
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the crystal structure of precisely spaced polyethylene-like polyphosphoesters†
Tobias
Haider‡
a,
Oksana
Suraeva‡
a,
Miriam L.
O'Duill
 b,
Julian
Mars
a,
Markus
Mezger
a,
Ingo
Lieberwirth
*a and
Frederik R.
Wurm
*a
b,
Julian
Mars
a,
Markus
Mezger
a,
Ingo
Lieberwirth
*a and
Frederik R.
Wurm
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de; lieberw@mpip-mainz.mpg.de
bSchool of Chemistry, NUI Galway, University Road, Galway H91 TK33, Ireland
First published on 5th May 2020
Abstract
Understanding polymer crystallization is important for polyethylene-like materials. A small fraction of monomers with functional groups within the polyethylene chain can act as crystallization “defects”. Such defects can be used to control the crystallization behavior in bulk and to generate functional anisotropic polymer crystals if crystallized from a dilute solution. Due to their geometry, phosphate groups cannot be incorporated in the polyethylene lamellae and thus control chain folding and crystal morphology. Herein, the synthesis and crystallization behavior for three different long-chain polyphosphates with a precise spacing of 20, 30, and 40 CH2-groups between each phosphate group are reported. Monomers were prepared by esterification of ethyl dichlorophosphate with respective tailor-made unsaturated alcohols. Acyclic diene metathesis (ADMET) polymerization and subsequent hydrogenation were used to receive polyethylene-like polyphosphoesters with molecular weights up 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1. Polymer crystallization was studied from the melt and dilute solution. Samples were characterized by differential scanning calorimetry (DSC), small-angle X-ray scattering (SAXS), wide-angle X-ray scattering (WAXS), transmission electron microscopy (TEM), and atomic force microscopy (AFM). A change in crystal structure from pseudo-hexagonal to orthorhombic was observed from the “C20” to the “C40” polymer. Melting points and lamellar thicknesses increased with the length of the aliphatic spacer from 51 °C (“C20”) to 62 °C (“C30”) and 91 °C (“C40”). Values for the long periods in bulk (3.1 nm for C20, 4.8 nm for C30, and 7.2 nm for C40) obtained by SAXS and TEM are in qualitative agreement. The thickness of the crystalline part obtained by AFM and TEM increased from about 1.0 nm (C20) to 2.0 nm (C30) to 2.9 nm (C40). Our systematic library of long-chain polyphosphates will allow designing anisotropic polymer colloids by crystallization from solution as functional and versatile colloid platform.
100 g mol−1. Polymer crystallization was studied from the melt and dilute solution. Samples were characterized by differential scanning calorimetry (DSC), small-angle X-ray scattering (SAXS), wide-angle X-ray scattering (WAXS), transmission electron microscopy (TEM), and atomic force microscopy (AFM). A change in crystal structure from pseudo-hexagonal to orthorhombic was observed from the “C20” to the “C40” polymer. Melting points and lamellar thicknesses increased with the length of the aliphatic spacer from 51 °C (“C20”) to 62 °C (“C30”) and 91 °C (“C40”). Values for the long periods in bulk (3.1 nm for C20, 4.8 nm for C30, and 7.2 nm for C40) obtained by SAXS and TEM are in qualitative agreement. The thickness of the crystalline part obtained by AFM and TEM increased from about 1.0 nm (C20) to 2.0 nm (C30) to 2.9 nm (C40). Our systematic library of long-chain polyphosphates will allow designing anisotropic polymer colloids by crystallization from solution as functional and versatile colloid platform.
Introduction
Semi-crystalline polymers make up more than 50% of all commodity polymers consumed, with polyethylene (PE) being the most produced synthetic polymer today.1 Tailoring the crystallinity as well as the size and shape of crystallites in polyethylene enables new possible applications. With so-called “defect engineering”, the crystallization of PE can be controlled by the synthesis of PE-derivatives with crystallization defects, i.e. side chains or bulky functional groups. Such “defects” can also be used for further chemical functionalization. Polymerization techniques that facilitate a precise distribution of the crystallization defects in the polymer backbone allow control over the crystal morphology including e.g. the lamellar thickness of the PE crystallites. Following this “defect engineering” approach, we present PE-like polyphosphates with distinctive spacing between the phosphate groups and elucidate the effect of the spacer length on the crystal structure and morphology of solution-grown polymer platelets and bulk-crystallized polymer crystals.The crystallization of PE results from the van-der-Waals forces between parallel ordered aliphatic polymer chains and yields lamellar crystals. Finally, these lamellae arrange to larger structures and form spherulites.2 Overall, PE crystallizes in an orthorhombic crystal structure.3 Side-chains or additional functional groups along the polymer backbone can affect the crystallization: any bulky alkyl side groups (e.g. branching in low-density polyethylene (LDPE) and linear low-density polyethylene (LLDPE)) might be expelled from the crystalline to the amorphous phase, reducing the overall crystallinity.4 At the same time, a high branch content reduces the thickness of the lamellae, resulting in lower melting points compared to defect-free, linear PE.5 The influence of branch length and the distribution of branching on the polymer chain on the overall crystal structure of precisely branched polyethylene have been studied by the group of Wagener.6–12
In contrast, functional groups incorporated into polyethylene can add new properties to the material: for instance, Mecking et al. recently reported ion-conducting PE-like polymers based on a sulfonate bearing polyester13 and a telechelic polyethylene with terminal carboxylic acids,14 respectively. Also, degradable PE mimics based on long-chain polyacetals15,16 and polyorthoesters17 had been reported. Different polymerization techniques enable the synthesis of PE-like polymers containing functional groups: long-chain polyesters, for instance, were obtained by polyesterification,18,19 ring-opening metathesis copolymerization,20 and acyclic diene metathesis (ADMET) polymerization.21 In this work, we used ADMET polymerization, a polycondensation, which uses α,ω-dienes to produce linear polymers and allows the installation of precise branches or other functionalities in PE-like materials.11
As mentioned above, functional groups act as crystallization defects in the polyethylene chain, which can be used to control the thickness of the crystal lamellae. Whether the defects are incorporated in the lamellar crystal segment or not depends on the size and the flexibility of the functional group. Fan et al. reported that the crystal structure of polyethylene derivatives with aryl ether defects in the main chain (with a precise spacing of 20 CH2 groups between each defect) was determined by the substitution pattern on the aromatic ring.22 For ortho-substituted polymers, the aromatic crystallization defects were urged into the amorphous phase, while the defect in the para-substituted polymer was incorporated into the crystal with a remaining orthorhombic crystal structure like polyethylene. Similarly, poly(1,3-adamantylene alkylene)s synthesized by ADMET polymerization also crystallized in an orthorhombic crystal structure.23 Due to the rigid adamantane defect, the black-folding of the polymer chain at adjacent reentry sites of the crystal lamellae is sterically impossible. In contrast, our group previously investigated the crystallization behavior of two different polyphosphates bearing a methyl and a phenyl side chain.24 All bonds in the phosphate group are flexible, thus only the size of the defect had an impact on crystallization. The phosphate group with a methyl side chain was incorporated into the polymer crystal, which was not the case for the polyphosphate with a bulky phenyl side chain. Polyphosphates are potentially enzymatically degradable and enable further functionalization trough variation of the side chain.25 Similarly to long-chain polyesters, long-chain polyphosphates were synthesized by ADMET, ring-opening metathesis (ROMP) polymerization or polytransesterification.26,27 Due to the formation of pyrophosphate groups in side-reactions during the polytransesterification, we used ADMET polymerization as a reliable technique that provides a precise spacing between the phosphate groups.
Here, we present the synthesis of three PE-like polyphosphates with precise alkyl spacing of 20, 30, and 40 CH2-units between each phosphate group. The phosphate groups are intended to act as crystallization defects, as they are expected to be expelled of the crystal lamellae. Thus, differences in e.g. lamellar thickness are expected to only rely on the length of the aliphatic spacer. We examined their influence on the thermal properties of the synthesized polymers by differential scanning calorimetry (DSC). Crystal structures and morphologies of the bulk polymers and solution-grown polymer platelets were determined by WAXS, SAXS, TEM, and AFM. The synthesized precise PE-like polyphosphates could, for example, represent a modular platform for anisotropic colloids with functional surfaces.
