
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemiluminescent self-reporting supramolecular transformations on macromolecular scaffolds†
Christina M.
Geiselhart
 ab,
Hatice
Mutlu
*ab,
Pavleta
Tzvetkova
c and
Christopher
Barner-Kowollik
*bde
ab,
Hatice
Mutlu
*ab,
Pavleta
Tzvetkova
c and
Christopher
Barner-Kowollik
*bde
aSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen 3, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: hatice.mutlu@kit.edu
bMacromolecular Architectures, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76128 Karlsruhe, Germany
cInstitute for Organic Chemistry and Institute for Biological Interfaces 4 – Magnetic Resonance, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dCentre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
eSchool of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
First published on 19th May 2020
Abstract
We introduce the synthesis of a self-reporting system with chemiluminescent output, which is regulated via dynamic supramolecular complex formation. By free radical polymerization and subsequent post-polymerization modification, a copolymer decorated with luminol and superbase (i.e. 1,5,7-triaza-bicyclo-[4.4.0]dec-5-ene (TBD)) moieties was synthesized, which in turn forms supramolecular host–guest-complexation with randomly methylated β-cyclodextrin (Me-β-CD). Upon hydrogen peroxide addition, the host–guest-interactions with the self-assembly are broken, and the luminol is oxidized to 3-aminophthalic acid (3-APA). Critically, no additional base, buffer or catalyst is required to generate the striking blue light emission of the polymeric system that is detectable by the naked eye. Thus, a fast and easy detection of reactive oxygen species (ROS), such as hydrogen peroxide, under mild conditions is established. The self-reporting system and its chemiluminescent properties are characterized by 1D and 2D NMR spectroscopy (particularly NOESY), dynamic light scattering (DLS), UV/Vis spectroscopy and chemiluminescence (CL) measurements.
Introduction
The ability of natural systems to sense and visibly report changes in damage in real time has inspired the development of numerous synthetic self-reporting systems.1–5 In fact, such systems have found applications in fields such as analytical chemistry, biology, medicine and diagnostics.4,6–8Between the different types of self-reporting phenomena, chemiluminescence (CL) has been recognized as a functional tool due to its high sensitivity and wide dynamic range without the requirement of sophisticated equipment.9–11 Besides common luminophores such as dioxetanes,12 peroxyoxalates4 or acridinium compounds,13 luminol has gained significant attention since its discovery by Albrecht in 1928,14,15 due to its low cost, beneficial properties and wide-ranging applications. Unfortunately, the quantum yield of luminol is rather low (≈0.01 in aqueous media and ≈0.09 in DMSO),16 therefore substantial efforts have been devoted to develop new luminol systems with higher CL emission. Indeed, the latter is achievable by substituting the aromatic ring of luminol with adequate molecules or by utilizing distinctive catalysts, such as organic frameworks17,18 or nanoparticles.19,20
Recently, we successfully implemented the organic superbase 1,5,7-triaza-bicyclo-[4.4.0]dec-5-ene (TBD) into the CL reaction of luminol.21 Without the requirement for additional additives (such as catalysts or enhancers), the CL of a luminol–TBD-mixture in organic media (DMSO) was readily triggered in the presence of hydrogen peroxide (H2O2). Indeed, the resulting CL of the system can be observed by the naked eye. Therefore, we submit that by fusing the properties of luminol and TBD within one polymeric backbone, the synthesis of polymeric self-reporting CL systems triggered by oxidants such as H2O2, would be feasible. Especially in the field of biomedicine and diagnostics, the fast and ready detection of H2O2 is critical, since its increased level is a sign for oxidative stress and diseased cells.22,23
