
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of conjugated polymers via cyclopentannulation reaction: promising materials for iodine adsorption†
Noorullah
Baig
ab,
Suchetha
Shetty
ab,
Saleh
Al-Mousawi
c and
Bassam
Alameddine
 *ab
*ab
aDepartment of Mathematics and Natural Sciences, Gulf University for Science and Technology (GUST), Kuwait. E-mail: alameddine.b@gust.edu.kw
bFunctional Materials Group – CAMB, GUST, Kuwait
cDepartment of Chemistry, University of Kuwait, Kuwait
First published on 3rd April 2020
Abstract
A new class of conjugated polymers is prepared by means of a versatile palladium-catalyzed cyclopentannulation reaction using a series of specially designed diethynyl aryl synthons with the commercially available 9,10-dibromoanthracene DBA monomer. The target polymers, CPP1–3, display high solubility and excellent chemical stability, which allow their structural and photophysical characterization by various instrumental analysis techniques such as gel permeation chromatography (GPC), and 1H- and 13C-nuclear magnetic resonance (NMR), Fourier transform infrared (FTIR), UV-vis absorption, and emission spectroscopy. GPC chromatograms of CPP1–3 display a high relative weight-average (Mw) molecular weight in the range of 15.8 to 34.3 kDa with a polydispersity index (Đ = Mw/Mn) of ∼2.5. Investigation of the iodine adsorption properties of CPP1–3 reveals their high uptake, namely ∼200 wt% for CPP2, whose sorption property was sustained even after its reuse several times.
Introduction
The design of new conjugated polymers has become one of the chief topics of interest for scientific endeavors because of the importance of this latter class of materials in diverse applications, namely, light-emitting diodes (LEDs),1–3 organic solar cells,4–7 field-effect transistors (FETs),8–16 sensors,17 optical switches,18–22 batteries,23 and thermoelectrics.24 In addition to the hitherto mentioned applications, conjugated microporous polymers (CMP) have become exceptionally eminent due to many reasons, notably their extended π-conjugation skeleton, high stability, synthesis versatility, large surface area, and excellent gas sorption properties.25The total world energy demand is increasing rapidly as a result of the rise in global population and economic growth. It is estimated that the energy consumption will reach 778 Etta Joule by 2035.26 In the meantime, the quest for a sustainable, efficient, low-emission energy source continues to be a global interdisciplinary research challenge.27 Wind power and solar energy are often considered potential alternatives for our current fossil-fuel based energy economy, but their sporadic nature of energy production leads to reliability concerns.28 Nuclear energy is considered to be a possible option for continuous energy production.29 Nevertheless, the successful mass production of power from nuclear fission is accompanied by the emission of several radioactive gases, such as, 14CO2, 85Kr, 3H, 123I, 125I, and 127–140I.30 In addition to the hitherto mentioned radioactive iodine, hydrogen iodide (HI) and alkyl halides are also considered hazardous due to their large heat release, involvement in metabolic processes, and long half-lives (e.g., t1/2 = 15.7 × 106 years for 129I).31,32 Consequently, in view of its threat to human health and for being a mutation source of plants and animals, there is a need for effective and longer-lasting solutions to capture and store the radioactive iodine species.33–35 Amongst the most efficient and cost-effective adsorption technologies, porous adsorbents, such as organic cages, silica gel, zeolites, activated carbon, and metal organic frameworks, have been tested for iodine capture.36–40
Various conjugated polymers have been reported with structures ranging from linear,14,41 hypercrosslinked,42 organic frameworks,25,43 to polycyclic aromatic hydrocarbons.44–48 Nevertheless, the rigidity and stiffness of these polymers make them poorly soluble, which can be circumvented by the introduction of aliphatic side chains in order to improve their solubility in common organic solvents. However, the insertion of long peripheral chains hampers microporosity since these latter block the intrinsic pores.49 An alternative strategy to improve the solubility of conjugated polymers would be the insertion of spiro-centers that result in the formation of contorted structures, which prevents any aggregation caused by π–π stacking.50–52 Herein we describe the synthesis of three conjugated organic polymers CPP1–3 from 9,10-dibromoanthracene DBA with various contorted aryl dialkyne comonomers 3a–cvia a versatile palladium-catalyzed cyclopentannulation reaction.53,54 It is worth mentioning that the target polymers were obtained in excellent yields and were found to be highly soluble in common organic solvents. Hence, CPP1–3 were characterized by GPC, NMR, FTIR, UV-vis absorption and emission spectroscopy. In addition, the target polymers were investigated for iodine uptake applications.
