DOI:
10.1039/D0PY00493F
(Paper)
Polym. Chem., 2020,
11, 4013-4021
Radical ring-opening polymerization of novel azlactone-functionalized vinyl cyclopropanes†
Received
3rd April 2020
, Accepted 29th May 2020
First published on 29th May 2020
Abstract
Azlactone-functionalized polymers are considered powerful materials for bioconjugation and many other applications. However, the limited number of azlactone monomers available and their multistage syntheses pose major challenges for the preparation of new reactive polymers from these monomers. In this article, we report the synthesis of a new class of azlactone monomers based on vinylcyclopropane (VCP). Furthermore, the (co)polymerization of the azlactone-functionalized VCPs has been successfully demonstrated to provide new azlactone polymers by using free-radical polymerization. The ability of the resulting amine-reactive polymers to be engaged in post-polymerization modifications was demonstrated using dansylcadaverine. These new azlactone-functionalized VCP monomers and polymers are potential candidates for the synthesis of innovative (bio)materials.
Introduction
In the last decade vinylcyclopropanes (VCP) have emerged as important intermediates for organic and materials chemistry.1,2 Owing to their unique structure which contains a vinyl group linked to a highly strained cyclopropane ring, VCP monomers easily undergo Radical Ring-Opening Polymerization (rROP) and display less polymerization shrinkage than other vinyl monomers such as methacrylates. This characteristic makes them attractive as low-shrinking components in dental adhesives,3–5 curing resins and (bio)material applications.6 Another interesting aspect of VCP monomers is that during rROP two distinct propagation pathways take place which give rise to poly(vinyl cyclopropane) (PVCP) with either unsaturated linear repeating units or saturated cyclic repeating units, or a mixture of both.7–9 The linear repeating units possess unsaturated bonds which have been successfully used for radical thiol–ene post-polymerization modifications.10 Therefore, the synthesis of PVCP with a high linear unit content within the polymer backbone is a topic of high interest. However, there are only a few examples of successful PVCP syntheses with predominantly linear repeating units, using Atom Transfer Radical Polymerization (ATRP).11,12 Nevertheless, according to previous studies, the formation of linear units in PVCP is strongly dependent upon the polymerization conditions as well as the monomer structure.9–12 Due to the uncertainty in the quantity of available unsaturated linear units within the polymer chains, the thiol–ene reaction cannot be considered an optimal choice for the post-polymerization modification of PVCP polymers. Fortunately, the PVCP chain can be modified not only using thiol–ene backbone modification, but also via the modification of the side-chains of PVCP. For example, Theato and coworkers successfully synthesized thermoresponsive PVCP polymers by using the post-polymerization modification of activated pentafluorophenyl ester-functionalized PVCP.13 Due to the high reactivity of PFP-activated esters toward amines, the PFP ester-functionalized PVCPs were readily reacted with different amines to afford thermoresponsive polymers. However, the reaction of PFP-activated esters with amines releases pentafluorophenol as a side-product.14 Extensive purification is thus necessary for the removal of this by-product and recovery of the final product. In contrast, the azlactone group can be considered as an excellent alternative for the preparation of activated esters. Owing to its high reactivity toward amines even in the absence of catalyst and without producing by-product, azlactone functionality has found wide use in organic, bioconjugation and (bio)macromolecular synthesis.15–20 Furthermore, the amine–azlactone reaction satisfied the requirements to be classified as a “click” reaction.21–24 Despite this potential, the synthesis and used of azlactone-functionalized polymers are limited by the availability of the corresponding monomers. Indeed, only three azlactone based monomers were reported in the literature namely isopropenyl azlactone,25 2-vinyl-4,4-dimethylazlactone,26 and 2-(p-vinylphenyl)-4,4-dimethylazlactone.27 To date, all azlactone-functionalized materials have primarily been obtained by incorporating the azlactone group into the polymer backbone via the radical (co)polymerization of azlactone monomers such as vinyl dimethylazlactone28–31 or styrene-bearing azlactones.27,32–35 Recently, Fontaine and coworkers demonstrated an efficient procedure to prepare azlactone-based linkers using a nucleophilic substitution reaction between a cesium carboxylate and a bromide-bearing azlactone molecule.36 As a straightforward and simple synthetic pathway, this process may prove to be a “magic toolbox” for the synthesis of monomers bearing azlactone groups.
In order to significantly enlarge the development of new azlactone-based monomers and hence new polymer structures, we proposed in the present work the synthesis of azlactone-functionalized VCP monomers. The advantages of this combination, VCP and azlactone functionality, are numerous such as an access to low-shrinking polymers, a rapid and modular synthesis of side-chain functionalized polymers and above all the fabrication of new platform chemically reactive to design new (bio)materials for dental adhesives and filling composites applications. To achieve our objective, two novel azlactone-functionalized VCP monomers were prepared and their free-radical (co)polymerization was performed. Thereafter, the ability of the azlactone to react with a fluorescent amine has also been demonstrated, showing the potential of these polymers for bioconjugation. To the best of our knowledge, this work represents the first example devoted to the synthesis and (co)polymerization of VCP monomers bearing an azlactone group.
Experimental
Materials
Methyl cyanoacetate (99%), dimethyl malonate (98%), cesium carbonate (Cs2CO3, 99%), cesium hydroxide monohydrate (CsOH·H2O, 99.95%), 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%), poly(ethylene glycol) methyl ether acrylate (PEGA,  ), dansylcadaverine (97%), tetrahydrofuran (THF, 99%), methanol (99%), N,N-dimethylformamide (DMF, 99.9%), chloroform (CHCl3, 99%), anhydrous 1,4-dioxane (99.8%), n-pentane (95%), diethylether (99%), sodium sulfate (Na2SO4), silica gel (SiO2) and Celite were purchased from Sigma-Aldrich. trans-1,4-Dibromo-2-butene was used as received from TCI Chemicals. 2-(1-Bromoethyl)-4,4-dimethyloxazol-5(4H)-one (referred to as bromo-azlactone) was prepared according to the literature.37
), dansylcadaverine (97%), tetrahydrofuran (THF, 99%), methanol (99%), N,N-dimethylformamide (DMF, 99.9%), chloroform (CHCl3, 99%), anhydrous 1,4-dioxane (99.8%), n-pentane (95%), diethylether (99%), sodium sulfate (Na2SO4), silica gel (SiO2) and Celite were purchased from Sigma-Aldrich. trans-1,4-Dibromo-2-butene was used as received from TCI Chemicals. 2-(1-Bromoethyl)-4,4-dimethyloxazol-5(4H)-one (referred to as bromo-azlactone) was prepared according to the literature.37
Analytical techniques
High resolution mass spectrometry (HR-MS).
Experiments were performed with a Synapt G2 HDMS quadrupole/time-of-flight (Manchester, UK). Samples were introduced at a 10 μl min−1 flow rate (capillary voltage +2.8 kV, sampling cone voltage: varied between +20 V and +60 V) under a desolvation gas (N2) flow of 100 L h−1 heated at 35 °C. Accurate mass experiments were performed using reference ions from PPG, PEG or CH3COONa internal standards. All the samples were dissolved in methanol doped with 3 mM ammonium acetate prior to analysis. Data analyses were conducted using MassLynx 4.1 programs provided by Waters.
Fourier
-
transformation infrared (FT
-
IR)
spectra were recorded using a PerkinElmer Spectrum Two FT-IR Spectrometer with an ATR accessory.
Nuclear magnetic resonance (NMR)
spectra were recorded by using Bruker AC 400 MHz.
Size exclusion chromatography (SEC).
Molar masses were determined by size exclusion chromatography (SEC). The apparatus was equipped with a 1260 infinity pump (Agilent Technologies), a 1260 infinity autosampler (Agilent Technologies), a 1260 infinity UV photodiode array detector (Agilent Technologies), a 1260 infinity RI detector (Agilent Technologies). The stationary phase was composed of 2 PSS-SDV Linear M columns and a precolumn at 40 °C. Mobile phase was THF at 1 mL min−1. Samples were solubilized in a mixture of THF and toluene (0.25 wt%) and filtered through 0.45 μm PTFE syringe filter (Agilent). Samples concentration was about 2.5 mg min−1. Polystyrene (PS) equivalent number-average and weight-average molar masses (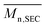 and
and 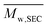 ) and dispersities (Đ) were calculated by means of PS calibration curve using PS standards from 0.86 to 526.0 kg mol−1 (Agilent, USA).
) and dispersities (Đ) were calculated by means of PS calibration curve using PS standards from 0.86 to 526.0 kg mol−1 (Agilent, USA).
Synthesis of methyl 1-cyano-2-vinylcyclopropanecarboxylate (1)
The methyl 1-cyano-2-vinylcyclopropanecarboxylate (1) was prepared according to the literature.38(1) was obtained as an oil product in 48% yield. HR-MS analysis (C8H9NO2): detected ion [M + Na]+, calculated value m/zcalc = 174.0525 Da and experimental value m/zexp = 174.0525 Da. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.82–5.18 (m, 3H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 3.82 (s, 3H, OCH3), 2.84–2.46 (m, 1H, CHring), 2.10–1.66 (m, 2H, CH2ring). 13C NMR (101 MHz, CDCl3) δ (ppm): 167.66 and 165.70 (COOCH3), 132.02 and 130.45 (CH2CH), 121.46 and 120.93 (CH2CH), 118.57 and 116.60 (CN), 53.59 and 53.44 (COOCH3), 35.83 and 33.87 (CHring), 23.94 and 22.70 (Cring), 20.97 and 20.18(CH2ring). FT-IR (cm−1): 2246 (ν(CN)), 1740 (ν(CO)ester), 1639 (ν(CC)).
CH), 3.82 (s, 3H, OCH3), 2.84–2.46 (m, 1H, CHring), 2.10–1.66 (m, 2H, CH2ring). 13C NMR (101 MHz, CDCl3) δ (ppm): 167.66 and 165.70 (COOCH3), 132.02 and 130.45 (CH2CH), 121.46 and 120.93 (CH2CH), 118.57 and 116.60 (CN), 53.59 and 53.44 (COOCH3), 35.83 and 33.87 (CHring), 23.94 and 22.70 (Cring), 20.97 and 20.18(CH2ring). FT-IR (cm−1): 2246 (ν(CN)), 1740 (ν(CO)ester), 1639 (ν(CC)).
Synthesis of dimethyl 2-vinylcyclopropane-1,1-dicarboxylate (2)
The dimethyl 2-vinylcyclopropane-1,1-dicarboxylate (2) was prepared according to the literature.38(1) was obtained as an colourless oil in 89% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.47–5.13 (m, 3H, CH2CH), 3.74 (s, 3H, OCH3), 2.58 (m, 1H, CHring), 1.73–1.55 (m, 2H, CH2ring). 13C NMR (101 MHz, CDCl3, δ ppm): 170.01 and 167.78 (COOCH3), 132.96 (CH2CH), 118.69 (CH2CH), 52.71 and 52.74 (COOCH3), 35.74 (Cring), 31.47 (CHring) and 20.60 (CH2ring). FT-IR (cm−1): 1730 (ν(CO)ester), 1639 (ν(CC)).
Synthesis of cesium 1-cyano-2-vinylcyclopropanecarboxylate (3)
The methyl 1-cyano-2-vinylcyclopropanecarboxylate (1) (2.00 g, 13.24 × 10−3 mol) and methanol (2.0 mL) were added into a round bottom flask and cooled to −10 °C under stirring and under argon atmosphere. A solution of cesium hydroxide monohydrate (CsOH·H2O, 2.25 g, 13.39 × 10−3 mol) in methanol (7.0 mL) was then added dropwise into the reaction. After addition, the reaction was continuously stirred at −10 °C for 2 h and then at room temperature for 24 h. The solvent was evaporated under reduced pressure at 40 °C until dryness. The crude product was then washed with diethyl ether and dried to obtain the final product as a yellow hygroscopic solid (3.35 g). Yield: 94%. HR-MS analysis (C7H6NO2−Cs+): detected ion [M + Cs]+, calculated value m/zcalc = 401.8502 Da and experimental value m/zexp = 401.8499 Da. 1H NMR (400 MHz, D2O) δ (ppm): 5.73 (m, 1H, CH2CH), 5.54–5.22 (m, 2H, CH2CH), 2.77–2.42 (m, 1H, CHring), 2.11–1.48 (m, 2H, CH2ring) (ESI, Fig. S1†). 13C NMR (101 MHz, D2O) δ (ppm): 173.03 and 171.0 (COO), 132.89 and 132.60 (CH2CH), 122.64 and 121.26 (CN), 119.72 and 119.15 (CH2CH), 32.88 and 31.87 (CHring), 23.71 and 23.06 (Cring), 22.21 and 20.81(CH2ring).
Synthesis of cesium 1-(methoxycarbonyl)-2-vinylcyclopropane-carboxylate (4)
The dimethyl 2-vinylcyclopropane-1,1-dicarboxylate (2) (2.00 g, 1.09 × 10−2 mol) and methanol (6.0 mL) were added into a round bottom flask and cooled to −10 °C under stirring and under argon atmosphere. A solution of cesium hydroxide monohydrate (CsOH·H2O, 1.88 g, 1.11 × 10−2 mol) in methanol (4.0 mL) was then added dropwise into the reaction. After addition the reaction was continuously stirred at −10 °C for 2 h and then at room temperature for 24 h. The solution was subsequently concentrated under a reduced atmosphere at 40 °C until dryness. The crude solid was then washed with diethyl ether, filtered and dried to obtain the final product (2.60 g) as white solid. Yield: 79%. HR-MS analysis (C8H9O4−Cs+): detected ion [M + Cs]+, calculated value m/zcalc = 434.8604 Da and experimental value m/zexp = 434.8602 Da. 1H NMR (400 MHz, D2O) δ (ppm): 5.58–5.16 (m, 3H, CH2CH), 3.79 (s, 3H, OCH3), 2.45 (m, 1H, CHring), 1.61–1.45 (m, 2H, CH2ring) (ESI, Fig. S2†).
Synthesis of azlactone-functionalized VCP (5)
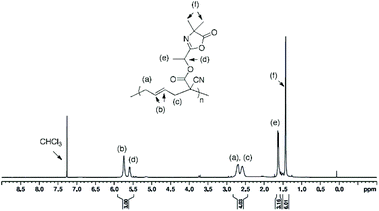
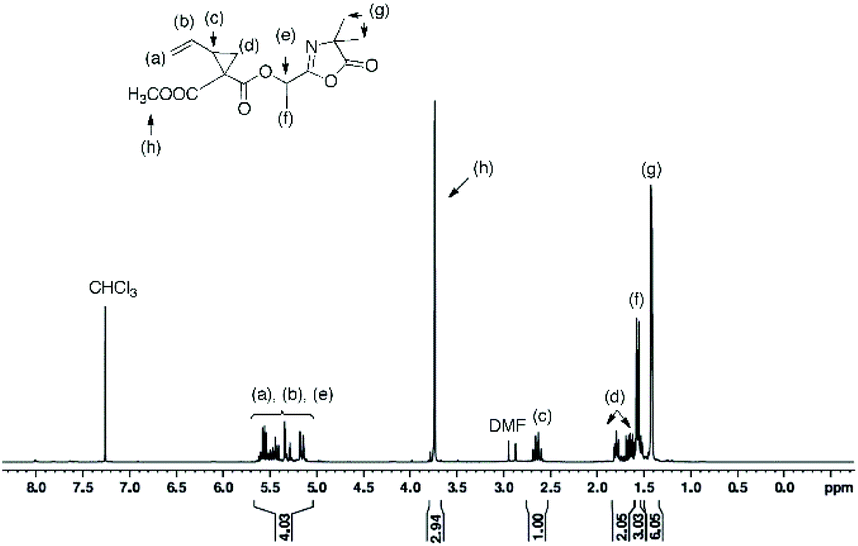
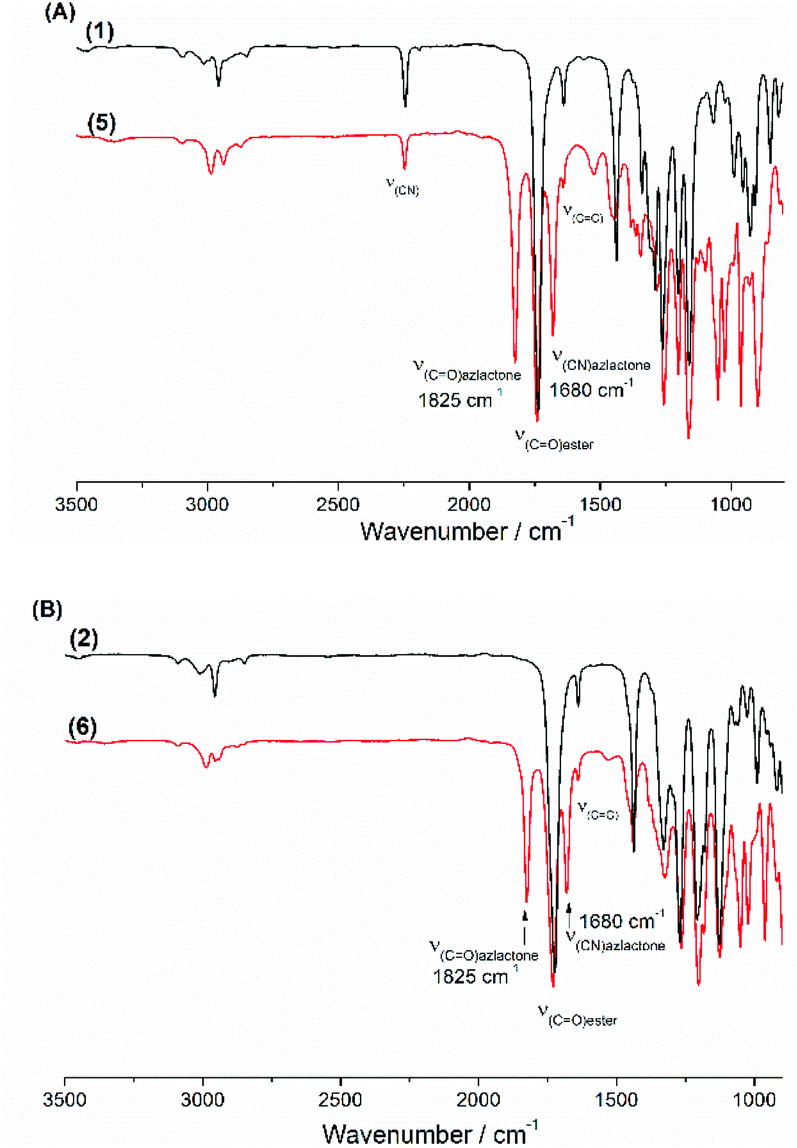
In a round bottom flask, cesium salt (3) (2.95 g, 10.97 × 10−3 mol) and DMF (10.0 mL) were added and stirred at 40 °C under argon. Then, a solution of 2-(1-bromoethyl)-4,4-dimethyloxazol-5(4H)-one (bromo-azlactone, 2.41 g, 10.95 × 10−3 mol) in DMF (4.0 mL) was added dropwise into the reaction. After addition was complete, the reaction was continuously stirred at 40 °C for 20 h. The reaction was then filtered to remove the white salt and subsequently concentrated under reduced atmosphere at 55 °C to remove the DMF. The crude product was then dissolved in chloroform, filtered through a short silica gel pad and concentrated under vacuum to obtain the final product as yellow viscous oil (2.57 g). Yield: 85%. HR-MS analysis (C14H16N2O4): detected ion [M + H]+, calculated value m/zcalc = 277.1183 Da and experimental value m/zexp = 277.1183 Da. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.78–5.35 (m, 4H, CH2CH, OCH(CH3)), 2.74–2.48 (m, 1H, CHring), 2.00–1.67 (m, 2H, CH2ring), 1.62 (3H, OCH(CH3)), 1.42–146 (6H, C(CH3)2) (Fig. 1). 13C NMR (101 MHz, CDCl3) δ (ppm): 180.09 (CO)azlactone, 160.62 (CN)azlactone, 166.32 (CO)ester, 131.76 and 131.66 (CH2CH), 121.32 and 121.27 (CH2CH), 116.00 (CN), 67.36 and 67.30 (OCH(CH3)), 65.57 (C(CH3)2), 34.37 and 34.31 (CHring), 24.51–24.22 (CH2ring and C(CH3)2), 21.13 and 21.09 (Cring), 16.71 and 16.68 (OCH(CH3)) (ESI, Fig. S3†). FT-IR (cm−1): 2246 (ν(CN)), 1825 (ν(CO)azlactone), 1730 (ν(CO)ester), 1680 (ν(CN)azlactone), 1639 (ν(CC)) (Fig. 3A).
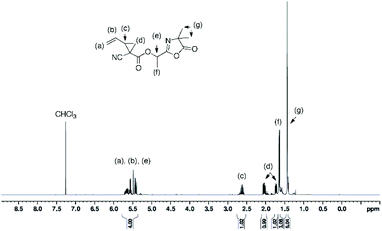 |
| | Fig. 1
1H NMR (400 MHz) spectrum of azlactone-functionalized vinylcyclopropane (5) in CDCl3. | |
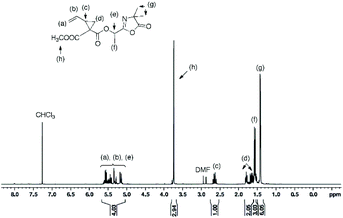 |
| | Fig. 2
1H NMR (400 MHz) spectrum of azlactone-functionalized vinylcyclopropane (6) in CDCl3. | |
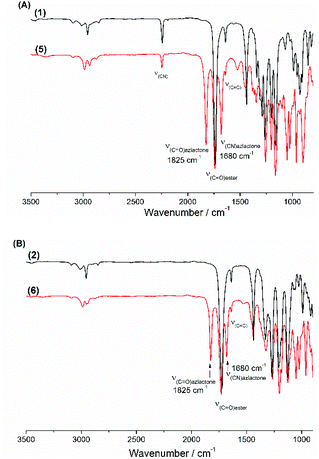 |
| | Fig. 3 Superposition of the FT-IR spectra of the VCP monomers before and after azlactone functionalization: (A) (1) and (5); (B) (2) and (6). | |
Synthesis of azlactone-functionalized VCP (6)
In a round bottom flask, cesium salt (4) (1.60 g, 5.30 × 10−3 mol) and DMF (6.0 mL) were added and stirred at 40 °C under argon. After, a solution of bromo-azlactone (1.16 g, 5.27 × 10−3 mol) in DMF (2.0 mL) was added dropwise into the reaction. After addition was complete, the reaction was continuously stirred at 40 °C for 19 h before being filtered to remove the white salt and subsequently concentrated under reduced atmosphere at 55 °C to remove the DMF. The crude product was then dissolved in chloroform, filtered through a short silica gel pad and then concentrated under vacuum to obtain the final product (1.34 g) as a viscous oil. Yield: 82%. HR-MS analysis (C15H19NO6): detected ion [M + H]+, calculated value m/zcalc = 310.1285 Da and experimental value m/zexp = 310.1285 Da. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.65–5.07 (m, 4H, CH2CH and OCH(CH3)), 3.74 (s, 3H, COOCH3), 2.68–2.58 (m, 1H, CHring), 1.83–1.50 (m, 5H, CH2ring, and OCH(CH3)), 1.46–1.42 (6H, C(CH3)2) (Fig. 2). 13C NMR (101 MHz, CDCl3) δ (ppm): 180.35 (CO)azlactone, 161.2 (CN)azlactone, 168.37–167.33 (CO)ester, 132.69 (CH2CH), 119.10 (CH2CH), 66.09 (OCH(CH3)), 65.46 (C(CH3)2)azlactone, 52.67 and 52.63 (COOCH3), 35.65 and 35.59 (CHring), 31.78 and 31.61 (CH2ring), 24.30 C(CH3)2, 21.01 and 20.78 (Cring), 16.61 (OCH(CH3)) (ESI, Fig. S4†). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1740 (ν(CO)ester), 1680 (ν(CN)azlactone), 1639 (ν(CC)) (Fig. 3B).
Free-radical polymerization of VCP monomers
As a general procedure, VCP–azlactone monomer (0.30 g, 1.09 × 10−3 mol), AIBN (5.60 mg, 3.41 × 10−5 mol), 1,4-dioxane (0.20 mL) and DMF (15.0 mg) as an internal reference were charged into a headspace vial equipped with a magnetic stir bar. The vial was then sealed using a headspace cap. After, the solution was deoxygenated by bubbling argon for 30 min and was then immersed in an oil bath at 80 °C to allow the polymerization to occur. The polymerization was quenched after 30 min by rapid cooling and exposure of the polymerization solution to air. The resulting product was then dissolved in chloroform and precipitated in diethyl ether. The final polymer was filtered and dried under vacuum to give a white solid.
Polymer (7) (Table 1, entry 2).
Isolated polymer yield: 105 mg, 85%. 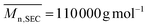 ; Đ = 2.25. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CHCH, OCH(CH3)), 2.84–2.44 (CH–CH2), 1.63 (OCH(CH3)), 1.43 (C(CH3)2) (Fig. 4). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1746 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S5†).
; Đ = 2.25. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CHCH, OCH(CH3)), 2.84–2.44 (CH–CH2), 1.63 (OCH(CH3)), 1.43 (C(CH3)2) (Fig. 4). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1746 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S5†).
 |
| | Fig. 4
1H NMR (400 MHz) spectrum of polymer (7) in CDCl3 (entry 2, Table 1). | |
Table 1 Polymerization results for azlactone-functionalized VCPsa
Polymer (8) (Table 1, entry 4).
Isolated polymer yield: 160 mg, 81%. 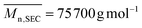 ; Đ = 1.6. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CHCH, OCH(CH3)), 3.71 (COOCH3), 2.84–2.44 (CH–CH2), 2.41–1.74 (CH2 and CH of cyclic units), 1.54 (OCH(CH3)), 1.42 (C(CH3)2) (Fig. 5). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1733 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S6†).
; Đ = 1.6. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CHCH, OCH(CH3)), 3.71 (COOCH3), 2.84–2.44 (CH–CH2), 2.41–1.74 (CH2 and CH of cyclic units), 1.54 (OCH(CH3)), 1.42 (C(CH3)2) (Fig. 5). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1733 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S6†).
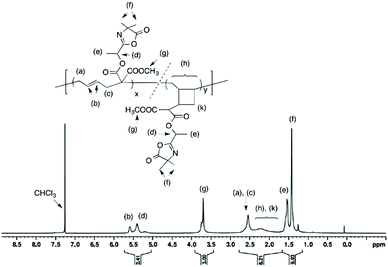 |
| | Fig. 5
1H NMR (400 MHz) spectrum of polymer (8) in CDCl3 (entry 4, Table 1). | |
Free-radical copolymerization of azlactone-functionalized VCP (5) and acrylate
In a general procedure, VCP–azlactone (5) monomer (0.20 g, 7.25 × 10−4 mol), poly(ethylene glycol) methyl ether acrylate (PEGA,  , 1.00 g, 2.08 × 10−3 mol), AIBN (4.3 mg, 2.62 × 10−5 mol) and 1,4-dioxane (1.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. The vial was then sealed with headspace cap. After, the solution was deoxygenated by bubbling argon for 30 min and then immersed in an oil bath at 80 °C to allow the polymerization to occur. The polymerization was quenched after 280 min by rapid cooling and exposure of the polymerization solution to air. The resulting product was then dissolved in chloroform and precipitated using diethyl ether. The final polymer was filtered and dried under vacuum to give a gum-like solid.
, 1.00 g, 2.08 × 10−3 mol), AIBN (4.3 mg, 2.62 × 10−5 mol) and 1,4-dioxane (1.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. The vial was then sealed with headspace cap. After, the solution was deoxygenated by bubbling argon for 30 min and then immersed in an oil bath at 80 °C to allow the polymerization to occur. The polymerization was quenched after 280 min by rapid cooling and exposure of the polymerization solution to air. The resulting product was then dissolved in chloroform and precipitated using diethyl ether. The final polymer was filtered and dried under vacuum to give a gum-like solid. 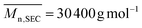 ; Đ = 2.26. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.14 (COOCH2CH2), 3.68–3.57 (OCH2CH2), 3.37 (CH3O), 1.58–1.39 (OCH(CH3) and C(CH3)2 of azlactone ring) (ESI, Fig. S7†). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1732 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S8†). The molar composition, determined by 1H NMR, of (5) and PEGA in this copolymer was 19 mol% and 81 mol% respectively.
; Đ = 2.26. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.14 (COOCH2CH2), 3.68–3.57 (OCH2CH2), 3.37 (CH3O), 1.58–1.39 (OCH(CH3) and C(CH3)2 of azlactone ring) (ESI, Fig. S7†). FT-IR (cm−1): 1825 (ν(CO)azlactone), 1732 (ν(CO)ester), 1680 (ν(CN)azlactone) (ESI, Fig. S8†). The molar composition, determined by 1H NMR, of (5) and PEGA in this copolymer was 19 mol% and 81 mol% respectively.
Reactivity of P(PEGA-co-(5)) with dansylcadaverine
Poly(PEGA-co-(5)) ( , Đ = 2.26, 0.152 g, 5 × 10−6 mol) and THF (2.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. Then, a solution of dansylcadaverine (11 mg, 3.28 × 10−5 mol) in THF (2.0 mL) was added into the reaction mixture and the resulting solution was stirred at 40 °C for 21 h under argon. The resulting product was subsequently precipitated in diethyl ether and then dried under vacuum to give a gum-like solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.28–7.69 (CH of aromatic ring) 4.14 (COOCH2CH2), 3.72–3.57 (OCH2CH2), 3.37 (CH3O) (ESI, Fig. S9†).
, Đ = 2.26, 0.152 g, 5 × 10−6 mol) and THF (2.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. Then, a solution of dansylcadaverine (11 mg, 3.28 × 10−5 mol) in THF (2.0 mL) was added into the reaction mixture and the resulting solution was stirred at 40 °C for 21 h under argon. The resulting product was subsequently precipitated in diethyl ether and then dried under vacuum to give a gum-like solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.28–7.69 (CH of aromatic ring) 4.14 (COOCH2CH2), 3.72–3.57 (OCH2CH2), 3.37 (CH3O) (ESI, Fig. S9†).
Results and discussion
Synthesis of azlactone-functionalized VCP monomers
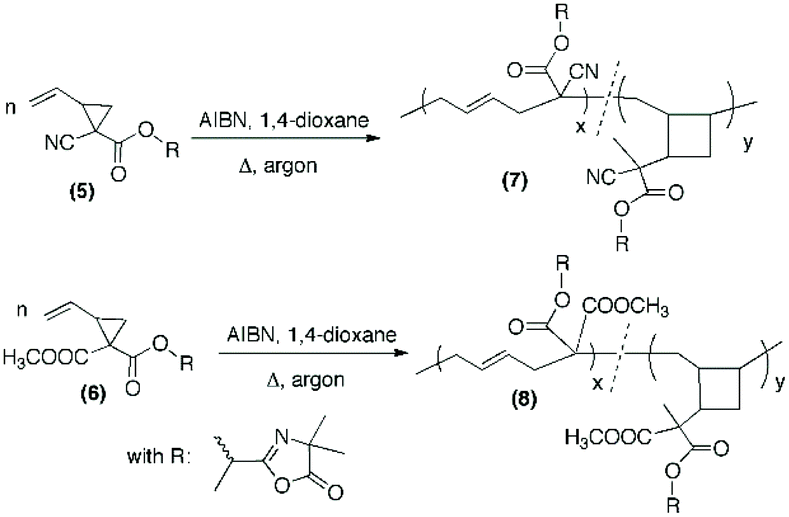
Azlactone-functionalized VCP monomers were successfully prepared in three steps, as outlined in Scheme 1. The starting precursors (1) and (2) were synthesized following a modified procedure involving the reaction of 1,4-dibromobut-2-ene and cesium carbonate (Cs2CO3) in THF with methyl cyanoacetate and dimethylmalonate, respectively.38 The resulting compounds (1) and (2) were subsequently hydrolyzed in methanol in the presence of cesium hydroxide (1 equiv.) to afford their corresponding cesium salts (3) and (4). The hydrolysis of (1) and (2) was monitored by 1H NMR spectroscopy by observing the decreasing intensity of the methyl moiety of the ester group –COOCH3 at 3.82 ppm or 3.74 ppm, respectively (ESI, Fig. S1 and S2†). In addition, the formation and purity of the cesium salts were confirmed by HR-MS analysis. The single hydrolysis of two methoxycarbonyl groups of VCP monomer (2) was already reported by Moszner et al. by using 1 equiv. of KOH solution.39 In the present study, we replaced KOH by CsOH to obtain the corresponding cesium carboxylate known for its higher nucleophilic reactivity than other alkaline metal carboxylates. This single hydrolysis selectivity can be explained firstly by the use of 1 equiv. of CsOH for 2 equiv. of methoxycarbonyl groups and secondly by sterically hindered cesium carboxylate which probably hinders the hydrolysis of the second methoxycarbonyl group.
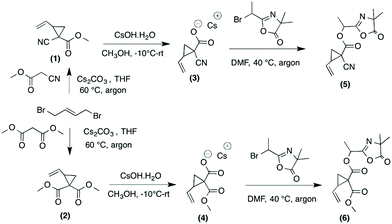 |
| | Scheme 1 Synthesis of azlactone-functionalized vinylcyclopropanes (5) and (6). | |
The cesium salt (3) was then reacted with 2-(1-bromoethyl)-4,4-dimethyloxazol-5(4H)-one37 (referred as bromo-azlactone) in N,N-dimethylformamide (DMF) at 40 °C yielding the azlactone-functionalized VCP monomer (5) (Scheme 1). After purification, the target compound was obtained with a good yield of 85%. Fig. 1 shows the 1H NMR spectrum of (5) which displays characteristic signals at 5.35–5.78 ppm due to the vinyl protons CH2CH– (labeled a and b) and at 1.42–1.46 ppm due to the gem dimethyl group –C(CH3)2 (labeled g) of the azlactone ring. Furthermore, the formation and purity of (5) were checked by HR-MS analysis (C14H16N2O4), showing an excellent agreement between [M + H]+ calculated value (m/zcalc = 277.1183 Da) and the experimental one (m/zexp = 277.1183 Da). These results demonstrate the successful synthesis of the azlactone-functionalized vinylcyclopropane (5). To expand the library of azlactone monomers, a second azlactone-functionalized VCP (6) (Scheme 1) was additionally synthesized in good yield (82%) using analogous conditions to give the cesium salt (4) followed by its subsequent reaction with bromo-azlactone at 40 °C in DMF. The successful synthesis of (6) was also confirmed by 1H NMR spectroscopy (Fig. 2) and HR-MS analysis. Additionally, the presence of the azlactone group in (5) and (6) was proven by FT-IR analysis by the addition of two novel stretching bands at 1680 cm−1 and 1825 cm−1 corresponding to ν(CN) and ν(CO) of the azlactone group, respectively (Fig. 3). Moreover, the successful synthesis and chemical structure of (5) and (6) were uphold by 13C NMR spectroscopy (Fig. S3 and S4 in ESI†).
Free-radical (co)polymerization of azlactone-functionalized VCP monomers
After the successful synthesis of the two azlactone-functionalized VCP monomers, the next step of this study was performing the polymerization of (5) and (6) using free-radical polymerization method. For this, the polymerization was carried out in 1,4-dioxane in the presence of 2,2′-azobis(2-methylpropionitrile (AIBN) as a thermal initiator (3 mol% relative to the monomer) at 70 °C and 80 °C (Table 1, Scheme 2). The results (Table 1) show that both monomers were successfully polymerized to give the corresponding polymers (7) and (8), respectively (Scheme 2). The 1H NMR spectra of the resulting polymers (7) and (8) (Fig. 4 and 5, respectively) show key signals corresponding to the structure of the polymers at 5.74 ppm due to the vinyl group (CHCH, labeled b) and a signal at 1.43 ppm due to the gem dimethyl –C(CH3)2 (labeled f) of the azlactone group. Furthermore, the presence of the azlactone group in the polymer backbone was confirmed by characteristic FT-IR bands at 1678 cm−1 (ν(CN)) and 1825 cm−1 (ν(CO)) (Fig. S5 and S6 in ESI†).
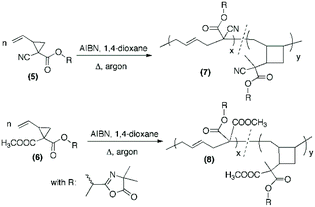 |
| | Scheme 2 Free-radical polymerization of (5) and (6) using AIBN as an initiator in 1,4-dioxane. | |
It should be noted that the radical polymerization of VCP monomers could follow different pathways to form either linear or cyclic units (Scheme 2). In order to investigate the effect of monomer structure and temperature of polymerization on polymer microstructure, the linear/cyclic ratio of the resulting polymers were determined by comparing the integral values at 2.48–2.84 ppm corresponding to the –CH2–CHCH–CH2– of the linear units (labeled a and c in Fig. 4 and 5) and at 1.74–2.41 ppm corresponding to the –CH2– and –CH– of the cyclic units (labeled h and k in Fig. 5). In the used conditions, the values for the linear/cyclic ratio confirmed that for both monomers the formation of the linear unit involving a ring-opening polymerization process was more favorable than the production of the cyclic unit (Table 1). These results are well correlated with previous reports regarding the polymerization of VCP monomers resulting in PVCP with predominantly unsaturated linear units.1,9 It was additionally recognized that lowering the temperature could reduce the polymerization rate but also significantly limit the cyclisation forming the cyclic unit within the polymer chains (entry 3 versus entry 4, Table 1). In particular, the monomer conversion of (6) was dramatically decreased from 66% to 16% by changing the temperature from 80 °C to 70 °C, while the percentage of cyclic units within the polymer chain decreased to 13% (entries 3 and 4, Table 1). Unexpectedly, the free-radical polymerization of (5) gave polymer (7) with a percentage of cyclic units lower than 5% at both temperatures (entries 1 and 2, Table 1). Regarding the chemical structure of (5) and (6), only one function (nitrile instead of methoxycarbonyl) distinguishes these two monomers but their polymerization behaviour was completely different. The steric factors play probably an important role in the mechanism of radical opening polymerization of VCP monomers. The formation of cycles within the polymer (8) backbone is probably favored to move both azlactone and sterically hindered methoxycarbonyl groups away from the polymer backbone. In comparison, the nitrile group, less sterically crowded leads only to a linear polymer (7).
The copolymerization of (5) with poly(ethylene glycol) methyl ether acrylate (PEGA, 480 g mol−1) at 80 °C resulted in a copolymer (P(PEGA-co-(5)), Scheme 3) ( , Đ = 2.26) which had no linear units in the polymer backbone, as indicated by the complete absence of 1H NMR signals at 5.74 ppm due to –CHCH– (ESI, Fig. S7†). The absence of linear units can be rationalized by the lower reactivity ratio value of the VCP monomer in comparison to (meth)acrylate. Accordingly, during copolymerization the propagating terminal radical of the VCP unit is rapidly transferred to (meth)acrylate, undergoing intramolecular cyclization to product six- and five-membered rings (Scheme 3) as reported by other studies using conventional radical polymerization.9,40–42 However, when copolymerization of VCP monomer and methyl methacrylate was performed using controlled rROP, Ata et al. shown that the copolymer was predominantly formed via 1,5 ring opening polymerization of VCP monomer instead cyclization.42 Our own results together with those of the literature indicate that the linear/cyclic ratio is strongly dependent on the monomer structure and its (co)polymerization conditions.
, Đ = 2.26) which had no linear units in the polymer backbone, as indicated by the complete absence of 1H NMR signals at 5.74 ppm due to –CHCH– (ESI, Fig. S7†). The absence of linear units can be rationalized by the lower reactivity ratio value of the VCP monomer in comparison to (meth)acrylate. Accordingly, during copolymerization the propagating terminal radical of the VCP unit is rapidly transferred to (meth)acrylate, undergoing intramolecular cyclization to product six- and five-membered rings (Scheme 3) as reported by other studies using conventional radical polymerization.9,40–42 However, when copolymerization of VCP monomer and methyl methacrylate was performed using controlled rROP, Ata et al. shown that the copolymer was predominantly formed via 1,5 ring opening polymerization of VCP monomer instead cyclization.42 Our own results together with those of the literature indicate that the linear/cyclic ratio is strongly dependent on the monomer structure and its (co)polymerization conditions.
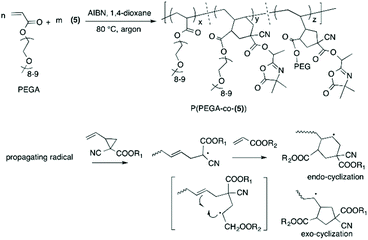 |
| | Scheme 3 Free-radical copolymerization of (5) and PEGA (Mn = 480 g mol−1) with AIBN in 1,4-dioxane at 80 °C and mechanism for the formation of cyclic monomer units. | |
Fig. S7 (ESI†) displays the 1H NMR spectrum of copolymer (P(PEGA-co-(5)). As one can observe, the resonances at δ = 1.4 ppm and 5.2 ppm indicate the presence of gem dimethyl–C(CH3)2 and COOCH(CH3) from the polymer of (5) respectively and the peak at δ = 3.3 ppm corresponds to the presence of CH3O– from poly(PEGA) part. The molar composition of polymer of (5) and P(PEGA) was calculated, based on the integration of CH3O– from poly(PEGA) part and that of COOCH(CH3) from the polymer of (5), to be 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :81, which is quite close to the feed composition (20:80). However, the peaks between 1.2 ppm and 2.5 ppm are too unresolved to allow the determination of the exact nature of ring structure.
:81, which is quite close to the feed composition (20:80). However, the peaks between 1.2 ppm and 2.5 ppm are too unresolved to allow the determination of the exact nature of ring structure.
Post-polymerization modification of PVCP bearing azlactone group
As mentioned in introduction, the primary goal of this work was the synthesis of a novel class of polymers based on azlactone-functionalized PVCP that are reactive with amines. To demonstrate the reactivity of the azlactone group toward amine-containing compounds, preliminary studies (not presented here) showed that the resulted products after post-polymerization modification of (7) or (8) with dansylcadaverine were insoluble in almost common organic solvents such as THF, acetone, dichloromethane, dioxane and DMF. This suggests the formation of π-stacking interactions between aromatic rings of dansylcadeverine when they were grafted along the polymer chains. The P(PEGA-co-(5)) copolymer was thus chosen for its high solubility toward polar solvents, that facilitates the characterization of forming products and subsequent reaction with bio-conjugates and additionally, azlactone groups are more accessible in VCP copolymer than in the corresponding VCP homopolymer. The P(PEGA-co-(5)) copolymer was thus reacted with dansylcadaverine (0.5 equivalent relative to the azlactone moiety), a fluorescent amine chosen as the model dye, in THF at 40 °C (Scheme 4). The efficiency of the coupling reaction was checked by SEC chromatography using a UV detector operating at 335 nm.28 By comparing the SEC traces before and after the coupling reaction, the SEC trace of the post-polymerization modified polymer shows an absorbance at 335 nm (red full line, Fig. 6). This absorbance corresponds to that of dansylcadaverine and demonstrates the successful grafting of dansylcadaverine to the backbone of the P(PEGA-co-(5)) copolymer via the aminolysis of the azlactone groups. However, we observed no significant shift of SEC chromatograms of copolymer before and after the reaction with dansylcadeverine while using RI detector (blue full and dashed lines, Fig. 6). This result was not surprising because the dispersity of P(PEGA-co-(5)) copolymer was high (Đ = 2.25). In addition, the 1H NMR spectrum of the resulting dansylcadaverine-functionalized copolymer (ESI Fig. S9†) displayed characteristics signals of the aromatic protons of dansylcadaverine around 7.0–8.5 ppm.43 Fig. S10† showed the superposition of FT-IR spectra of the copolymer P(PEGA-co-(5)) before and after the reaction with dansylcadaverine. As we can see, the FT-IR spectrum of substituted polymer exhibits a new small peak at 1525 cm−1 and a shoulder at 1667 cm−1 related respectively to N–H bending vibration and CO stretching vibration of amide function. Concerning the aromatic C–H stretching bands from naphthalene core of dansylcadaverine, usually observed around 2800–3000 cm−1, were overlapped with CH2 stretching vibration of PEG side chain. The large band at 3570 cm−1 is attributed to N–H stretching vibration. All of these results confirmed the grafting of dansylcadaverine to copolymer backbone via the reactivity of azlactone group without using catalyst.
 |
| | Scheme 4 Reaction of P(PEGA-co-(5)) copolymer with dansylcadaverine in THF at 40 °C. | |
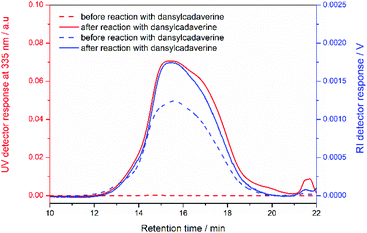 |
| | Fig. 6 Overlaid SEC traces from UV detector operating at 335 nm and RI detector of P(PEGA-co-(5)) before and after post-polymerization modification with dansylcadaverine. | |
Conclusions
In summary, we have described a simple strategy to synthesize novel azlactone-functionalized vinylcyclopropane monomers using a nucleophilic substitution reaction involving a cesium VCP salt with a bromide-containing compound bearing an azlactone group. Furthermore, the (co)polymerization of the novel azlactone-functionalized VCP monomers and their subsequent post-polymerization modification with a fluorescent amine was successfully performed. In addition, this report has proven that the formation of linear and cyclic units is dependent on both the monomer structure and polymerization conditions, in particular the temperature in the present study. We believe that such azlactone-functionalized VCP monomers could be powerful tools for the synthesis of novel classes of reactive polymers and (bio)materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This manuscript is a tribute to the 50 years anniversary of the French Polymer Group-GFP.
References
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479–2490 RSC.
- N. Moszner, Macromol. Symp., 2004, 217, 63–75 CrossRef CAS.
- Y. Catel, P. Fassler, U. Fischer, C. Pesch, C. Pruvost, S. Tauscher and N. Moszner, Polym. Int., 2017, 66, 1410–1417 CrossRef CAS.
- P. P. Contreras, P. Tyagi and S. Agarwal, Polym. Chem., 2015, 6, 2297–2304 RSC.
- Y. F. Shi, H. Schmalz and S. Agarwal, Polym. Chem., 2015, 6, 6409–6415 RSC.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1993, 26, 1818–1824 CrossRef CAS.
- N. Moszner, F. Zeuner, T. Volkel and V. Rheinberger, Macromol. Chem. Phys., 1999, 200, 2173–2187 CrossRef CAS.
- F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 265–276 CrossRef CAS.
- D. H. S. Ntoukam, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 122, 109319 CrossRef.
- N. K. Singha, A. Kavitha, P. Sarker and S. Rimmer, Chem. Commun., 2008, 3049–3051 RSC.
- D. F. Chen, B. M. Boyle, B. G. McCarthy, C. H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 13268–13277 CrossRef CAS PubMed.
- N. D. H. Seuyep, G. A. Luinstra and P. Theato, Polym. Chem., 2013, 4, 2724–2730 RSC.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- Y. Prai-in, K. Tankanya, B. Rutnakornpituk, U. Wichai, V. Montembault, S. Pascual, L. Fontaine and M. Rutnakornpituk, Polymer, 2012, 53, 113–120 CrossRef CAS.
- V. Delplace, S. Harrisson, H. T. Ho, A. Tardy, Y. Guillaneuf, S. Pascual, L. Fontaine and J. Nicolas, Macromolecules, 2015, 48, 2087–2097 CrossRef CAS.
- M. Le Bohec, K. B. Kenzo, S. Pioge, S. Mura, J. Nicolas, N. Casse, G. Forcher, L. Fontaine and S. Pascual, Polym. Chem., 2019, 10, 1968–1977 RSC.
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655–3677 CrossRef CAS.
- M. Pantin, J. Caille, F. Boeda, L. Fontaine, M. S. M. Pearson-Long and P. Bertus, Eur. J. Org. Chem., 2019, 7359–7366 CrossRef CAS.
- T. H. Ho, M. Levere, J. C. Soutif, V. Montembault, S. Pascual and L. Fontaine, Polym. Chem., 2011, 2, 1258–1260 RSC.
- M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66–80 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- H. T. Ho, M. E. Levere, D. Fournier, V. Montembault, S. Pascual and L. Fontaine, Aust. J. Chem., 2012, 65, 970–977 CrossRef CAS.
- Y. Iwakura, F. Toda and Y. Torii, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2649–2657 CrossRef CAS.
- L. D. Taylor, H. S. Kolesinski, A. C. Mehta, L. Locatell and P. S. Larson, Makromol. Chem., Rapid Commun., 1982, 3, 779–782 CrossRef CAS.
- R. C. Fazio, J. M. Grasshoff and L. D. Taylor, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 329–331 CrossRef CAS.
- H. T. Ho, M. E. Levere, S. Pascual, V. Montembault, N. Casse, A. Caruso and L. Fontaine, Polym. Chem., 2013, 4, 675–685 RSC.
- D. C. Tully, M. J. Roberts, B. H. Geierstanger and R. B. Grubbs, Macromolecules, 2003, 36, 4302–4308 CrossRef CAS.
- J. L. Grace, M. Amado, J. C. Reid, A. G. Elliott, C. B. Landersdorfer, N. P. Truong, K. Kempe, M. A. Cooper, T. P. Davis, V. Montembault, S. Pascual, L. Fontaine, T. Velkov, J. F. Quinn and M. R. Whittaker, J. Mater. Chem. B, 2019, 7, 6796–6809 RSC.
- M. E. Levere, H. T. Ho, S. Pascual and L. Fontaine, Polym. Chem., 2011, 2, 2878–2887 RSC.
- C. Lucchesi, A. Arbore, S. Pascual, L. Fontaine, C. Maignan and G. Dujardin, Carbohydr. Res., 2010, 345, 844–849 CrossRef CAS PubMed.
- C. Lucchesi, S. Pascual, G. Dujardin and L. Fontaine, React. Funct. Polym., 2008, 68, 97–102 CrossRef CAS.
- C. Lucchesi, S. Pascual, A. Jouanneaux, G. Dujardin and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3677–3686 CrossRef CAS.
- A. Laquievre, N. S. Allaway, J. Lyskawa, P. Woisel, J. M. Lefebvre and D. Fournier, Macromol. Rapid Commun., 2012, 33, 848–855 CrossRef CAS PubMed.
- H. T. Ho, A. Benard, G. Forcher, M. Le Bohec, V. Montembault, S. Pascual and L. Fontaine, Org. Biomol. Chem., 2018, 16, 7124–7128 RSC.
- H. T. Ho, F. Leroux, S. Pascual, V. Montembault and L. Fontaine, Macromol. Rapid Commun., 2012, 33, 1753–1758 CrossRef CAS PubMed.
- A. P. Dieskau, M. S. Holzwarth and B. Plietker, J. Am. Chem. Soc., 2012, 134, 5048–5051 CrossRef CAS PubMed.
- N. Moszner, F. Zeuner and V. Rheinberger, Macromol. Rapid Commun., 1997, 18, 775–780 CrossRef CAS.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1994, 27, 3982–3985 CrossRef CAS.
- T. Takahashi, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 739–749 CrossRef CAS.
- S. Ata, D. Mal and N. K. Singha, RSC Adv., 2013, 3, 14486–14494 RSC.
- Y. Lee, S. Hanif, P. Theato, R. Zentel, J. Lim and K. Char, Macromol. Rapid Commun., 2015, 36, 1089–1095 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR spectra VCP monomers and copolymers, FT-IR spectra of VCP polymers and copolymer before and after the reaction with dansylcadaverine. See DOI: 10.1039/d0py00493f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Véronique
Montembault
a,
Véronique
Montembault
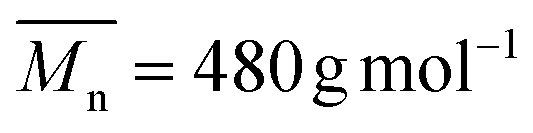 ), dansylcadaverine (97%), tetrahydrofuran (THF, 99%), methanol (99%), N,N-dimethylformamide (DMF, 99.9%), chloroform (CHCl3, 99%), anhydrous 1,4-dioxane (99.8%), n-pentane (95%), diethylether (99%), sodium sulfate (Na2SO4), silica gel (SiO2) and Celite were purchased from Sigma-Aldrich. trans-1,4-Dibromo-2-butene was used as received from TCI Chemicals. 2-(1-Bromoethyl)-4,4-dimethyloxazol-5(4H)-one (referred to as bromo-azlactone) was prepared according to the literature.37
), dansylcadaverine (97%), tetrahydrofuran (THF, 99%), methanol (99%), N,N-dimethylformamide (DMF, 99.9%), chloroform (CHCl3, 99%), anhydrous 1,4-dioxane (99.8%), n-pentane (95%), diethylether (99%), sodium sulfate (Na2SO4), silica gel (SiO2) and Celite were purchased from Sigma-Aldrich. trans-1,4-Dibromo-2-butene was used as received from TCI Chemicals. 2-(1-Bromoethyl)-4,4-dimethyloxazol-5(4H)-one (referred to as bromo-azlactone) was prepared according to the literature.37
 and
and  ) and dispersities (Đ) were calculated by means of PS calibration curve using PS standards from 0.86 to 526.0 kg mol−1 (Agilent, USA).
) and dispersities (Đ) were calculated by means of PS calibration curve using PS standards from 0.86 to 526.0 kg mol−1 (Agilent, USA).

 ; Đ = 2.25. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CH
; Đ = 2.25. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CH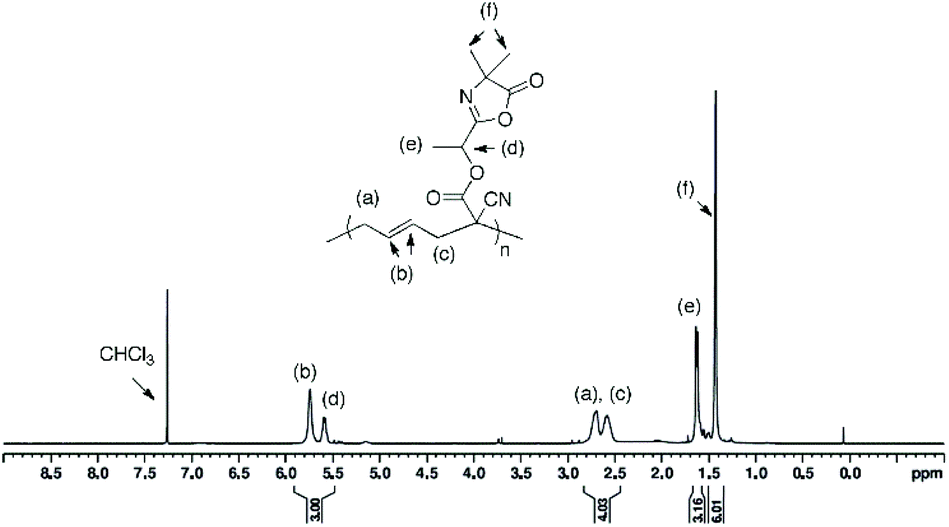

 ; Đ = 1.6. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CH
; Đ = 1.6. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.74–5.03 (CH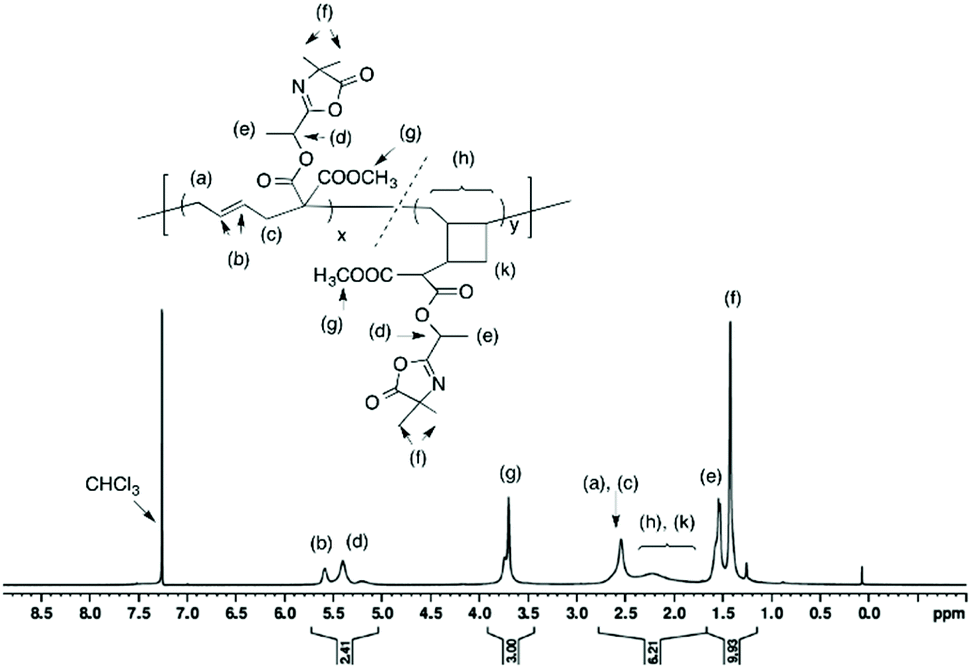
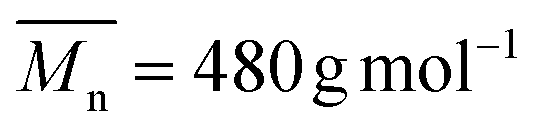 , 1.00 g, 2.08 × 10−3 mol), AIBN (4.3 mg, 2.62 × 10−5 mol) and 1,4-dioxane (1.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. The vial was then sealed with headspace cap. After, the solution was deoxygenated by bubbling argon for 30 min and then immersed in an oil bath at 80 °C to allow the polymerization to occur. The polymerization was quenched after 280 min by rapid cooling and exposure of the polymerization solution to air. The resulting product was then dissolved in chloroform and precipitated using diethyl ether. The final polymer was filtered and dried under vacuum to give a gum-like solid.
, 1.00 g, 2.08 × 10−3 mol), AIBN (4.3 mg, 2.62 × 10−5 mol) and 1,4-dioxane (1.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. The vial was then sealed with headspace cap. After, the solution was deoxygenated by bubbling argon for 30 min and then immersed in an oil bath at 80 °C to allow the polymerization to occur. The polymerization was quenched after 280 min by rapid cooling and exposure of the polymerization solution to air. The resulting product was then dissolved in chloroform and precipitated using diethyl ether. The final polymer was filtered and dried under vacuum to give a gum-like solid.  ; Đ = 2.26. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.14 (COOCH2CH2), 3.68–3.57 (OCH2CH2), 3.37 (CH3O), 1.58–1.39 (OCH(CH3) and C(CH3)2 of azlactone ring) (ESI, Fig. S7†). FT-IR (cm−1): 1825 (ν(C
; Đ = 2.26. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.14 (COOCH2CH2), 3.68–3.57 (OCH2CH2), 3.37 (CH3O), 1.58–1.39 (OCH(CH3) and C(CH3)2 of azlactone ring) (ESI, Fig. S7†). FT-IR (cm−1): 1825 (ν(C , Đ = 2.26, 0.152 g, 5 × 10−6 mol) and THF (2.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. Then, a solution of dansylcadaverine (11 mg, 3.28 × 10−5 mol) in THF (2.0 mL) was added into the reaction mixture and the resulting solution was stirred at 40 °C for 21 h under argon. The resulting product was subsequently precipitated in diethyl ether and then dried under vacuum to give a gum-like solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.28–7.69 (CH of aromatic ring) 4.14 (COOCH2CH2), 3.72–3.57 (OCH2CH2), 3.37 (CH3O) (ESI, Fig. S9†).
, Đ = 2.26, 0.152 g, 5 × 10−6 mol) and THF (2.0 mL) were charged into a headspace vial equipped with a magnetic stir bar. Then, a solution of dansylcadaverine (11 mg, 3.28 × 10−5 mol) in THF (2.0 mL) was added into the reaction mixture and the resulting solution was stirred at 40 °C for 21 h under argon. The resulting product was subsequently precipitated in diethyl ether and then dried under vacuum to give a gum-like solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.28–7.69 (CH of aromatic ring) 4.14 (COOCH2CH2), 3.72–3.57 (OCH2CH2), 3.37 (CH3O) (ESI, Fig. S9†).
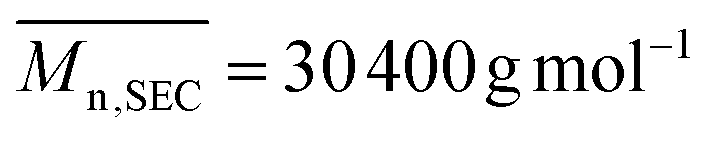 , Đ = 2.26) which had no linear units in the polymer backbone, as indicated by the complete absence of 1H NMR signals at 5.74 ppm due to –CH
, Đ = 2.26) which had no linear units in the polymer backbone, as indicated by the complete absence of 1H NMR signals at 5.74 ppm due to –CH