In order to construct the self-reporting CL system with relevance to the aforementioned applications, we expanded our superbase driven luminol concept further by constructing supramolecular assemblies based on a luminol–TBD–polymer with tailor-made supramolecular host-molecules. Host–guest binding is analogous to the way many biomolecule–substrate interactions occur,24,25 thus supramolecular assemblies based on diverse host–guest interactions are of key interest and find widespread applications in biomedicine, drug delivery, catalysis, nanotechnology molecular recognition or sensor technology.26–30 Wild cyclodextrins (CD) as well as their chemically modified variants have proven to be able to encapsulate biomolecules (such as amino acids) to form supramolecular self-assemblies.31–33 Especially the guanidine-functional group in the essential amino acid L-arginine enables selective host–guest inclusion complexes with β-CD.34,35 We thus consider a possible inclusion of the guanidine-functionality in TBD with β-CD. In addition, CDs are efficient boosters for the CL of luminol.36–38
Synthetically, the luminol–superbase–polymers were obtained via a post-polymerization modification (PPM) approach. In fact, PPMs have been proven to be a useful tool to synthesise highly functionalized polymers, especially since conventional polymerization processes do not allow the direct polymerization of monomers decorated with multiple functional groups.39–41 In addition to synthetic polymers, commercially available polymers (e.g. polybutadiene42) can be decorated with a broad variety of functionalities to obtain innovative macromolecules with tailor-made properties. Among the different reactions enabling PPMs (copper-catalyzed azide–alkyne cycloaddition (CuAAC),43 thiol–ene addition,44 Michael addition,45 or electrophilic cascade reactions42), active esters, such as pentafluorophenyl (PFP) esters,46–48 are considerably attractive functional units to deliver well-defined multi-functionalized polymers. Especially the possibility to modify active PFP ester groups within a copolymer backbone in a precise and orthogonal manner is highly beneficial.49
Herein, via free radical polymerization (FRP) of two different active PFP ester monomers in the presence of methyl methacrylate (MMA), and subsequent orthogonal PPM, we are able to synthesize the luminol–superbase copolymers P1 and P2, as illustrated in Scheme 1. To evidence the concept of a supramolecular self-reporting CL-system with tuneable supramolecular complexation properties and to gain insights into the structural influence of the employed superbase, an acyclic (G3 in Scheme 1A) and a cyclic (namely TBD, Scheme 1B) guanidine derivative were evaluated. In-depth characterization of the supramolecular self-reporting CL-systems were performed using NMR (1D and 2D) spectroscopy, DLS and UV/Vis spectroscopy. Furthermore, CL measurements were utilized to reveal the efficiency as self-reporting systems.
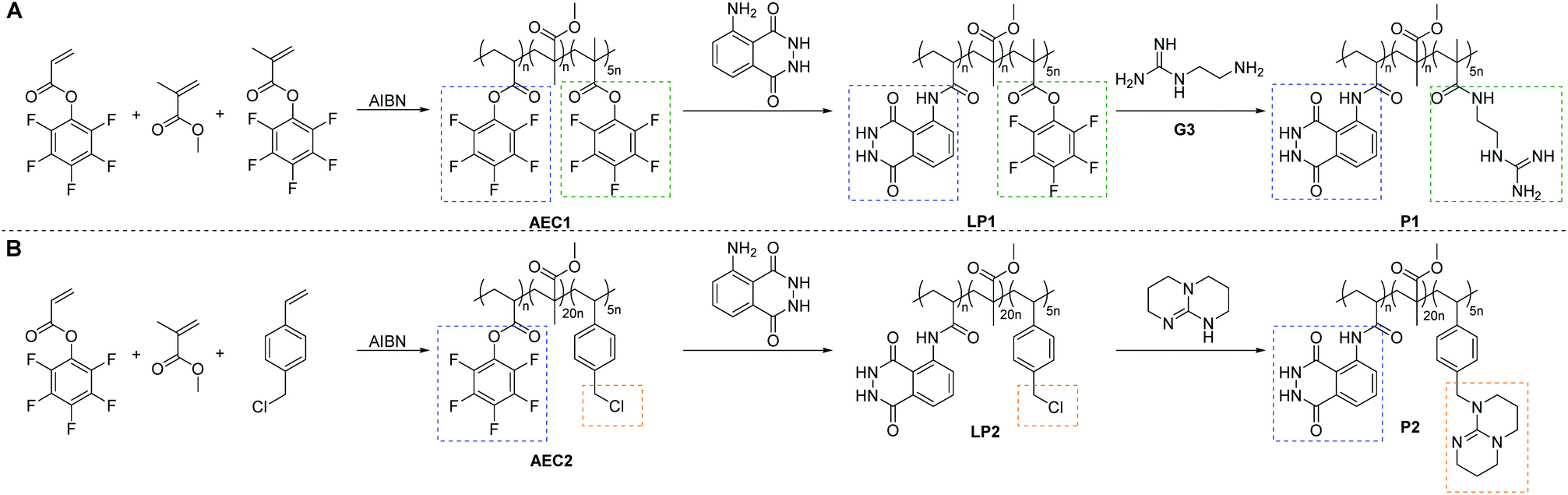 | ||
| Scheme 1 Synthetic route to the luminol–superbase polymers P1 (A) and P2 (B) via free radical polymerization and subsequent post-polymerization modification with luminol and the respective superbase. | ||
Results and discussion
Synthesis of a luminol–superbase copolymer
In order to afford the targeted copolymer system, it was of critical importance to synthesize adequate luminol-, guanidine- and TBD-derivative monomers. Thus, in Scheme S1 (refer to the ESI†), we depict an overview of the synthesized monomers. While an efficient synthesis of similar guanidine-50–52 and luminol-49,53,54 derivative monomers is reported in the literature, none of the attempts to reproduce these approaches was successful (refer to Scheme S1 in the ESI†). Indeed, the latter was evidenced with a test polymerization experiment, in which a controlled reversible addition–fragmentation chain transfer (RAFT) polymerization was attempted for monomer G2 (for more details refer to section B4 in the ESI†). The 1H NMR spectra of the polymerization mixture illustrated in Fig. S1 in the ESI,† show resonances only attributable to the unreacted G2. Therefore, an orthogonal post-polymerization modification (PPM) approach was explored in order to synthesize the targeted copolymer systems P1 and P2 (Scheme 1). Initially, a random copolymer was synthesized via FRP using MMA, PFP methacrylate (PFPMA) and PFP acrylate (PFPA), respectively, as monomers (refer to Scheme 1A). Based the high reactivity of acrylate PFP-ester derivatives toward aromatic amines compared to their own methacrylate analogue,49 the PFPA moiety reacts selectively in an orthogonal manner with luminol, while the PFPMA moiety should be substituted with the superbase derivative. Furthermore, according to the results of the previously reported small molecule study,21 in which an excess of 5.0 eq. of superbase (i.e. TBD) is key for an efficient CL of luminol, the FRP was conducted with a PFPA/PFPMA ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.
:5.
On the one hand, the synthesis of the luminol–guanidine–copolymer P1 (displayed in Scheme 1A) was confirmed via 1D NMR analysis of the purified copolymer. Critically, all relevant resonances assignable to the respective protons of P1 were detected in the 1H NMR spectrum (Fig. S2 in the ESI†). In addition, the 19F NMR spectrum (Fig. S2 in the ESI†) confirmed that all active PFP ester moieties were substituted by the luminol and guanidine-functionalities. The resonance at −75 ppm is associated with the TFA utilized during the monomer synthesis. On the other hand, P1 is not soluble in typical organic solvents employed for SEC (such as THF or DMAc), thus the apparent number average molecular weight and the dispersity values of P1 could not be determined via SEC.
In order to obtain the luminol–TBD–copolymer P2 with efficient CL to be detected by the naked eye, the monomer choice employed for the PPM approach of P1 was slightly altered. Thus, a copolymer of MMA, PFPA and 4-vinyl benzylchloride (VBC) was synthesized via FRP (refer to Scheme 1B). Subsequently, P2 was obtained by the orthogonal PPM of the PFPA moiety with luminol and substitution of VBC with TBD, respectively. The successful synthesis of P2 was confirmed by 1D NMR spectroscopy, as shown in Fig. S3 in the ESI.† Importantly, the resonances of the NH-protons of luminol (i, h′) and TBD (w), as well as the resonances of h, m, n, and o of P1 in Fig. S2,† were intense compared to resonance of methylene spacer (i.e., t), which resulted from the electrostatic interactions in the delocalized charge of each (de-)protonated species, and furthermore with the increased pH value of the environment that is basic due to excess of guanidine units. Similar to P1, the measurement of the apparent number average molecular weight of P2 remained elusive via conventional SEC eluents.
Whereas the definition of size and thus molecular weight, measured by dynamic light scattering (DLS) differs from that of SEC, due to the inherent limitation of intensity-biased detection, we have validated the hydrodynamic radii (Rh) of P1 and P2via DLS analysis. While the average radii of P1 was 4.4 nm at a copolymer concentration of 1 mg mL−1 (red trace in Fig. 1), the corresponding value for P2 (blue trace) is close to twice the size of P1, e.g. 10.6 nm. These results are in accordance with the SEC results of the active ester copolymers AEC1 and AEC2 (refer to Scheme 1), which in contrast to P1 and P2 were soluble in typical SEC eluents (i.e. THF). As shown in Fig. S4 in the ESI,† the apparent average molecular weight of AEC2 (Mn = 14600 Da) was twice the size of AEC1 (Mn = 7720 Da). Despite the different average molecular weights, the number of repeating units and thus the number of active species incorporated into each polymer remains constant, allowing for a comparison of the resulting chemiluminescent properties.