Experimental section
Materials
All available reagents were purchased from Sigma Aldrich, Alfa Aesar, Acros Organics or TCI and were used without further purification unless otherwise stated. Deuterated solvents were purchased from Sigma Aldrich.Instrumentation and characterization techniques
Thin-layer chromatography (TLC) was performed using Merck aluminum-foil baked plates coated with Kieselgel 60 F245. The products were visualized using UV fluorescence (254 nm) or potassium permanganate stain. Flash column chromatography was performed over Merck silica gel C60 (40–60 μm) using eluent systems as described for each experiment. Size exclusion chromatography (SEC) measurements were performed in THF on an Agilent Technologies 1260 instrument consisting of an autosampler, pump, and column oven. The column set consists of 3 columns: SDV 106 Å, SDV 104 Å, and SDV 500 Å (PSS Standards Service GmbH, Mainz, Germany), all of 300 × 8 mm and 10 μm average particle size were used at a flow rate of 1.0 mL min−1 and a column temperature of 30 °C. The injection volume was 100 μL. Detection was accomplished with an RI detector (Agilent Technologies). The data acquisition and evaluation were performed using PSS WINGPC UniChrom (PSS Polymer Standards Service GmbH, Mainz, Germany). Calibration was carried out by using polystyrene provided by PSS Polymer Standards Service GmbH (Mainz, Germany). For nuclear magnetic resonance (NMR) analysis 1H, 13C and 31P NMR spectra of the monomers were recorded on a Bruker AVANCE III 300, 400, 500, or 700 MHz spectrometer. All spectra were measured in CDCl3 at 298 K. The spectra were calibrated against the solvent signal and analyzed using MestReNova 14.1.0. (Mestrelab Research S.L). The thermal properties of the synthesized polymers have been measured by differential scanning calorimetry (DSC) on a Mettler Toledo DSC 823 calorimeter. Three scanning cycles of heating/cooling were performed in a nitrogen atmosphere (30 mL min−1) with a heating and cooling rate of 10 °C min−1. The heating rate was 10 °C min−1 in a range of temperatures between −100 and 180 °C. Wide-angle X-ray scattering (WAXS) measurements on powder diffractometers with Cu radiation (wavelength 1.5418 Å) were performed using a Philips PW1820 for poly(1)-H and a Rigaku SmartLab for poly(2)-H and poly(3)-H. Small-angle X-ray scattering (SAXS) experiments were performed on a home-built instrument.28 The crystal morphology, thickness, and crystal structure were determined using an FEI Tecnai F20 transmission electron microscope operated at an acceleration voltage of 200 kV. Bright-field (BF) and energy-filtered transmission electron microscopy (EFTEM) techniques were used for measurements. AFM measurements were performed using a Dimension Icon FS with tapping mode. For the measurements, one droplet of the dispersion containing the solution-grown crystals was dropped onto a freshly cleaved mica substrate, and excess liquid was blotted off with the edge of a filter paper.Sample preparation
Both solution- and melt-crystallization methods were used for sample preparation. To prepare solution-grown crystals, the polymer was dissolved in hot n-octane at a concentration of 0.05 wt%. The solution was kept in a temperature-controlled oil bath, whereby the change of the temperature was controlled by the change of the oil bath. After full dissolution, the solution was slowly cooled down to room temperature for crystallization. Afterwards, one droplet of the dispersion was dropped onto a carbon-coated grid for further TEM measurement.For the melt-grown crystals, the samples were annealed in the oven at the temperature 5 degrees below the melting point for 2 days and slowly cooled down to room temperature. For TEM examination the melt crystallized bulk samples were prepared using ultramicrotomy. The samples were embedded in epoxy resin and subsequently sectioned at room temperature using a Leica ultracut UCT. To decrease the compression of the sample, a 35° DiATOME ultrasonic oscillating diamond knife was used for sectioning. The thin sections were collected on the copper grids and subsequently RuO4 stained for 24 h.
Synthetic procedures
Monomer synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 5.06–4.87 (m, 4H, CH2CH–), 4.18–3.94 (m, 6H, –OPO3–CH2–), 2.06–1.98 (m, 4H, CH–CH2–), 1.70–1.63 (m, 4H, –OPO3–CH2–CH2–), 1.38–1.27 ppm (m, 27H). 13C NMR (176 MHz, CDCl3, 298 K): δ = 139.27, 114.25, 67.78, 63.76, 33.91, 30.41, 29.54, 29.23, 29.03, 25.56, 16.30 ppm. 31P NMR (283 MHz, CDCl3, 298 K): δ = −0.71 ppm.
CH–), 5.06–4.87 (m, 4H, CH2CH–), 4.18–3.94 (m, 6H, –OPO3–CH2–), 2.06–1.98 (m, 4H, CH–CH2–), 1.70–1.63 (m, 4H, –OPO3–CH2–CH2–), 1.38–1.27 ppm (m, 27H). 13C NMR (176 MHz, CDCl3, 298 K): δ = 139.27, 114.25, 67.78, 63.76, 33.91, 30.41, 29.54, 29.23, 29.03, 25.56, 16.30 ppm. 31P NMR (283 MHz, CDCl3, 298 K): δ = −0.71 ppm.
5-(Benzyloxy)pentyl-4-methylbenzenesulfonate (2a). 2a was synthesized following a literature procedure:29 5-Benzyloxypentanol (3.85 mL, 20 mmol) was dissolved in 40 mL anhydrous CH2Cl2 (0.5 M) at room temperature and triethylamine (4.18 mL, 30 mmol), tosyl chloride (4.19 g, 22 mmol) and 4-dimethylamino pyridine (122 mg, 1 mmol) were added successively. The reaction was stirred at room temperature for 16 h, after which it was diluted with CH2Cl2 (100 mL), washed with NaHCO3(aq.), water and brine. The organic layer was dried over MgSO4, filtered and concentrated at reduced pressure. Purification by silica flash column chromatography (eluent: 20% ethyl acetate in petroleum ether 40/60) afforded 5.82 g (84% yield) of the title compound as a pale-yellow solid. NMR data matched that recorded in the literature:301H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 8.0 Hz, 2H), 7.37–7.27 (m, 7H), 4.50–4.45 (m, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.42 (t, J = 6.5 Hz, 2H), 2.41 (s, 3H), 1.72–1.49 (m, 5H), 1.46–1.35 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 144.6, 138.4, 133.0, 129.7, 128.2, 127.7, 127.5, 127.4, 72.7, 70.4, 69.8, 28.9, 28.5, 22.0, 21.5.
Hexadec-15-enyloxymethyl-benzene (2b). 2b was synthesized following a literature procedure:29 The Grignard reagent 1-undecene-11-methylmagnesium bromide was synthesized by refluxing 11-bromo-1-undecene (4.7 mL, 21 mmol) and one bead of iodine over Mg turnings (613 mg, 25.2 mmol) in anhydrous THF (32 mL) for 2 h, after which the reaction was allowed to cool to room temperature. The Grignard solution was then cooled to −78 °C and 5-(benzyloxy)pentyl-4-methylbenzenesulfonate 1 (2.3 g, 6.6 mmol) in anhydrous THF (6 mL) was added dropwise, followed by Li2CuCl4 (0.1 M in THF, 1.9 mL, 0.19 mmol). The reaction was warmed to room temperature and stirred overnight, after which it was quenched with NH4Cl(aq.) and extracted with ethyl acetate. The combined organic fractions were washed with water, NaHCO3(aq.) and brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (eluent: petroleum ether 40/60 to 20% ethyl acetate in petroleum ether 40/60) afforded 2.2 g (95% yield) of the title compound as a yellow oil. When the reaction was scaled up to 20 mmol, 5.61 g (85% yield) of the title compound were isolated, in addition to 0.79 g (15% yield) of compound 2c; 1H NMR (300 MHz, CDCl3) δ 7. 41–7.31 (m, 5H), 5.89 (ddt, J = 17.0, 10.0, 6.5 Hz, 1H), 5.16–4.93 (m, 2H), 4.57 (s, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.15–2.09 (m, 2H), 1.74–1.65 (m, 2H), 1.37 (m, 22H); 13C NMR (75 MHz, CDCl3) δ 139.2, 138.8, 128.4, 127.6, 127.5, 114.2, 72.9, 70.6, 33.9, 29.9, 29.8, 29.7, 29.6, 29.6, 29.3, 29.1, 26.3. APCI MS: m/z = 643.3 [2M + Na]+.
Hexadec-15-en-1-ol (2c). 2c was synthesized following a modified literature procedure:29 Hexadec-15-enyloxymethyl-benzene 2b (3.0 g, 9.1 mmol) was dissolved in CH2Cl2 (45 mL) and cooled to −78 °C. BCl3 (1 M in DCM, 20 mL, 20 mmol) was added dropwise, the reaction was brought to room temperature and stirred for 30 min. (Caution: The addition of BCl3 was straightforward on a small scale, however in this larger scale reaction large amounts of HCl gas were released.) The mixture was then cooled to 0 °C and quenched very carefully with H2O. The crude reaction mixture was extracted with CH2Cl2, and the combined organics were washed with H2O and brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (eluent: 10% acetone in petroleum ether 30/40) afforded 2.1 g (97% yield) of the title compound as a yellow solid. NMR data matched that recorded in the literature:311H NMR (250 MHz, CDCl3) δ 5.81 (ddt, J = 17.0, 10.0, 6.5 Hz, 1H), 5.14–4.85 (m, 2H), 3.64 (t, J = 6.5 Hz, 2H), 2.08–2.00 (m, 2H), 1.63–1.49 (m, 2H), 1.41–0.89 (m, 22H); 13C NMR (75 MHz, CDCl3) δ 139.3, 114.2, 62.9, 33.9, 32.8, 29.7, 29.7, 29.6, 29.5, 29.2, 29.0, 25.8.
Ethyl di(hexadec-15-en-1-yl) phosphate (2). 2 was synthesized following a literature procedure:32 Ethyl dichlorophosphate (0.22 mL, 1.8 mmol) was dissolved in CH2Cl2 (12 mL) and cooled to 0 °C. 2c (961 mg, 4 mmol) and pyridine (0.32 mL, 4 mmol) were added successively and the reaction was stirred overnight at room temperature. The crude mixture was diluted with Et2O and washed with 10% HCl. The organic fraction was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was filtered over neutral alumina (eluting with large amounts of CH2Cl2) to afford 550 mg (53% yield) of the title compound as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 5.81 (ddt, J = 17.0, 10.0, 6.5 Hz, 2H), 4.95 (dd, J = 19.0, 13.5 Hz, 4H), 4.16–3.99 (m, 6H), 2.07–2.00 (m, 4H), 1.77–1.62 (m, 4H), 1.40–1.21 (m, 47H); 13C NMR (75 MHz, CDCl3) δ 139.0, 114.1, 67.6 (d, J = 6.0 Hz), 63.5 (d, J = 6.0 Hz), 33.7, 30.2 (d, J = 7.0 Hz), 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.1, 29.1, 29.0, 28.9, 25.4, 16.1 (d, J = 6.5 Hz); 31P NMR (121 MHz, CDCl3) δ −0.83. APCI MS: m/z = 571.1 [M + H]+.
2-Octadecyn-1-ol (3a). 3a was synthesized following a modified literature procedure:33 To a solution of 3-tetrahydropyranyloxy-1-propyne (2.81 mL, 20 mmol) in anhydrous THF (20 mL, 1.0 M) at 0 °C was added nBuLi (1.6 M in Hexanes, 14.4 mL, 24 mmol) dropwise. A solution of 1-bromooactadecane (7.67 g, 23 mmol) in anhydrous DMPU/Hexanes (40 mL/5 mL) was added at 0 °C. The reaction was allowed to warm to room temperature and stirred for 1.5 h, after which it was quenched with NH4Cl(aq.) and extracted with petroleum ether 30/40. The combined organic fractions were washed with H2O, dried over MgSO4, filtered and concentrated in vacuo. The crude reaction mixture was re-dissolved in methanol (50 mL), conc. HCl (1.0 mL) was added and the reaction was stirred at room temperature overnight. The reaction was poured into ice-cold water and extracted with diethyl ether. The organics were dried over MgSO4, filtered and concentrated in vacuo. Recrystallization from hot CH2Cl2 afforded 5.92 g (83% yield) of the title compound as a white solid. NMR data matched that recorded in the literature:331H NMR (250 MHz, CDCl3) δ 4.24 (t, J = 2.5 Hz, 2H), 2.29–2.13 (m, 2H), 1.54–1.45 (m, 3H), 1.40–1.22 (m, 29H), 0.88 (t, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 86.9, 78.4, 51.6, 32.1, 29.7–29.9 (m), 29.7, 29.5, 29.3, 29.0, 28.8, 22.8, 18.9, 14.3.
20-Henicosyn-1-ol (3b). 3b was synthesized following a slightly modified literature procedure:34 To freshly distilled ethylene diamine (24 mL) at 0 °C was added NaH (60% in mineral oil, 2.4 g, 60 mmol) and the mixture was stirred at room temperature for 1 h. The reaction was slowly warmed to 60 °C and stirred for 2 h. The deep blue mixture was cooled to 45 °C and 2-octadecyn-1-ol 3a (3.7 g, 12 mmol) was slowly added. After addition, the reaction was heated to 70 °C and stirred overnight. The mixture was then cooled to 0 °C, diluted with water and neutralized with conc. HCl. The crude product was extracted into CH2Cl2 and the combined organic layers were washed with 1 M HCl and brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (gradient: petroleum ether 40/60 to 20% acetone in petroleum ether 40/60) followed by recrystallization afforded 3.15 g (85% yield) of the title compound as a white, fluffy solid. 1H NMR (300 MHz, CDCl3) δ 3.64 (t, J = 6.5 Hz, 2H), 2.17 (td, J = 7.0, 2.5 Hz, 2H), 2.12 (br, 1H, OH), 1.93 (t, J = 2.5 Hz, 1H), 1.59–1.47 (m, 4H), 1.34–1.23 (m, 30H); 13C NMR (75 MHz, CDCl3) δ 85.0, 68.2, 63.2, 32.8, 29.8 (m), 29.8, 29.7, 29.6, 29.3, 28.9, 28.6, 25.9, 18.5.
20-Henicosen-1-ol (3c). To a solution of 3b (1.03 g, 3.3 mmol) in ethanol (80 mL) at 0 °C was added Lindlar's catalyst (6 mg, 1 mol%). The reaction was stirred under an atmosphere of H2 (balloon) for 3 h, after which the flask was purged with N2 and the reaction was filtered over Celite. The solvent was removed in vacuo and the crude product was recrystallized from hot CH2Cl2 to afford 1.02 g (98% yield) of the title compound as a white solid. 1H NMR (300 MHz, CDCl3) δ 5.81 (ddt, J = 17.0, 10.0, 6.5 Hz, 1H), 5.07–4.85 (m, 2H), 3.64 (t, J = 6.5 Hz, 2H), 2.07–2.00 (m, 2H), 1.85 (br, 1H, OH), 1.59–1.52 (m, 2H), 1.40–1.25 (m, 30H), 0.90–0.82 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 139.4, 114.2, 63.2, 34.0, 32.9, 29.8 (m), 29.8, 29.7, 29.6, 29.3, 29.1, 25.9. APCI MS: m/z = 313.1 [M-H2O + H]+.
Ethyl di(henicos-20-en-1-yl) phosphate (3). 3 was synthesized following a literature procedure:32 Ethyl dichlorophosphate (0.11 mL, 0.94 mmol) was dissolved in CH2Cl2 (10 mL) and cooled to 0 °C. 3c (610 mg, 1.96 mmol) and pyridine (0.16 mL, 2.96 mmol) were added successively and the reaction was stirred overnight at room temperature. The crude mixture was diluted with diethyl ether and washed with 10% HCl. The organic fraction was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was filtered over neutral alumina (eluting with large amounts of CH2Cl2) to afford 260 mg (39% yield) of the title compound as a white solid. 1H NMR (300 MHz, CDCl3) δ 5.77 (tt, J = 16.5, 7.0 Hz, 2H), 4.97–4.86 (m, 4H), 4.09–3.96 (m, 6H), 2.01–1.96 (m, 4H), 1.66–1.61 (m, 4H), 1.42–1.07 (m, 60H), 0.86–0.82 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 139.2, 114.1, 67.7 (d, J = 6.0 Hz), 63.6 (d, J = 6.0 Hz), 33.9, 30.4 (d, J = 7.0 Hz), 29.8–29.7 (m), 29.6, 29.2, 29.0, 25.5, 16.2 (d, J = 6.5 Hz); 31P NMR (121 MHz, CDCl3) δ = –0.74. APCI MS: m/z = 711.3 [M + H]+.
Polymer synthesis
1 H NMR (300 MHz, CDCl3) δ = 5.37 (m, J = 6.7, 5.3 Hz, 2H), 4.06 (m, 6H), 1.98 (m, 4H), 1.67 (m, 4H), 1.53–1.03 (m, 47H). 13C NMR (75 MHz, CDCl3) δ = 130.33, 118.16, 67.68, 63.60, 32.62, 30.31, 29.96, 28.84, 25.46, 16.17. 31P NMR (121 MHz, CDCl3): δ = −0.70 ppm.
1 H NMR (300 MHz, CDCl3) δ 5.37 (m, 2H), 4.07 (m, 6H), 2.14–1.85 (m, 4H), 1.67 (m, 4H), 1.52–1.01 (m, 59H), 0.85 (m, 8H). 31P NMR (121 MHz, CDCl3): δ = −0.70 ppm.
Results and discussion
Monomer synthesis
To vary the distance between the phosphate groups in PE-like materials, three α,ω-diene monomers were synthesized by esterification of ethyl dichlorophosphate with linear unsaturated alcohols containing a terminal double bond and a different number of methylene groups (Scheme 1). The chain length of the alcohol determines the spacer length between two phosphate groups in the polymer. For example, the polymerization of monomer 1 with 22 carbons gives the “C20 polymer”. In this way, polymers with a precise spacing of 20, 30, and 40 CH2-groups between each phosphate group were prepared. The synthesis of monomer 1 with the shortest alkyl chain was previously reported by our group,37 using commercially available 10-undecen-1-ol. The unsaturated alcohols with 16 or 21 methylene groups were synthesized according to Scheme 1. 5-Benzyloxypentanol was protected with tosyl chloride (2a). Subsequent Li2CuCl4-catalysed cross-coupling of tosylate 2a with undec-10-enylmagnesium bromide elongated the carbon backbone by nine methylene units (2b). Selective removal of the benzyl ether with BCl3 gave unsaturated alcohol 2c bearing 16 carbon atoms. For the synthesis of 3c with 21 carbon atoms, the nucleophilic substitution of 1-bromooctadecane (stearyl bromide) with 3-tetrahydropyranyloxy-1-propyne was performed followed by acidic hydrolysis of the THP protecting group to give internal alkyne 3a. In the next step, an alkyne zipper reaction with NaH and ethylene diamine catalyzed isomerization to the terminal alkyne. Selective reduction from the alkyne to the alkene with Lindlar's catalyst yielded the unsaturated alcohol 3c with the fully saturated alcohol as a side product (ca. 30%), which could not be removed by column chromatography or recrystallization; in the following, the mixture was used for further syntheses.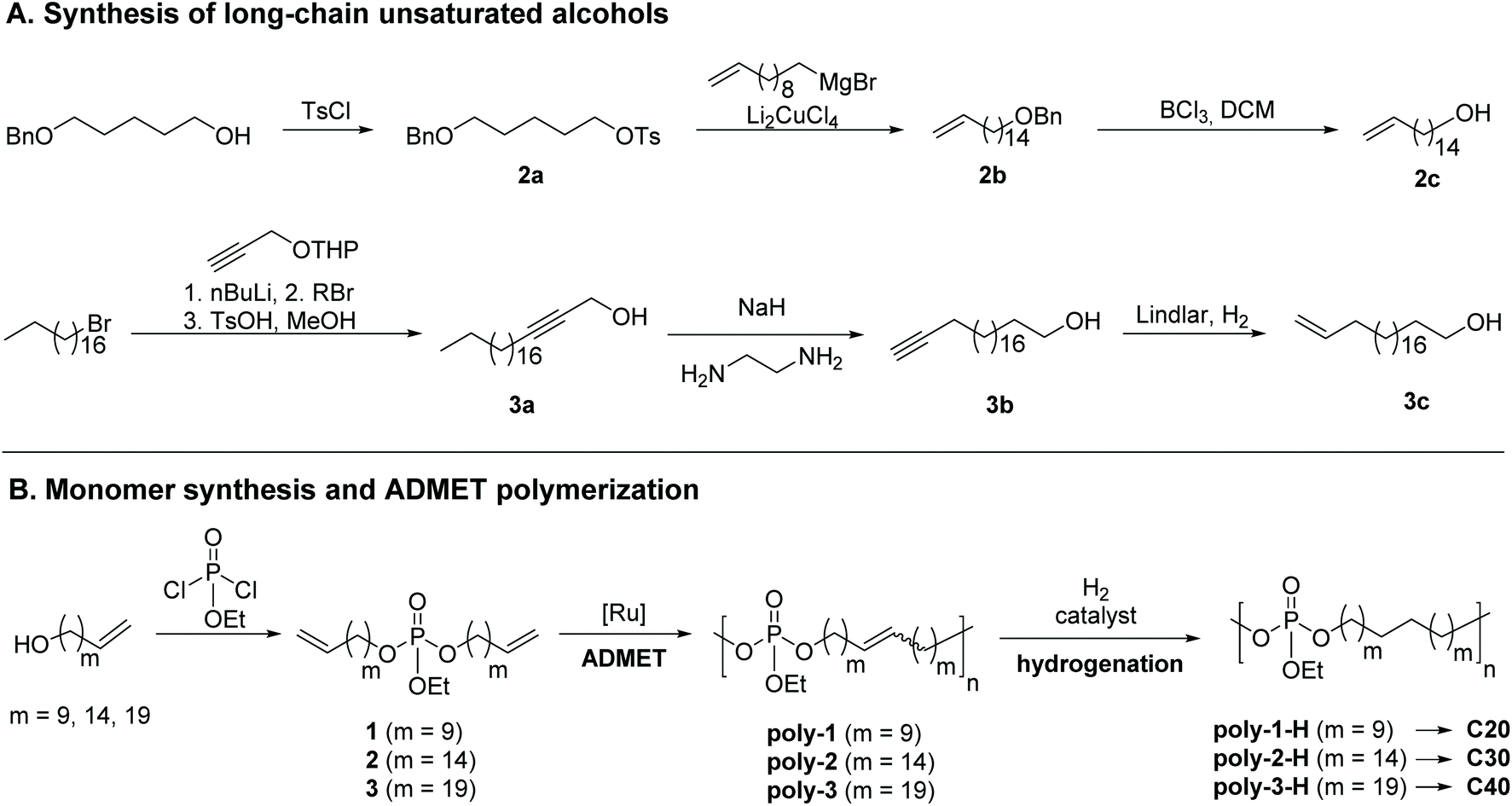 | ||
| Scheme 1 Synthesis of PE-like polyphosphoesters with different lengths of the aliphatic spacers between the phosphate groups. (A) Synthesis of long-chain alcohols for C30 and C40 polymers. (B) Synthesis of phosphate diene monomers and their ADMET polymerization and hydrogenation. | ||
The esterification reactions of ethyl dichlorophosphate with the respective unsaturated alcohols were performed in the presence of triethylamine as an HCl scavenger to give monomers 1–3. While monomer 1 was obtained as an oil with low viscosity, 2 appeared honey-like and 3 as a solid wax. All monomers were analyzed by NMR spectroscopy (the 1H, 13C, and 31P NMR spectra including the assignment of all peaks can be found in the ESI, Fig. S13–S21†).
ADMET polymerization and hydrogenation
ADMET polymerization was carried out with 1st generation Grubbs catalyst. In order to obtain high precision polymers with defined spacing between two defect groups, the 1st generation Grubbs catalyst is beneficial to the more reactive 2nd generation Grubbs catalyst and the Hoveyda–Grubbs catalysts, as it disfavors olefin isomerization.38 Still, olefin isomerization can occur at elevated temperatures while using 1st generation Grubbs catalyst,39 so care was taken to never exceed 85 °C during polymerization. Monomer 1 was polymerized in bulk at 65 to 85 °C for 48 h at reduced pressure to remove evolving ethylene. In contrast, ADMET polymerization of monomers 2 and 3 was carried out for 48 h in solution with 1-chloronaphthalene as a high-boiling solvent to enable agitation during the polymerization. The amount of solvent was kept low (concentration of polymer ca. 750 mg mL−1) to prevent cyclization.40 The obtained honey-like C20 polymer, poly(1), revealed an apparent molecular weight Mw of ca. 23100 g mol−1 (by SEC, Mw/Mn = 2.5). Both polymers poly(2) and poly(3) had a waxy appearance and apparent Mws of ca. 15400 and 12100 g mol−1, respectively (Table 1). In the 1H NMR spectra, the resonances of the terminal olefins at 5.8 and 4.9 ppm vanished (cf. Fig. S22, S23 and S26†) and new signals at 5.4 ppm were detected, which were assigned to the internal double bonds of the polymer. The resonances in the 31P NMR spectra remained unchanged at 0.7 ppm (Fig. S25 and S28†).