Experimental
Materials
All the reactions were carried out under an inert atmosphere using dry argon. All chemical reagents were used without further purification as purchased from Aldrich, Merck, and HiMedia unless otherwise specified. The triptycene nonaflate 2c was synthesized following the literature.55 The solvents, namely, hexane, DCM, THF, benzene, toluene, diisopropylamine, DMSO, and acetone, were dried and deoxygenated by bubbling with argon gas for 30 minutes. Thin-layer chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 and revealed using a UV lamp.Instruments
NMR (1H: 600 MHz, 13C: 150 MHz) spectra were recorded on a Bruker BioSpin GmbH 600 MHz spectrometer using CD2Cl2/CDCl3 as a solvent with the chemical shifts (δ) given in ppm and referred to tetramethylsilane (TMS). Electron impact high-resolution mass spectra (EI-HRMS) were recorded on a Themo Fisher DFS analyzer with a standard PFK (perfluorokerosene) as lock mass. The analysed data were converted to accurate mass employing X-Calibur accurate mass calculation software. UV-Vis spectra were recorded on a Shimadzu UV1800 spectrophotometer. Photoluminescence (PL) spectra were recorded on an Agilent G9800 Cary Eclipse Fluorescence spectrophotometer. An Agilent Gel Permeation Chromatography (GPC/SEC) system equipped with two columns (PL mixed-C) and calibrated against twelve monodisperse polystyrene (PS) standards, using THF as the eluent at a flow rate of 1.0 mL min−1, was employed to determine the relative weight-average (Mw), number-average (Mn) molecular weights, and polydispersity index (Đ = Mw/Mn) of all the reported polymers. FT-IR spectra were recorded on an FT/IR-6300 type A instrument using a KBr matrix.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v). The reaction mixture was refluxed overnight and the solvent was then evaporated under reduced pressure. The resulting residue was extracted with ethyl acetate from an aqueous solution of 10% HCl (80 mL) and the organic layer was washed with a brine solution followed by deionized water (100 mL × 2). The desired product was isolated by silica gel column chromatography, using ethyl acetate/hexane (10:90 v/v) as the eluent. Pale yellow solid (182 mg, 54%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 7.50 (d, 4H, J = 8.4 Hz, ArH), 7.43 (d, 4H, J = 8.4 Hz, ArH), 1.37 (s, 18H, –CH3). 13C NMR (150 MHz, CD2Cl2, ppm): δ 151.56, 131.15, 125.41, 120.31, 88.72, 34.66, 30.89. EI-HRMS: m/z calculated for M˙+ C22H26 290.2035 found 290.2037.
:1, v/v). The solution was refluxed overnight and the solvent was evaporated under reduced pressure. The resulting mixture was dissolved in DCM and extracted with a saturated solution of NaHCO3 (50 mL × 2). The combined organic layer was washed with deionized water (100 mL × 3), concentrated, and the product was precipitated by adding acetone. The precipitate was isolated by filtration under reduced pressure over a Millipore® filter and washed exhaustively with acetone. Green solid (183 mg, 74%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 7.67 (d, 2H, J = 6.6 Hz, ArH), 7.49 (d, 2H, J = 8.6 Hz, ArH), 7.44 (s, 8H, ArH) 7.34 (t, 2H, J = 7.2 Hz, ArH), 7.30 (d, 8H, J = 2.3 Hz, ArH), 1.35 (s, 18H, –CH3), 1.25 (s, 18H, –CH3). 13C NMR (150 MHz, CD2Cl2, ppm): δ 151.28, 150.44, 141.49, 139.85, 138.88, 138.77, 134.78, 132.68, 130.86, 130.39, 128.95, 128.35, 126.37, 125.74, 125.67, 125.47, 35.20, 35.03, 31.79, 31.62. EI-HRMS: m/z calculated for M˙+ C58H58 754.4538 found 754.4539.
:1 DMF/toluene mixture in a Schlenk tube under argon. After 4 days of reaction, the solvent was evaporated under reduced pressure and the resulting residue was dissolved in CHCl3 and extracted with a saturated solution of NaHCO3 (50 mL × 2). The organic layer was washed with deionized water (100 mL × 3), concentrated, and precipitated by adding acetone. The green solid was filtered and washed with water, methanol, and acetone and then dried under vacuum. Green solid (53 mg, 86%). 1H NMR (600 MHz, CDCl3, ppm): δ 8.10–7.88 (m, 4H, ArH), 7.77 (bd, 2H, ArH), 7.53–7.34 (bm, 11H, ArH) and 1.43–1.36 (br, 15H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 153.77, 149.75, 140.86, 139.74, 138.19, 137.82, 136.12, 134.26, 132.14, 130.53, 130.50, 130.04, 129.33, 128.26, 126.40, 125.82, 125.11, 119.75, 46.45, 34.60, 31.37, 26.73; GPC traces: Mw = 15800 Da, Mn = 6600 Da, Đ = 2.4; FTIR (KBr, cm−1): 2960, 1626, 1436; UV-vis: (THF, 10−6 M), λmax [nm] = 340 and 450.
:1 DMF/toluene degassed solution mixture in a Schlenk tube under argon for 4 days. Green solid (100 mg, 97%). 1H NMR (600 MHz, CDCl3, ppm): δ 7.86–7.64 (m, 4H, ArH), 7.33–6.96 (bm, 21H, ArH), 1.43–1.38 (br, 9H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 150.22, 149.53, 148.96, 141.77, 140.54, 138.81, 138.12, 137.56, 136.71, 133.65, 131.87, 130.51, 130.22, 129.83, 127.82, 127.67, 127.02, 125.75, 125.02, 123.89, 119.95, 66.08, 34.58, 31.40; GPC traces: Mw = 34300 Da, Mn = 12800 Da, Đ = 2.6; FTIR (KBr, cm−1): 2964, 1682, 1428, 730; UV-vis: (THF, 10−6 M), λmax [nm] = 340 and 450.
:1 DMF/toluene solution mixture in a Schlenk tube under argon for 4 days. Green solid (105 mg, 91%). 1H NMR (600 MHz, CDCl3, ppm): δ 8.04 (br, 4H, ArH), 7.58–6.86 (bm, 17H, ArH), 5.75 (br, 2H, triptycene-CH), 1.44 (br, 9H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 154.72, 149.84, 141.53, 140.43, 138.64, 137.19, 132.57, 132.09, 129.79, 129.71, 129.04, 128.23, 125.30, 125.12, 124.81, 120.87, 51.94, 31.52, 30.93; GPC traces: Mw = 32200 Da, Mn = 11600 Da, Đ = 2.7; FTIR (KBr, cm−1): 2960, 1432; UV-vis: (THF, 10−6 M), λmax [nm] = 395, 445 and 470.
:3 v/v). The reaction mixture was refluxed overnight and the solvent was then evaporated under reduced pressure. The resulting residue was extracted with ethyl acetate from an aqueous solution of 10% HCl (80 mL) and the organic layer was washed with a brine solution followed by deionized water (100 mL × 2). The desired product was isolated by silica gel column chromatography, using ethyl acetate/hexane (10:90 v/v) as the eluent. Pale yellow solid (182 mg, 54%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 7.50 (d, 4H, J = 8.4 Hz, ArH), 7.43 (d, 4H, J = 8.4 Hz, ArH), 1.37 (s, 18H, –CH3). 13C NMR (150 MHz, CD2Cl2, ppm): δ 151.56, 131.15, 125.41, 120.31, 88.72, 34.66, 30.89. EI-HRMS: m/z calculated for M˙+ C22H26 290.2035 found 290.2037.
:1, v/v). The solution was refluxed overnight and the solvent was evaporated under reduced pressure. The resulting mixture was dissolved in DCM and extracted with a saturated solution of NaHCO3 (50 mL × 2). The combined organic layer was washed with deionized water (100 mL × 3), concentrated, and the product was precipitated by adding acetone. The precipitate was isolated by filtration under reduced pressure over a Millipore® filter and washed exhaustively with acetone. Green solid (183 mg, 74%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 7.67 (d, 2H, J = 6.6 Hz, ArH), 7.49 (d, 2H, J = 8.6 Hz, ArH), 7.44 (s, 8H, ArH) 7.34 (t, 2H, J = 7.2 Hz, ArH), 7.30 (d, 8H, J = 2.3 Hz, ArH), 1.35 (s, 18H, –CH3), 1.25 (s, 18H, –CH3). 13C NMR (150 MHz, CD2Cl2, ppm): δ 151.28, 150.44, 141.49, 139.85, 138.88, 138.77, 134.78, 132.68, 130.86, 130.39, 128.95, 128.35, 126.37, 125.74, 125.67, 125.47, 35.20, 35.03, 31.79, 31.62. EI-HRMS: m/z calculated for M˙+ C58H58 754.4538 found 754.4539.
:1 DMF/toluene mixture in a Schlenk tube under argon. After 4 days of reaction, the solvent was evaporated under reduced pressure and the resulting residue was dissolved in CHCl3 and extracted with a saturated solution of NaHCO3 (50 mL × 2). The organic layer was washed with deionized water (100 mL × 3), concentrated, and precipitated by adding acetone. The green solid was filtered and washed with water, methanol, and acetone and then dried under vacuum. Green solid (53 mg, 86%). 1H NMR (600 MHz, CDCl3, ppm): δ 8.10–7.88 (m, 4H, ArH), 7.77 (bd, 2H, ArH), 7.53–7.34 (bm, 11H, ArH) and 1.43–1.36 (br, 15H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 153.77, 149.75, 140.86, 139.74, 138.19, 137.82, 136.12, 134.26, 132.14, 130.53, 130.50, 130.04, 129.33, 128.26, 126.40, 125.82, 125.11, 119.75, 46.45, 34.60, 31.37, 26.73; GPC traces: Mw = 15800 Da, Mn = 6600 Da, Đ = 2.4; FTIR (KBr, cm−1): 2960, 1626, 1436; UV-vis: (THF, 10−6 M), λmax [nm] = 340 and 450.
:1 DMF/toluene degassed solution mixture in a Schlenk tube under argon for 4 days. Green solid (100 mg, 97%). 1H NMR (600 MHz, CDCl3, ppm): δ 7.86–7.64 (m, 4H, ArH), 7.33–6.96 (bm, 21H, ArH), 1.43–1.38 (br, 9H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 150.22, 149.53, 148.96, 141.77, 140.54, 138.81, 138.12, 137.56, 136.71, 133.65, 131.87, 130.51, 130.22, 129.83, 127.82, 127.67, 127.02, 125.75, 125.02, 123.89, 119.95, 66.08, 34.58, 31.40; GPC traces: Mw = 34300 Da, Mn = 12800 Da, Đ = 2.6; FTIR (KBr, cm−1): 2964, 1682, 1428, 730; UV-vis: (THF, 10−6 M), λmax [nm] = 340 and 450.
:1 DMF/toluene solution mixture in a Schlenk tube under argon for 4 days. Green solid (105 mg, 91%). 1H NMR (600 MHz, CDCl3, ppm): δ 8.04 (br, 4H, ArH), 7.58–6.86 (bm, 17H, ArH), 5.75 (br, 2H, triptycene-CH), 1.44 (br, 9H, –CH3). 13C NMR (150 MHz, CDCl3, ppm): δ 154.72, 149.84, 141.53, 140.43, 138.64, 137.19, 132.57, 132.09, 129.79, 129.71, 129.04, 128.23, 125.30, 125.12, 124.81, 120.87, 51.94, 31.52, 30.93; GPC traces: Mw = 32200 Da, Mn = 11600 Da, Đ = 2.7; FTIR (KBr, cm−1): 2960, 1432; UV-vis: (THF, 10−6 M), λmax [nm] = 395, 445 and 470.
Results and discussion
Synthesis of comonomers
Scheme 1 shows the synthesis of the dialkyne comonomers 3a–ci.e. 9,9-dimethylfluorene 3a, spiro bifluorene 3b, and triptycene 3c. The aforementioned building blocks were prepared by reacting 1-(tert-butyl)-4-ethynylbenzene 1a with either the dibromo derivatives 2a and 2b or nonaflate synthon 2cvia a conventional palladium-catalyzed Sonogashira cross-coupling reaction.56 Comonomers 3a–c were isolated in very good yields (∼66–100%) and have excellent solubility in common organic solvents, namely, dichloromethane, chloroform, tetrahydrofuran, and toluene. It is noteworthy that the formation of 3a–c was confirmed by 1H- and 13C-NMR spectroscopy, as well as EI-HRMS (see Fig. 3 and Fig. S1–3, S9–11, and S18–20 in the ESI†). | ||
| Scheme 1 Synthesis of comonomers 3a–c. | ||
Fig. 1 shows the representative 1H- and 13C-NMR spectra of synthon 3a which confirm the presence of all the desired peaks. All the aromatic protons and carbons of 3a were detected in the ranges of 7.4–7.7 ppm and 120.8–154.6 ppm, respectively. The 1H NMR spectrum of 3a shows the characteristic aliphatic peaks of the methyl groups of fluorene and t-butyl moieties at 1.5 and 1.3 ppm, respectively (cf. peaks labeled a and b in Fig. 1). The 13C NMR spectrum displays two peaks at 90.4 and 89.9 ppm, which are attributed to the characteristic sp carbons of 3a (cf. peaks labeled e and f in Fig. 1). In addition, all the aliphatic peaks in the range of 47.4–27.3 ppm are in full agreement with the desired synthon 3a. Similarly, 1H- and 13C-NMR spectra of comonomers 3b and 3c display the characteristic chemical shifts, and thus, confirm the formation of the desired products (see Fig. S1–3 and S9–11 in the ESI†). Furthermore, electron-impact high-resolution mass spectrometry (EI-HRMS) of 3a–c reveal the formation of all the desired building blocks in high purity, as can be observed from the isotopic distribution (see Fig. S18–20 in the ESI†).
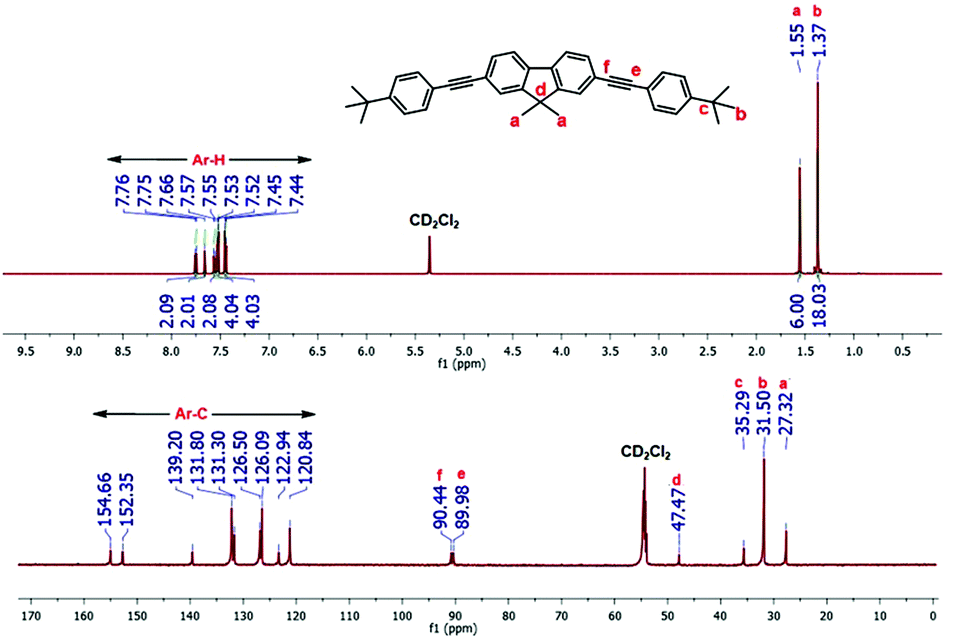 | ||
| Fig. 1 1H (top) and 13C (bottom) NMR of 3a recorded in CD2Cl2. | ||
In order to prove the feasibility of the cyclopentannulation reaction, a model test was carried out by reacting 9,10-dibromoanthracene DBA with two equivalents of 1,2-bis(4-(tert-butyl)phenyl)ethyne TBPE in a 1:1 DMF/toluene mixture at 130 °C (Scheme 2) affording the prototypical target CPM in 74% yield.
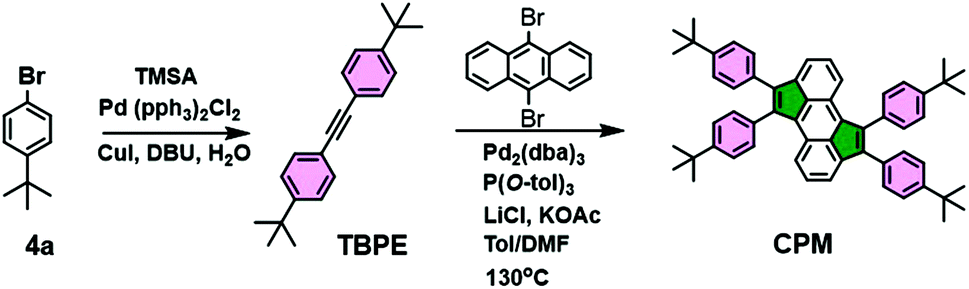 | ||
| Scheme 2 Synthesis of prototypical monomer CPM. | ||
The structure of CPM was confirmed by 1H- and 13C-NMR spectroscopy, high-resolution mass spectroscopy (HRMS), and hydrogen carbon-13 correlation 2D-NMR spectroscopy. Fig. 2 shows the 1H NMR spectra of CPM where all the characteristic aromatic protons are revealed at 7.67 ppm (doublet, labeled a), 7.50 ppm (doublet, labeled b), and 7.34 ppm (triplet, labeled c). The aliphatic t-butyl protons are detected at 1.35 ppm and 1.25 ppm. 13C-NMR spectral analysis corroborates the above findings, and thus, further confirms the formation of the desired monomer CPM by revealing the presence of all the peaks at the expected chemical shifts. Formation of CPM is also supported by the 2D hydrogen carbon-13 correlation spectra which also confirm the sole formation of the desired product with the absence of any trace amount of side products (see Fig. S5, S13, and S17 in the ESI†).
 | ||
| Fig. 2 1H NMR of CPM recorded in CD2Cl2. | ||
The analysis of CPM by electron-impact high-resolution mass spectrometry (EI-HRMS) confirms its formation in high purity, as revealed by the experimentally determined isotopic patterns when compared to the calculated ones (Fig. 3).
 | ||
| Fig. 3 EI-HRMS spectrum of CPM; inset: calculated (down) and measured (up) isotopic patterns of C58H58˙+. | ||
Synthesis of copolymers CPP1–3
As could be observed from Scheme 3, the conjugated polymers CPP1–3 were synthesized from the cyclopentannulation reaction of DBA with monomers 3a–c following the same conditions which were applied to prepare the prototypical monomer CPM described in Scheme 2. The polymerization was carried out by the reaction of a 0.1 M solution of 3a–c with an equimolar amount of DBA in the presence of a pd2(dba)3 catalyst, a p(o-tol)3 ligand, a KOAc base and LiCl in a 1:1 DMF/toluene solvent mixture at 130 °C over four days. CPP1–3 target polymers were isolated in excellent yields (86–97%) and were found to be highly soluble in common organic solvents such as CHCl3, DCM, and THF. The high purity of CPP1–3 was confirmed by GPC analysis, and 1H- and 13C-NMR, FTIR, UV-vis absorption and emission spectroscopy (see Fig. 4 and Fig. S6–8, S14–16, and S22–23 in the ESI†).
 | ||
| Scheme 3 Synthesis of conjugated polymers CPP1–3. | ||
 | ||
| Fig. 4 Normalized GPC chromatogram of CPP2. | ||
The GPC results of CPP1–3 reveal that the average weight molar mass (Mw) ranges from 32.2 to 15.8 kDa, while the average number molecular mass (Mn) varies between 11.6 and 6.6 kDa, thus yielding polydispersity values (Đ) varying from 2.4 to 2.8 (Fig. 4 and Table 1, entry 1–3).
| Entry | Product | M w (g mol−1) | M n (g mol−1) | Đ |
|---|---|---|---|---|
| 1 | CPP1 | 15800 |
6600 | 2.4 |
| 2 | CPP2 | 34300 |
12800 |
2.7 |
| 3 | CPP3 | 32200 |
11600 |
2.8 |
Fig. 5 illustrates the comparative FT-IR absorption spectra of comonomer 3c and its corresponding polymer CPP3. The characteristic stretching and bending vibrations of the aliphatic C–H groups in 3c are observed at 2967 and 1440 cm−1, respectively. In addition, 3c divulges the characteristic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibrations at 2209 cm−1, which completely disappears in the FT-IR absorption spectrum of the corresponding polymer CPP3. The stretching and bending vibrations of the aliphatic C–H groups in CPP3 are detected at 2960 and 1432 cm−1, respectively. It is noteworthy that the FT-IR spectrum of CPP3 discloses more pronounced aromatic stretching vibrations for C–C (1400–1600 cm−1) and C–H (2960–3050 cm−1), when compared to those recorded for monomer 3c. This clearly indicates the full conversion of the ethynylene moieties into the corresponding conjugated polymers.
C stretching vibrations at 2209 cm−1, which completely disappears in the FT-IR absorption spectrum of the corresponding polymer CPP3. The stretching and bending vibrations of the aliphatic C–H groups in CPP3 are detected at 2960 and 1432 cm−1, respectively. It is noteworthy that the FT-IR spectrum of CPP3 discloses more pronounced aromatic stretching vibrations for C–C (1400–1600 cm−1) and C–H (2960–3050 cm−1), when compared to those recorded for monomer 3c. This clearly indicates the full conversion of the ethynylene moieties into the corresponding conjugated polymers.
 | ||
| Fig. 5 Comparative FT-IR spectra of 3c and CPP3. | ||
The photophysical properties of the target polymers were measured by means of UV-Vis absorption and fluorescence spectroscopy. CPP1 and CPP2 display similar features with a strong UV absorption band at 340 nm, whereas CPP3, i.e. the polymer that contains triptycene units, discloses a strong absorption band at 395 nm, thus revealing a red-shift by 55 nm. Interestingly, the emission spectra of polymers CPP1–3 show two peak maxima, the first ranging from ∼403 to 433 nm, while the second being observed between 540 and 564 nm (Fig. 6).
 | ||
| Fig. 6 Normalized UV-VIS absorption (left) and emission (right) spectra of CPP1–3 (CM = 10−6 M in THF). | ||
Iodine uptake studies
Target polymers CPP1–3 offer several advantages, namely, their contorted structures, excellent solubility, and long-range conjugation. Whilst the former property could be explained by cyclopentannulation reaction, the last two properties could be attributed to the introduction of the specially designed polycondensed aromatic hydrocarbon comonomers 3a–c, whose spiro centers prevent the target polymers CPP1–3 from aggregation and improve the overall polymer porosity. Therefore, the specially designed graphite-like structures of CPP1–3 prompted us to explore their iodine adsorption and desorption properties. Vapor iodine adsorption experiments were carried out gravimetrically according to a protocol reported in the literature.57,58CPP1–3 target compounds were exposed to excess iodine vapors by taking a 20 mg sample of each polymer in an open glass vial, which was in turn placed inside a sealed glass vessel that contained solid iodine at 80 °C under atmospheric pressure. The iodine uptake capacity of CPP1–3 was monitored gravimetrically as shown in Table 2. It was observed that after one hour of iodine exposure, ∼65% by weight was adsorbed by CPP2 while half of this amount i.e. ∼30 wt% was recorded for CPP1 and CPP3 (Table 2, entry 3). Subsequently, the iodine uptake by CPP2 reached ∼190 wt% after 24 hours, whereas an average of 135 wt% was recorded for CPP1 and CPP3 for the same exposure period (Table 2, entry 9). The adsorption values showed little increase when the polymers were kept for 72 hours, thus, suggesting the saturation of CPP1–3, reaching maximum iodine uptake capacities of 153, 200, and 140 wt%, respectively (Table 2, entry 10). It is noteworthy that the iodine uptake capacity of the prototypical monomer CPM is just 32 wt% after 72 hours of exposure to iodine vapors, which is far lower than that of its corresponding polymers CPP1–3.The 200 wt% iodine uptake value of CPP2 is superior to those of polymers reported in the literature, and notable among these are nitrogen-rich triptycene-based materials (NTP, 180 wt%),59 calix[4]arene-based 2D macromolecules (CX4-NS, 114 wt%),60 porphyrin and pyrene-based conjugated microporous polymers (Por-Py-CMP, 130 wt%),61 nitrogen-containing materials (NRPPs, 192 wt%),57 JLUE covalent organic polymer (JLUE-COP-3, up to 90.29%),62 conjugated microporous polymers with thiophene units (SCMPs, 222 wt%),34 hierarchically porous adamantane-based macromolecules (202 wt%),63 triazine-based covalent frameworks (TTPT, 177wt%),64 fluorine-enriched polymers (FCMP-600@1–4, up to 141 wt%)65 and many others.66,67
The iodine adsorbed by CPP1–3 could be released by simple heating of these latter polymers in air at 125 °C. The complete 100% iodine desorption efficiency for the iodine-loaded polymers (I2@CPP1–3) was recorded at different time intervals (Fig. 7 and Fig. S24 and S26 in the ESI†). The reusable adsorption efficiency of the polymers was investigated using CPP2, which showed the maximum uptake capacity for iodine. For this, we first heated a sample of CPP2 fully loaded with iodine vapors (I2@CPP2) at 125 °C for 24 hours, in order to ensure the complete release of the adsorbate from the polymer backbone. The reactivated CPP2 was then exposed to iodine vapors and its uptake was recorded gravimetrically using the procedure described above, revealing an uptake pattern similar to that of a freshly prepared polymer (Table 2). It is worth mentioning that CPP1–3 undergo a color change from green/brown to black upon exposure to iodine vapors and return to their original color when heated to 125 °C in order to release the adsorbate (Fig. 7, inset).
 | ||
| Fig. 7 CPP2 gravimetric adsorption (left) and desorption (right, heated at 125 °C in air) of iodine as a function of time. Inset: crucible photographs showing the color change after iodine adsorption. | ||
Iodine desorption results were further supported by immersing CPP1–3 in ethanol, an excellent solvent for dissolving iodine but not the target polymers (Fig. 8 and Fig. S25 and S27 in the ESI†). The iodine extraction in ethanol was studied by recording the UV-visible absorbance spectra at different time intervals (Fig. 8). A noticeable increase with time in the intensity of the absorbance maxima that correspond to iodine, i.e. ∼227 nm (due to I2), ∼290 nm and ∼359 nm (due to polyiodide ions), was observed which confirms the adsorbate release from CPP2 under ambient conditions. The amount of iodine released increased rapidly within the first 20 minutes and reached equilibrium after 45 minutes. The color of the solution changed from colorless to yellow (Fig. 8) which further confirms the iodine release in ethanol. These experimental observations strongly suggest that CPP1–3 can be employed as sorbent materials for efficient iodine vapor uptake. Moreover, once the polymers are loaded with iodine (I2@CPP1–3), they can be easily regenerated either by heating or soaking in ethanol, which makes the recycling process quite practical.
 | ||
| Fig. 8 UV-Vis absorption spectra upon immersion of I2@CPP2 in ethanol. Inset: photographs of the solutions showing a color change upon immersion in ethanol. | ||
Conclusion
We report the synthesis of three new conjugated polymers CPP1–3via typical palladium-catalyzed cyclopentannulation reaction conditions. The target polymers, which contain contorted monomers, namely, dimethyl fluorene (CPP1), spirobifluorene (CPP2), and triptycene (CPP3), were isolated in excellent yields and found to have high relative weight-average molecular weights (Mw) in the range of 15.8–34.3 kDa and a polydispersity index (Đ = Mw/Mn) varying between 2.4 and 2.8. Iodine vapor sorption studies of CPP1–3 reveal a high % by weight uptake that reaches 200 wt% for CPP2 with the possibility to regenerate the polymer either by heating in ambient air or soaking in ethanol. This paves the way towards using these promising materials as versatile recyclable iodine gas adsorbents for applications in the field of environmental remediation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was partially supported by Kuwait Foundation for the Advancement of Sciences (KFAS) under project code PN18-14SC-03. We would like to thank the general facilities projects GS01/01, GS01/03, GS01/05, GS03/01, and GS03/08 at Kuwait University.References
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D.-K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, J. Am. Chem. Soc., 2018, 140, 1622–1626 CrossRef CAS PubMed.
- A. Ichige, H. Saito, J. Kuwabara, T. Yasuda, J.-C. Choi and T. Kanbara, Macromolecules, 2018, 51, 6782–6788 CrossRef CAS.
- M. J. Sung, S. Yoon, S.-K. Kwon, Y.-H. Kim and D. S. Chung, ACS Appl. Mater. Interfaces, 2016, 8, 31172–31178 CrossRef CAS PubMed.
- J. Yang, P. Cong, L. Chen, X. Wang, J. Li, A. Tang, B. Zhang, Y. Geng and E. Zhou, ACS Macro Lett., 2019, 8, 743–748 CrossRef.
- K. Aoshima, M. Nomura and A. Saeki, ACS Omega, 2019, 4, 15645–15652 CrossRef CAS PubMed.
- H. Huang, H. Bin, Z. Peng, B. Qiu, C. Sun, A. Liebman-Pelaez, Z.-G. Zhang, C. Zhu, H. Ade, Z. Zhang and Y. Li, Macromolecules, 2018, 51, 6028–6036 CrossRef CAS.
- J. Yuan, J. Ouyang, V. Cimrová, M. Leclerc, A. Najari and Y. Zou, J. Mater. Chem. C, 2017, 5, 1858–1879 RSC.
- S.-H. Kang, A. Jeong, H. R. Lee, J. H. Oh and C. Yang, Chem. Mater., 2019, 31, 3831–3839 CrossRef CAS.
- Y. Du, H. Yao, L. Galuska, F. Ge, X. Wang, H. Lu, G. Zhang, X. Gu and L. Qiu, Macromolecules, 2019, 52, 4765–4775 CrossRef CAS.
- L. Zhang, Z. Wang, C. Duan, Z. Wang, Y. Deng, J. Xu, F. Huang and Y. Cao, Chem. Mater., 2018, 30, 8343–8351 CrossRef CAS.
- S. Shin, F. Menk, Y. Kim, J. Lim, K. Char, R. Zentel and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 6088–6094 CrossRef CAS PubMed.
- I. Choi, S. Yang and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 17218–17225 CrossRef CAS PubMed.
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 CrossRef CAS.
- T. M. Swager, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- T. Bura, S. Beaupre, M.-A. Legare, J. Quinn, E. Rochette, J. T. Blaskovits, F.-G. Fontaine, A. Pron, Y. Li and M. Leclerc, Chem. Sci., 2017, 8, 3913–3925 RSC.
- A. Marrocchi, A. Facchetti, D. Lanari, S. Santoro and L. Vaccaro, Chem. Sci., 2016, 7, 6298–6308 RSC.
- A. Hirose, K. Tanaka, R. Yoshii and Y. Chujo, Polym. Chem., 2015, 6, 5590–5595 RSC.
- B. Alameddine, N. Baig, S. Shetty and S. Al-Mousawi, Polymer, 2019, 178, 121589 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, Mater. Today Chem., 2018, 10, 213–220 CrossRef CAS.
- K. Aravindu, E. Cloutet, C. Brochon, G. Hadziioannou, J. Vignolle, F. Robert, D. Taton and Y. Landais, Macromolecules, 2018, 51, 5852–5862 CrossRef CAS.
- Z. Wang, C. Wang, Y. Fang, H. Yuan, Y. Quan and Y. Cheng, Polym. Chem., 2018, 9, 3205–3214 RSC.
- B. Alameddine, R. Sobhana Anju, S. Shetty, N. Baig, S. Al-Mousawi and F. Al-Sagheer, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3565–3572 CrossRef CAS.
- C.-F. Yao, K.-L. Wang, H.-K. Huang, Y.-J. Lin, Y.-Y. Lee, C.-W. Yu, C.-J. Tsai and M. Horie, Macromolecules, 2017, 50, 6924–6934 CrossRef CAS.
- X. Yan, M. Xiong, J.-T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z.-Y. Wang, Z.-F. Yao, J.-Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- A. Rafiee and K. R. Khalilpour, in Polygeneration with Polystorage for Chemical and Energy Hubs, ed. K. R. Khalilpour, Academic Press, 2019, pp. 331–372, DOI:10.1016/B978-0-12-813306-4.00011-2.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- B. K. Sovacool, Util. Policy, 2009, 17, 288–296 CrossRef.
- D. Banerjee, A. J. Cairns, J. Liu, R. K. Motkuri, S. K. Nune, C. A. Fernandez, R. Krishna, D. M. Strachan and P. K. Thallapally, Acc. Chem. Res., 2015, 48, 211–219 CrossRef CAS PubMed.
- H. Ma, J.-J. Chen, L. Tan, J.-H. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed.
- X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- K. Kosaka, M. Asami, N. Kobashigawa, K. Ohkubo, H. Terada, N. Kishida and M. Akiba, Water Res., 2012, 46, 4397–4404 CrossRef CAS PubMed.
- F. Baig, K. Rangan, S. M. Eappen, S. K. Mandal and M. Sarkar, CrystEngComm, 2020, 22, 751–766 RSC.
- M. Janeta, W. Bury and S. Szafert, ACS Appl. Mater. Interfaces, 2018, 10, 19964–19973 CrossRef CAS PubMed.
- X. Zhang, P. Gu, X. Li and G. Zhang, Chem. Eng. J., 2017, 322, 129–139 CrossRef CAS.
- A. Azadbakht, A. R. Abbasi and N. Noori, J. Inorg. Organomet. Polym. Mater., 2016, 26, 479–487 CrossRef CAS.
- T. Hasell, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 14920–14923 CrossRef CAS PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- Y. Xie, Y. Gong, M. Han, F. Zhang, Q. Peng, G. Xie and Z. Li, Macromolecules, 2019, 52, 896–903 CrossRef.
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00550.
- Y. Yano, N. Mitoma, H. Ito and K. Itami, J. Org. Chem., 2020, 85, 4–33 CrossRef PubMed.
- W. Wang, H. Chen, H. Y. Zhu, Y. L. Huang and J. W. Yang, Chem. – Asian J., 2017, 12, 3016–3026 CrossRef PubMed.
- I. C.-Y. Hou, Y. Hu, A. Narita and K. Müllen, Polym. J., 2017, 50, 3 CrossRef.
- B. Alameddine, N. Baig, S. Shetty, S. Al-Mousawi and F. Al-Sagheer, Polymer, 2018, 154, 233–240 CrossRef CAS.
- Z. Zeng, X. Shi, C. Chi, J. T. Lo, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- D. Reinhard, W.-S. Zhang, F. Rominger, R. Curticean, I. Wacker, R. Schröder Rasmus and M. Mastalerz, Chem. – Eur. J., 2018, 24, 11433–11437 CrossRef CAS PubMed.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesana-Gandara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 CrossRef CAS PubMed.
- X. Zhu, S. R. Bheemireddy, S. V. Sambasivarao, P. W. Rose, R. T. Guzman, A. G. Waltner, K. H. DuBay and K. N. Plunkett, Macromolecules, 2016, 49, 127–133 CrossRef CAS.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, J. Am. Chem. Soc., 2017, 139, 5801–5807 CrossRef CAS PubMed.
- Z. Zhu and T. M. Swager, Org. Lett., 2001, 3, 3471–3474 CrossRef CAS PubMed.
- B. Alameddine, N. Baig, S. Shetty, S. Al-Mousawi and F. Al-Sagheer, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 931–937 CrossRef CAS.
- Y. H. Abdelmoaty, T.-D. Tessema, F. A. Choudhury, O. M. El-Kadri and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2018, 10, 16049–16058 CrossRef CAS PubMed.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 CrossRef PubMed.
- R. M. Weiss, A. L. Short and T. Y. Meyer, ACS Macro Lett., 2015, 4, 1039–1043 CrossRef CAS.
- D. Shetty, T. Skorjanc, J. Raya, S. K. Sharma, I. Jahovic, K. Polychronopoulou, Z. Asfari, D. S. Han, S. Dewage, J.-C. Olsen, R. Jagannathan, S. Kirmizialtin and A. Trabolsi, ACS Appl. Mater. Interfaces, 2018, 10, 17359–17365 CrossRef CAS PubMed.
- K. C. Park, J. Cho and C. Y. Lee, RSC Adv., 2016, 6, 75478–75481 RSC.
- H. Guan, D. Zou, H. Yu, M. Liu, Z. Liu, W. Sun, F. Xu and Y. Li, Front. Mater., 2019, 6(12) DOI:10.3389/fmats.2019.00012.
- D. Chen, Y. Fu, W. Yu, G. Yu and C. Pan, Chem. Eng. J., 2018, 334, 900–906 CrossRef CAS.
- T. Geng, W. Zhang, Z. Zhu, G. Chen, L. Ma, S. Ye and Q. Niu, Polym. Chem., 2018, 9, 777–784 RSC.
- G. Li, C. Yao, J. Wang and Y. Xu, Sci. Rep., 2017, 7, 13972 CrossRef PubMed.
- M. Ansari, A. Alam, R. Bera, A. Hassan, S. Goswami and N. Das, J. Environ. Chem. Eng., 2019 DOI:10.1016/j.jece.2019.103558 , In Press.
- M. Liu, C. Yao, C. Liu and Y. Xu, Sci. Rep., 2018, 8, 14071 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00286k |
| This journal is © The Royal Society of Chemistry 2020 |