 | ||
| Fig. 1 DLS traces of Me-β-CD, the luminol–superbase–copolymers (P1 and P2) and the supramolecular complexes before (C1, C2) and after (C1*, C2*) the addition of H2O2. All traces were recorded in DMF at 20 °C with the concentration of 1 mg mL−1. | ||
Supramolecular assembly
Having established the synthesis of the copolymers P1 and P2, the self-assembly behaviour of the incorporated guanidine moieties in the presence of Me-β-CD as host-molecule was investigated. To prove the suitability of Me-β-CD for the complexation of the guanidine moieties, test reactions with luminol and G3 itself with Me-β-CD were conducted. The cross resonances in the nuclear Overhauser effect spectroscopy (NOESY) results (illustrated in Fig. S5 in the ESI†) evidence the encapsulation of the guanidine-moieties. While there are cross resonances between luminol and Me-β-CD in the absence of G3, no cross resonances are detected in the presence of G3, thus supporting the encapsulation properties.For the complexation reaction, the respective luminol–superbase–copolymer was dissolved in DMSO, succeeded by the addition of Me-β-CD to obtain the supramolecular assemblies of C1 and C2 as depicted in Scheme 2. The key indication for a successful host–guest-complexation comes from 1H NMR spectra (refer to Fig. 2A and B). For C1 (Fig. 2A), a broadening of the resonances e and m at 8.5 ppm and of the resonances i and o at 7.8 ppm suggest interactions of the guanidine- and luminol-functionality with the Me-β-CD. Similarly, the resonances of the luminol- (i, h′) and TBD-protons (u, v, w) are broadening in the presence of Me-β-CD (Fig. 2B). It should be noted that due to solubility issues of P1 and P2, different deuterated solvents were used for the NMR analysis of C1 and C2. Whereas the spectra of P1/C1 were recorded in DMF-d7, the spectra of P2/C2 were analysed in DMSO-d6, resulting in (de-)protonated luminol- and guanidine moieties in P1/C1 and P2/C2.
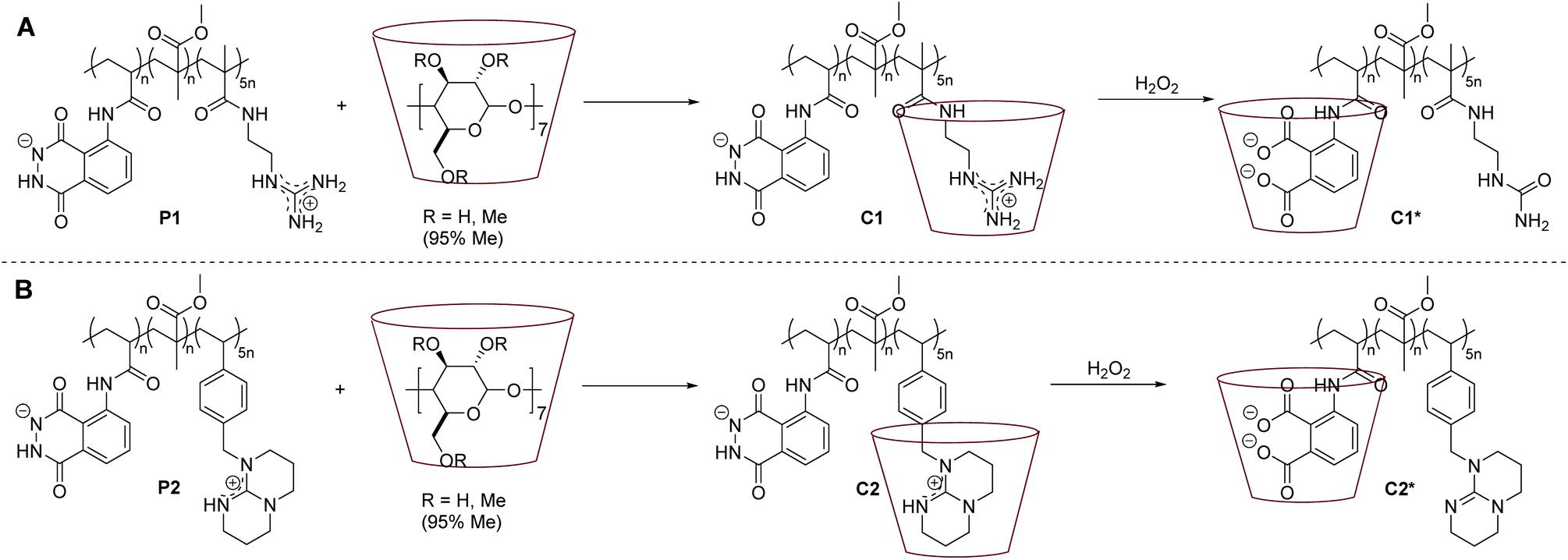 | ||
| Scheme 2 General reaction pathway of the host–guest-complexation reaction to obtain C1 (A) and C2 (B) (the chemical structures of each compound is depicted in solution, i.e. DMF or DMSO, thus P1 and P2 are presented in their (de-)protonated species). Subsequently, the oxidation process is displayed, triggered by the addition of H2O2 resulting in the host–guest-complexes C1* (A) and C2* (B). | ||
 | ||
| Fig. 2 A: 1H NMR spectra of P1 and C1 in DMF-d7. B: 1H NMR spectra of P2 and C2 in DMSO-d6. C: NOESY spectrum of C1 in DMF-d7. D: NOESY spectrum of C2 in DMSO-d6. The 1H NMR spectra were recorded at ambient temperature, the NOESY spectra at 300 K. | ||
Further insight into the complexation process is obtained from NOESY analysis. The recorded spectra of C1 and C2 are displayed in Fig. 2C and D. As expected, there are cross-resonances (i.e. NOEs) at 8.5 ppm (green and orange circles in Fig. 2C and D), assigned to the anticipated dominant dipolar interaction between the Me-β-CD annular protons and the guanidine derivative (either acyclic or cyclic one). Additional cross-resonances at 7.5 ppm (blue circles in Fig. 2C and D) are detectable, which arise from the interactions of the Me-β-CD with the luminol moiety both for C1 and C2. The cross-resonance at 4.5 ppm can be assigned to a complexation of the MMA moieties or further superbase functionalities by the Me-β-CD (black circles in Fig. 2C and D). Although only a supramolecular assembly of the superbase-functionality with the host-molecule was expected, the complexation of the superbase-, luminol- and MMA-moieties by the Me-β-CD in the polymer backbone clearly evidences the competence of the luminol–superbase–polymers to form supramolecular assemblies.