| Polymer | No. of CH2 groups | M n /g mol−1 | M w /g mol−1 | M w/Mna | T g /°C | T m /°C | ΔHb/J g−1 | Crystallinityc/% |
|---|---|---|---|---|---|---|---|---|
| a Determined by SEC in THF. b Determined by DSC. c Relative to 100% crystalline PE (ΔHm = −293 J g−1). n.d. not determined. d Due to insolubility in the SEC eluent THF. | ||||||||
| poly(1) | 20 | 9300 | 23100 |
2.5 | −61 | 14 | −35 | n.d. |
| poly(2) | 30 | 6000 | 15400 |
2.6 | n.d. | n.d. | n.d. | n.d. |
| poly(3) | 40 | 4500 | 12100 |
2.7 | n.d. | n.d. | n.d. | n.d. |
| poly(1)-H | 20 | 9900 | 23100 |
2.3 | −47 | 51 | −71 | 24 |
| poly(2)-H | 30 | 5900 | 15200 |
2.6 | −39 | 62 | −105 | 36 |
| poly(3)-H | 40 | n.d.d | n.d.d | n.d.d | −38 | 91 | −119 | 41 |
To obtain PE-like materials, we performed hydrogenation of the polymers with either Pd/C or the Fischer carbene derivative of Grubbs catalyst 1st generation.36 The disappearance of the double bond signal at 5.4 ppm in the 1H NMR spectra after the reaction confirmed the complete hydrogenation of the polymers (Fig. S29, S32 and S35†). For poly(3)-H, a signal at 0.9 ppm in the range of –CH3 groups is detectable, most likely indicating the presence of ethyl dihenicosyl phosphate, a side product during monomer synthesis. Molecular weights of poly(1)-H and poly(2)-H were determined by SEC in THF vs. polystyrene standards and are following the values of the respective unsaturated polymers (cf. Fig. S37 and S38). SEC measurements of poly(3)-H in THF were not possible due to the insolubility of the hydrogenated polymer in the solvent for SEC.
Solid-state characterization
In contrast to the oily or waxy unsaturated polymers, all hydrogenated polymers were solid at room temperature. All polymers showed a brittle deformation behavior and with an increasing length of the aliphatic spacer, the polymeric materials became harder. By differential scanning calorimetry (DSC), the melting points and the crystallinity of polymers poly(1)-H to poly(3)-H were determined. Theoretically, the lamellae thickness of the PE-like crystallite is expected to increase with an increase in the length of the aliphatic chain (equals a decrease in the number of crystallization defects) resulting in higher melting points according to the Gibbs–Thompson equation. As expected, the melting points increased from 51 °C for poly(1)-H to 62 °C for poly(2)-H up to 91 °C (poly(3)-H) (Fig. 1). At the same time, the melting enthalpies ΔHm increased from −71 to −105 and −119 J g−1 (Table 1). By comparing ΔHm to ΔH of theoretical 100% crystalline polyethylene (ΔHm = 293 J g−1), the crystallinity of the synthesized polymers was estimated.41 Values for the semi-crystalline polyphosphates ranged from 24% to 41% (Table 1). Glass transition temperatures (Tg) were below room temperature, ranging from −47 °C to −38 °C. In the DSC thermogram of poly(3)-H, an additional melting process at 80.5 °C was visible, which overlapped with the main melting peak at 91 °C. The pre-melting peak might be explained either by the presence of polymorphism or by co-crystallization of long-chain impurities that could not be entirely removed during monomer synthesis and polymer work-up. Additionally, melting and recrystallization cannot be excluded as a reason for the additional melting peak. In general, the melting endotherms broadened from the C20 to the C40 polymer, indicating a larger distribution in crystallite sizes for poly(3)-H and poly(2)-H compared to poly(1)-H. This may be explained by an increasing molar mass distribution from C20 to C40 as well as a decreasing weight average molecular weight (Mw).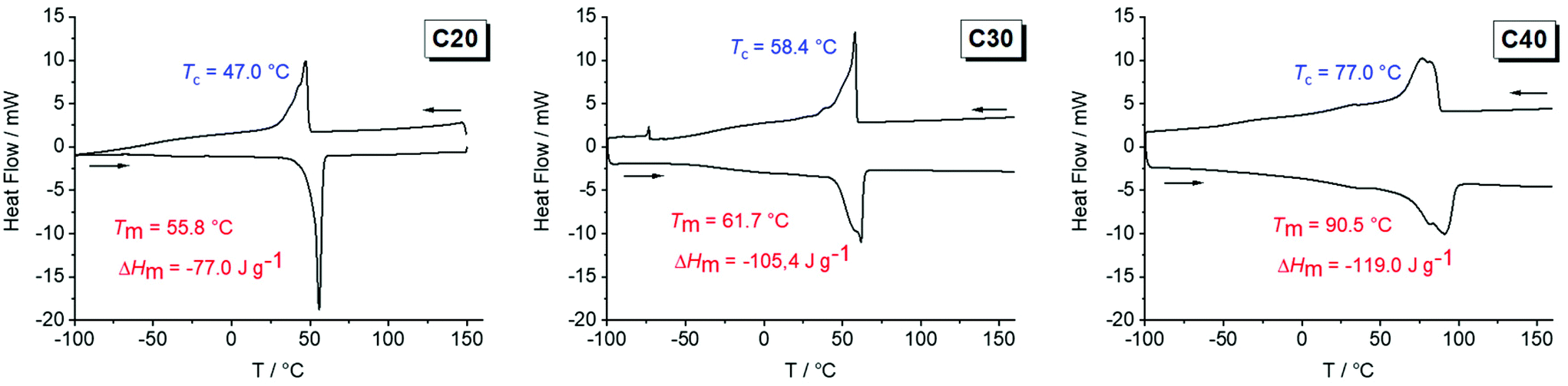 | ||
| Fig. 1 DSC thermograms of poly(1)-H (left), poly(2)-H (middle) and poly(3)-H (right) (exo up, heating and cooling rate 10 K min−1 (second run)). | ||
The crystal morphologies of the polyphosphates were investigated by wide and small-angle X-ray scattering, atomic force microscopy, and transmission electron microscopy. Scheme 2 summarizes the information which is provided by each method. As all hydrogenated polyphosphates were party crystalline, the bulk material consists of crystalline and an amorphous regions. Both, solution-grown and melt-grown crystals were studied. By SAXS and TEM the thickness of the long period, including both regions, can be determined from the melt-crystallized polymers. The thickness of the lamellar crystal can be obtained by AFM (solution-grown crystals) and TEM (melt-grown crystals), while the crystal structure within the lamellae is measured by WAXS (melt-grown crystals), yielding the lattice constants.
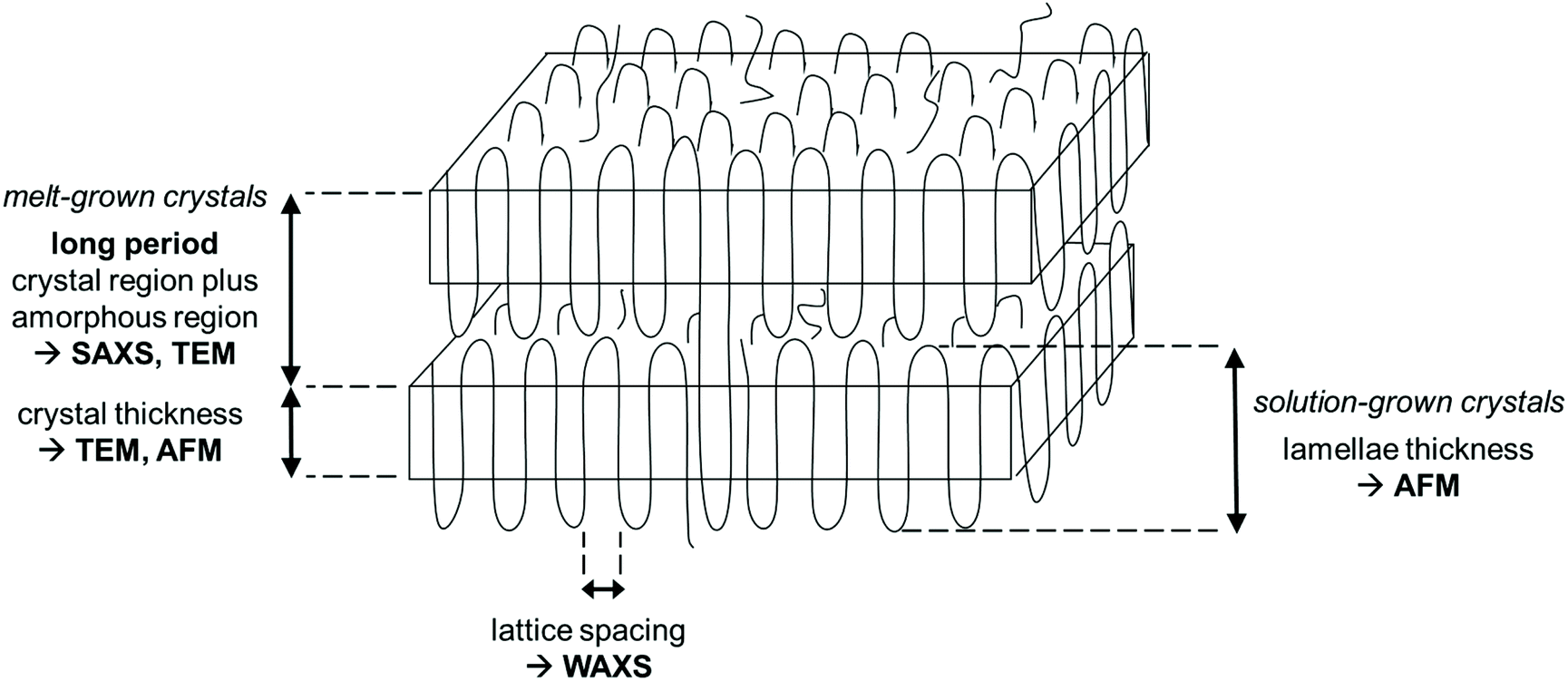 | ||
| Scheme 2 General schematic representation of polymer crystallization and information about the morphologies of semi-crystalline polymers in the bulk or of solution-grown crystals obtained by SAXS, WAXS, AFM, and TEM. | ||
To investigate the crystal structure of the different polymers, X-ray diffraction (XRD) measurements were performed. The WAXS patterns indicated a change in the crystal structure, as the length of the aliphatic spacer was increased (Fig. 2). The XRD diffractogram of poly(1)-H revealed a single peak at 21.5°, confirming a pseudo-hexagonal crystal structure.42 In contrast, the crystal structure of poly(3)-H was found to be orthorhombic with two distinct reflections at 21.6° and 23.8°, similar to linear polyethylene.43 For poly(2)-H, two overlapping reflections at 21.2° and 23.1° indicate a transition from the pseudo-hexagonal and the orthorhombic crystal structure. Comparing the different polymers, the intensity of the amorphous halo increases with the number of defects in the polyethylene chain from poly(3)-H to poly(1)-H, which is in agreement with the literature.5
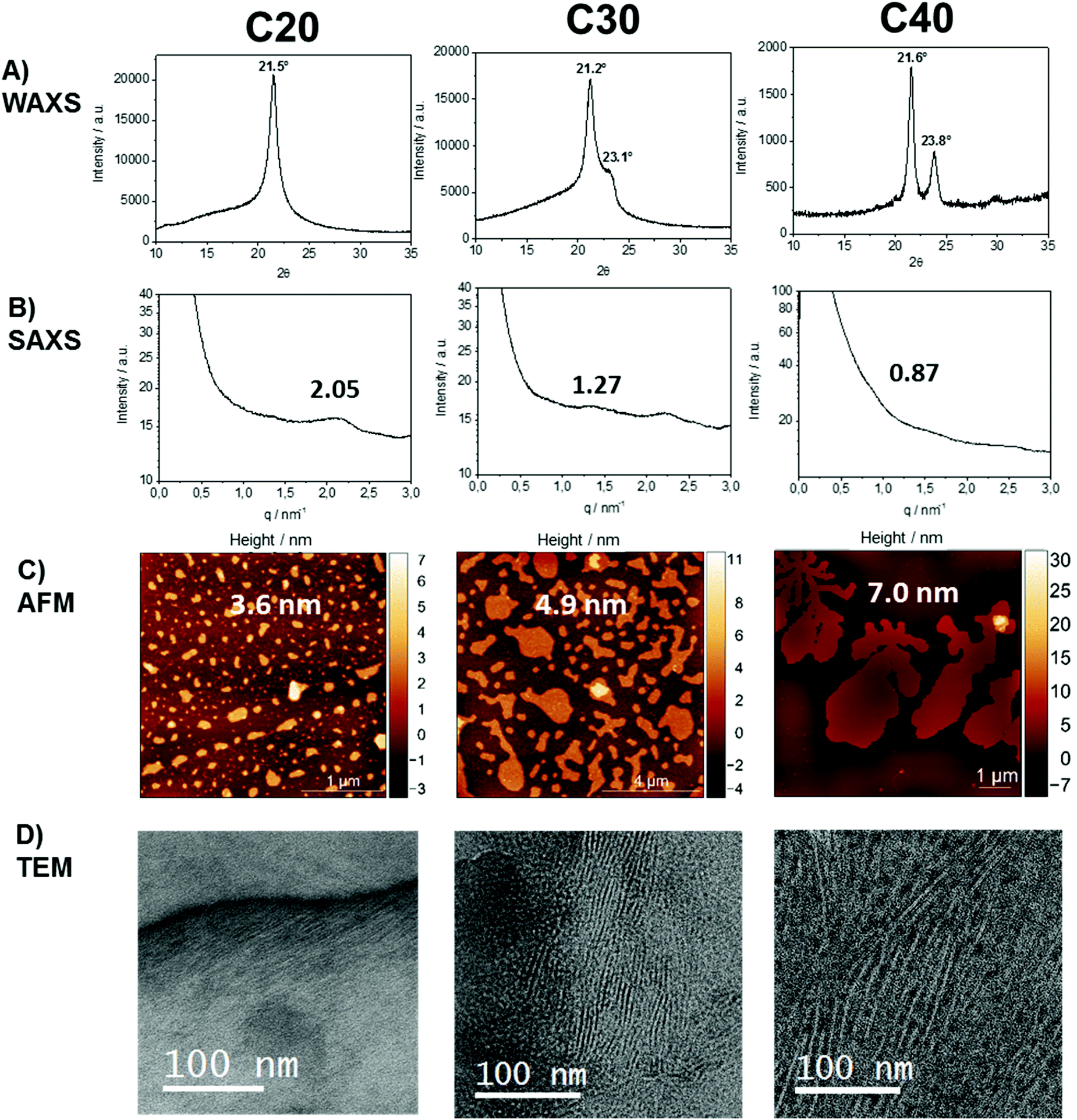 | ||
| Fig. 2 Solid-state characterization of polyethylene-like PPEs. (A) Wide and (B) small X-ray diffractograms; (C) atomic force microscopy images; (D) and corresponding lamellar morphologies visualized by TEM of poly(7)-H (left), poly(8)-H (middle) and poly(9)-H (right). The micrographs display a cross-section perpendicular to the lamellae, so the crystal- and amorphous thickness of the crystals is visualized. WAXS, SAXS, and TEM data were obtained from bulk polymer samples, AFM measurement from solution-grown crystals. | ||
SAXS measurements show peaks at scattering vectors of 2.05, 1.27, and 0.87 nm−1 for the polymers with 20, 30, and 40 CH2, respectively (Fig. 2B). Values for the long period D = 2pi/q0 were estimated from the scattering vector q0 at the peak maximum. For the polymers poly(1)-H with 20 CH2 groups and poly(3)-H with 40 CH2-groups, the long period was 3.1 nm and 7.2 nm, respectively. In combination with the crystallinity extracted from the DSC data, the crystal thickness can be calculated from the long period obtained from SAXS (Table 2). Additionally, the topography of solution-grown crystals was measured by AFM. From these measurements, the lamellar thickness was extracted, yielding thicknesses of 3.6 nm for poly(1)-H (20 CH2), 4.9 nm for poly(2)-H, and 7.0 nm for poly(3)-H. Remarkably, these thicknesses of solution-growth polymers correlate well with the long period of bulk polymers obtained by SAXS (Fig. 3).
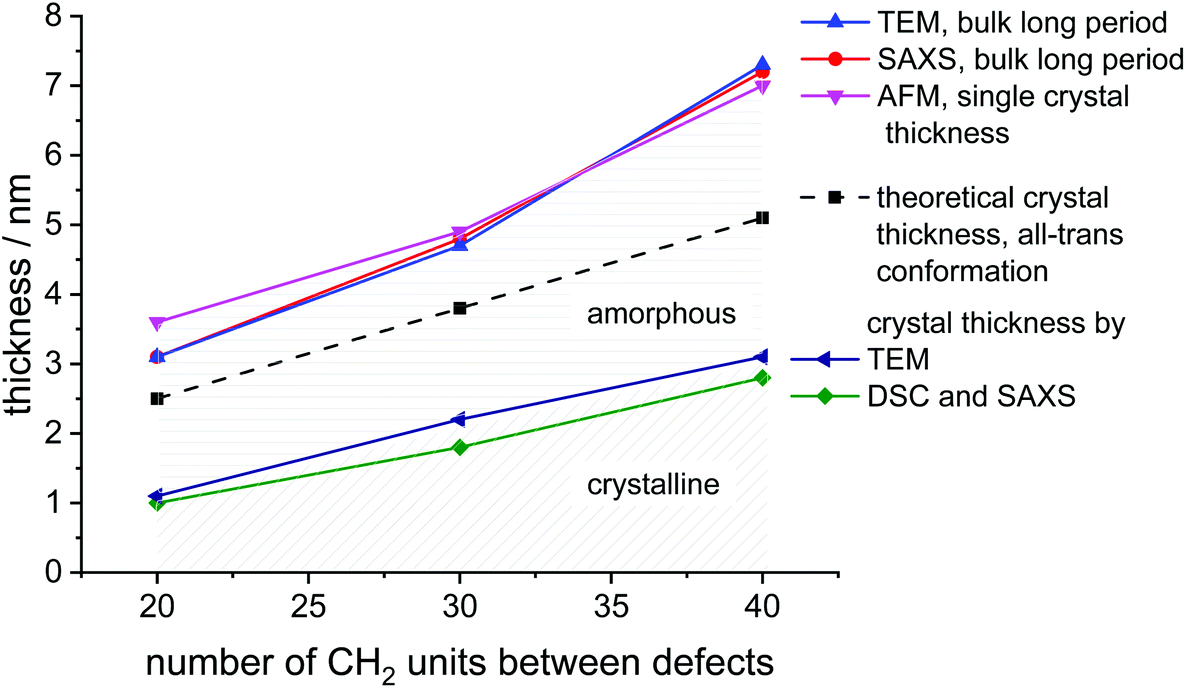 | ||
| Fig. 3 Graphical representation of crystal parameters for PE-like PPEs determined by different methods (data listed in Table 2). | ||
| Method | C20 | C30 | C40 |
|---|---|---|---|
| Theoretical crystal thickness | 2.5 | 3.8 | 5.1 |
| All-trans formation, nm | |||
| SAXS | 3.1 | 4.8 | 7.2 |
| Bulk long period, nm | |||
| TEM | 3.1 | 4.7 | 7.3 |
| Bulk long period, nm | |||
| AFM | 3.6 | 4.9 | 7.0 |
| Single crystals thickness, nm | |||
| Thickness of crystalline part from DSC and SAXS, nm | 1.0 | 1.8 | 2.8 |
| Thickness of crystalline part from TEM, nm | 1.1 | 2.2 | 3.1 |
In order to visualize the lamellar structure of the different polymers, additional TEM examinations have been performed (Fig. 2D). Here, the bulk crystallized polymer was sectioned to achieve a cross-section perpendicular to the crystals. Due to the RuO4 staining, the amorphous regions show a darker contrast compared to the crystal region. The micrographs allow determining the crystal thickness as well as the long period. All values obtained by WAXS, SAXS, AFM, and TEM measurements (Fig. 2) are listed in Table 2. Theoretically, a fully crystalline polyethylene segment of 20 CH2 groups in an all-trans conformation would have a length of 2.5 nm. Accordingly, 30 CH2 groups stretch to 3.8 nm and 40 CH2 groups to 5.1 nm. The theoretical crystal thickness is displayed in Fig. 3 (dashed line). TEM measurements of stained sections of annealed polymer samples provided the crystal thickness for the bulk crystallization with 1.1, 2.2, and 3.1 nm for polymers with 20, 30, and 40 CH2-groups, respectively. As these values are lower than the theoretical numbers, it is likely that despite some incorporated defects, also a considerable part of the CH2-groups was necessary for the formation of the reentry of the chain to the crystal. This correlates well as the crystallinity of the PPEs was calculated to be between 26 and 41% (assuming ΔHm for PE) and increase with increasing spacer length.
The combination of complementary methods, including WAXS, SAXS, AFM, and TEM helps to understand the crystallization of all three polymers and to elucidate the differences between their crystal structures and morphologies. Theoretically, an addition of every 20 CH2 groups in aliphatic segment in the ideal case would lead to an increase of the lamellae thickness by 2.5 nm. However, the obtained difference was determined as 4.1 nm instead of 2.5 nm and cannot be explained by the difference in the length of polymer chain segments alone. The combination of the density of crystal packing and the amorphous/loop region might be a reason for the inconsistent values. Also, the data indicates, that there is not necessarily a perfect arrangement with an adjacent reentry model, but instead a part of polymer chain segments might be expelled to the amorphous phase during the bulk crystallization, yielding a considerable amount of random reentry.44 By extrapolation of the crystal thickness data to shorter aliphatic length, we can theoretically evaluate the minimal number of CH2-units that are necessary to form crystalline parts in the polymer. From Fig. S48† we obtain a spacer length of 9 CH2-units. For polymers with less CH2 units between the defects, the length of the aliphatic part would not be enough for both loop formation and PE crystallization.
There are different models of polymer chain arrangement depending on the polymer structure and crystallization conditions. In bulk, polymer chains are rather folding according to a random reentry or the so-called “switchboard” model.45,46 This model was first proposed by Flory and recently confirmed for a number of semi-crystalline polymers,47 consists of chains randomly folding back into the same lamella or participating in adjoining lamellae. However, for solution-grown single monolayer polymer crystals, the most preferable chain-folding model is the adjacent reentry. This model is characterized by a sharp phase boundary between the crystal and the amorphous phase.48 The position of reentry of the chains is the adjacent neighbor with only a few exceptions due to multiple nucleations and chain-end defects. Fig. 3 summarizes the measured thickness ratios of the crystal lamellae graphically. The theoretical thickness of an all-trans configuration is indicated as the dashed line. It is noticeable that the measured lamellar thickness is greater than the theoretically achievable value. In determining the theoretical thickness, however, we have disregarded the phosphate groups, which could at least be the first attempt at an explanation. However, this does not explain the increase in thickness difference found in the C40 polymer. This observation applies both to bulk and solution-grown crystals. Hence, the increased thickness of the long period of the crystals is a strong indication, that even the solution grown crystals still contain some part of random reentry folds. As has been shown by NMR44,49–51 and AFM52 solution grown crystals can have up to 10%, whereas melt grown crystals exhibit up to 35% of random reentry folds. In the case of the PPEs synthesized in this work, we can therefore assume that a certain amount of random reentry folding is also present in the solution-grown crystals. Hence, several segments are expelled from the crystal phase and contribute to a thickening of the amorphous phase. This explains, why even solution-grown crystals exhibit lamellar thicknesses larger than expected for the respective defect distance. In general, the obtained values for lamellar thickness from AFM and SAXS techniques show only minor differences, although the crystals were prepared following different procedures in bulk and solution. Thus, we can claim that the phosphate defects confine the lamellae thickness, regardless of the way of crystallization.
Summary
We report on a “defect engineering” approach using PE-like polyphosphates with a varying number of phosphate defects in the polymer backbone to control the structure and lamellar thickness of polymer crystals. Three different α,ω-diene monomers with identical phosphate groups but different aliphatic spacer lengths were synthesized for acyclic diene metathesis polymerization. Linear polyphosphates with 20, 30, or 40 CH2-groups between each phosphate group were prepared. The polymers were crystallized both in bulk and from solution. With the phosphate side chain being identical for all three polymers, differences in the crystal structure and morphology and thermal properties relied only on the length of the aliphatic spacers. Melting temperatures increased with the increasing length of the aliphatic spacer segment up to 91 °C for the polymer with 40 methylene groups. A change from a pseudo-hexagonal to an orthorhombic crystal structure was observed by WAXS with a decrease of phosphate defects in the polymer chains, i.e. increasing similarity to polyethylene. A combination of WAXS, SAXS, AFM, and TEM revealed an increase in lamellar and crystal thickness with the increasing length of the aliphatic spacer. Following this approach, different functionalities could be added to the polymers by varying the side chain of the phosphate group in the future. The synthesized PE-like polyphosphates with precisely engineered lamellar crystals thicknesses show potential for applications where distinct spacing on a nanometer scale is advantageous, e.g. for electronics, or as highly functional and anisotropic polymer colloids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. H., O. S., I. L., and F. R. W. thank the German Federal Ministry for Education and Research (BMBF) for their support of the program “Research for sustainable development (FONA)”, “PlastX – Plastics as a systemic risk for social-ecological supply systems” (grant number: 01UU1603A). M.O'D. thanks the Royal Society for a Chemistry Researcher Mobility Grant. We thank Michael Steiert (MPIP) for XRD measurements, Christine Rosenauer (MPIP) for SEC measurements, and Dr Rüdiger Berger (MPIP) for AFM measurements. We thank Dr Hisaschi Tee (MPIP) for synthetic assistance. Open Access funding provided by the Max Planck Society.References
- Plastics – the Facts 2018, https://www.plasticseurope.org/application/files/6315/4510/9658/Plastics_the_facts_2018_AF_web.pdf, (accessed 04.10.2019).
- C. Vasile, Handbook of Polyolefins, CRC Press, Boca Raton, 2000 Search PubMed.
- M. D. Lechner, K. Gehrke and E. H. Nordmeier, Makromolekulare Chemie, Springer Spektrum, Berlin, Heidelberg, 2014 Search PubMed.
- R. G. Alamo and L. Mandelkern, Macromolecules, 1989, 22, 1273–1277 CrossRef CAS.
- S.-D. Clas, R. D. Heyding, D. C. McFaddin, K. E. Russell, M. V. Scammell-Bullock, E. C. Kelusky and D. St-Cyr, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 1271–1286 CrossRef CAS.
- B. Inci, I. Lieberwirth, W. Steffen, M. Mezger, R. Graf, K. Landfester and K. B. Wagener, Macromolecules, 2012, 45, 3367–3376 CrossRef CAS.
- G. Rojas, B. Inci, Y. Wei and K. B. Wagener, J. Am. Chem. Soc., 2009, 131, 17376–17386 CrossRef CAS PubMed.
- K. Matsui, S. Seno, Y. Nozue, Y. Shinohara, Y. Amemiya, E. B. Berda, G. Rojas and K. B. Wagener, Macromolecules, 2013, 46, 4438–4446 CrossRef CAS.
- J. C. Sworen, J. A. Smith, J. M. Berg and K. B. Wagener, J. Am. Chem. Soc., 2004, 126, 11238–11246 CrossRef CAS PubMed.
- G. Rojas, E. B. Berda and K. B. Wagener, Polymer, 2008, 49, 2985–2995 CrossRef CAS.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- S. Hosoda, Y. Nozue, Y. Kawashima, K. Suita, S. Seno, T. Nagamatsu, K. B. Wagener, B. Inci, F. Zuluaga, G. Rojas and J. K. Leonard, Macromolecules, 2011, 44, 313–319 CrossRef CAS.
- L. Yan, C. Rank, S. Mecking and K. I. Winey, J. Am. Chem. Soc., 2020, 142, 857–866 CrossRef CAS PubMed.
- L. Yan, M. Häußler, J. Bauer, S. Mecking and K. I. Winey, Macromolecules, 2019, 52, 4949–4956 CrossRef CAS.
- X. Zhang, X. Zuo, P. Ortmann, S. Mecking and R. G. Alamo, Macromolecules, 2019, 52, 4934–4948 CrossRef CAS.
- C. Samir, S. Florian and M. Stefan, Macromol. Rapid Commun., 2012, 33, 1126–1129 CrossRef PubMed.
- T. Haider, O. Shyshov, O. Suraeva, I. Lieberwirth, M. von Delius and F. R. Wurm, Macromolecules, 2019, 52, 2411–2420 CrossRef CAS PubMed.
- M. G. Menges, J. Penelle, C. Le, F. de Ten Hove, A. M. Jonas and K. Schmidt-Rohr, Macromolecules, 2007, 40, 8714–8725 CrossRef CAS.
- C. L. F. De Ten Hove, J. Penelle, D. A. Ivanov and A. M. Jonas, Nat. Mater., 2004, 3, 33–37 CrossRef PubMed.
- M. P. F. Pepels, M. R. Hansen, H. Goossens and R. Duchateau, Macromolecules, 2013, 46, 7668–7677 CrossRef CAS.
- P. Ortmann and S. Mecking, Macromolecules, 2013, 46, 7213–7218 CrossRef CAS.
- S.-F. Song, Y.-T. Guo, R.-Y. Wang, Z.-S. Fu, J.-T. Xu and Z.-Q. Fan, Macromolecules, 2016, 49, 6001–6011 CrossRef CAS.
- J. Friebel, C. P. Ender, M. Mezger, J. Michels, M. Wagner, K. B. Wagener and T. Weil, Macromolecules, 2019, 52, 4483–4491 CrossRef CAS PubMed.
- Y.-R. Zheng, H. T. Tee, Y. Wei, X.-L. Wu, M. Mezger, S. Yan, K. Landfester, K. Wagener, F. R. Wurm and I. Lieberwirth, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef CAS.
- S. Majumder, H. Busch, P. Poudel, S. Mecking and G. Reiter, Macromolecules, 2018, 51, 8738–8745 CrossRef CAS.
- H. Weiss, J. Mars, H. Li, G. Kircher, O. Ivanova, A. Feoktystov, O. Soltwedel, M. Bier and M. Mezger, J. Phys. Chem. B, 2017, 121, 620–629 CrossRef CAS PubMed.
- K. Y. Hostetler, J. R. Beadle, J. Ruiz, M. R. Almond, G. R. Painter, T. A. Riley and P. Francom, US Pat., US7994143B2, 2010 Search PubMed.
- E. M. van Oosten, A. A. Wilson, D. C. Mamo, B. G. Pollock, B. H. Mulsant, S. Houle and N. Vasdev, Can. J. Chem., 2010, 88, 1222–1232 CrossRef CAS.
- Y.-S. Hon, Y.-C. Wong, C.-P. Chang and C.-H. Hsieh, Tetrahedron, 2007, 63, 11325–11340 CrossRef CAS.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- W. Oppolzer, R. N. Radinov and E. El-Sayed, J. Org. Chem., 2001, 66, 4766–4770 CrossRef CAS PubMed.
- S. E. Denmark and S.-M. Yang, J. Am. Chem. Soc., 2002, 124, 2102–2103 CrossRef CAS PubMed.
- H. D. Maynard and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 4137–4140 CrossRef CAS.
- P. Ortmann, F. P. Wimmer and S. Mecking, ACS Macro Lett., 2015, 4, 704–707 CrossRef CAS.
- K. N. Bauer, H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2016, 49, 3761–3768 CrossRef CAS.
- S. E. Lehman, J. E. Schwendeman, P. M. O'Donnell and K. B. Wagener, Inorg. Chim. Acta, 2003, 345, 190–198 CrossRef CAS.
- P. A. Fokou and M. A. R. Meier, J. Am. Chem. Soc., 2009, 131, 1664–1665 CrossRef CAS PubMed.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- H. Busch, E. Schiebel, A. Sickinger and S. Mecking, Macromolecules, 2017, 50, 7901–7910 CrossRef CAS.
- A. Cankaya, M. Steinmann, Y. Bülbül, I. Lieberwirth and F. R. Wurm, Polym. Chem., 2016, 7, 5004–5010 RSC.
- C. W. Bunn, Trans. Faraday Soc., 1939, 35, 482–491 RSC.
- Y.-L. Hong, T. Koga and T. Miyoshi, Macromolecules, 2015, 48, 3282–3293 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1962, 84, 2857–2867 CrossRef CAS.
- P. J. Flory, in Structural Orders in Polymers, ed. F. Ciardelli and P. Gusti, Permagon Press, New York, 1981 Search PubMed.
- E. B. Trigg, M. J. Stevens and K. I. Winey, J. Am. Chem. Soc., 2017, 139, 3747–3755 CrossRef CAS PubMed.
- J. D. Hoffman and J. I. Lauritzen Jr., J. Res. Natl. Bur. Stand., Sect. A, 1961, 65, 297 CrossRef PubMed.
- Y.-L. Hong, W. Chen, S. Yuan, J. Kang and T. Miyoshi, ACS Macro Lett., 2016, 5, 355–358 CrossRef CAS.
- S. Wang, S. Yuan, W. Chen, Y. Zhou, Y.-L. Hong and T. Miyoshi, Macromolecules, 2018, 51, 8729–8737 CrossRef CAS.
- S. Wang, S. Yuan, K. Wang, W. Chen, K. Yamada, D. Barkley, T. Koga, Y.-L. Hong and T. Miyoshi, Macromolecules, 2019, 52, 4739–4748 CrossRef CAS.
- Z. Ma, P. Yang, X. Zhang, K. Jiang, Y. Song and W. Zhang, ACS Macro Lett., 2019, 8, 1194–1199 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Analytical and spectral characterization data. See DOI: 10.1039/d0py00272k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |