 Most prodrugs do not provide facile spectroscopic readouts upon activation; thus, liquid chromatography with tandem mass spectrometry (LC-MS-MS) or inductively coupled plasma mass spectrometry (ICP-MS) are traditionally used to quantify prodrug versus free drug in tissue samples of interest. These methods require tissue harvesting and thus are inherently invasive, ex vivo techniques. The development of non-invasive, safe, “turn-ON/OFF” MRI probes that enables real-time correlation of prodrug activation could offer powerful new research tools and clinical readouts for prodrug development.
Toward addressing this challenge, we describe herein a novel “pro-ORCA” design concept wherein the release of covalently conjugated ORCAs from a BASP scaffold is shown to induce a large (∼30-fold) change in magnetic relaxivity that correlates with prodrug activation. Our proof-of-principle pro-ORCA system is based on BASPs that feature both nitroxide ORCAs (for MRI imaging) and the Top2 poison doxorubicin (DOX) covalently conjugated to the same BASP through chemically equivalent linkers that cleave in response to either external or endogenous triggers (Fig. 1). Linker cleavage events, i.e., pro-ORCA and prodrug activation, cause the nitroxide and DOX to diffuse away from the BASP. The former leads to a ∼30-fold change in transverse relaxivity and a concomitant change in MRI contrast, while the latter leads to DOX-induced cell death, enabling MRI-based, real-time monitoring of prodrug activation in a simple, modular format. This basic pro-ORCA concept (Fig. 1) is shown to apply to multiple widely-used triggering mechanisms, variable release kinetics, and in vitro and in vivo models, offering a promising new concept for MRI-guided prodrug activation.
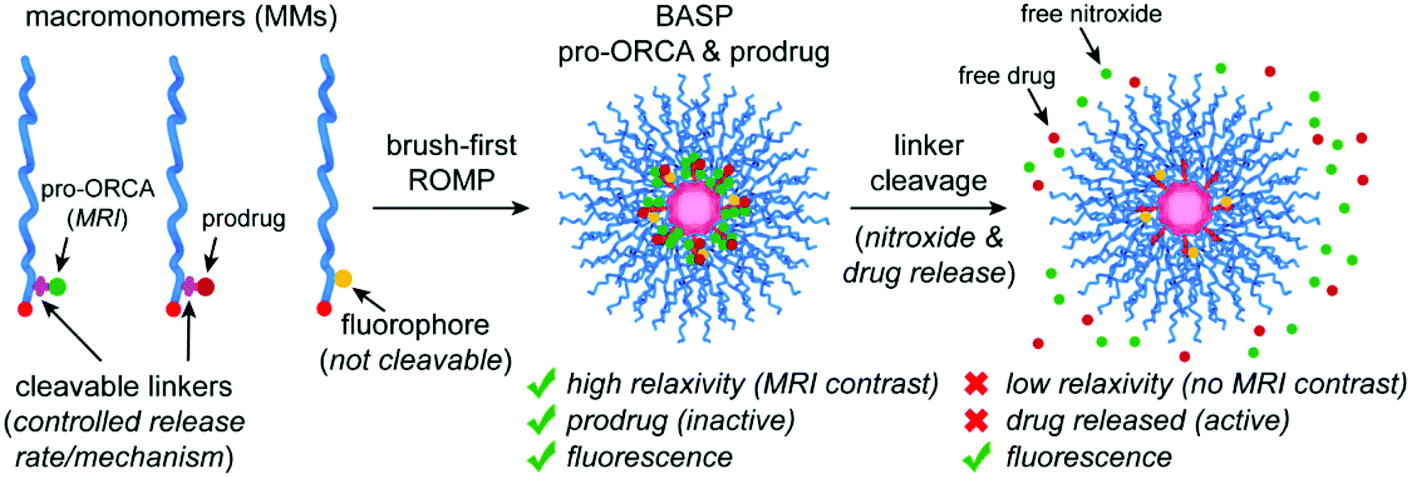 | ||
| Fig. 1 Design concept for brush-arm star polymer (BASP) pro-ORCAs. Macromonomers (MMs) featuring a pro-ORCA nitroxide (for MRI) and a prodrug (therapeutic) conjugated through cleavable linkers are copolymerized with a non-cleavable fluorophore-conjugated MM via the brush-first ring-opening metathesis polymerization (ROMP) method. The resulting BASP, which carries both the pro-ORCA and prodrug on the same polymer, displays high transverse relaxivity (r2) enabling T2-weighted MRI. Moreover, the prodrug is therapeutically inactive. Upon cleavage of the linkers that connect the nitroxide and drug to the BASP, i.e., pro-ORCA and prodrug activation, the nitroxide relaxivity drops significantly leading to a change of MRI contrast while the drug becomes therapeutically active. Thus, MRI signal changes (in this case, return of negative T2 contrast back to baseline) can be correlated with prodrug activation. Meanwhile, the fluoroscence signal should not change significantly before and after nitroxide and drug release, providing a constant imaging handle to visualize the BASP. | ||
Experimental section
Relaxivity and in vitro measurements by MRI
Phantom MRI data were acquired in a 12 cm outer diameter birdcage transceiver for imaging in a 20 cm bore Bruker 7T Avance III MRI scanner. For relaxivity determination, samples at varying concentrations in PBS buffer were used. For in vitro measurements, samples were prepared as described below, and loaded into a 384-well clear polystyrene plate (Thermo Scientific Nunc), which had been pre-cut in half to optimally fit the coil. Unused wells were filled with PBS buffer. 2 mm slices were imaged through the samples with the field of view of 5 × 5 cm; and, the data matrices were 256 × 256 points. Longitudinal (r1) and transverse (r2) relaxivity measurements were acquired using multi-spin multi-echo (MSME) sequences (flip angle = 180°). r1; TE = 12 ms, TR = 300, 350, 400, 450, 500, 600, 800, 1000, 1200, 1500, 3000, 5000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms. r2; TR = 5000 ms, TE = 12, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, 168, 280, 192, 204, 216, 228, 240, 252, 264, 276, 288, 300, 312, 324, 336, 348, 360 ms. Custom routines written in Matlab (Mathworks, Natick, MA) were used to reconstruct the images and compute relaxation time constants by fitting image intensity data to exponential decay curves.
000 ms. r2; TR = 5000 ms, TE = 12, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, 168, 280, 192, 204, 216, 228, 240, 252, 264, 276, 288, 300, 312, 324, 336, 348, 360 ms. Custom routines written in Matlab (Mathworks, Natick, MA) were used to reconstruct the images and compute relaxation time constants by fitting image intensity data to exponential decay curves.
For in vitro measurements, cells (A549, MM.1S, and KMS11) were plated at 10000 cells per well in a 96 well plate and incubated overnight. PC2 (17 mg BASP mL−1, 107 μM DOX) was then added, and cells were incubated for another 2 h. Excess PC2 was removed via media wash, and fresh media was added. Cells were then either exposed to UV for 30 min (+UV) or not (−UV). Cells were then incubated for pre-determined time points (2 h, 6 h, 24 h, or 48 h), then subject to either MRI or viability assay (CellTiter-Glo, following standard operating procedure). For cells that were imaged by MRI, the media was removed. The wells were then filled with PBS (50 μL), mixed with 1% Triton X-100, transferred to a 384 well plate, and imaged using the MRI protocol described above.
Cell culture
Human multiple myeloma cells (MM.1S and KMS11, ATCC) were cultured in RPMI media (Thermo Fisher Scientific) and were supplemented with 10% fetal bovine serum (FBS, VWR), 1% penicillin/streptomycin (Thermo Fisher Scientific), and 1% glutamine (Thermo Fisher Scientific). Human lung adenocarcinoma cells (A549, ATCC) were cultured in RPMI media, which was supplemented with 10% FBS and 1% penicillin/streptomycin. Cell lines were authenticated by short tandem repeat DNA profiling and were confirmed to be mycoplasma negative, using the MycoAlert Mycoplasma Testing Kit (Lonza). All cells were housed in 5% CO2 humidified atmosphere at 37 °C.In vitro cell viability
MM.1S, KMS11, and A549 cells were plated at 10000 cells per well overnight in a 96 well plate. The media was then replaced with fresh media containing BASPs at various concentrations. The plate was incubated for 48 h unless otherwise stated; cell viability was then determined using the CellTiter-Glo assay (Promega).
For viability assays involving DOX-PC or chex-PC, cells were plated at 10000 cells per well in a 96 well plate and incubated overnight. Cells were incubated for 2 h with DOX-PC or chex-PC at various concentrations. Excess DOX-PC was removed via media wash, and fresh media was added. Cells were then either exposed to UV for 30 min (+UV) or not (−UV). Viability was evaluated at 48 h with CellTiter-Glo.
Animal usage
All animal studies were conducted under federal, state, and local guidelines in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals with approval from the Massachusetts Institute of Technology Committee on Animal Care. MR in vivo tumor imaging was performed at the Brain and Cognitive Science Complex at MIT. IVIS imaging was performed at the David H. Koch Institute for Integrative Cancer Research at MIT. NCR/NU mice (female, 8–12 weeks old, Taconic; n = 3 mice per group) were used for imaging studies.In vivo therapeutic efficacy of DOX-MHC-BASP
A549 cells were cultured following the protocol described above to a final concentration of 20%. Cells were then harvested, mixed with Matrigel and sterile pH 7.4 PBS buffer (1:1), filtered through sterile 0.2 μm filters, and injected subcutaneously (2.0 × 106 cells) into the hind flank of NCR-NU mice. Tumor growth was monitored for 2–4 weeks until appropriate cumulative diameters (∼1 cm, measured by a digital caliper) were achieved. Tumor-bearing mice were then randomized into groups of n = 3 and injected intratumorally with 50 μL of DOX-M at varying concentrations (1, 2.5, 5, 10 mg BASP mL−1). Tumor growth was then accessed via caliper measurements for 25 days.
In vivo MRI instrumentation
In vivo MRI was acquired using a Bruker 9.4T Biospec MRI scanner using a cross coil volume transmitter and surface receiver configuration. The tumor region was localized in the focal spot of the surface coil, with the animal restrained in order to allow unhindered breathing while minimizing motion around the hind leg and tumor. Axial T1 weighted images (T1WIs) were collected using a RARE pulse sequence with TR = 721.1 ms; TE(eff) = 11.8 ms; RARE factor = 4; FOV = 30 × 30 mm2; 256 × 256 matrix and 4 averages over 12 slices of 1 mm thickness and 0 mm gap, with a total scan time of 2 min 18 s. Axial T2 weighted images (T2WIs) were collected using a RARE pulse sequence with TR = 4000 ms; TE(eff) = 48 ms; RARE factor = 8; FOV = 30 × 30 mm2; 256 × 256 matrix and 2 averages over 12 slices of 1 mm thickness and 0 mm gap, with a total scan time of 3 min 12 s. Images were analyzed using either ImageJ or custom routines written in Matlab (Mathworks, Natick, MA).In vivo MRI in tumor-bearing mice
Tumor-bearing NCR-NU mice were generated as described above. MRI and NIRF images were acquired for each animal (n = 3 mice per group) before injections. BASPs were prepared, passed through a sterile 0.2 μm filter, and administered directly into the tumor via intratumoral injections (50 μL of 70 mg mL−1 BASPs, F, M, or S). Tumor imaging was done at pre-determined time points.In vivo MRI data analysis
T 2-weighted images were inverted before analysis was performed so that high T2 regions would appear bright. Signal intensities pre- and post-injection were compared using only slices in which tumors and muscle were clearly visible. Using ImageJ software (v.1.52i), a region of interest (ROI) around each component was manually drawn. The average signal intensities and areas of the ROIs were measured; these data were then normalized against the signal intensity of the muscle tissue. This process was repeated for all relevant slices for a given organ; the sum of these signal intensities was then calculated and divided for the total area, affording the volume-averaged signal intensity. SNR variations were acquired via normalization against the earliest time point. A detailed step-by-step analysis is included in the ESI.†Ex vivo fluorescence imaging
For the 24 h time point, mice were euthanized and their tumors were harvested (n = 3 mice per group). Tumor slices were mounted with DAPI staining and imaged with an upright Carl Zeiss microscope with an HXP 120C light source at 20× magnification (DAPI λex/λem = 365/445 nm; Cy5.5 λex/λem = 640/690 nm; DOX λex/λem = 470/525 nm).Results and discussion
We initially targeted photoresponsive linkers to establish the pro-ORCA concept for MRI-correlated prodrug activation. Photocleavable linkers based on ortho-nitrobenzyl (ONB) derivatives have been widely used in the context of protecting group chemistry, drug delivery, and materials applications,73–79 and while such linkers may have translational limitations, they offer powerful research tools for triggering specific, light-induced biological responses. DOX was chosen as the therapeutic payloads due to its clinical utility, its established mechanisms of action (in both prodrug and free drug forms), and its inherent fluorescence, the latter of which provides a useful secondary imaging handle for our proof-of-concept studies.59–61,67,80 We have previously shown that conjugation of DOX to BASPs via an ONB linker deactivates the drug (i.e., it is a prodrug), providing no detectable release or cytotoxicity in cell culture over 72 h in the absence of 365 nm light.59–61,67 Here, we reasoned that this “on/off” nature of light-induced release would simplify our system by enabling a binary readout upon irradiation. A previously reported MM with DOX59 and a new MM with chex conjugated via chemically identical ONB linkers (Scheme 1A, see ESI† for full synthetic details) were prepared; their structures were confirmed by electron paramagnetic resonance spectroscopy (EPR; Fig. S1†), matrix-assisted laser desorption ionization time-of-flight mass spectroscopy (MALDI-ToF MS; Fig. S2†), and nuclear magnetic resonance (NMR) spectroscopy (Fig. S3†) where appropriate. Four BASPs comprising either 100% chex (chex-PC) or 100% DOX (DOX-PC) or different chex:DOX ratios (1:1 and 27:1; PC1 and PC2, respectively, Table 1) were synthesized via the brush-first ring-opening metathesis polymerization (ROMP) strategy using an acetal-based bis-norbornene cross-linker (AXL) and Grubbs 3rd-generation bis-pyridine complex (G3) (Scheme 1B).59–62,66 Leveraging the efficiency of ROMP and the branched MM design,59–66 different chex:DOX ratios can be conveniently realized by altering the stoichiometry of MM feed ratios. Gel permeation chromatography (GPC) and dynamic light scattering (DLS) analyses confirmed high MM-to-brush and brush-to-BASP conversions and consistent sizes and size distributions for these four particles; the hydrodynamic diameters (Dh) were all ∼22 ± 5 nm (Fig. S4a† and Table 1), highlighting the payload-agnostic nature of BASP synthesis. A cy5.5-based MM (Cy-MM) was also incorporated into BASPs that were intended to be deployed for in vivo studies; the cy5.5 dye is non-releasable, providing an independent imaging handle for the BASP scaffold that enables validation of the pro-ORCA design (vide infra). EPR spectroscopy revealed the characteristic broadening associated with conjugation of chex to BASPs (Fig. S4b†).64–66 Moreover, large per-chex r2 relaxivity enhancements compared to the model nitroxide 3-CP (∼30-fold), similar to those reported previously upon conjugation of chex to BASPs,64–66 were observed (Table 1). This result was encouraging, as it confirmed that chex-conjugated BASPs display high r2 values when cleavable pro-ORCAs linkers are used (previous work has exclusively used non-releasable chex linkers) and in the presence of a second payload such as DOX bound to the same polymer.
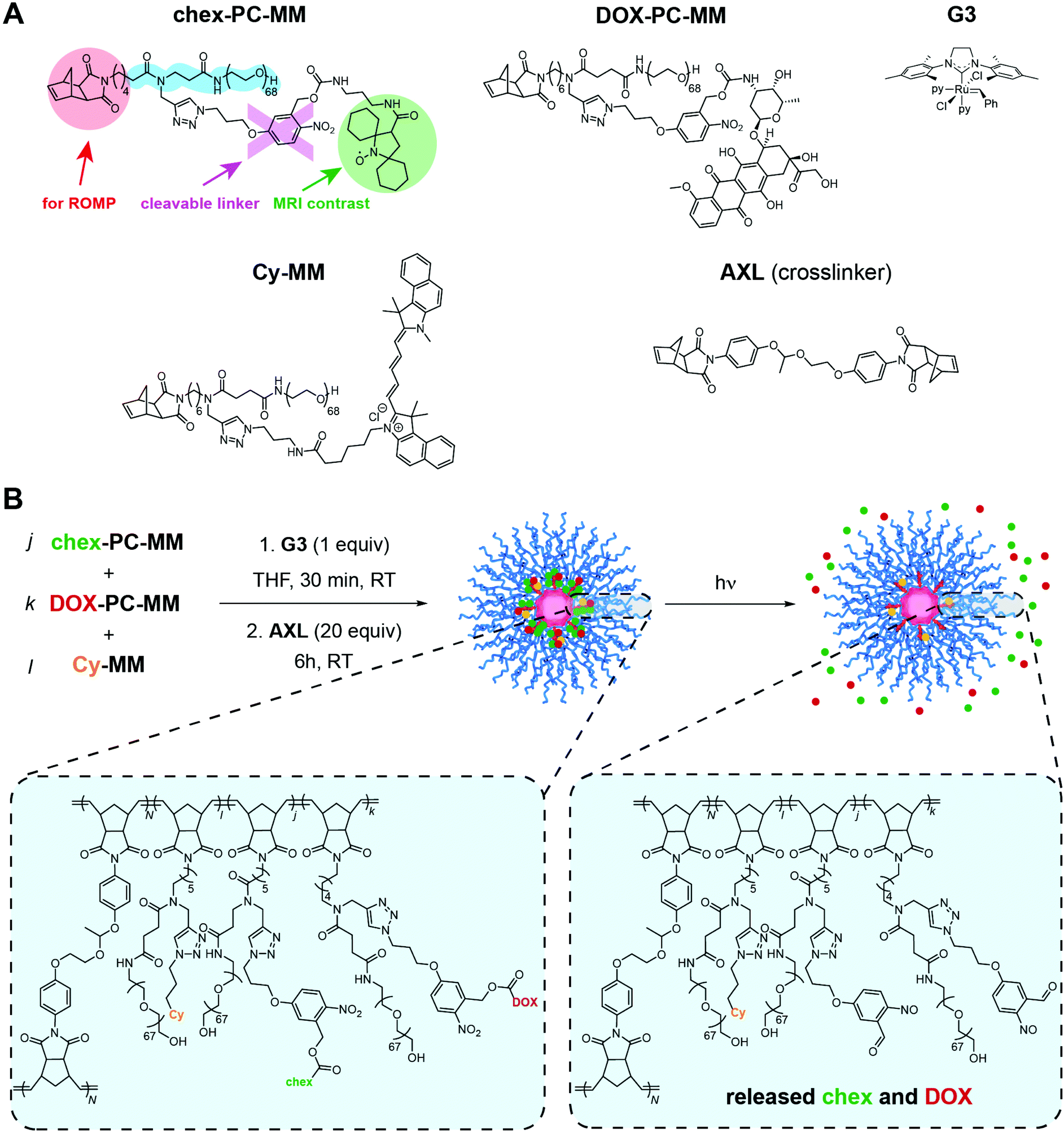 | ||
| Scheme 1 Photoactivatable BASP pro-ORCA + prodrug design. (A) Chemical structures of MMs chex-PC-MM, DOX-PC-MM, and Cy-MM. The latter two MMs were reported previously, while chex-PC-MM was newly synthesized to enable photocontrolled release of chex. Grubbs 3rd generation bis-pyridine complex (G3, py = pyridine) and acetal-based crosslinker AXL structures are also shown. (B) Brush-first ROMP was conducted by exposing mixtures of the MMs listed above to G3 to generate short bottlebrush polymers. Subsequent addition of AXL induces cross-linking to provide photocleavable BASP “reporter” theranostic agents (e.g., PC1 and PC2). Exposure to light (365 nm) releases chex and DOX from the BASP, inducing concomitant changes in MRI signal and therapeutic efficacy. | ||
| Sample name | Linker | chex:G3 | DOX:G3 | cy:G3 | D h/nm | r 1/mM−1 s−1 | r 2/mM−1 s−1 |
|---|---|---|---|---|---|---|---|
| PC = photocleavable; HC = hydrolytically cleavable (ester-based linker); G3 = Grubbs 3rd generation bis-pyridine complex; Dh = hydrodynamic diameter as measured by dynamic light scattering; per-nitroxide r1 and r2 values; S, M, and F refer to the ester linker used to form the corresponding BASP: slow-, medium-, and fast-releasing. | |||||||
| DOX-PC | PC | 0 | 7 | 0 | 22 ± 5 | — | — |
| chex-PC | PC | 7 | 0 | 0 | 21 ± 4 | 0.31 | 5.05 |
| PC1 | PC | 3.5 | 3.5 | 0 | 22 ± 4 | 0.25 | 5.43 |
| PC2 | PC | 6.75 | 0.25 | 0 | 22 ± 6 | 0.29 | 4.91 |
| DOX-S | HC, S | 0 | 7 | 0 | 21 ± 3 | — | — |
| chex-S | HC, S | 7 | 0 | 0 | 20 ± 4 | 0.27 | 5.02 |
| S | HC, S | 6 | 1 | 0.07 | 21 ± 5 | 0.29 | 5.01 |
| DOX-M | HC, M | 0 | 7 | 0 | 21 ± 4 | — | — |
| chex-M | HC, M | 7 | 0 | 0 | 20 ± 5 | 0.22 | 4.37 |
| M | HC, M | 6 | 1 | 0.07 | 20 ± 5 | 0.21 | 4.69 |
| DOX-F | HC, F | 0 | 7 | 0 | 20 ± 3 | — | — |
| chex-F | HC, F | 7 | 0 | 0 | 18 ± 5 | 0.32 | 4.94 |
| F | HC, F | 6 | 1 | 0.07 | 21 ± 5 | 0.26 | 4.85 |
| C | N/A | N/A | N/A | N/A | 20 ± 4 | — | — |
| 3-CP51 | N/A | N/A | N/A | N/A | N/A | 0.15 | 0.17 |
To examine light-induced pro-ORCA and pro-drug activation, DOX-PC, chex-PC, and PC1 (1:1 chex:DOX) were exposed to 365 nm light (see ESI† for details). Samples of the reactions were taken at various timepoints and were analyzed by liquid chromatography-MS (LC-MS). The release behaviors were consistent for the three BASPs (Fig. 2A) with maximal release occurring with ∼30 min of light exposure regardless of the payload (i.e., DOX, chex, or 1:1 DOX:chex). As such, an exposure duration of 30 min was used for subsequent experiments.
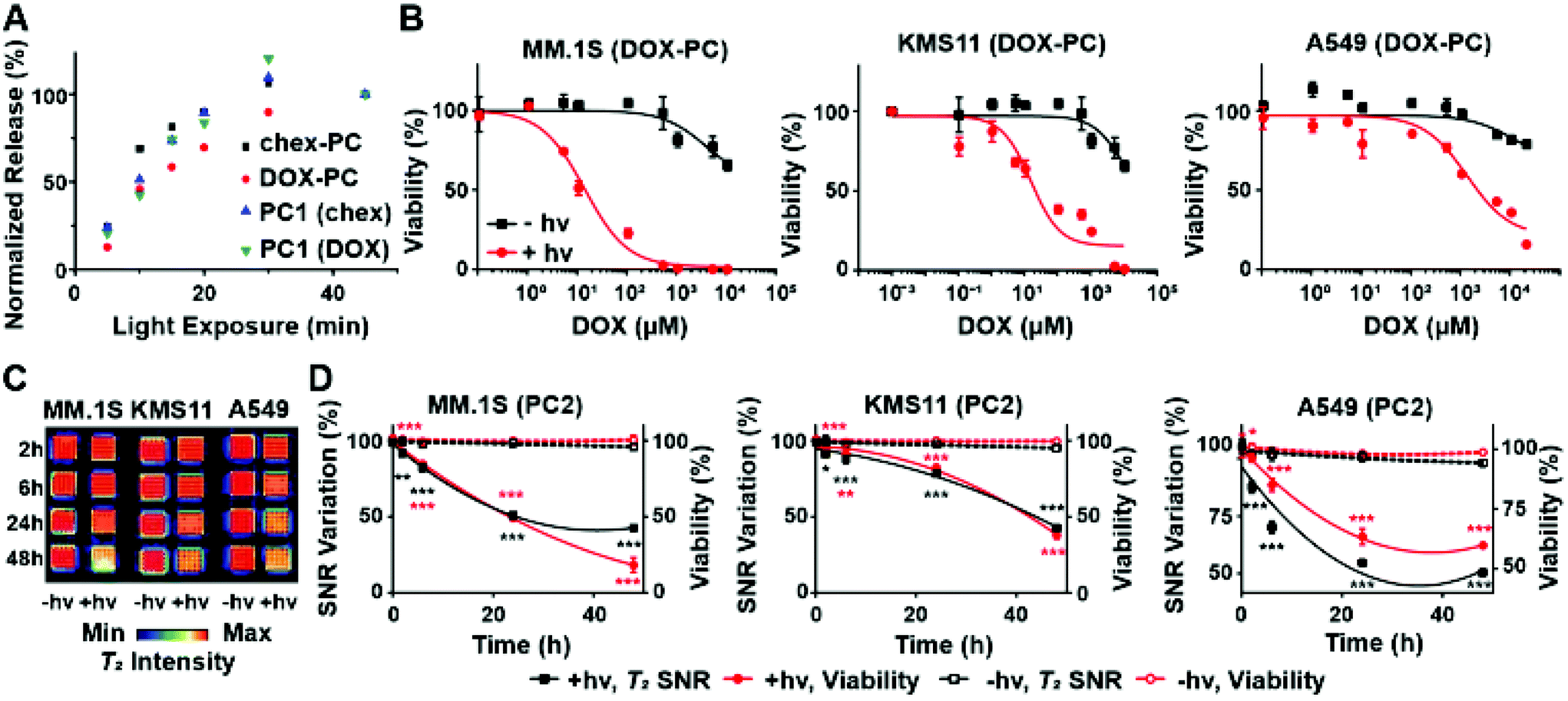 | ||
| Fig. 2 In vitro investigations of photocleavable (PC)-BASP pro-ORCAs and prodrugs. (A) Chex and DOX release from PC-BASPs as a function of irradiation (365 nm) time as determined by liquid chromatography-mass spectrometry (LC-MS). (B) In vitro toxicity of DOX-PC with (red curves) or without (black curves) light-triggered release as examined upon addition to different cancer cell lines (multiple myeloma – MM.1S and KMS11; lung adenocarcinoma – A549). Data are presented as mean ± SEM (n = 3). (C) T2-Weighted MRI of cancer cells 2, 6, 24, and 48 h after addition of PC2. False-color inverse-contrast was applied for visualization. (D) MRI signal quantification, revealing changes in SNR that track with incubation time post-UV. Data are presented as mean ± SEM (n = 3); statistical analyses between pre- and post-light exposure (±hυ) for T2 MRI SNR variation (black) and cell viability (red) were performed using 2-tailed student t-test (n.s.: not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). | ||
To confirm that photoinduced DOX prodrug activation induces cell death, DOX-PC was incubated with suspended multiple myeloma (MM.1S and KMS11) and adherent lung adenocarcinoma (A549) cells for 3 h to provide sufficient time for uptake. Excess DOX-PC was then removed by media washing, and the cells were exposed to 365 nm light for 30 min. After incubation for an additional 48 h, cell viability was assessed using the CellTiter-Glo kit (Fig. 2B). Consistent with our expectations, significantly greater toxicity was observed with all cell lines following exposure to 365 nm light when compared to its absence; note that exposure of cells to 365 nm light in the absence of DOX-PC induced no observable toxicity (99+% viability in all 3 cell lines). Moreover, irradiation of cells exposed to chex-PC rather than DOX-PC under the same conditions led to no meaningful effects at similar concentrations (Fig. S5†). These results confirm the low/no in vitro toxicities imparted by the pro-ORCA alone, the polymer components of the BASP, or from 365 nm light. Only light-triggered DOX prodrug activation induces significant cell death.
Motivated by these results, we examined the use of PC2 for correlation of DOX prodrug activation with changes in MRI contrast in vitro. PC2, which has a chex:DOX ratio of 27:1, was designed to maximize the MRI signal at practical concentrations (i.e., high chex loading) and to still provide sufficient amounts of DOX to induce toxicity following 365 nm light exposure. Cells were incubated with PC2 (17 mg PC2 mL−1; 107 μM of conjugated DOX) following the same procedure as described above. The cells were imaged using a 7T MRI scanner (Fig. 2C). MRI SNR was measured as a function of time. In support of our design concept, these SNR values correlated with cell viability (Fig. 2D). Upon irradiation, a diminished T2 MRI signal was observed as the molar transverse relaxivity of the nitroxide decreases by ∼30-fold upon release (Table 1, r2 of water-soluble chex analogue 3-CP = 0.17 mM−1 s−1 compared to PC2, which has a per-chex r2 of 4.91 mM−1 s−1). Substantial SNR changes were observed in all three irradiated cell lines (Fig. 2D, black solid curves) when compared to non-irradiated cells (Fig. 2D, black dotted curves). For instance, in the case of MM.1S a 57 ± 3% decrease in the T2 signal was observed from irradiated cells as compared to 5 ± 2% for non-irradiated cells. Finally, the MRI SNR data correlated with cytotoxicity (Fig. 2D, red solid curves) at all time points up to 48 h, indicating that chex and DOX are cleaved and diffuse away from the BASP at similar rates and that the latter was therapeutically active. Altogether, these results demonstrate proof-of-principle for the pro-ORCA concept, showing that light-induced prodrug activation correlates with MRI signal in vitro (Fig. 2D).
Having established proof-of-concept for our pro-ORCA design, we sought to further demonstrate its generally using more translationally-relevant ester-based prodrugs.81 Three ester linkers were designed and used to prepare pro-ORCAs and DOX prodrugs (Scheme 2A): one that was previously reported to display excellent in vivo efficacy when conjugated to DOX60,61 (referred to herein as “medium”) and two novel linkers with steric and electronic properties tuned to afford relatively “fast” and “slow” hydrolysis kinetics in vitro. These linkers utilize the same release mechanism: hydrolytic cleavage of the aromatic ester (rate-determining step), followed by a 1,6-elimination73 to release free DOX. Control over their hydrolysis rate is predictable and tunable using rational structural modifications. Using these 3 linkers, 6 different MMs were synthesized: DOX-S-MM; DOX-M-MM;60DOX-F-MM; chex-S-MM; chex-M-MM; and chex-F-MM where S, M, and F correspond to the linker structure: slow, medium and fast, respectively (Scheme 2A, see ESI† for synthetic details). To validate the structures of these MMs (Fig. S6–18†), EPR spectroscopy (Fig. S6, S9, and S12†), NMR spectroscopy (Fig. S7, S10, S13, S15 and S17†), and MALDI-ToF MS (Fig. S8, S11, S14, S16, and S18†) were deployed where appropriate.
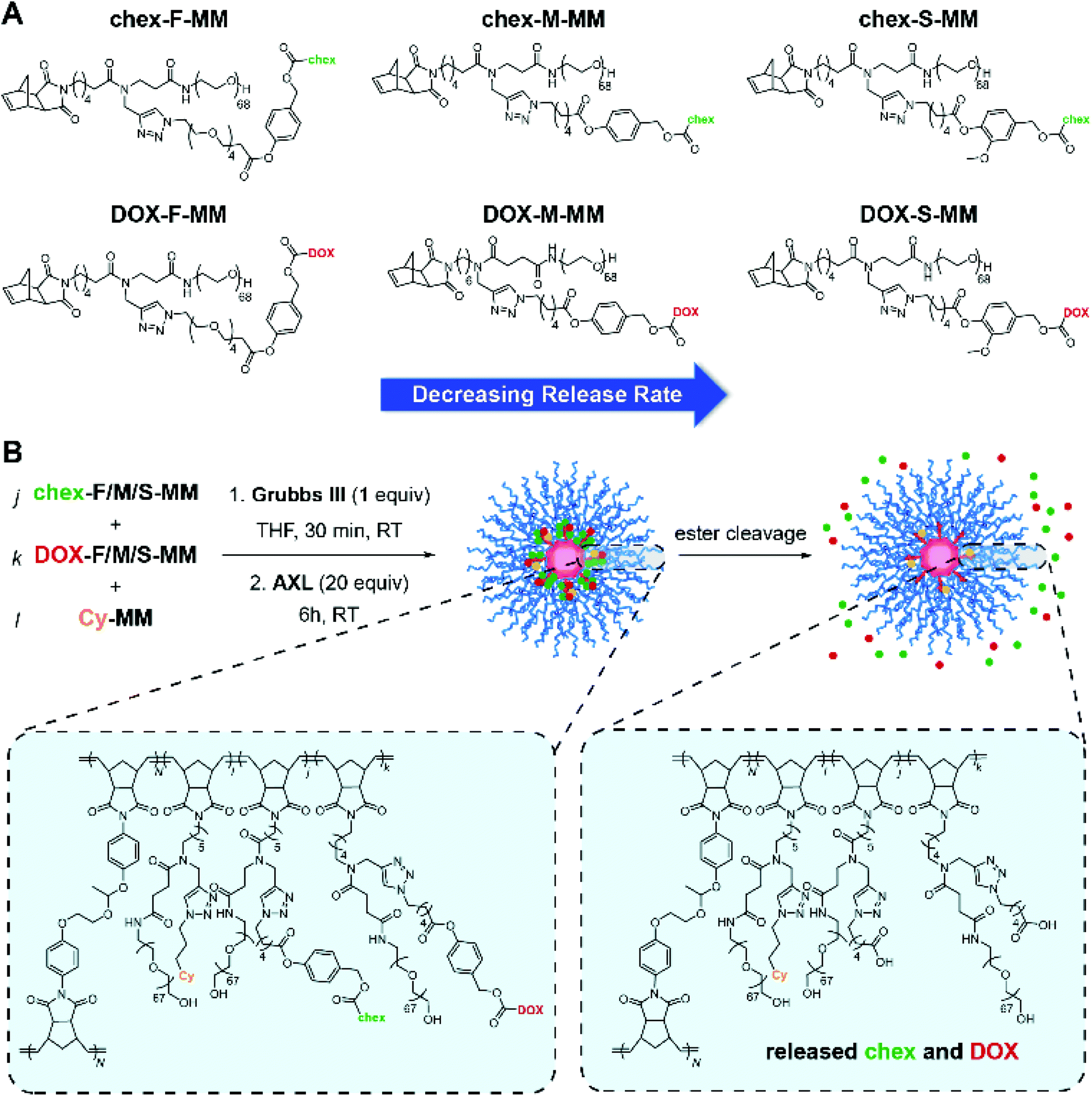 | ||
| Scheme 2 Hydrolytically labile ester-based BASP pro-ORCAs and prodrugs. (A) Chemical structures of 6 synthesized MMs featuring chex or DOX conjugated via slow, medium, or fast releasing linkers. (B) Brush-first ROMP was used to synthesize BASP “reporter” theranostic agents. Here, release of chex and DOX via ester hydrolysis leads to concomitant MRI and efficacy changes that correlate with the measured hydrolysis rates. | ||
To confirm their relative release rates, the MMs were incubated in PBS (pH 7.4) buffer; the amounts of released DOX or chex were quantified as a function of time via LC-MS. The pseudo-first order rate constants for release of DOX from DOX-F-MM, DOX-M-MM, and DOX-S-MM were 21.26 × 10−3 h−1, 8.95 × 10−3 h−1, and 3.62 × 10−3 h−1, respectively (Fig. 3A and Table S1†) while the pseudo-first order rate constants for release of chex from chex-F-MM, chex-M-MM, and chex-S-MM were 8.23 × 10−3 h−1, 4.31 × 10−3 h−1, and 1.63 × 10−3 h−1, respectively (Fig. S15 and Table S1†). Although these rate constants are not identical across DOX and chex pairs due to differences in the physical properties (e.g., hydrophobicity) of chex and DOX, the DOX:chex release rate ratios were similar: 2.58, 2.08, and 2.22 for the F, M, and S pairs, respectively, which enables correlation of DOX prodrug and pro-ORCA activation (vide infra). We note that these in vitro release kinetics studies were conducted using MMs rather than BASPs to ease sample handling and analysis. Though release from BASPs is generally much slower than from MMs using the same linker, trends in release kinetics across different linkers are preserved.62 Next, these 6 MMs were used to prepare 6 different singly-loaded BASPs (DOX-F, DOX-M, DOX-S, chex-F, chex-M, chex-S) (Scheme 2B). GPC and DLS results confirmed that the BASP sizes (∼21 nm) were independent of the payload (DOX or chex) or linker (F, M, or S); EPR spectroscopy and MRI-measured per-chex r2 values for the three BASP pro-ORCAs showed that the magnetic properties of chex were not meaningfully affected by the linkers (Fig. S20–22† and Table 1).
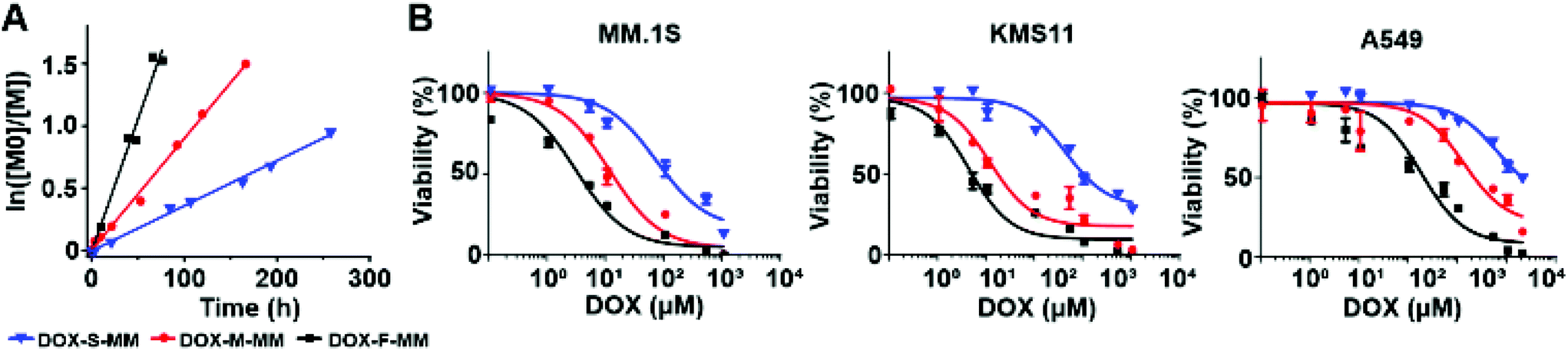 | ||
| Fig. 3 In vitro investigations of hydrolytically labile MMs and BASPs based on ester linkers. (A) DOX release kinetics from DOX-S-MM (blue), DOX-M-MM (red), and DOX-F-MM (black) in neutral PBS buffer as a function of incubation time as determined by LC-MS. Note: release assays were conducted for MMs rather than BASPs for ease of analysis. Though release is generally much slower from BASPs compared to MMs using the same linker, trends in release rates as a function of linker are preserved. (B) In vitro toxicity of DOX-S (blue), DOX-M (red), and DOX-F (black) in different cell lines. Data are presented as mean ± SEM (n = 3). | ||
Cell viability assays were conducted with the same 3 cell lines used above following 48 h incubation with these 6 BASPs. As expected, negligible toxicity was observed for the pro-ORCAs lacking DOX (Fig. S23†). In contrast, the toxicity of the DOX prodrug BASPs increased with the rate of DOX release (Fig. 3B); in MM.1S cells, DOX-F, DOX-M, and DOX-S exhibited IC50 values of 3.1 μM, 17.4 μM, and 113.3 μM DOX, respectively. Given that these BASPs have similar sizes and compositions (Table 1) and, thus, expectedly similar cellular internalization rates, this toxicity trend is likely due to varying levels of DOX prodrug activation and release within the 48 h incubation period. Moreover, the DOX-F viability curve approaches that of free DOX (IC50 of 2.9 μM for MM.1S), suggesting that the majority of DOX may be released from this material in this timeframe (Fig. S24†). Next, viability assessments of MM.1S and A549 were performed as a function of exposure time to each DOX-containing BASP using a fixed dose of 0.5 mg mL−1 BASP (83–86 μM DOX; Fig. S25†). Validating our prodrug linker design, the toxicity toward both cell lines followed the same trend: DOX-F > DOX-M > DOX-S.
In order to inform subsequent studies of MRI-guided monitoring of prodrug activation, we studied the in vivo toxicity of DOX-M using a subcutaneous lung adenocarcinoma (A549) model. A549 cells were injected into the hind flank of BALB/c mice (4 groups of n = 3 mice). Once the tumors reached ∼1 cm in diameter, the mice were randomized and each received a single 50 μL intratumoral dose (day 0) of 0 (blank), 1, 5, or 10 mg mL−1 of DOX-M. Intratumoral administration was selected to maximize tumor MRI SNR and preclude variables of tumor accumulation and pharmacokinetics, thereby simplifying imaging studies for this proof-of-concept work (vide infra). It should be noted that intratumoral drug delivery is a clinically-viable strategy for cancer therapy;82–85 ∼200 ongoing clinical trials leverage this method of administration. Moreover, intratumoral administration of BASPs has not been demonstrated before. In our study, significant tumor growth inhibition was observed over the 25 d period of observation following the administration of DOX-M when compared to the control group (Fig. S26†). Moreover, a dose-dependent response was observed; the 10 mg mL−1 (1.7 mM) DOX-M dose provided gradual tumor shrinkage that we reasoned could allow for facile comparisons between different ester linkers in a “reporter” theranostic study.
Based on the above results, a new set of 3 pro-ORCA + prodrug BASPs – F, M, and S – that each contained both DOX and chex attached to the same polymer (DOX:chex ratio of 1:6 to maximize MRI sensitivity) conjugated via the respective fast-, medium-, and slow-release ester linkers, as well as 1 mol% of non-releasable cy5.5, were synthesized and characterized by GPC, DLS, EPR, and in vitro MRI (Table 1, Fig. S20–22†). The same subcutaneous A549 murine model was employed to correlate DOX prodrug activation from F, M, and S with MRI SNR changes in vivo. Each mouse was intratumorally administered either a pro-ORCA-based BASP (F, M, or S) or a non-cleavable chex-containing BASP66 as a control (C); note that every animal received 50 μL of a 70 mg mL−1 solution, which corresponded to ∼1.7 mM of conjugated DOX, matching the dose used for DOX-M (vide supra). T2-Weighted MR images of the tumors were taken at pre-determined time points, beginning at 30 min after BASP injection (Fig. 4A). Inversion of image intensity was performed for subsequent analysis in order to aid visualization. In the inverted T2-weighted images, signal decreases upon pro-ORCA activation; the image appears brightest in the areas where the pro-ORCA (F, M, or S) concentration is the highest. Importantly, SNR variations that correlated with the release kinetics of chex from the corresponding MMs were observed while no changes were observed for C (Fig. 4B), which confirmed the ability of this approach to monitor pro-ORCA activation kinetics in vivo using MRI. For instance, 24 h post-injection decreases in inverse T2-weighted signal of 80 ± 7%, 49 ± 14%, and 9 ± 1% were observed for F, M, and S, respectively; the constant signal observed for non-cleavable BASP C suggests that these BASPs remain in the tumor environment and that there is an insignificant amount of BASP-nitroxide degradation (e.g., via reduction) throughout the timeframe of this study (Fig. 4B and Fig. S27†).66 To support our findings that these MRI SNR changes correlated with DOX release, the tumors of mice that had been administered F, M, and S were extracted for ex vivo fluorescence imaging to correlate the intratumoral locations of DOX (via its inherent fluorescence) with those of the BASPs (via cy5.5 fluorescence) (Fig. 4C). We stress that fluorescence is used here as a supporting tool to validate our MRI measurements for this proof-of-concept study; most drugs are not inherently fluorescent, motivating our development of pro-ORCAs. Gratifyingly, the DOX (green) and BASP (red) signals from the micrographs of tumors treated with the slow-releasing BASP (S) displayed good signal co-localization, suggesting that DOX was still conjugated to the BASP in its prodrug form and in agreement with MRI SNR variations. In contrast, significant separations of DOX (green) and BASP (red) were observed for the fast (F) as well as the medium (M) BASPs, suggesting that DOX release had occurred at sufficiently earlier time points with these constructs. It is, however, difficult to directly compare F and M, as the fluorescence of DOX is susceptible to alteration by several biological processes.86,87 Nonetheless, these results support the fact that T2 MRI signal changes induced by pro-ORCA activation correlate with DOX prodrug activation in this system, establishing an example of controlled nitroxide release for correlation of drug release in vivo.
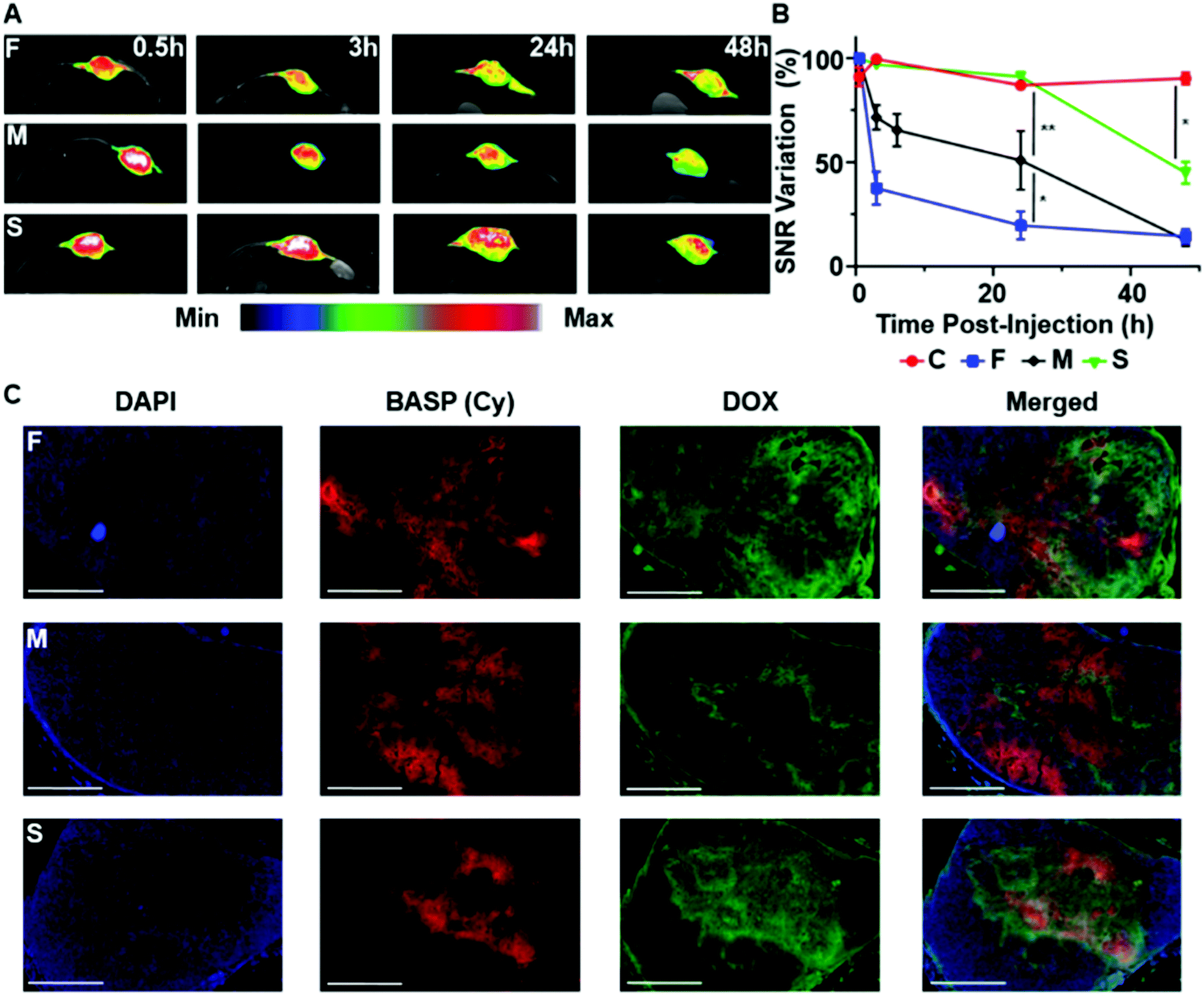 | ||
| Fig. 4 In vivo examination of hydrolytically labile BASP “reporter” theranostic agents for monitoring drug release in real time. (A) In vivo T2-weighted MR images at various time points after intratumoral injection of F, M, or S into mice bearing subcutaneous A549 tumors (n = 3 mice per group). False-color inverted T2-weighted images of the tumor region are overlaid for visualization. These inverted images were used for subsequent analysis. (B) Quantifcation of the changes in contrast over time in mice administered F, M, S, or non-cleavable control BASP (C)52 (n = 3 mice per group). Volume-averaged signal intensity across the whole tumor was calculated (see ESI† for full description). Data are presented as the mean ± SEM; statistical analyses were performed using one-way ANOVA (n.s.: not significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). (C) Representative fluorescence micrographs of tumors from mice administered F, M, or S 24 h post-injection (scale bar: 2 mm). Blue channel: DAPI; red channel: BASP (Cy); green channel: DOX. | ||
Conclusions
Herein, we introduced design principles and provided in vitro and in vivo proofs-of-concept for stimuli-responsive pro-ORCAs that enable real-time, non-invasive monitoring of prodrug activation in biological systems using a clinically viable imaging technique (MRI). Specifically, chex ORCAs were conjugated to BASPs through cleavable linkers generating “pro-ORCAs”. When attached to the BASP, chex has a large r2 value that, when combined with the high density of chex on each BASP, leads to MRI contrast on par with clinically used metal-based contrast agents; cleavage of the linker that connects chex to the BASP leads to a ∼30-fold decrease in r2 and concomitant loss of MRI contrast. By combining this pro-ORCA concept with DOX prodrugs conjugated to the same BASP using the same linkers, simultaneous pro-ORCA and prodrug activation occurs, which leads to changes in MRI contrast that correlate with drug release. This concept was demonstrated using 4 different linkers: 1 photocleavable and 3 hydrolytically cleavable linkers with different rates of hydrolysis, in both in vitro and in vivo settings. Regarding the latter, real-time monitoring of drug release via changes in T2 MRI signals within a tumor was demonstrated. Altogether, this work establishes a promising platform technology for monitoring prodrug activation in vitro and in vivo. We note that while the subject of this study was the real-time visualization of prodrug activation in biological environments via MRI, the demonstrated linker “plug-and-play” concept can potentially be expanded in the future to reporters of cell apoptosis or immunogenic cell death. Moreover, this approach could potentially be expanded to other therapeutic entities and a range of imaging modalities, providing valuable research tools as well as clinically translatable systems for personalized medicine.Author contributions
H. V.-T. N, A. D., P. P. G., I. M. G., and J. A. J conceived the idea. H. V.-T. N, A. D., P. H., N. G., C. M., M. P. A., O. Z., W. W., and Y. J. performed the experiments. H. V.-T. N, A. D., P. P. G., and J. A. J. wrote the manuscript. All authors discussed the results and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH-NCI (1R01CA220468-01 for J. A. J., P. P. G., A. R., A. J.), the NIH (U01-NS090451 for A. J.), and the National Science Foundation (Graduate Research Fellowship for H. V.-T. N.) for support of this research. A. D. acknowledges support from the Fondation Française pour la Recherche contre le Myélome et les Gammapathies, the Philippe Foundation, and the Multiple Myeloma Research Foundation. P. H. acknowledges support from a Wellcome Trust-MIT Postdoctoral Fellowship. P. P. G. acknowledges the generous support of the Charles W. and Jennifer C. Johnson Clinical Investigator Fund as well as the Kathryn Fox Samway Foundation. This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. We thank the Koch Institute Swanson Biotechnology Center for technical support.Notes and references
- M. A. Hamburg and F. S. Collins, N. Engl. J. Med., 2010, 363, 301–304 CrossRef CAS PubMed.
- I. S. Chan and G. S. Ginsburg, Annu. Rev. Genomics Hum. Genet., 2011, 12, 214–244 CrossRef PubMed.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS PubMed.
- R. Kojima, D. Aubel and M. Fussenegger, Curr. Opin. Chem. Biol., 2015, 28, 29–38 CrossRef CAS PubMed.
- X. Chen, S. S. Gambhir and J. Cheon, Acc. Chem. Res., 2011, 44, 841 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS PubMed.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- A. M. Nystrom and K. L. Wooley, Acc. Chem. Res., 2011, 44, 969–978 CrossRef CAS PubMed.
- M. Elsabahy, G. S. Heo, S.-M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavia-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- H. Chen, D. Sulejmanovic, T. Moore, D. C. Colvin, B. Qi, O. T. Mefford, J. C. Gore, F. Alexis, S.-J. Hwu and J. N. Anker, Chem. Mater., 2014, 26, 2105–2112 CrossRef CAS PubMed.
- Y. Li, T.-Y. Lin, Y. Luo, Q. Liu, W. Xiao, W. Guo, D. Lac, H. Zhang, C. Feng, S. Wachsmann-Hogiu, J. H. Walton, S. R. Cherry, D. J. Rowland, D. Kukis, C. Pan and K. S. Lam, Nat. Commun., 2014, 5, 4712 CrossRef CAS PubMed.
- K. Y. Choi, E. J. Jeon, H. Y. Yoon, B. S. Lee, J. H. Na, K. H. Min, S. Y. Kim, S.-J. Myung, S. Lee, X. Chen, I. C. Kwon, K. Choi, S. Y. Jeong, K. Kim and J. H. Park, Biomaterials, 2012, 33, 6186–6193 CrossRef CAS PubMed.
- C. Grange, S. Geninatti-Crich, G. Esposito, D. Alberti, L. Tei, B. Bussolati, S. Aime and G. Camussi, Cancer Res., 2010, 70, 2180–2190 CrossRef CAS PubMed.
- M. Kumar, M. Yigit, G. Dai, A. Moore and Z. Medarova, Cancer Res., 2010, 70, 7553–7561 CrossRef CAS PubMed.
- S.-M. Lee, Y. Song, B. J. Hong, K. W. MacRenaris, D. J. Mastarone, T. V. O'Halloran, T. J. Meade and S. T. Nguyen, Angew. Chem., Int. Ed., 2010, 49, 9960–9964 CrossRef CAS PubMed.
- S. Kaida, H. Cabral, M. Kumagai, A. Kishimura, Y. Terada, M. Sekino, I. Aoki, N. Nishiyama, T. Tani and K. Kataoka, Cancer Res., 2010, 70, 7031–7041 CrossRef CAS PubMed.
- K. Kim, J. H. Kim, H. Park, Y.-S. Kim, K. Park, H. Nam, S. Lee, J. H. Park, R.-W. Park, I.-S. Kim, K. Choi, S. Y. Kim, K. Park and I. C. Kwon, J. Controlled Release, 2010, 146, 219–227 CrossRef CAS PubMed.
- J. H. Lee, K. Lee, S. H. Moon, Y. Lee, T. G. Park and J. Cheon, Angew. Chem., Int. Ed., 2009, 48, 4174–4179 CrossRef CAS PubMed.
- T. Schluep, J. Hwang, I. J. Hildebrandt, J. Czernin, C. H. J. Choi, C. A. Alabi, B. C. Mack and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11394–11399 CrossRef CAS PubMed.
- Z. Medarova, W. Pham, C. Farrar, V. Petkova and A. Moore, Nat. Med., 2007, 13, 372–377 CrossRef CAS PubMed.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S.-F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- J. Cheon and J.-H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS PubMed.
- T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC.
- A. Kulkarni, P. Rao, S. Natarajan, A. Goldman, V. S. Sabbisetti, Y. Khater, N. Korimerla, V. Chandrasekar, R. A. Mashelkar and S. Sengupta, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2104–E2113 CrossRef CAS PubMed.
- R. Wang, L. Zhou, W. Wang, X. Li and F. Zhang, Nat. Commun., 2017, 8, 14702 CrossRef PubMed.
- S. Bhuniya, S. Maiti, E.-J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469–4474 CrossRef CAS PubMed.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- S.-Y. Li, L.-H. Liu, L. Rong, W.-X. Qiu, H.-Z. Jia, B. Li, F. Li and X.-Z. Zhang, Adv. Funct. Mater., 2015, 25, 7317–7326 CrossRef CAS.
- X. Guo, L. Wang, K. Duval, J. Fan, S. Zhou and Z. Chen, Adv. Mater., 2018, 30, 1705436 CrossRef PubMed.
- X. Li, M. Bottini, L. Zhang, S. Zhang, J. Chen, T. Zhang, L. Liu, N. Rosato, X. Ma, X. Shi, Y. Wu, W. Guo and X.-J. Liang, ACS Nano, 2019, 13, 176–186 CrossRef CAS PubMed.
- M. H. Lee, E.-J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380–163887 CrossRef CAS PubMed.
- C. Kaittanis, T. M. Shaffer, A. Ogirala, S. Santra, J. M. Perez, G. Chiosis, Y. Li, L. Josephson and J. Grimm, Nat. Commun., 2014, 5, 3384 CrossRef PubMed.
- S. Santra, S. D. Jativa, C. Kaittanis, G. Normand, J. Grimm and J. M. Perez, ACS Nano, 2012, 6, 7281–7294 CrossRef CAS PubMed.
- Y. Onuki, I. Jacobs, D. Artemov and Y. Kato, Biomaterials, 2010, 31, 7132–7138 CrossRef CAS PubMed.
- B. L. Viglianti, A. M. Ponce, C. R. Michelich, D. Yu, S. A. Abraham, L. Sanders, P. S. Yarmolenko, T. Schroeder, J. R. MacFall, D. P. Barboriak, O. M. Colvin, M. B. Bally and M. W. Dewhirst, Magn. Reson. Med., 2006, 56, 1011–1018 CrossRef CAS PubMed.
- A. S. Thakor, J. V. Jokerst, P. Ghanouni, J. L. Campbell, E. Mittra and S. S. Gambhir, J. Nucl. Med., 2016, 57, 1833–1837 CrossRef CAS PubMed.
- V. C. Pierre, M. J. Allen and P. Caravan, J. Biol. Inorg. Chem., 2014, 19, 127–131 CrossRef CAS PubMed.
- Y. X. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- Y. X. Wang, World J. Gastroenterol., 2015, 21, 13400–13402 CrossRef CAS PubMed.
- S. Swaminathan, T. D. Horn, D. Pellowski, S. Abul-Ezz, J. A. Bornhorst, S. Viswamitra and S. V. Shah, N. Engl. J. Med., 2007, 357, 720–722 CrossRef CAS PubMed.
- I. A. Mendichovszky, S. D. Marks, C. M. Simcock and O. E. Olsen, Pediatr. Radiol., 2008, 38, 489–496 CrossRef PubMed.
- U.S. Food, & Drug Administration (FDA), 2015; https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-strengthens-warnings-and-changes-prescribing-instructions-decrease (accessed Dec 16, 2019).
- F. Hyodo, K. H. Chuang, A. G. Goloshevsky, A. Sulima, G. L. Griffiths, J. B. Mitchell, A. P. Koretsky and M. C. Krishna, J. Cereb. Blood Flow Metab., 2008, 28, 1165–1174 CrossRef CAS PubMed.
- Z. Zhelev, R. Bakalova, I. Aoki, K.-I. Matsumoto, V. Gadjeva, K. Anzai and I. Kanno, Chem. Commun., 2009, 1, 53–55 RSC.
- N. Bye, O. E. Hutt, T. M. Hinton, D. P. Acharya, L. J. Waddington, B. A. Moffat, D. K. Wright, H. X. Wang, X. Mulet and B. W. Muir, Langmuir, 2014, 30, 8898–8906 CrossRef CAS.
- F. Caglieris, L. Melone, F. Canepa, G. Lamura, F. Castiglione, M. Ferro, L. Malpezzi, A. Mele, C. Punta, P. Franchi, M. Lucarini, B. Rossi and F. Trotta, RSC Adv., 2015, 5, 76133–76140 RSC.
- L. Huang, C. Yan, D. Cui, Y. Yan, X. Liu, X. Lu, X. Tan, X. Lu, J. Xu, Y. Xu and R. Liu, Macromol. Biosci., 2015, 15, 788–798 CrossRef CAS.
- J. M. W. Chan, R. J. Wojtecki, H. Sardon, A. L. Z. Lee, C. E. Smith, A. Shkumatov, S. Gao, H. Kong, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2017, 6, 176–180 CrossRef CAS.
- S. Garmendia, D. Mantione, S. Alonso-de Castro, C. Jehanno, L. Lezama, J. L. Hedrick, D. Mecerreyes, L. Salassa and H. Sardon, Polym. Chem., 2017, 8, 2693–2701 RSC.
- M. Dharmarwardana, A. F. Martins, Z. Chen, P. M. Palacios, C. M. Nowak, R. P. Welch, S. Li, M. A. Luzuriaga, L. Bleris and B. S. Pierce, Mol. Pharm., 2018, 15, 2973–2983 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodriguez-Rodriguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- K.-A. Hansen and J. P. Blinco, Polym. Chem., 2018, 9, 1479–1516 RSC.
- O. U. Akakuru, M. Z. Iqbal, M. Saeed, C. Liu, T. Paunesku, G. Woloschak, N. S. Hosmane and A. Wu, Bioconjugate Chem., 2019, 30, 2264–2286 CrossRef CAS PubMed.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896–5899 CrossRef CAS PubMed.
- A. X. Gao, L. Liao and J. A. Johnson, ACS Macro Lett., 2014, 3, 854–857 CrossRef CAS PubMed.
- J. C. Barnes, P. M. Bruno, H. V.-T. Nguyen, L. Liao, J. Liu, M. T. Hemann and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 12494–12501 CrossRef CAS PubMed.
- M. R. Golder, J. Liu, J. N. Andersen, M. V. Shipitsin, F. Vohidov, H. V.-T. Nguyen, D. C. Ehrlich, S. J. Huh, B. Vangamudi, K. D. Economides, A. M. Neenan, J. C. Ackley, J. Baddour, S. Paramasivan, S. W. Brady, E. J. Held, L. A. Reiter, J. K. Saucier-Sawyer, P. W. Kopesky, D. E. Chickering, P. Blume-Jensen and J. A. Johnson, Nat. Biomed. Eng., 2018, 2, 707 CrossRef PubMed.
- H. V.-T. Nguyen, N. M. Gallagher, F. Vohidov, Y. Jiang, K. Kawamoto, H. Zhang, J. V. Park, Z. Huang, M. F. Ottaviani, A. Rajca and J. A. Johnson, ACS Macro Lett., 2018, 7, 472–476 CrossRef CAS PubMed.
- H. V.-T. Nguyen, A. Detappe, N. M. Gallagher, H. Zhang, P. Harvey, C. Yan, C. Mathieu, M. R. Golder, Y. Jiang, M. F. Ottaviani, A. Jasanoff, A. Rajca, I. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, ACS Nano, 2018, 12, 11343–11354 CrossRef CAS PubMed.
- M. A. Sowers, J. R. McCombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 5460 CrossRef.
- H. V.-T. Nguyen, Q. Chen, J. T. Paletta, P. Harvey, Y. Jiang, H. Zhang, M. D. Boska, M. F. Ottaviani, A. Jasanoff, A. Rajca and J. A. Johnson, ACS Cent. Sci., 2017, 3, 800–811 CrossRef CAS PubMed.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. Xia, A. C. Durrell, D. A. Tirrell and R. H. Grubbs, Macromolecules, 2010, 43, 10326–10335 CrossRef CAS PubMed.
- K. Kawamoto, M. Zhong, K. R. Gadelrab, L.-C. Cheng, C. A. Ross, A. Alexander-Katz and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 11501–11504 CrossRef CAS.
- Y. Shibuya, H. V.-T. Nguyen and J. A. Johnson, ACS Macro Lett., 2017, 6, 963–968 CrossRef CAS.
- Y. Shibuya, R. Tatara, Y. Jiang, Y. Shao-Horn and J. A. Johnson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 448–455 CrossRef CAS.
- G. Gil Alvaradejo, H. V.-T. Nguyen, P. Harvey, N. M. Gallagher, D. Le, M. F. Ottaviani, A. Jasanoff, G. Delaittre and J. A. Johnson, ACS Macro Lett., 2019, 8, 473–478 CrossRef PubMed.
- A. Rajca, Y. Wang, M. Boska, J. T. Paletta, A. Olankitwanit, M. A. Swanson, D. G. Mitchell, S. S. Eaton, G. R. Eaton and S. Rajca, J. Am. Chem. Soc., 2012, 134, 15724–15727 CrossRef CAS PubMed.
- J. A. Barltrop, J. Plant and P. Schofield, Chem. Commun., 1966, 22, 822–823 RSC.
- A. Patchornik, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromolecules, 2007, 40, 3589–3598 CrossRef CAS.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- W. E. Georgianna, H. Lusic, A. L. McIver and A. Deiters, Bioconjugate Chem., 2010, 21, 1404–1407 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- H. Yu, J. Li, D. Wu, Z. Qiu and Y. Zhang, Chem. Soc. Rev., 2010, 39, 464–473 RSC.
- Z. Chen, N. Li, L. Chen, J. Lee and J. J. Gassensmith, Small, 2016, 12, 4563–4571 CrossRef CAS PubMed.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Guan, J. Med. Chem., 1999, 42, 3657–3667 CrossRef CAS PubMed.
- I. Sagiv-Barfi, D. K. Czerwinski, S. Levy, I. S. Alam, A. T. Mayer, S. S. Gambhir and R. Levy, Sci. Transl. Med., 2018, 10, eaan4488 CrossRef PubMed.
- S. Voulgaris, M. Partheni, M. Karamousiz, P. Dimopoulos, N. Papadakis and H. Kalofonos, Am. J. Clin. Oncol., 2002, 25, 60–64 CrossRef PubMed.
- A. Marabelle, L. Tselikas, T. de Baere and R. Houot, Ann. Oncol., 2017, 28, xii33–xii43 CrossRef CAS PubMed.
- M. A. Aznar, N. Tinari, A. J. Rullan, A. R. Sanchez-Paulete, M. E. Rodriguez-Ruiz and I. Melero, J. Immunol., 2017, 198, 31–39 CrossRef CAS.
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- P. Mohan and N. Rapoport, Mol. Pharm., 2010, 7, 1959–1973 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00558d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |