DOI:
10.1039/D0PY00555J
(Paper)
Polym. Chem., 2020,
11, 5534-5541
Aqueous copper-mediated reversible deactivation radical polymerization (RDRP) utilizing polyetheramine derived initiators†‡
Received
16th April 2020
, Accepted 5th July 2020
First published on 3rd August 2020
Abstract
Copper-mediated reversible deactivation radical polymeriation (Cu-RDRP) in aqueous media has been employed to synthesize temperature-responsive block copolymers, utilizing both hydrophobic and hydrophilic amide functional macroinitiators derived from polyetheramines (Jeffamines™). The in situ and rapid diproportionation of Cu(I)Br/Me6TREN in water is exploited for the efficient homopolymerization of N-isopropyl acrylamide (NIPAM) and dimethyl acrylamide (DMA), at near full conversions (>99%), with low dispersity (Đ < 1.18) and with a range of molar masses. The Jeffamine™-derived macroinitiators were used for both the synthesis of homopolymer and for one-pot chain extensions and block copolymerizations (i.e. Jeffamine™-PNIPAM-b-PDMA). The obtained polymers exhibit controlled thermoresponsive aggregation behaviour which varies depending on the hydrophilicity/hydrophobicity of the macroinitiators and the composition of the block copolymers. Thermal analysis and dynamic light scattering (DLS) give an insight into the effect of these macroinitiators on the thermoresponsive aggregation behaviour of the synthesized polymers.
Introduction
Polyetheramines (known by trademarks Jeffamine™ from Huntsman Corporation and Baxxodur™ from BASF) represent a family of polyethers with varying ratios of propylene oxide (PO) and ethylene oxide (EO) repeating units with primary amine functional terminal group(s).1,2 The versatile nature of these materials has attracted significant interest owning to their flexible backbone, tuneable thermoresponsive behaviour, low viscosity, controllable reactivity, hydrogen bonding properties, primary amine functionality and broad range of molar masses.3–5 Additionally, their low cost and wide industrial use as additives in coatings, foams, encapsulation strategies and composites, render them a polyfunctional family of products.6 Among these versatile characteristics of polyetheramines, their thermoresponsive nature has been particularly exploited previously.7–10 For example, Jeffamine M-2005 has been used in combination with hydroxypropyl methyl cellulose for the synthesis of thermoresponsive copolymers with micellar structures, which exhibited cytocompatibility, thus showing potential for drug delivery applications.11 In 2007, Lecommandoux and co-workers reported the synthesis of Jeffamine-b-poly(É©-glutamic acid) double hydrophilic block copolymers via ring-opening polymerization of γ-benzyl-L-glutamate N-carboxyanhydride, with an –NH2 terminated Jeffamine macroinitiator.12 The copolymers showed the ability to self-assemble to generate thermoresponsive micelles which were used for the specific absorption and /or release of small molecules.
Due to the increasing development of controlled radical polymerization, researchers have combined the properties of polyetheramines with Cu-mediated Reversible Deactivation Radical Polymerization techniques (i.e. Atom Transfer Radical Polymerization (ATRP) and Single Electron Transfer-Living Radical Polymerization (SET-LRP)). This is due to the fact that Cu-RDRP has been considered as one of the most robust strategies due to successful implementation under various conditions (e.g. in the presence of oxygen/air,13–15 in various reaction media16–18) and also due to the wide of range of materials that can be synthesized (i.e. polyacrylates, polymethacrylates, polystyrene, polyacrylamides) with excellent control over their (macro)molecular characteristics.19–25 In this context, Armes and colleagues used Jeffamine precursors for the synthesis of Y-shaped block copolymers via ATRP.26 Amongst other approaches,27,28 Jeffamine-derived macroinitiators have also been used for the synthesis of amphiphilic triblock terpolymers with thermo- and pH-responsive self-assembly.29 The above-mentioned approaches provide significant developments for the incorporation of polyetheramines into controlled radical polymerization, although limitations including prolonged reaction times, high temperatures and use of organic media have been present to date.
One of the main advantages of Cu-RDRP is its successful implementation in aqueous media, with excellent control over the macromolecular characteristics of the synthesized polymers.15,18,19,21,30,31 In the presence of multidentate aliphatic amine ligands (e.g. tris [2-(dimethylamino)ethyl]amine, Me6TREN) and in aqueous media, Cu(I) is highly unstable towards rapid disproportionation, leading to Cu(II) and nascent, highly active Cu(0). Furthermore, these tertiary amine ligands stabilize Cu(II) leading to a highly controlled polymerization of water-soluble monomers within impressively short reaction times, with this approach being compatible with both homopolymerizations and in situ chain extensions,19,22,32 as well as the synthesis of sophisticated architectures as reported by Becer and colleagues.31 This surprising lack of termination within such rapid reaction times, which is not in accordance with classical free radical kinetics, has recently been elucidated by Ballard and Asua by invoking a probability density function explanation.33
Although the use of aqueous media provides many benefits, it can be disadvantageous if products are hydrolytically unstable. Thus, the choice of initiator is important, since the usual ester-derived initiators used in Cu-RDRP are prone to hydrolysis in systems with excess of water which can lead to degradation and decomposition.30 Thus compounds with greater hydrolytic stability, such as amide-based initiators are desirable. In this context, Sawamoto and colleagues reported the synthesis of poly(dimethylacrylamide) utilizing 2-bromo-2-methylpropionamide-based initiators, obtaining controlled molecular weights but with high dispersities (Đ ∼1.6).34 Matyjaszewski reported the polymerization of (meth)acrylamides, using haloamide-based initiator for efficient for the synthesis of block copolymers, with dispersity of ∼1.3–1.6.35 In 2006, Haddleton reported the use of initiators containing bromo-2-methylpropionamide for the synthesis of various poly(methacrylates). Although good control over the molecular weights was attained, elevated temperatures and prolonged reaction times (>7 hours) were required.36 Wooley and co-workers utilized an amino acid-based initiator (L-valine-derived α-haloamide) for the synthesis of tert-butyl acrylate-b-styrene copolymers, with control over the molecular weights, dispersities of ≥1.20 and at temperatures of ∼55–90 °C.37 These initiators could be considered as efficient alternatives to the conventionally employed, but hydrolytically unstable, ester-derived compounds. However, higher temperatures and prolonged reaction times were required, leading to (relatively) high dispersity values.
In this context, we envisaged that the use of Jeffamines as macroinitiators could address the limitations of Cu-RDRP in aqueous media due to their increased hydrolytic stability. Consequently, both hydrophilic and hydrophobic Jeffamine-derived macroinitiators were synthesized and used for the aqueous Cu-RDRP of various acrylamides under mild reaction conditions (e.g. at 0 °C) (Scheme 1). Rapid polymerization rates were observed and the polyacrylamide products exhibited low dispersities and controlled molecular weights at near-quantitative conversions (>99%), even when higher degrees of polymerization (e.g. DPn = 320) were targeted. Moreover, the high end-group fidelity which was attained allowed for in situ chain extensions and block co-polymerizations. This provided access to well-defined amphiphilic block copolymers with thermo-switchable behaviour, dependent on the ratio between the hydrophilic and hydrophobic segments (Scheme 1). The thermally-induced aggregation behaviour of these materials was studied by UV/Vis and DLS, and the thermal stability of the copolymers was investigated by TGA and DSC.
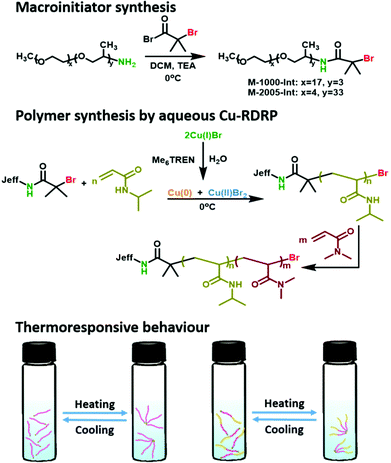 |
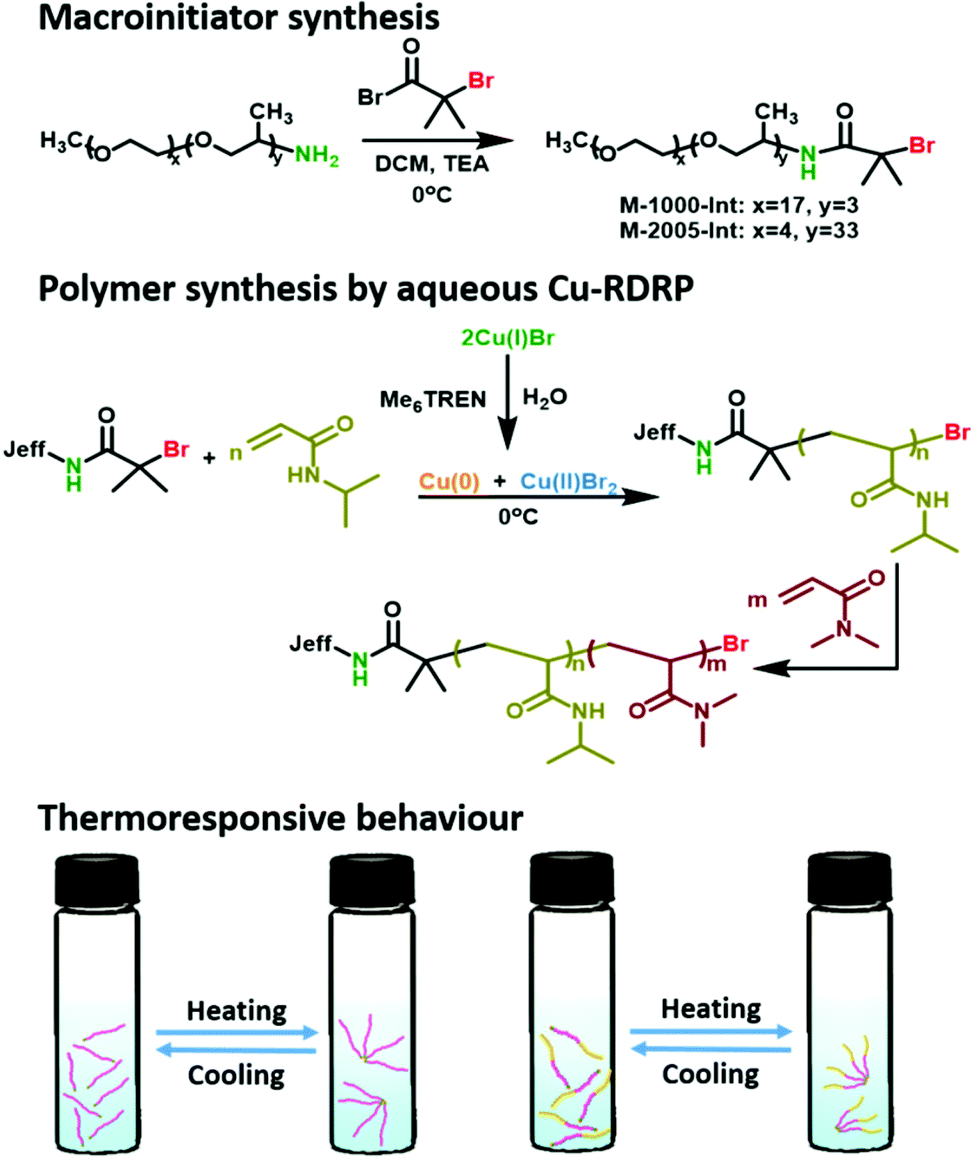
| | Scheme 1 Schematic representation for the synthesis of Jeffamine-based macroinitiators and NIPAM-based polymers using aqueous Cu-mediated RDRP. | |
Results and discussion
Macroinitiator synthesis
The macroinitiators were derived from two commercially available polyetheramines, M-1000 and M-2005, and prepared using a one-step amidation reaction. The polyetheramines were selected based on their differing hydrophobicity, which depends on the ratio of the PO (propylene oxide) and EO (ethylene oxide) units in the polymer. The products (colourless oils) were characterized by 1H NMR, 13C NMR, SEC and MALDI-TOF MS. The signals arising from the –CH3 groups (h) from the α-bromoisobutyryl bromide, in the 1H NMR spectrum, are at approximately 1.8–1.9 ppm for both the hydrophilic (M-1000-Int) and the hydrophobic macroinitiators (M-2005-Int), Fig. S2 and S6.‡ The ratios of PO/EO were confirmed to be 3/17 ratio for the M-1000-Int and 33/4 for the hydrophobic M-2005-Int. SEC analysis revealed narrow mono-modal molecular weight distributions (Đ < 1.31) as shown in Fig. S1a‡ (M-1000-Int) and Fig. S5‡ (M-2005-Int). Additionally, MALDI-TOF MS of both macroinitiators confirmed the ω-bromine end group in both cases (Fig. S3 and S7‡). It is noted that the analysis of primary amines by MALDI-TOF can be challenging when DCTB is used as matrix due to reactions occurring between the sample and the matrix.38 Indeed, when the same Jeffamine sample was analysed using two different matrices (DHB and DCTB) the obtained spectra exhibited differences (Fig. S1b‡). Additional peaks were observed in the spectra where DCTB was used, whilst the spectra with DHB only contained the intact ions as expected. Additional peaks in the DCTB spectra are representative of a noted artefact/reaction product which occurs when using the DCTB matrix with primary or secondary amines.
Homopolymerization of N-isopropyl acrylamide with DPn = 20–320 using the hydrophilic macroinitiator (M-1000-Int)
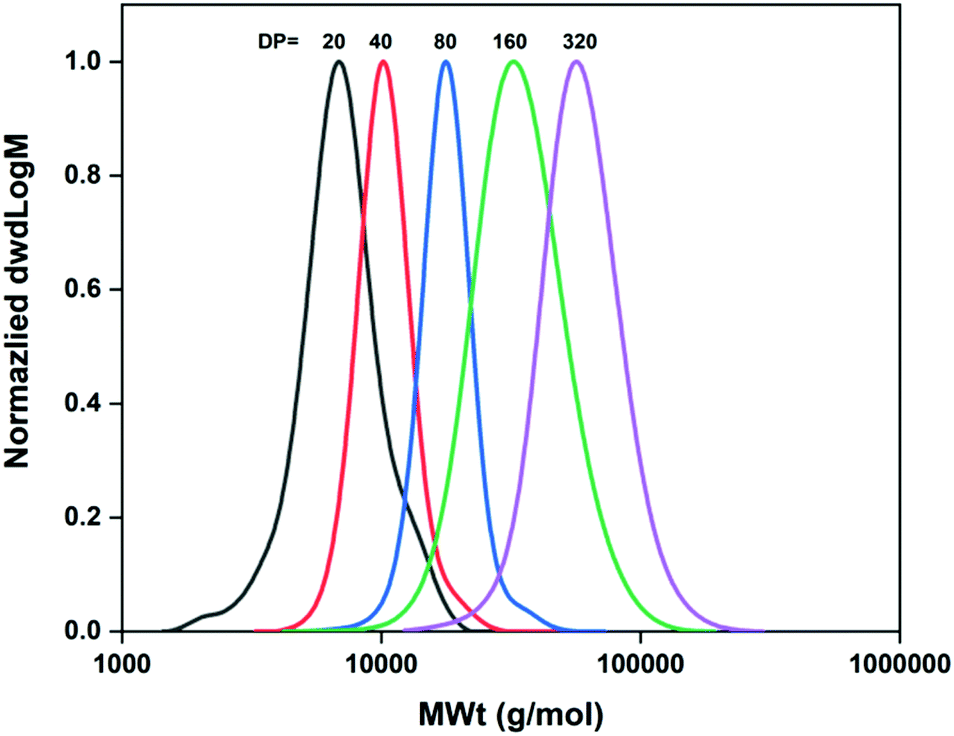
For the synthesis of the different polyacrylamides, Cu-RDRP in water was used as previously described in the literature.19,21 An essential step for a successful aqueous Cu-RDRP is the efficient pre-disproportionation of Cu(I)Br/Me6TREN into Cu(0) and Cu(II), which occurs prior to polymerization. The polymerisation conditions were first optimised using NIPAM as the monomer. Initially, for the synthesis of PNIPAM with targeted DPn = 20, a Cu(I)Br/Me6TREN molar ratio of [0.1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[0.1] was employed relative to the macroinitiator (Table 1, entry 1). It was found that the monomer conversion was limited to 62% and the dispersity values were as high as Đ ∼1.7. Based on this, we hypothesized that the copper equivalents used were not sufficient for successful deactivation as has previously been reported,19 thus leading us to examine different [Cu(I)Br]:[Me6TREN] ratios, in order to achieve better control and higher monomer conversions. Indeed, when [Cu(I)Br]:[Me6TREN] = 0.4:0.4 with respect to 1 eq. of macroinitiator was used (Table 1, entry 3) near full conversion and low dispersity was obtained, after 15 minutes of the polymerization. However, some deviations between the theoretical and experimental (SEC-derived) Mn values were observed, which were also evident for higher loadings of copper and ligand, probably indicating loss of efficiency for the M-1000-Int macroinitiator, to some extent. Based on the results illustrated in Table 1 (entries 1–5), best control over the polymerization of NIPAM was achieved when [I]:[Cu(I)Br]:[Me6TREN]:[NIPAM] = [1]:[0.4]:[0.4]:[20] was used, resulting in near-quantitative monomer conversion (>99%), symmetrical and mono-modal SEC-traces, and dispersity values as low as Đ = 1.15. Based on kinetic studies, the polymerization exhibited very fast rates, reaching 99% monomer conversion after 7 minutes (Fig. S9 and S10‡). It should be noted that when higher equivalents of Cu(I)Br were used (Table 1, entry 4), the monomer conversion was as low as 53%, probably due to higher deactivation. When higher DPs were targeted (DPn = 40, 80, 160, 320), the overall concentration of Cu(I)Br and ligand was lower, thus higher concentrations of those components were used (Table 1, entries 7–10). As a result, for the higher molar masses targeted, full conversions (>99%) were obtained within 15–30 minutes (Table 1) with DMF-SEC analysis demonstrating symmetrical, mono-modal polymer peak distributions with Đ ≤ 1.18 (Table 1, entries 7–10 and Fig. 1).
:[0.1] was employed relative to the macroinitiator (Table 1, entry 1). It was found that the monomer conversion was limited to 62% and the dispersity values were as high as Đ ∼1.7. Based on this, we hypothesized that the copper equivalents used were not sufficient for successful deactivation as has previously been reported,19 thus leading us to examine different [Cu(I)Br]:[Me6TREN] ratios, in order to achieve better control and higher monomer conversions. Indeed, when [Cu(I)Br]:[Me6TREN] = 0.4:0.4 with respect to 1 eq. of macroinitiator was used (Table 1, entry 3) near full conversion and low dispersity was obtained, after 15 minutes of the polymerization. However, some deviations between the theoretical and experimental (SEC-derived) Mn values were observed, which were also evident for higher loadings of copper and ligand, probably indicating loss of efficiency for the M-1000-Int macroinitiator, to some extent. Based on the results illustrated in Table 1 (entries 1–5), best control over the polymerization of NIPAM was achieved when [I]:[Cu(I)Br]:[Me6TREN]:[NIPAM] = [1]:[0.4]:[0.4]:[20] was used, resulting in near-quantitative monomer conversion (>99%), symmetrical and mono-modal SEC-traces, and dispersity values as low as Đ = 1.15. Based on kinetic studies, the polymerization exhibited very fast rates, reaching 99% monomer conversion after 7 minutes (Fig. S9 and S10‡). It should be noted that when higher equivalents of Cu(I)Br were used (Table 1, entry 4), the monomer conversion was as low as 53%, probably due to higher deactivation. When higher DPs were targeted (DPn = 40, 80, 160, 320), the overall concentration of Cu(I)Br and ligand was lower, thus higher concentrations of those components were used (Table 1, entries 7–10). As a result, for the higher molar masses targeted, full conversions (>99%) were obtained within 15–30 minutes (Table 1) with DMF-SEC analysis demonstrating symmetrical, mono-modal polymer peak distributions with Đ ≤ 1.18 (Table 1, entries 7–10 and Fig. 1).
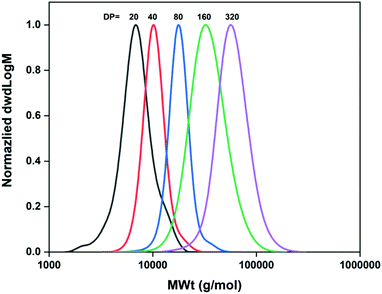 |
| | Fig. 1 Molecular weight distributions of the M-1000-Int-pNIPAMx (DPn = 20–320) synthesized under optimized conditions as measured by DMF-SEC. | |
Table 1 Optimization of the homopolymerizations of NIPAM with DPn = 20–320 using the hydrophilic macroinitiator (M-1000-Int)
| Entry |
[I]:[Cu(I)Br]:[Me6TREN]:[M] |
M
n,theo (g mol−1) |
M
n, SEC
(g mol−1) |
Đ
|
Conv.b (%NMR) |
|
Determined by DMF-SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
Conversion was calculated via1H NMR using D2O as the solvent.
Reaction was left to commence for 15 minutes.
Reaction was left to commence for 30 minutes.
|
| 1c |
1:0.1:0.1:20 |
2500 |
5500 |
1.70 |
62 |
| 2c |
1:0.4:0.2:20 |
2800 |
5200 |
1.12 |
73 |
| 3c |
1:0.4:0.4:20 |
3400 |
6400 |
1.15 |
>99 |
| 4c |
1:0.8:0.4:20 |
2300 |
4200 |
1.17 |
53 |
| 5c |
1:0.8:0.6:20 |
3400 |
5700 |
1.22 |
>99 |
| 6c |
1:0.8:0.8:20 |
3400 |
5700 |
1.33 |
>99 |
| 7c |
1:0.8:0.4:40 |
5600 |
10000 |
1.07 |
>99 |
| 8c |
1:0.8:0.4:80 |
10100 |
17000 |
1.06 |
>99 |
| 9d |
1:0.8:0.4:160 |
19000 |
31000 |
1.18 |
>99 |
| 10d |
1:0.8:0.4:320 |
37000 |
55000 |
1.15 |
>99 |
Homopolymerization of N-isopropyl acrylamide with DPn = 20–320 utilizing the hydrophobic initiator (M-2005-Int)
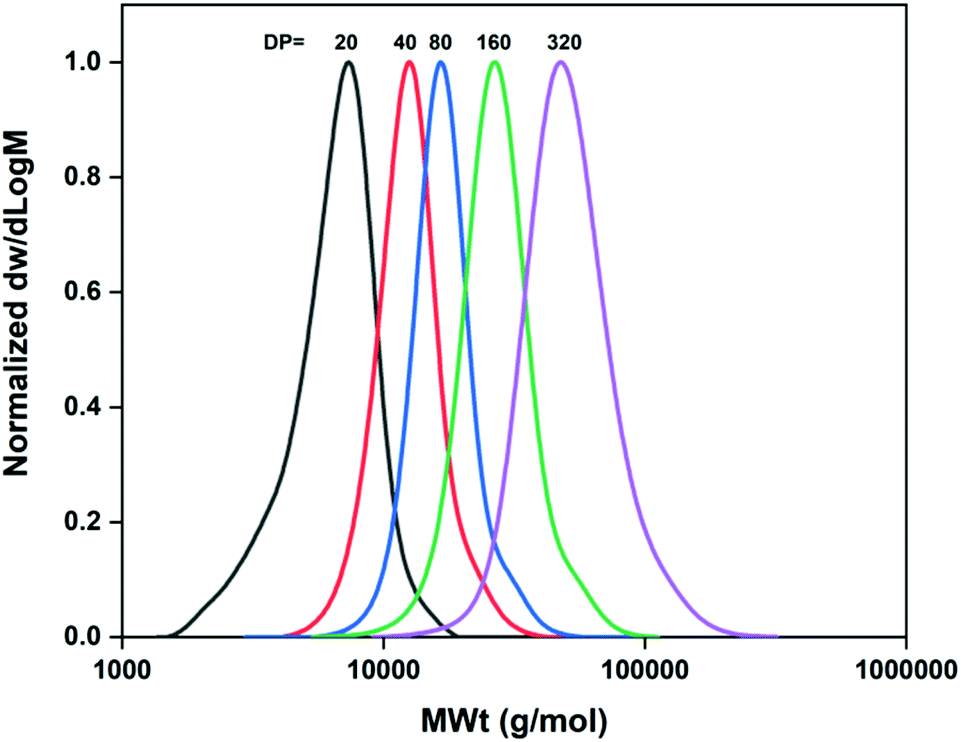
Following the same strategy as in the case of M-1000-Int, initially the hydrophobic initiator M-2005-Int was used to prepare PNIPAM homopolymers with various targeted DPs. As previously, the ratio [Cu(I)Br]/[Me6TREN] = [0.4]/[0.4] with respect to 1 eq. of initiator exhibited the best results for the synthesis of PNIPAM20. A narrow and symmetrical molecular weight distribution (Đ = 1.16) was achieved with full monomer conversion (Table 2, entry 3) in short reaction times (∼15 min). Again, Mn,SEC values higher than the theoretical were observed, probably indicating some loss of initiator efficiency and/or not sufficient deactivation, leading to imperfect control over the molecular weights. However, this did not hinder the ability to afford higher molar masses, targeting DPn from 40 to 320, under optimized conditions (Table 2, entries 7–10). As shown in Table 2, by tuning the ratio of Cu(I)Br:Me6Tren, good control on molecular weights and low dispersity was achieved at >99% conversions (Table 2 and Fig. 2).
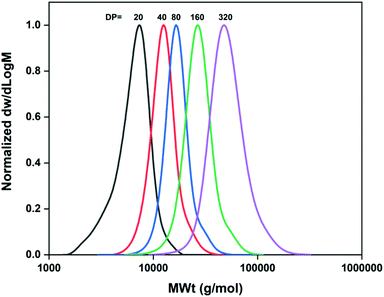 |
| | Fig. 2 Molecular weight distributions of the M-2005-Int-pNIPAMx (DPn = 20–320) synthesized under optimized conditions as measured by DMF-SEC. | |
Table 2 Optimization of the homopolymerizations of NIPAM with DPn = 20–320 using the hydrophobic macroinitiator (M-2005-Int)
| Entry |
[I]:[Cu(I)Br]:[Me6TREN]:[M] |
M
n,theo (g mol−1) |
M
n, SEC
(g mol−1) |
Đ
|
Conv.b (%NMR) |
|
Determined by DMF SEC analysis and expressed as molecular weight equivalents to PMMA narrow molecular weight standards.
Conversion was calculated via1H NMR using D2O as the solvent.
Reaction was left to commence for 15 minutes.
Reaction was left to commence for 30 minutes.
|
| 1c |
1:0.1:0.1:20 |
2900 |
4300 |
1.20 |
30 |
| 2c |
1:0.4:0.2:20 |
3200 |
4300 |
1.35 |
44 |
| 3c |
1:0.4:0.4:20 |
4500 |
5800 |
1.16 |
>99 |
| 4c |
1:0.8:0.4:20 |
3300 |
4500 |
1.38 |
50 |
| 5c |
1:0.8:0.6:20 |
4500 |
5000 |
1.32 |
>99 |
| 6c |
1:0.8:0.8:20 |
4500 |
5200 |
1.40 |
>99 |
| 7c |
1:0.8:0.4:40 |
6700 |
12000 |
1.09 |
>99 |
| 8c |
1:0.8:0.4:80 |
11200 |
16000 |
1.08 |
>99 |
| 9d |
1:0.8:0.4:160 |
20100 |
25000 |
1.12 |
>99 |
| 10d |
1:0.8:0.4:320 |
38000 |
47000 |
1.17 |
>99 |
Chain extension and block copolymerization via aqueous Cu-RDRP
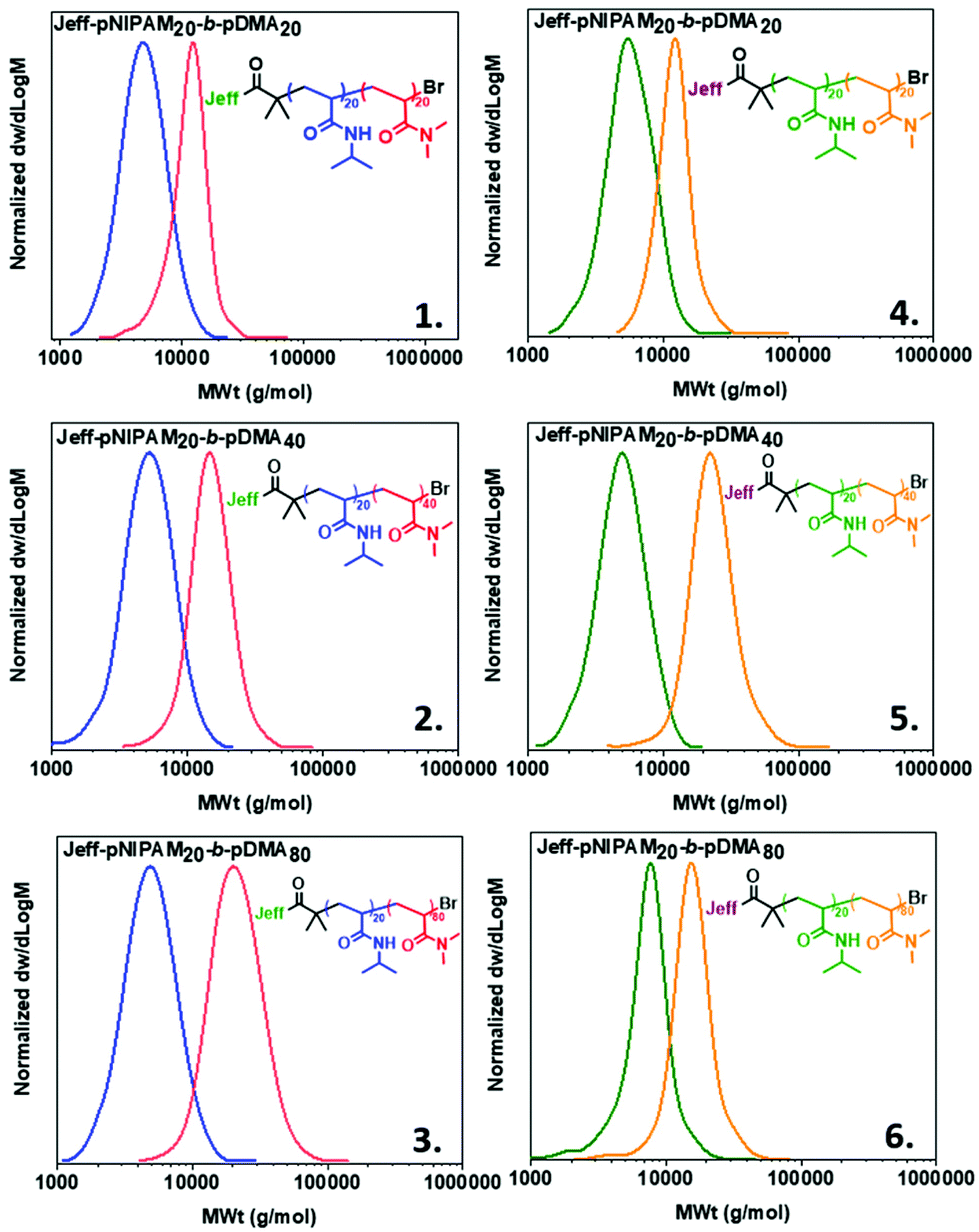
Apart from examining the capacity of our Jeffamine-derived macroinitiators at homopolymers of different molar masses, we were interested in examining the ability to conduct in situ chain extensions, aiming to investigate the extent of end-group fidelity that can be achieved. Initially, the M-1000-Int initiated PNIPAM with targeted DPn = 20 was chain-extended with NIPAM (20 eq.) to give Jeff-PNIPAM20-b-PNIPAM20 (Table S1, entry 1 and Fig. S13‡) at high conversions and dispersity values as low as Đ ∼1.08. Similar results were obtained with the use of M-2005-In. (Table S1 and Fig. S14‡). In this context, a series of block copolymers consisting of different PNIPAM/poly(N,N-dimethyl acrylamide) (PDMA) ratios were prepared via sequential monomer addition, using both the M-1000-Int and M-2005-Int initiators (Table S1‡ and Fig. 3). For the formation of these copolymers, PNIPAM (DPn = 20) was synthesized as described above, and upon reaching high conversion (7–15 minutes) a second aliquot of the deoxygenated second monomer (dissolved in water) was transferred into the reaction vessel via a nitrogen purged syringe. After 30 min, samples were taken from the polymerization solution for 1H NMR and DMF-SEC analysis, revealing a clear shift to higher molecular weights. In all cases, near-quantitative conversions (Table S1‡), and low dispersity (Đ ≤ 1.20) were observed (Fig. 3), verifying our system's potential to afford block copolymers while using both the hydrophilic and hydrophobic Jeffamine-derived macroinitiators.
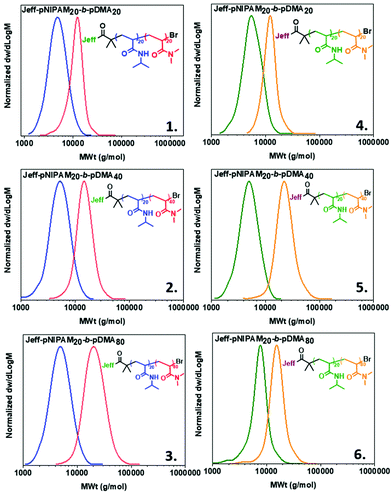 |
| | Fig. 3 Molecular weight distributions of block copolymers utilizing the M-1000-Int ((1–3) and the M-2005-Int macroinitiators (4–6). Conditions for the synthesis of Jeff-PNIPAM20:[Jeff]:[Cu(I)Br]:[Me6TREN]:[NIPAM] = [1]:[0.4]:[0.4]:[20] at 0 °C. | |
Investigation of the thermoresponsive aggregation behaviour
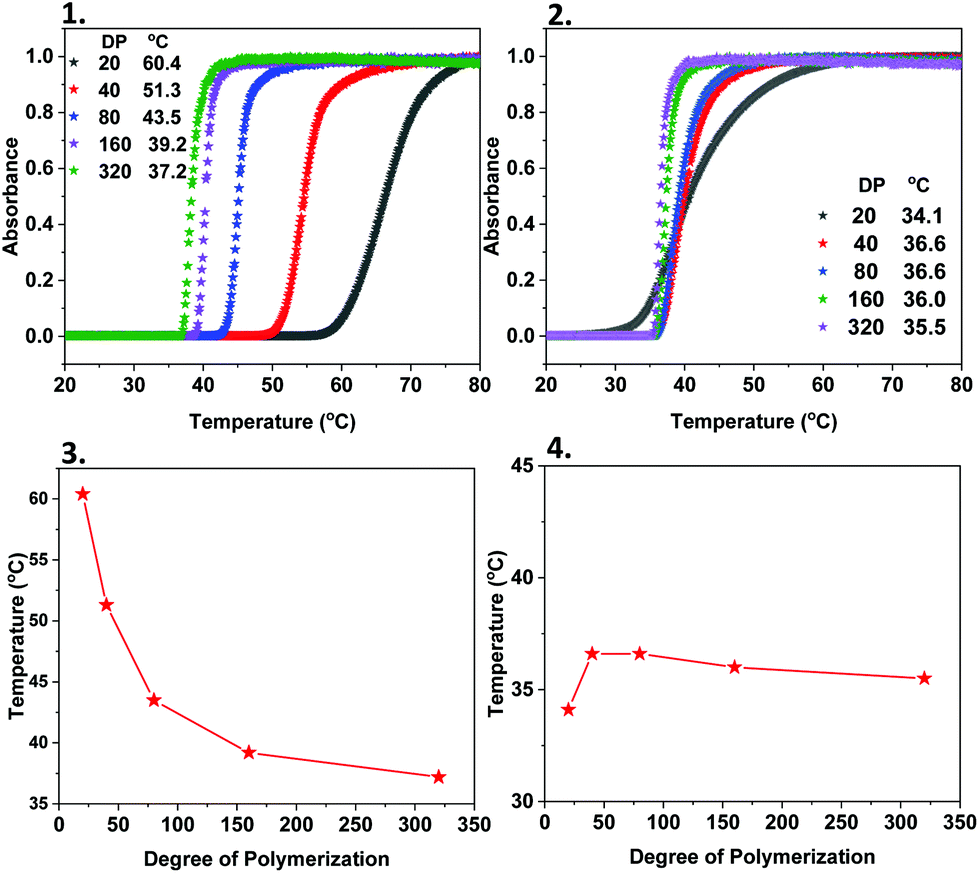
PNIPAM is a widely-investigated thermoresponsive material with a lower critical solution temperature (LCST) of 32 °C. Based on this, we anticipated that both the homopolymers and the PNIPAM-containing copolymers which are initiated by M-1000-Int and M-2005-Int, would exhibit different thermoresponsive behaviour than expected (i.e. when using a small organic molecule as initiator). Initially, the cloud point (Tcp) of PNIPAM40 by the water-soluble initiator (WSI) 2,3-dihydroxypropyl 2-bromo-2-methylpropanoate was examined and compared with PNIPAM40 initiated by M-1000-Int and M-2005-Int. Measurements were carried out using UV/Vis spectroscopy and the transmitted intensity recorded as a function of heating and cooling for cycles, with Tcp defined as being the onset temperature of transmittance (Fig. 4). It was observed that the cloud points of PNIPAM were different when the different initiators were used, while in all cases the polymer concentration was the same. Although the PNIPAM40 initiated by WSI had a Tcp ∼45 °C, when the hydrophobic M-2005-Int macroinitiator was used the Tcp decreased to ∼37 °C, while in the case of the hydrophilic M-1000-Int, the Tcp exhibited an increase up to ∼51 °C (Fig. S15 and S16‡). These differences were attributed to the increased hydrophobicity, and respectively hydrophilicity occurring from the two Jeffamine-derived macroinitiators. Apart from PNIPAM40, the Tcp of PNIPAM with different DPs exhibited differences when the hydrophilic macroinitiator was used, depending on the chain length of the polymers. These were attributed to the amphiphilic nature of M-1000-Int-P(NIPAM)x. Specifically, it was found that the cloud point decreases with increasing chain length, from ∼60 °C for DPn = 20 to ∼37 °C for DPn = 320 (Fig. 4(1) and (3)) and this is likely due to the easier assembly of short polymer chains into micelles.39
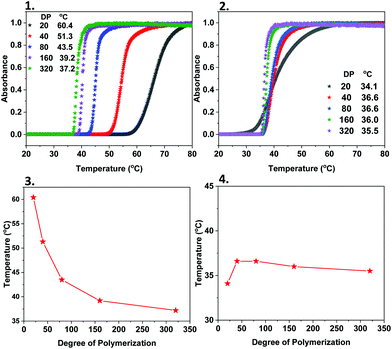 |
| | Fig. 4 Cloud point measurements of Jeff-P(NIPAM)x (DPn = 20–320) using (1) the hydrophilic M-1000-Int initiator and (2) the hydrophobic M-2005-Int and dependence of the cloud point temperature (Tcp) of (3) M-1000-Int-P(NIPAM)x and (4) M-2005-Int-P(NIPAM)x on the degree of polymerization. | |
Although we observed some differences in cloud point when the hydrophilic macroinitiator was used for the different DPs of PNIPAM, the same trends were not observed in the case of the hydrophobic M-2005-Int. The Tcp of M-2005-Int-P(NIPAM)40 (∼36 °C) did not change significantly with an increase (or decrease) of the chain length, and this was anticipated due to the already hydrophobic nature of both the polymers and the macroinitiator (Fig. 4(2) and (4)). It should be noted that in this case, aggregation of the polymers was observed visually (polymer solution going from clear to opaque) between 25 °C and 60 °C for DPn = 20–80.
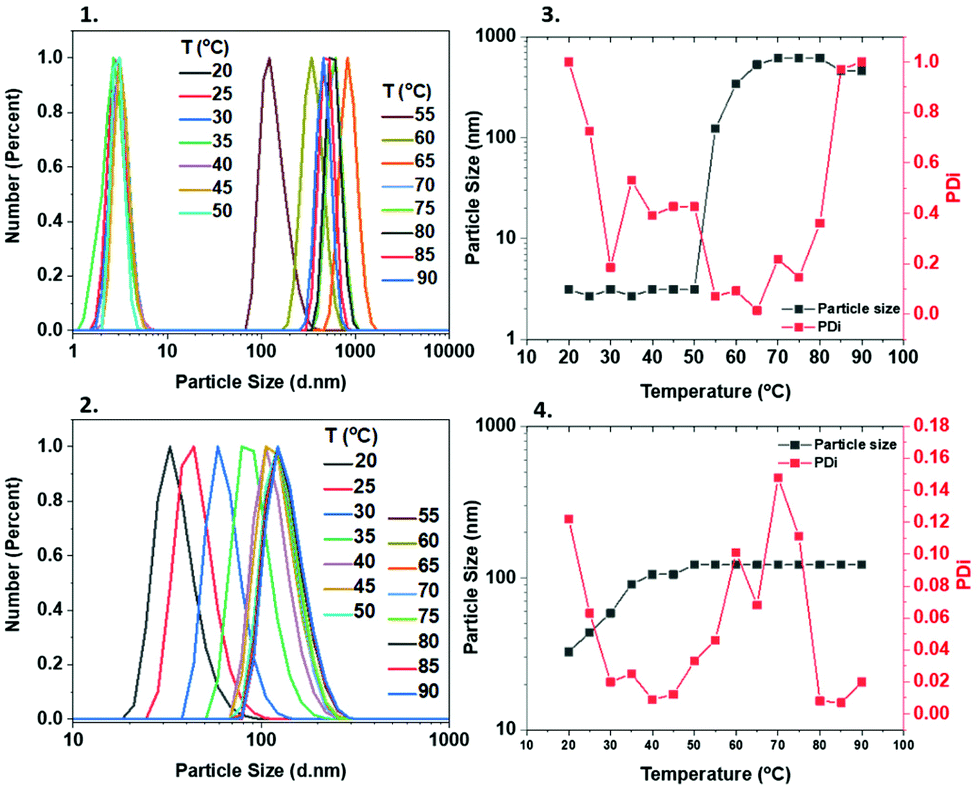
Particle size studies
In order to obtain more information about the temperature-dependent aggregation behaviour of these polymers, dynamic light scattering (DLS) measurements were carried out, with all samples being measured at a range of temperatures (20–90 °C). When the M-1000-Int-PNIPAM20 was measured from 20–50 °C, the particle size was found to be ∼3 nm. Above ∼50 °C, it was rapidly increased, reaching >600 nm at 70–80 °C (Fig. 5(1) and (3)), indicating the formation of large (compared to lower temperatures) aggregates, possibly attributed to higher hydrophobic interactions between the PNIPAM chains. These observations can be correlated with the Tcp of M-1000-Int-PNIPAM20, which was found to be ∼60 °C due to the increased hydrophilic content (macroinitiator). In the case of M-2005-Int-PNIPAM20, the particle size exhibited less significant differences at varying temperatures (Fig. 5(2) and (4)), while consistently smaller aggregates (up to ∼120 nm) with narrower size distributions were observed for elevated temperatures (above 50 °C). Apart from the PNIPAM homopolymers, the particle size properties of the PNIPAM/PDMA copolymers were also investigated at different temperatures. For this purpose, DLS measurements were carried out from 20 to 90 °C for the Jeff-PNIPAM20-b-PDMA80 copolymers (using both the hydrophilic and hydrophobic Jeffamine-macroinitiators). Same as previously, when temperatures below the Tcp of PNIPAM20 were applied, the size of the aggregates was as low as ∼5 nm, while at temperatures above 65 °C, a rapid increase (∼160 nm) was observed when the hydrophilic macroinitiator was used (Fig. S17‡). For the Jeff-PNIPAM20-b-PDMA80 with the hydrophobic M-2005-Int, increase on the particle size was observed at lower temperatures (>35 °C), whilst aggregates smaller than when M-1000-Int was used (from ∼5 to ∼20 nm) were observed (Fig. S18‡). In summary, the use of the hydrophilic initiator led to the formation of bigger aggregates, which is expected due to the hydrophilic macroinitiator which is responsible for the amphiphilic nature of the copolymer and allows for interactions with the solvent. Conversley, the use of the hydrophobic M-2005-Int led to the formation of smaller particles/aggregates, which is expected due to the higher hydrophobic content which restricts interaction with the solvent leading to more “packed” aggregates.
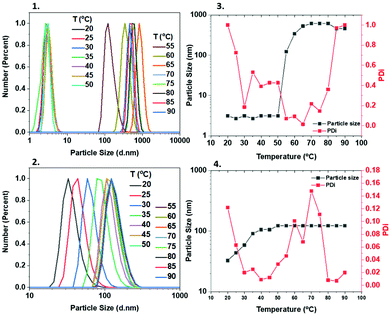 |
| | Fig. 5 DLS characterization of the Jeff-poly(NIPAM)20 using the hydrophilic (1 and 3) and hydrophobic (2 and 4) Jeffamine-derived macroinitiators. | |
Thermal analysis
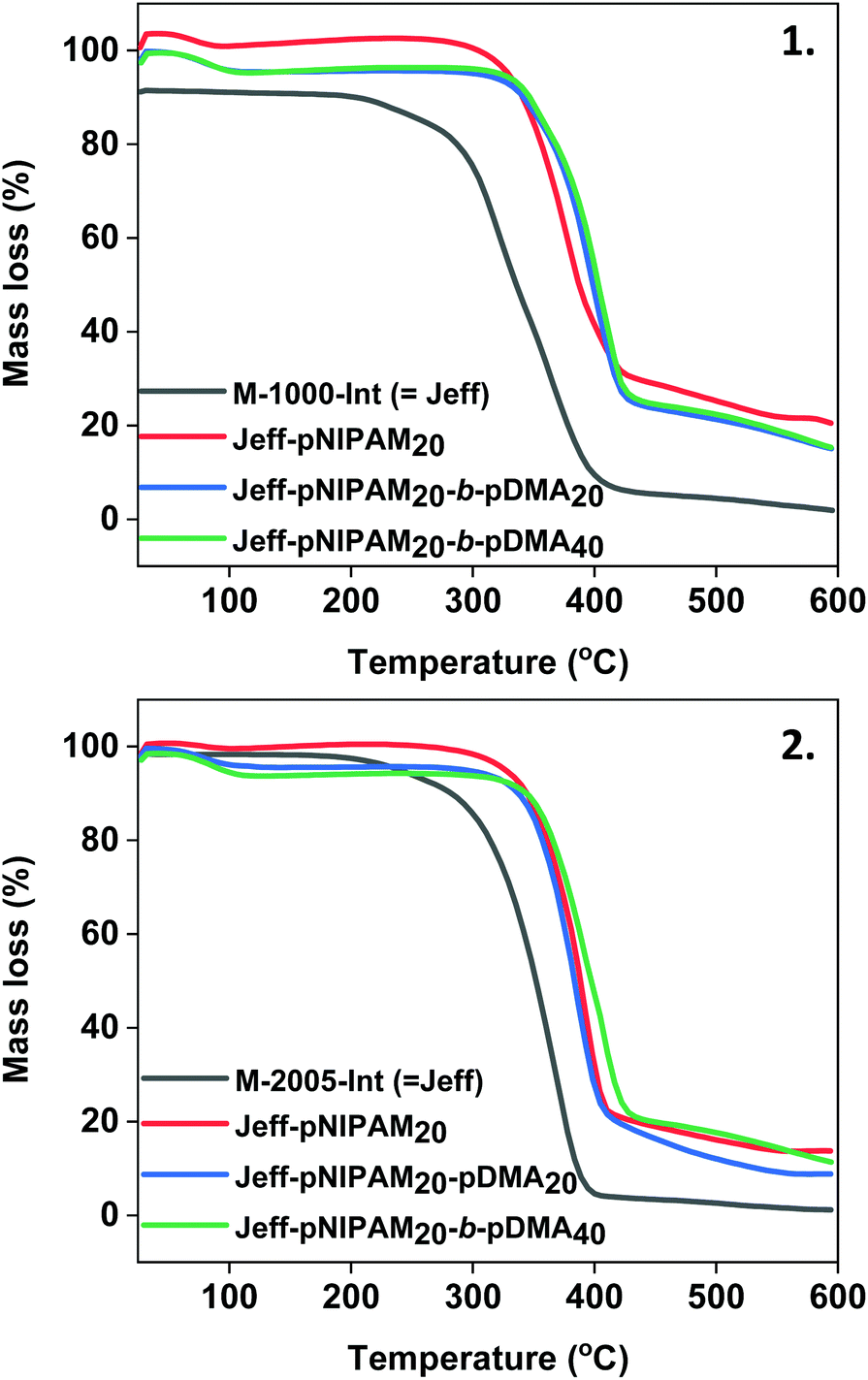
Thermal analyses (TGA) was carried out in order to investigate the decomposition temperatures and physical properties of the Jeffamine-initiated polymers. Initially, PNIPAM20 initiated by WSI was examined, showing a decomposition temperature at 225 °C, with 79.6%, mass loss (Fig. S19‡). On the other hand, the M-1000-Int-PNIPAM20 exhibited degradation at 258 °C with 70.5% mass loss, indicating that the incorporation of the hydrophilic Jeffamine macroinitiator leads to an increase on the decomposition temperature, thus providing higher thermal stability (Fig. 6(1), red). Further increase on the thermal stability was observed with the incorporation of a second PDMA block, with the Jeff-PNIPAM20-b-PDMAx copolymers showing a decomposition temperature at ∼300 °C (with 79.5% and 79.1% mass loss) (Fig. 6(1), blue & green).
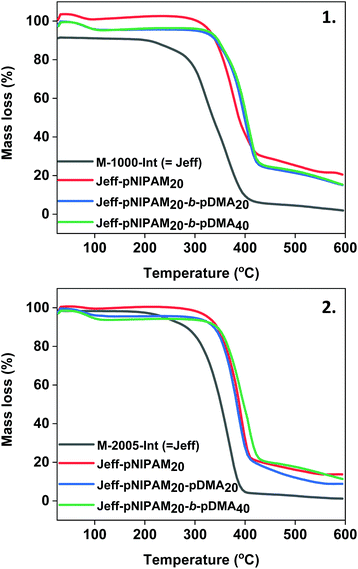 |
| | Fig. 6 TGA plots for the polyacrylamides synthesized using (1) the hydrophilic M-1000-Int macroinitiator and (2) the hydrophobic M-2005-Int macroinitiator. | |
A similar behaviour was observed for the homopolymer and block copolymers initiated by the hydrophobic M-2005-Int (heating from 25 °C to 600 °C). When the WSI-initiated PNIPAM20 was compared with the M-2005-Int-PNIPAM20, the decomposition temperature increased by ∼40 °C with 86.4% mass loss (Fig. 6(2) and Fig. S20‡). However, it should be noted that the mass loss of the hydrophobic macroinitiator is higher (95%) with relatively lower decomposition temperature (from 166 °C), compared with the derivative PNIPAM-containing polymers. In all cases, the use of the Jeffamine macroinitiators provided higher thermal stability for the synthesized polyacrylamides. To examine the effect of the macroinitiators on the glass transition temperature of the synthesized polyacrylamides, differential scanning calorimetry (DSC) was employed. The Tg of the WSI-initiated PNIPAM20 and WSI-initiated PNIPAM40 was 136.0 °C and 137.1 °C, respectively (Fig. S23 and Table S2‡). The Tg of the two Jeffamine-macroinitiators was 27.7 °C for the hydrophilic M-1000-Int and −69.9 °C for the hydrophobic M-2005-Int (Table S2 and Fig. S21, S22‡), with this difference on the Tg of the two macroinitiators occurring from the different EO/PO ratios. When the M-1000-Int-PNIPAM40 was measured, compatibility between the hydrophilic macroinitiator and the polymer was observed (Fig. S23‡), while the Tg (104.8 °C) was found to be lower than the Tg of analogous WSI-PNIPAM, due to the effect of the Jeffamine-macroinitiator (Table S2‡). A similar observation was made for the hydrophobic macroinitiator and the synthesized PNIPAM40, with the M-2005-Int-pNIPAM40 having a Tg at 115.7 °C (Fig. S23‡). When the block copolymers Jeff-PNIPAM20-b-PDMAx were measured, their glass transition temperatures were found to be within the range 90.6–114.9 °C (the Tg of the WSI-initiated PDMA40 was found at 120.8 °C), again showing compatibility between the three segments that comprise the diblocks (Fig. S24 and S25‡). It should be noted that different glass transition temperatures were observed depending on the DP of the PDMA block, with higher DPs of PDMA resulting in higher glass transition temperatures.
Conclusion
In summary, the use of polyetheramines (Jeffamines) as macroinitiators for the synthesis of well-defined polyacrylamides via aqueous Cu-RDRP is reported. By using both hydrophilic and hydrophobic Jeffamine-derived macroinitiator, homo- and block copolymers were synthesized with narrow, mono-modal molecular weight distributions at near-quantitative conversions. By leveraging the pre-disproportionation of Cu(I)/Me6Tren in water, rapid polymerization rates (<1 hour) were achieved at low temperature (0 °C). The use of Jeffamine-initiated polyacrylamides exhibited thermoresponsive aggregation behaviour dependent on the hydrophobicity/hydrophilicity of the macroinitiator. Thermal analyses revealed that the use of these polyetheramine-based macroinitiators enhances the thermal stability of the synthesized polyacrylamides, while a clear effect of the macroinitiators was observed on the glass transition temperatures of the synthesized homo- and block co-polymers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We appreciate financial support from the University of Warwick. We are grateful for the Polymer Characterization RTP, Dr D. Lester and Ms D. Coursari for providing use of SEC, DLS and TGA equipment. James S. Town thanks EPSRC for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1.
Notes and references
- I. Krakovský and N. K. Székely, Eur. Polym. J., 2011, 47, 2177–2188 CrossRef.
- R. Mladenova, M. Ignatova, N. Manolova, T. Petrova and I. Rashkov, Eur. Polym. J., 2002, 38, 989–999 CrossRef CAS.
- B.-H. Jiang, Y.-J. Peng and C.-P. Chen, J. Mater. Chem. A, 2017, 5, 10424–10429 RSC.
- H. Zhong, Z. Qiu, D. Sun, D. Zhang and W. Huang, J. Nat. Gas Sci. Eng., 2015, 26, 99–107 CrossRef CAS.
- C. Zhao, K. Tong, J. Tan, Q. Liu, T. Wu and D. Sun, Colloids Surf., A, 2014, 457, 8–15 CrossRef CAS.
- G. Mocanu, D. Mihaï, V. Dulong, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 87, 1440–1446 CrossRef CAS.
- G. Mocanu, Z. Souguir, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 89, 578–585 CrossRef CAS PubMed.
- D. J. Overstreet, R. Huynh, K. Jarbo, R. Y. McLemore and B. L. Vernon, J. Biomed. Mater. Res., Part A, 2013, 101, 1437–1446 CrossRef PubMed.
- N. d. N. Marques, R. d. C. Balaban, S. Halila and R. Borsali, Carbohydr. Polym., 2018, 184, 108–117 CrossRef CAS PubMed.
- V. Dulong, G. Mocanu, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 87, 1522–1531 CrossRef CAS.
- A. Lu, E. Petit, S. Li, Y. Wang, F. Su and S. Monge, Int. J. Biol. Macromol., 2019, 135, 38–45 CrossRef CAS PubMed.
- W. Agut, A. Brûlet, D. Taton and S. Lecommandoux, Langmuir, 2007, 23, 11526–11533 CrossRef CAS PubMed.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- A. Marathianos, E. Liarou, A. Anastasaki, R. Whitfield, M. Laurel, A. M. Wemyss and D. M. Haddleton, Polym. Chem., 2019, 10, 4402–4406 RSC.
- E. Liarou, Y. Han, A. M. Sanchez, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 2041–6520 Search PubMed.
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC.
- C. Waldron, Q. Zhang, Z. Li, V. Nikolaou, G. Nurumbetov, J. Godfrey, R. McHale, G. Yilmaz, R. K. Randev, M. Girault, K. McEwan, D. M. Haddleton, M. Droesbeke, A. J. Haddleton, P. Wilson, A. Simula, J. Collins, D. J. Lloyd, J. A. Burns, C. Summers, C. Houben, A. Anastasaki, M. Li, C. R. Becer, J. K. Kiviaho and N. Risangud, Polym. Chem., 2014, 5, 57–61 RSC.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914–8924 CrossRef CAS.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- Y. Cai, Y. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728–9737 CrossRef CAS.
- F. Azzam, L. Heux, J.-L. Putaux and B. Jean, Biomacromolecules, 2010, 11, 3652–3659 CrossRef CAS PubMed.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3947–3957 CrossRef CAS.
- C. Pottier, G. Morandi, V. Dulong, Z. Souguir, L. Picton and D. Le Cerf, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2606–2616 CrossRef CAS.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 10468–10482 CrossRef CAS PubMed.
- R. Aksakal, M. Resmini and C. R. Becer, Polym. Chem., 2016, 7, 171–175 RSC.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 6421–6432 CrossRef CAS.
- N. Ballard and J. M. Asua, ACS Macro Lett., 2020, 9, 190–196 CrossRef CAS.
- M. Senoo, Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 8005–8009 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromol. Rapid Commun., 2000, 21, 190–194 CrossRef CAS.
- A. Limer and D. M. Haddleton, Macromolecules, 2006, 39, 1353–1358 CrossRef CAS.
- S. Venkataraman and K. L. Wooley, Macromolecules, 2006, 39, 9661–9664 CrossRef CAS PubMed.
- X. Lou, B. F. M. de Waal, J. L. J. van Dongen, J. A. J. M. Vekemans and E. W. Meijer, J. Mass Spectrom., 2010, 45, 1195–1202 CrossRef CAS PubMed.
- S. Won, D. J. Phillips, M. Walker and M. I. Gibson, J. Mater. Chem. B, 2016, 4, 5673–5682 RSC.
Footnotes |
| † Raw data files for this paper can be found at https://wrap.warwick.ac.uk/138913. |
| ‡ Electronic supplementary information (ESI) available: Additional NMR, SEC TGA/DSC data. See DOI: 10.1039/d0py00555j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
James
Town
,
James
Town