Supplementary to NMR analysis, DLS was used as a basis for assessing the inclusion process. Indeed, the comparison of the DLS traces of the individual components (P1, P2 and Me-β-CD) with C1 and C2 (refer to Fig. 1) support the formation of host–guest-complexes. As evident from the single DLS measurements depicted in Fig. S6 in the ESI,† only one distribution is obtained for C1 and C2, respectively. If the encapsulation did not take place with the aid of Me-β-CD, the DLS data should reveal two distinguishable distributions, one associated with the non-encapsulated polymer derivative, the other one exclusively for Me-β-CD. According to the literature, the diameter of the supramolecular complexes should lie between the diameters of the individual components prior to the complex formation.55 As illustrated in Fig. 1, the diameters of C1 and C2 lie in between the diameters of Me-β-CD and the corresponding luminol–superbase–polymer. The results also reveal that the supramolecular interactions between TBD and Me-β-CD have a higher influence for the hydrodynamic diameter of the host–guest-complexes than the interactions between guanidine and Me-β-CD, respectively. In case of the luminol–TBD–system, the hydrodynamic diameter of C2 is 51% lower than the one of P2, while the difference in the luminol–guanidine system is only 21%.
Chemiluminescence properties
In the presence of ROS, the luminol is oxidized to the 3-aminophthalic acid and a strong blue light is emitted. Accordingly, the formation of the oxidized luminol in C1* and C2* can be clearly detected in the UV/Vis spectra in Fig. 3. Prior to the oxidation, there are two main bands at 360 nm and 300 nm (grey and red lines), and an additional small band at close to 260 nm appears in the C1 spectrum. Post the addition of an oxidizing agent (i.e. H2O2), the bands at 360 nm and 300 nm decrease, while a peak at 260 nm arises. By comparison with commercially available 3-aminophthalic acid, the band at 260 nm can be clearly assigned to the oxidized luminol derivative. Eventually, a difference in the oxidation process between C1 and C2 is detectable, since the addition of 0.1 mL H2O2 (1 mol L−1) to C2 results in a more drastic change in the absorption behaviour in comparison to the addition of the same amount of H2O2 to C1 (blue lines in Fig. 3). In order to determine the effect of H2O2 to the supramolecular complexes C1 and C2, the samples were analysed via DLS and NOESY. The DLS results of C1 depicted in Fig. 1 confirm the exclusion of the host–guest complex. The apparent hydrodynamic diameter of C1* decreases in the presence of H2O2 to the size of the single Me-β-CD, and no trace of the polymer could be detected. Likely the exclusion may have led to an aggregation of the polymer and Me-β-CD, which may prevent the detection of the oxidized polymer. Furthermore, the NOESY spectrum illustrated in Fig. 5A evidences the de-complexation of the oxidized luminol–guanidine–polymer and the Me-β-CD. There are no visible cross-resonances between Me-β-CD and the oxidized luminol- or the oxidized guanidine-functionality in the range of 9–7 ppm. Merely a cross-resonance at 10.5 ppm (grey box), associated with the interactions between the Me-β-CD and H2O2 can be identified. However, the CL measurements of C1 only reveals a very-weak emission, as depicted in Fig. 4 (red line). | ||
| Fig. 3 UV/Vis spectra of P1/C1 and P2/C2 (c(P/C) = 3.25 × 10−6 mmol mL−1) before and after addition of H2O2 (1 mol L−1). All spectra were recorded in DMSO at ambient temperature. | ||
 | ||
| Fig. 4 CL emission of C1, C2 and P2 (c = 3.25 × 10−4 mmol mL−1) in DMSO at ambient temperature, triggered by 0.1 mL H2O2 (1 mol L−1). | ||
 | ||
| Fig. 5 NOESY spectra of A: C1 + H2O2 in DMF-d7, and B: C2 + H2O2 in DMSO-d6. All spectra were recorded at 300 K. | ||
Similarly, the CL properties of C2 were investigated. The DLS results clearly exhibit an alternation of the host–guest interactions after the addition of H2O2 (refer to Fig. 1 and Fig. S6 in the ESI†). Upon addition of 0.1 mL H2O2 (1 mol L−1), the hydrodynamic size of C2* is 47% larger than the one of C2 and close to the size of P2 (7% difference). Presumably, in contrast to the luminol–guanidine–polymer, the new complex C2* is formed as depicted in Scheme 2 with host–guest interactions between the oxidized luminol and Me-β-CD, in accordance with the results of the NOESY spectrum of C2* displayed in Fig. 5B. As in the spectrum of C1*, there is a cross-resonance at 10.5 ppm (grey box), which can be assigned to interactions between H2O2 and Me-β-CD or the polymer backbone, and there is no detectable cross-resonance between Me-β-CD and TBD, respectively. The cross-resonance in the aromatic region (blue box) is indicating interactions between the oxidized luminol and Me-β-CD, thus supporting the DLS results. The CL emission of C2 is higher than the one for C1 (∼760 times), as pictured in Fig. 4 (blue line). Consequently, the light output is visible by the naked eye, enabling a fast and easy detection of the oxidized luminol in the presence of ROS (i.e. H2O2). Additionally, the CL emission of P2 without Me-β-CD has been recorded (green line in Fig. 4) in order to evaluate the effect of Me-β-CD on the CL reaction of luminol. As expected, a (∼140 times) higher CL emission is obtained for C2 compared to P2, underpinning the enhancing effect of Me-β-CD.
A plausible explanation for the immense difference in the CL emission between C1 and C2 lies in the structural difference of the superbase tethered to the polymer backbone. As reported in the literature,21 the type of the superbase plays a critical role in the CL reaction of luminol, not only as catalyst but also as co-reactant. While the non-cyclic guanidine-derivative in P1/C1 is transformed to the urea functionality in the presence of an oxidant (e.g. H2O2), TBD in P2/C2 remains intact due to its bulkiness. Therefore, the generated urea derivative acts no longer as a base or catalyst, thus hindering the CL reaction of luminol.
Conclusions
Within the present work, the successful synthesis of a luminol–superbase–copolymer is reported, which enables supramolecular (dis)assembly combined with a strong CL output. The incorporation of the organic superbase TBD and luminol in the same polymeric backbone allows for the formation of host–guest-interactions with Me-β-CD, cleaving under oxidative conditions (e.g. H2O2) and subsequently emitting a striking blue light as a self-reporting function for the decomplexation process. We postulate that such self-reporting chemiluminescent polymeric materials can be used to map reactive oxidative stress in the human body without the need for an external trigger. Furthermore, our study can serve as inspiration for the development of artificial materials for the sensing of critical situations (damages or structural changes) in polymeric materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BioInterfaces in Technology and Medicine (BIFTM) and Science and Technology of Nanosystems (STN) programs. Further, C. B.-K. acknowledges key support from the Queensland University of Technology (QUT) and the Australian Research Council (ARC) in the form of the Laureate Fellowship underpinning his photochemical research program. C. M. G. acknowledges the German Academic Exchange Service (DAAD) for additional funding. The authors thank the Institut für Biologische Grenzflächen I for the access to the DLS, UV/Vis, and CL instruments. H. M. acknowledges continued support from the Karlsruhe Institute of Technology (KIT).Notes and references
- R. Merindol, G. Delechiave, L. Heinen, L. H. Catalani and A. Walther, Nat. Commun., 2019, 10, 528 CrossRef CAS PubMed.
- S. Shree, M. Dowds, A. Kuntze, Y. K. Mishra, A. Staubitz and R. Adelung, Mater. Horiz., 2020, 7, 598 RSC.
- J. V. Araujo, O. Rifaie-Graham, E. A. Apebende and N. Bruns, in Bio-inspired Polymers, The Royal Society of Chemistry, 2017, p. 354 Search PubMed.
- L. Delafresnaye, F. R. Bloesser, K. B. Kockler, C. W. Schmitt, I. M. Irshadeen and C. Barner-Kowollik, Chem. – Eur. J., 2020, 26, 114 CrossRef CAS PubMed.
- T. H. Lee, Y. K. Song, S. H. Park, Y. I. Park, S. M. Noh and J. C. Kim, Appl. Surf. Sci., 2018, 434, 1327 CrossRef CAS.
- M. Theerasilp and D. Crespy, Polym. Chem., 2020, 11, 1462 RSC.
- Y. Liu, Q. Pei, L. Chen, Z. Li and Z. Xie, J. Mater. Chem. B, 2016, 4, 2332 RSC.
- C. Calvino and C. Weder, Small, 2018, 14, 1802489 CrossRef PubMed.
- O. Green, T. Eilon, N. Hananya, S. Gutkin, C. R. Bauer and D. Shabat, ACS Cent. Sci., 2017, 3, 349 CrossRef CAS PubMed.
- C. Zhao, H. Cui, J. Duan, S. Zhang and J. Lv, Anal. Chem., 2018, 90, 2201 CrossRef CAS PubMed.
- L. Mi, Y. Sun, L. Shi and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 7879 CrossRef CAS PubMed.
- S. Gnaim, O. Green and D. Shabat, Chem. Commun., 2018, 54, 2073 RSC.
- K. K. Krzymiński, A. D. Roshal, P. B. Rudnicki-Velasquez and K. Żamojć, Luminescence, 2019, 34, 512 CrossRef PubMed.
- T. Martin, J. Fleissner, W. Milius and J. Breu, Z. Anorg. Allg. Chem., 2020, 646, 162 CrossRef CAS.
- W. Gu, H. Wang, L. Jiao, Y. Wu, Y. Chen, L. Hu, J. Gong, D. Du and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3534 CrossRef CAS PubMed.
- P. Reschiglian, G. Zomer, J. W. Hastings, F. Berthold, A. Lundin, A. M. G. Campana, R. Niessner, T. K. Christopolous, C. Lowik and B. Branchini, Chemiluminescence and Bioluminescence: Past, Present and Future, Royal Society of Chemistry, 2010 Search PubMed.
- H. Tan, Y. Zhao, X. Xu, Y. Sun, Y. Li and J. Du, Microchim. Acta, 2019, 187, 42 CrossRef PubMed.
- D. Li, S. Zhang, X. Feng, H. Yang, F. Nie and W. Zhang, Sens. Actuators, B, 2019, 296, 126631 CrossRef CAS.
- M. Iranifam, N. R. Hendekhale and H. A. J. Al Lawati, Anal. Methods, 2018, 10, 429 RSC.
- S. Mohammad Beigi, F. Mesgari, M. Hosseini, M. Aghazadeh and M. R. Ganjali, Anal. Methods, 2019, 11, 1346 RSC.
- C. M. Geiselhart, C. W. Schmitt, P. Jöckle, H. Mutlu and C. Barner-Kowollik, Sci. Rep., 2019, 9, 14519 CrossRef PubMed.
- P. Wang, F. Zhou, K. Guan, Y. Wang, X. Fu, Y. Yang, X. Yin, G. Song, X.-B. Zhang and W. Tan, Chem. Sci., 2020, 11, 1299 RSC.
- Y. Zhang, M. Dai and Z. Yuan, Anal. Methods, 2018, 10, 4625 RSC.
- J. W. Steed, D. R. Turner and K. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry, Wiley, 2007 Search PubMed.
- Z. Feng, T. Zhang, H. Wang and B. Xu, Chem. Soc. Rev., 2017, 46, 6470 RSC.
- T.-G. Zhan and K.-D. Zhang, in Handbook of Macrocyclic Supramolecular Assembly, ed. Y. Liu, Y. Chen and H.-Y. Zhang, Springer Singapore, Singapore, 2019, p. 1 Search PubMed.
- S. van Dun, C. Ottmann, L.-G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960 CrossRef CAS PubMed.
- R. W. Chakroun, A. Sneider, C. F. Anderson, F. Wang, P.-H. Wu, D. Wirtz and H. Cui, Angew. Chem., Int. Ed., 2020, 59, 4434 CrossRef CAS PubMed.
- Y. Liu, A. D. Gill, Y. Duan, L. Perez, R. J. Hooley and W. Zhong, Chem. Commun., 2019, 55, 11563 RSC.
- M. Xu, F. Huo and C. Yin, Sens. Actuators, B, 2017, 240, 1245 CrossRef CAS.
- M. N. Roy, A. Roy and S. Saha, Carbohydr. Polym., 2016, 151, 458 CrossRef CAS PubMed.
- A. Roy, S. Saha and M. N. Roy, Fluid Phase Equilib., 2016, 425, 252 CrossRef CAS.
- M. Kundu, S. Saha and M. N. Roy, J. Mol. Liq., 2017, 240, 570 CrossRef CAS.
- R. Kataky, P. M. Kelly, D. Parker and A. F. Patti, J. Chem. Soc., Perkin Trans. 2, 1994, 2381 RSC.
- S. Saha, T. Ray, S. Basak and M. N. Roy, New J. Chem., 2016, 40, 651 RSC.
- R. Maeztu, G. Tardajos and G. González-Gaitano, J. Phys. Chem. B, 2010, 114, 2798 CrossRef CAS PubMed.
- R. Maeztu, G. González-Gaitano and G. Tardajos, J. Phys. Chem. B, 2010, 114, 10541 CrossRef CAS PubMed.
- R. Maeztu, G. González-Gaitano, G. Tardajos and P. Stilbs, J. Lumin., 2011, 131, 662 CrossRef CAS.
- D. H. S. Ntoukam, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 122, 109319 CrossRef.
- J. Zhuang, B. Zhao and S. Thayumanavan, ACS Macro Lett., 2019, 8, 245 CrossRef CAS PubMed.
- J. Lee, S. Han, M. Kim and B.-S. Kim, Macromolecules, 2020, 53, 355 CrossRef CAS.
- C. M. Geiselhart, J. T. Offenloch, H. Mutlu and C. Barner-Kowollik, ACS Macro Lett., 2016, 5, 1146 CrossRef CAS.
- J. A. Martín-Illian, S. Royuela, M. M. Ramos, J. L. Segura and F. Zamora, Chem. – Eur. J., 2020, 26, 1 CrossRef.
- A. K. Williams, J. Tropp, E. R. Crater, N. Eedugurala and J. D. Azoulay, ACS Appl. Polym. Mater., 2019, 1, 309 CrossRef CAS.
- S.-I. Matsuoka, Y. Kamijo and M. Suzuki, Polym. J., 2017, 49, 423 CrossRef CAS.
- S. Noree, V. Tangpasuthadol, S. Kiatkamjornwong and V. P. Hoven, J. Colloid Interface Sci., 2017, 501, 94 CrossRef CAS PubMed.
- Y. Pinyakit, T. Palaga, S. Kiatkamjornwong and V. P. Hoven, J. Mater. Chem. B, 2020, 8, 454 RSC.
- H. Gaballa, S. Lin, J. Shang, S. Meier and P. Theato, Polym. Chem., 2018, 9, 3355 RSC.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569 CrossRef CAS.
- N. J. Treat, D. Smith, C. Teng, J. D. Flores, B. A. Abel, A. W. York, F. Huang and C. L. McCormick, ACS Macro Lett., 2012, 1, 100 CrossRef CAS PubMed.
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021 CrossRef CAS PubMed.
- K. E. S. Locock, T. D. Michl, N. Stevens, J. D. Hayball, K. Vasilev, A. Postma, H. J. Griesser, L. Meagher and M. Haeussler, ACS Macro Lett., 2014, 3, 319 CrossRef CAS.
- L. Xiao, Y. Chen and K. Zhang, Macromolecules, 2016, 49, 4452 CrossRef CAS.
- G. N. Chen, R. E. Lin, H. S. Zhuang, Z. F. Zhao, X. Q. Xu and F. Zhang, Anal. Chim. Acta, 1998, 375, 269 CrossRef CAS.
- A. Bertrand, M. Stenzel, E. Fleury and J. Bernard, Polym. Chem., 2012, 3, 377 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00332h |
| This journal is © The Royal Society of Chemistry 2020 |