
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Turning natural δ-lactones to thermodynamically stable polymers with triggered recyclability†
Linnea
Cederholm
,
Peter
Olsén
,
Minna
Hakkarainen
 and
Karin
Odelius
*
and
Karin
Odelius
*
Wallenberg Wood Science Center, WWSC, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, 100 44 Stockholm, Sweden. E-mail: hoem@kth.se
First published on 14th April 2020
Abstract
To extend the use of naturally occurring substituted δ-lactones within the polymer field, their commonly low ceiling temperature and thereby challenging equilibrium behavior needs to be addressed. A synthetic strategy to control the polymerization thermodynamics was therefore developed. This was achieved by copolymerizing δ-decalactone (δDL) with either ε-decalactone (εDL) or ε-caprolactone (εCL) at room temperature (RT), with diphenyl phosphate (DPP) as catalyst. The thermodynamic stability of PδDL-co-εDL and PδDL-co-εCL increased with increased comonomer ratio in the feed, to 10% and 30% monomeric δDL, respectively, at 110 °C. This is in contrast to the PδDL homopolymer, which under the same conditions depolymerized to 70% monomeric δDL at equilibrium. The copolymers’ macromolecular structure, originating from the copolymerization kinetics, was found to be the crucial factor to mitigate δDLs equilibrium behavior. To close the loop, designing materials for a circular economy, the recycling of PδDL-co-εDL was demonstrated, by reaction with benzyl alcohol (BnOH) as an external nucleophile, leading to cyclic monomers or dimers with BnOH at high yield.
Introduction
Nature is an amazing chemical plant that has, throughout the history of humanity, served us as a source of energy, organic compounds, materials, food etc.1 For thousands of years we have had to rely on what this factory has given us, and we have learned to process and modify its output in a way that serves our needs. However, over the last century society has gone through a drastic transformation, from being dependent on biobased resources to surrender into the dominance of petroleum as a source of both chemicals and materials, as well as energy.2 Today, the tide is once again turning. Increased environmental awareness has brought us to the middle of a new overthrowing transformation. From a polymer synthesis perspective, the surge towards a sustainable society implicates polymerization of non-toxic, renewable monomers at conditions consuming low energy amounts and with a minimized use of solvents and purification. Moreover, the importance of considering the materials end-of-life and recyclability, when designing new polymeric materials, cannot be over-emphasized.From an environmental perspective, bulk ring-opening polymerization (ROP) of lactones contributes by being an atom economic, solvent free and catalyzed reaction, yielding aliphatic polyesters. Aliphatic polyesters are hydrolytically degradable and potentially biodegradable.3–6 In addition, ROP is an equilibrium reaction, hence having a built-in reversibility which opens for the possibility of chemical recycling. Lactones can be man-made, like lactide (LA) and ε-caprolactone (εCL), but nature also serves us with a large library of bio-derived natural lactones. These lactones often contribute to the taste and/or smell of e.g. flowers and milk. Many of the naturally occurring lactones are five- (γ) or six-membered (δ) rings, and have traditionally been utilized by the food- and fragrance industry. Lately, the bio-derived natural lactones have received academic attention as monomers for ROP,7–11 but so far, they are not utilized commercially by the polymer industry.
ROP is an equilibrium reaction, meaning that both ring-opening and ring-closing reactions take place simultaneously throughout the reaction. Whether the equilibrium favors monomer or polymer formation is determined by the thermodynamic state of the system. For small cyclic monomers, like γ- and δ-lactones, the low ring-strain makes the enthalpic driving force for ROP low, compared to the entropic decrease upon polymerization. A general trend is therefore that decreasing the temperature pushes the equilibrium towards polymer formation since a lower temperature decreases the entropic contribution.7,12–17 At the same time, the equilibrium can be reversely shifted towards monomer, simply by an increase in temperature, which opens up for polymers that could be chemically recycled back to the original cyclic monomers in a straight forward and easy manner. However, the challenge lies in understanding how the system's features dictate the equilibrium behavior, and learning how to control it. Another consequence of this thermodynamic behavior is that for many γ- and δ-lactones only low to moderate conversions can be reached at conventional operating conditions (ambient pressure, temperatures ≥ RT). This generates a challenge both in the polymerization (e.g. difficulties in reaching high molecular weights and a large quantity of remaining unreacted monomers) and processing steps (e.g. by depolymerization caused by the low thermodynamic stability).
One way to enable polymerization of a monomer with a low ability to homopolymerize is by utilizing copolymerization18–21 with a second monomer that has more favorable thermodynamics and thereby very different equilibrium behavior under specific conditions (illustrated in Fig. 1). The thermodynamically favored monomer, with the equilibrium strongly driven towards polymer, may then act as a continuous capping agent for the thermodynamically disfavored monomer, locking it into the polymeric structure. Of course, the copolymerization kinetics is of great significance here. If there are no transesterification reactions taking place, the conversion of the thermodynamically disfavored monomer into polymer would rely on capping by the thermodynamically favored monomer, which can only take place as long as there are more favored monomers available in the reaction mixture at the end of the polymerization. This has been demonstrated for γ-butyrolactone (γBL) during copolymerization with εCL20,22 or β-propiolactone,23 and for α-bromo- γ-butyrolactone24,25 (αBrγBL) when copolymerized with εCL or LLA. δ-Lactones are, compared to γ-lactone analogues, in general more prone to homopolymerize. However, they still have the issues of moderate monomer conversion and the resulting polymers being very sensitive to heat. A parallel may be drawn to polyoxymethylene (POM), a thermally unstable polyacetal which, when heated, depolymerizes to formaldehyde, if not end-capped or copolymerized.26,27 In fact, this thermodynamic instability is used as a water free way to synthesize formaldehyde in situ, underlining how the equilibrium behavior can be utilized for chemical recovery.
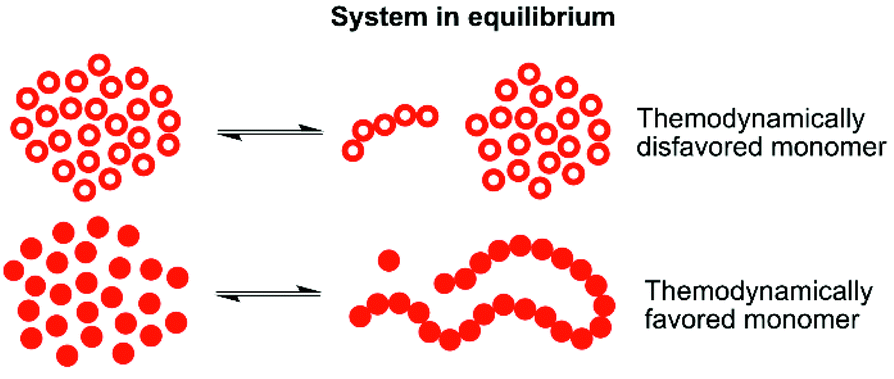 | ||
| Fig. 1 Illustration of two different cyclic monomers with very different equilibrium behavior under the given conditions. The thermodynamically disfavored monomer has a low equilibrium conversion, resulting in a higher concentration of free monomer in the polymer mixture. Opposite, the thermodynamically favored monomer has a high equilibrium conversion, leading to a low free monomer concentration in the polymer mixture. | ||
δ-Decalactone (δDL) is a δ-lactone that, throughout the years, has received academic attention as a biobased monomer used in copolymerization together with e.g. δ-dodecalactone,28 PEG and ω-pentadecalactone,29 lactide,30 methylene diphenyl diisocyanate,10 1,4 butylene oxide,14 δ-valerolactone31 and maltohepotaose.32 δDL occurs naturally in fruits33 and milk,34 but can also be produced enzymatically on large scale from fatty acids and essential oils.35,36 Recently, it was shown that catalytic transfer hydrogenation37 could be a new and interesting course to synthesize the monomer, which also increases the potential of establishing δDL as a biobased platform monomer. Moreover, the polymer of δDL is amorphous, hence, the ROP thermodynamics will not change due to crystallization.38,39 It is therefore a good model to study the equilibrium behavior of the family δ-lactones. Since ROP of δDL is favored by a low polymerization temperature, the reaction is preferably performed at room temperature (or lower), which is also desired form an environmental perspective utilizing an effective catalyst. Organocatalytic ROP has gained large attention in the last 10–15 years by generating high rate and selectivity at low temperatures40–44 and DPP is one of the organocatalysts that has been successfully used for ROP of δ-lactones with high control.14,45–47
In this work, our aim was to open up for a broader utilization of natural lactones within the polymer field and their subsequent recycling via depolymerization. For that purpose, a simple one-pot polymerization strategy for δ-lactones was developed in order to circumvent the polymerization thermodynamics, enabling controlled synthesis, use, processing and depolymerization. δDL was used as a model, and ring-opening copolymerized (ROcP) with two different ε-lactones with more favorable polymerization thermodynamics but different kinetic behavior, ε-decalactone and ε-caprolactone, and the thermodynamic stability of the copolymers were studied.
Experimental
Materials
All chemicals were used as received without any further purification. δ-Decalactone (δDL) (Sigma Aldrich, ≥98%), ε-decalactone (εDL) (Sigma Aldrich, ≥99%) and ε-caprolactone (εCL) (Sigma Aldrich, 97%) were used as monomers for ring-opening co-polymerization (ROcP). 1,4-Benzenedimethanol (Sigma Aldrich, 99%) was used as initiator (I) with diphenyl phosphate (DPP) (Sigma Aldrich, 99%) as catalyst. The was monitored as a function of time by addition of trimethylamine (TEA) (Sigma Aldrich, >99.5%) to drawn aliquots to deactivate the catalyst. Chloroform (Fischer Scientific, 99.8%) was used for polymer purification by liquid–liquid extraction. ZnCl2 (Sigma Aldrich, ≥98%, anhydrous), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (Sigma Aldrich, 98%), acetic acid (Merck, 100%) and benzyl alcohol (BnOH) (Sigma Aldrich, 99.8%) were used in recycling experiments. All NMR-analyses were carried out with deuterated chloroform (CDCl3) (VWR, 99.8%) as solvent and internal reference.Polymerizations
All polymerizations were performed on a scale of 5 g of monomer, with a constant molar content [I]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[cat]:[M]tot of 1:5:100. A bifunctional initiator was chosen to enable purity assessment of the system by size exclusion chromatography. Homopolymerizations of δDL, εDL and εCL, as well as ring-opening copolymerizations (ROcP) of δDL with εDL or εCL varying the amount of comonomer between 2.5, 5.0, 10 and 20 mol% were carried out. Experimental details for all ROP and ROcP are presented in Table 1. As an example of a typical procedure, ROcP of δDL with 20 mol% εCL was performed by weighing the desired amount of the initiator (I) 1,4-benzenedimethanol (43 mg, 0.31 mmol, 1 equiv.) into a 25 mL round bottom flask equipped with a magnetic stirrer. Thereafter, δDL (4.3 g, 25 mmol, 80 equiv.) together with εCL (0.72 g, 6.3 mmol, 20 equiv.) was added and the initiator was left to dissolve. The reaction was started by the addition of DPP (0.39 g, 1.6 mmol, 5 equiv.), and it was let to proceed in bulk at room temperature (RT). The polymerization kinetics were studied by 1H-NMR and SEC analysis of aliquots withdrawn at regular time intervals quenched by the addition of TEA. When the reaction reached its monomer equilibrium conversion, the reaction was heated to 110 °C by immersing the flask into a thermostatic oil bath. The polymerization kinetics were followed as previously described, until a new monomer equilibrium conversion was reached. The monomer conversion was calculated according to eqn (S1)–(S9), from the chemical shifts assigned in Fig. S2, in ESI.†
:[cat]:[M]tot of 1:5:100. A bifunctional initiator was chosen to enable purity assessment of the system by size exclusion chromatography. Homopolymerizations of δDL, εDL and εCL, as well as ring-opening copolymerizations (ROcP) of δDL with εDL or εCL varying the amount of comonomer between 2.5, 5.0, 10 and 20 mol% were carried out. Experimental details for all ROP and ROcP are presented in Table 1. As an example of a typical procedure, ROcP of δDL with 20 mol% εCL was performed by weighing the desired amount of the initiator (I) 1,4-benzenedimethanol (43 mg, 0.31 mmol, 1 equiv.) into a 25 mL round bottom flask equipped with a magnetic stirrer. Thereafter, δDL (4.3 g, 25 mmol, 80 equiv.) together with εCL (0.72 g, 6.3 mmol, 20 equiv.) was added and the initiator was left to dissolve. The reaction was started by the addition of DPP (0.39 g, 1.6 mmol, 5 equiv.), and it was let to proceed in bulk at room temperature (RT). The polymerization kinetics were studied by 1H-NMR and SEC analysis of aliquots withdrawn at regular time intervals quenched by the addition of TEA. When the reaction reached its monomer equilibrium conversion, the reaction was heated to 110 °C by immersing the flask into a thermostatic oil bath. The polymerization kinetics were followed as previously described, until a new monomer equilibrium conversion was reached. The monomer conversion was calculated according to eqn (S1)–(S9), from the chemical shifts assigned in Fig. S2, in ESI.†
| Name | Mco | [MδDL]:[Mco]:[I]:[cat] |
Time (days) | Conversiona (%) |
M
n,theob (kDa) |
M n`c (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| a Monomer equilibrium conversion calculated according to (S1), (S3) and (S8) in ESI.† b Theoretical molecular weight calculated according to (S2), (S4) and (S9) in ESI.† c Data obtained by CHCl3 SEC utilizing polystyrene standards. Chromatograms are presented in Fig. S1 in ESI.† d A broader and bifunctional molecular weight distribution was observed (Fig. S1 in ESI†). This might be related to the long reaction time (25 days), since the dispersity of the PδDL-co-εDL copolymers did not increase with increased amount of εDL. | |||||||
| PδDL | 100:0:1:5 |
4 | 86 | 14.2 | 9.6 | 1.3 | |
| PεDL | εDL | 0:100:1:5 |
25 | 97 | 16.2 | 10.2d | 1.6d |
| PεCL | εCL | 0:100:1:5 |
0.08 | 99 | 12.0 | 23.6 | 1.1 |
| PδDL-co-2.5εDL | εDL | 97.5:2.5:1:5 |
5 | 87 | 15.4 | 10.8 | 1.3 |
| PδDL-co-5εDL | εDL | 95:5:1:5 |
5 | 87 | 15.3 | 10.0 | 1.3 |
| PδDL-co-10εDL | εDL | 90:10:1:5 |
5 | 87 | 15.1 | 9.7 | 1.3 |
| PδDL-co-20εDL | εDL | 80:20:1:5 |
7 | 90 | 15.6 | 10.1 | 1.3 |
| PδDL-co-2.5εCL | εCL | 97.5:2.5:1:5 |
5 | 88 | 15.1 | 9.5 | 1.4 |
| PδDL-co-5εCL | εCL | 95:5:1:5 |
5 | 88 | 15.1 | 9.4 | 1.4 |
| PδDL-co-10εCL | εCL | 90:10:1:5 |
5 | 89 | 14.8 | 11.5 | 1.3 |
| PδDL-co-20εCL | εCL | 80:20:1:5 |
5 | 91 | 14.5 | 12.0 | 1.3 |
To evaluate the role of the comonomer as a pure end-capping agent, two experiments with sequential addition of the monomers were also carried out with the feed ratio [MδDL]:[Mco]:[cat]:[I] = 90:10:5:1. δDL was first homopolymerized at RT until monomer equilibrium conversion was reached. Due to the high viscosity of PδDL which prevented magnetic stirring, the comonomer was added to the flask and mixed briefly with a glass rod, where after it was left at room temperature without stirring. Aliquots were withdrawn at regular time intervals, for 1H NMR and SEC analysis, and the catalyst was deactivated with TEA (1.2 equiv. to catalyst). The reaction was let to proceed until at least 50% of the comonomer conversion was reached (3 equivalents per initiator). The flask was then immersed into a thermostatic oil bath at 110 °C, and the kinetics were followed as before, until a new monomer equilibrium conversion was reached.
Feedstock recycling of PδDL-co-εDL
To recycle PδDL-co-εDL back to cyclic monomers, three different strategies were screened: (i) Thermolysis, (ii) Chemolysis with ZnCl2 and (iii) Chemolysis with an external nucleophile. All experiments were performed on PδDL-co-20εDL, synthesized as described above. For (i) and (ii) the same batch of PδDL-co-20εDL (Mn = 12600 Da, Đ = 1.2) was used. DPP was removed from the polymer by three liquid–liquid extractions in CHCl3 and water. CHCl3 was removed using a rotary evaporator, and the polymer was dried at RT under vacuum for 1 week. For (iii) a second batch of PδDL-co-20εDL (Mn = 12500 Da, Đ = 1.2) was used.
Thermolysis
In a typical experiment, a sealed 5 mL round bottom flask, containing 100 mg PδDL-co-20εDL, was heated under nitrogen atmosphere at 220 °C for 6 h. The flask was let to cool down to RT before being opened. CDCl3 was added, and the solution was analyzed by 1H NMR.Chemolysis, ZnCl2
PδDL-co-20εDL (2 g) and ZnCl2 (50 mg, 0.03 equiv. to repeating unit) was added to a Schlenk-tube equipped with a magnetic stirrer. The reaction mixture was heated to 120 °C under a nitrogen atmosphere for 8 h. Samples were taken at regular time intervals, quenched by adding CDCl3 at RT. The samples were analyzed by 1H NMR.Chemolysis, external nucleophile
The experiments were carried out with varying amount of BnOH as external nucleophile, and all set-up preparation was performed inside a glove box. As an example of a typical procedure, PδDL-co-20εDL (1.0 g, 1 equiv. of repeating unit) was added to a vial equipped with a magnetic stirrer, without removing the catalyst (DPP). Then BnOH (0.13 g, 0.2 equiv. to repeating unit) was added as external nucleophile, and the vial was sealed with a Teflon-septum cap before being transferred out from the glove box. The reaction was carried out at 150 °C, and was studied by 1H-NMR and SEC analysis of aliquots withdrawn at regular time intervals quenched by the addition of TEA. The final raw product was obtained by adding TEA directly to the reaction before removing it from the oil bath. The same experiments were also carried out with δDL monomer instead of the copolymer as starting material.TBD were also evaluated as catalyst. PδDL-co-20εDL (2.0 g, 1 equiv. of monomer units), purified by CHCl3–water liquid–liquid extraction and dried, was added to a Schlenk-tube equipped with a magnetic stirrer. Then BnOH (0.26 g, 0.2 equiv.) and TBD (40 mg, 0.025 equiv.) were added. The reaction was carried out at 150 °C, and was studied by 1H-NMR and SEC analysis of aliquots withdrawn at regular time intervals quenched by the addition of acetic acid.
Characterization
000 g mol−1 were used for calibration.
Results and discussion
To extend the use of naturally occurring δ-lactones within the polymer field, and to gain control over their equilibrium behavior, δDL was ring-opening copolymerized (ROcP) with more thermodynamically favored monomers, ε-decalactone (εDL) and ε-caprolactone (εCL). These comonomers were utilized to achieve more thermodynamically stable, yet, recyclable copolymers. The experimental procedure is illustrated in Fig. 2, where the temperature change in the systems are distinguished by color (grey = RT, yellow = 110 °C). As demonstrated in previous work, understanding the kinetic behavior of a system can be a powerful tool in macromolecular design.14,46,48,49 The two different comonomers were therefore selected to, together with δDL, yield systems of different kinetic behavior, which would subsequently generate different macromolecular structures. The differences in macromolecular structures are expected to translate to different equilibrium behaviors at 110° C, that in turn are reflected in the thermodynamic stability.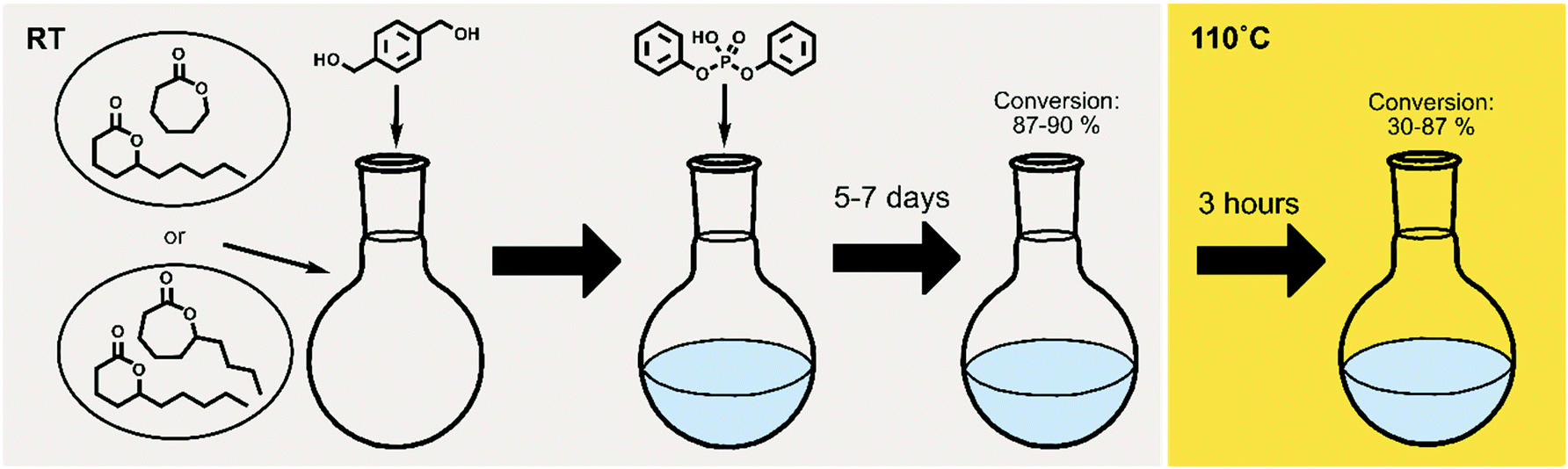 | ||
| Fig. 2 Scheme of the experimental procedure. ROcP was performed with either εDL or εCL as comonomer, and with different feed ratios. The reaction was carried out in RT (lt grey) for 5–7 days, until equilibrium conversion was established. The temperature was then raised to, and kept at, 110 °C (yellow) until the system had reached its new equilibrium. | ||
Homopolymerization of δDL, εDL and εCL
Like all equilibrium reactions, the thermodynamic behavior of ROP is determined by the relationship between the enthalpic and entropic changes during polymerization, together with temperature at which the reaction is performed. This is described by (1), where ΔHp is the change in enthalpy, ΔSp the change in entropy, T the absolute temperature and ΔGp the change in Gibb's free energy:| ΔGp = ΔHp − TΔSp | (1) |
Since ROP is an equilibrium reaction, the monomer conversion is very much dependent on the thermodynamic features of the system, which was first formulated by Dainton and Ivin50,51 followed by Tobolsky and Eisenberg.52–54 With Flory's assumption, that the reactivity of the propagating chain is independent of the length of the macromolecular chain, (1) can be rewritten in terms of standard polymerization enthalpy 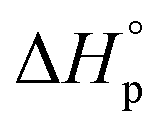 and entropy
and entropy 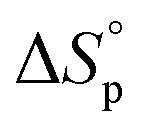 and the monomer concentration [M], according to (2) where R denotes the gas constant:
and the monomer concentration [M], according to (2) where R denotes the gas constant:
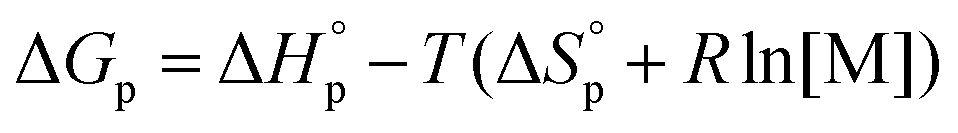 | (2) |
For ROP of small cyclic monomers, the ROP is in general driven by a decreased 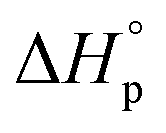 due to release of ring strain. Meanwhile, the change in
due to release of ring strain. Meanwhile, the change in 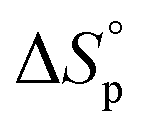 is commonly negative, similar to most other polymerization reactions.50 As a consequence of polymerization, the total entropy of the system decreases as the monomer is consumed. As appears from (2), at a constant temperature, the term
is commonly negative, similar to most other polymerization reactions.50 As a consequence of polymerization, the total entropy of the system decreases as the monomer is consumed. As appears from (2), at a constant temperature, the term 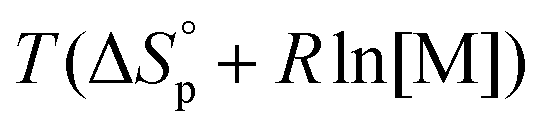 decreases upon polymerization due to a decreased monomer concentration. Hence, if
decreases upon polymerization due to a decreased monomer concentration. Hence, if 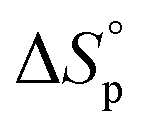 < 0, this results in a more and more negative
< 0, this results in a more and more negative 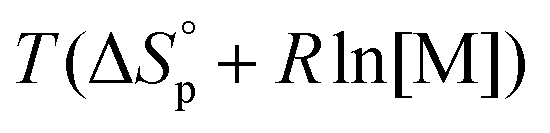 term, which may overrule the enthalpic driving force of ROP. This is also illustrated in Fig. 3. As long as ΔGp < 0, the forward reaction towards polymer is favored (red in Fig. 3). At some point in the conversion of monomers to polymers, the slope of G will invert, ΔGp>0, and the reverse reaction towards monomer is favored (green in Fig. 3). In the transition between these two scenarios, there is a minimum point where ΔGp = 0, and the reaction is at equilibrium (blue in Fig. 3). The monomer conversion at this G-minimum is called the equilibrium monomer conversion [M]eq. What (2) also reveals is that [M]eq is decreasing with increasing temperature, for a specific polymerization. Hence, these thermodynamic features have to be taken into consideration in order to understand the ROP behavior of different lactones and their chemical recyclability back to the monomer form.
term, which may overrule the enthalpic driving force of ROP. This is also illustrated in Fig. 3. As long as ΔGp < 0, the forward reaction towards polymer is favored (red in Fig. 3). At some point in the conversion of monomers to polymers, the slope of G will invert, ΔGp>0, and the reverse reaction towards monomer is favored (green in Fig. 3). In the transition between these two scenarios, there is a minimum point where ΔGp = 0, and the reaction is at equilibrium (blue in Fig. 3). The monomer conversion at this G-minimum is called the equilibrium monomer conversion [M]eq. What (2) also reveals is that [M]eq is decreasing with increasing temperature, for a specific polymerization. Hence, these thermodynamic features have to be taken into consideration in order to understand the ROP behavior of different lactones and their chemical recyclability back to the monomer form.
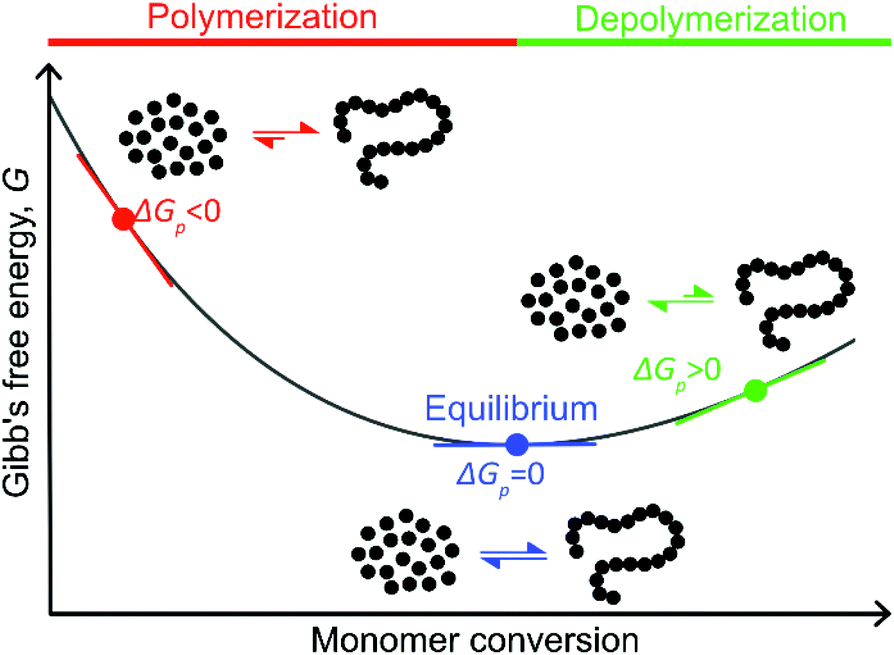 | ||
| Fig. 3 Illustration of how Gibb's free energy changes with monomer conversion during ROP. As the monomer conversion increases the slope of Gibb's free energy changes. The reaction is at equilibrium as the slope is zero, which corresponds to the minimum of the curve. | ||
Polymerization thermodynamic parameters determined for monomers of different ring-size, in-ring-functionality and substitutions can be found in literature.15,55,56 The ROP thermodynamics of δ-lactones are substantially less favorable than the thermodynamics of the corresponding ε-lactones (e.g. δ-valerolactone vs. ε-caprolactone).56 We therefore studied the effect of copolymerizing δDL with two different ε-lactones, εDL and εCL, on the equilibrium behavior. εDL is, like δDL, a naturally occurring monomer,35 and has successfully been copolymerized together with lactide and 2,2-dimethyltrimethylene carbonate.57–60
Polymerization of δDL at RT with DPP as catalyst proceeded in a controlled manner, with a linear relationship between molecular weight and conversion (Fig. 4). After 24 h, the monomer conversion was approximately 80% with a dispersity of 1.2 (Fig. 4). After an additional 4 days, the conversion had only increased to 86%, meanwhile the dispersity started to increase (Đ = 1.3). This is indicative for a reaction approaching its equilibrium, and the system was close to the G minimum (ΔGp close to zero) as illustrated in Fig. 3. The reaction vessel was then immersed into a thermostatic oil bath of 110 °C. Due to the changed thermodynamic features of the system, the reaction had been pushed upwards the G-slope, and is found in the green area in Fig. 3 where ΔG > 0. Hence, in the strive towards G-minimum and the systems new equilibrium, depolymerization to cyclic monomers occurs and the monomer conversion started to decrease rapidly. After only 40 min the monomer conversion was reduced to 38%, and after 3 h the reaction reached a new plateau at 30% conversion. Hence, at 110 °C, δDL could be classified as a thermodynamically disfavored monomer according to Fig. 1.
 | ||
| Fig. 4 ROP kinetic and thermodynamic behavior with DPP (5 equiv.) as catalyst and 1,4-benzenedimethanol (1 equiv.) as initiator for (a and b) δDL (100 equiv.) (c) εDL (100 equiv.) and (d) εCL (100 equiv.). Plots a, c and d present the conversion (circle) and ln([M]0/[M]) (triangle) as functions of time. Plot (b) show the dependence of Mn (circles) and dispersity (triangles) on the conversion. Lt grey and yellow areas represents data from the system at RT and 110 °C. | ||
As indicated by the relationship between the molecular weight and conversion (Fig. 4), depolymerization took place through the same, but reverse, path as ROP, i.e. the polymer chain “unzipped” from the propagating end. However, at 110 °C the dispersity started to increase with time, which is indicative of transesterification, chain scission or backbiting.61 These mechanistic details imply that the depolymerization can be prevented by simply capping the PδDL chain with a more thermodynamically favored monomer. This will hinder unzipping of the chain as the thermodynamic features of the system (e.g. temperature) are changed. However, if δDL would be copolymerized with a second monomer in a one-step reaction, then macromolecular architecture and, hence, equilibrium behavior, would be very much dictated by the relative copolymerization rate.
εDL and εCL were therefore homopolymerized at the same conditions as δDL, and the apparent rate constant of polymerization (kappp) for respective monomer was calculated from the slope of the curve of ln(([M]0 − [M]eq)/([M] − [M]eq)) against reaction time (Fig. 4). The reaction rate of εDL (kappp = 0.0065 h−1) was found to be 10 times lower as compared to δDL (kappp = 0.093 h−1), which agrees with previous observations that compared the kinetics of six- and seven-membered lactone analogues.45,47 Contrary to δDL, for which an instant decrease in conversion was observed as the temperature was raised to 110 °C, the conversion of εDL only increased from 94% to 99% under the same conditions. Hence, εDL is thermodynamically favored but kinetically unfavored compared to δDL. On the other hand, the polymerization of εCL was about 10 times faster (kappp = 1.2 h−1) than that of δDL and 200 times faster than εDL. These observations can be related to the nucleophilicity of the propagating end, where a primary alcohol in general is more reactive than a secondary alcohol due to induction, but also to the effect of substitution on the rate of cyclization, where an increased degree of substitution results in an increased ring-closing rate.62,63 At the same time, the conversion of εCL was stable around 99% also when temperature was raised to 110 °C. εCL is therefore both thermodynamically and kinetically favored compared to δDL. Although it is important to note that homopolymerization kinetics usually differ from the kinetics during copolymerization. These results indicate that copolymerization of δDL with εDL or εCL should result in two systems with very different kinetic behaviors, yielding polymers with clearly different macromolecular structures.
Copolymerization of δDL with εDL and εCL
The ROcP was performed at RT with DPP as catalyst, with δDL and varying ratio of comonomer (εDL or εCL) in the monomer feed: 2.5, 5, 10 and 20%. After 5–7 days at RT, as the monomer equilibrium conversion was reached (87–91%, Table 1), the temperature was raised to 110 °C and kept isothermal for 3 h, while the depolymerization towards the systems new G-minimum and equilibrium was studied (Fig. S6–S9 in ESI†). The monomer conversion at 110 °C after 40 min is presented as a function of mol% comonomer in the monomer feed in Fig. 5, and it is clear that the conversion was positively affected by the addition of εDL. When 20 mol% εDL was added in the feed (PδDL-co-20εDL), the monomer conversion after 40 min at 110 °C was increased from 38% to 90% compared to the homopolymerized PδDL, i.e. by 136%. εCL also had a positive effect on the thermodynamic stability of the copolymer, although not as substantial as εDL. With 20 mol% εCL (PδDL-co-20εCL), the conversion after 40 min at 110 °C was increased by 89% (from 38% to 72%) compared to PδDL. Two experiments with sequential addition of 10 mol% comonomers were also carried out, where both εDL and δDL had a positive effect on the thermodynamic stability of the copolymer (Fig. 5). This supports the theory that the comonomer acts as a capping agent, preventing depolymerization by unzipping to take place. However, the sequential addition was not more effective than then one-step copolymerization, which could be due to difficulties in mixing the comonomer with the highly viscous PδDL prepolymer, and was therefore not considered further.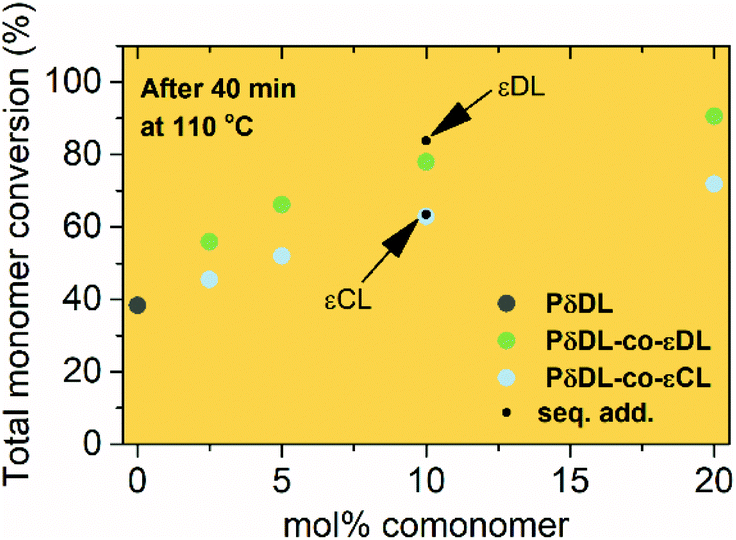 | ||
| Fig. 5 The plot shows how the total monomer conversion of PδDL-co-εDL (green) and PδDL-co-εCL (blue) depends on the feed mol% of the comonomer after 40 min at 110 °C. The circles represent one-step copolymerization and the black points represent copolymerization by sequential addition. The grey circle represents δDL homopolymer. | ||
The copolymerization kinetics of PδDL-co-20εDL (determined by 2D HSQC NMR, details on pages S3–S5 in ESI†) revealed how the reaction rate of εDL was much slower as compared to δDL, and how half of the εDL monomers were left unreacted as δDL was reaching its equilibrium conversion (Fig. 6a). When the reaction temperature was raised to 110 °C, the δDL conversion started to decline slightly at that same time as the conversion of unreacted εDL started to increase. In contrast, for PδDL-co-20εCL, the reaction rate of the two monomers was more similar to each other, with a slight preference towards εCL (Fig. 6b). However, the main difference lied in that εCL was fully converted to polymer whereas δDL reached its equilibrium conversion at 85%. As the temperature was raised to 110 °C, the instant decrease in δDL conversion was very clear, while εCL remained at a full conversion. Hence, as predicted, the kinetic behavior of the thermodynamically favored comonomer had a significant effect on the thermodynamic behavior of the copolymers.
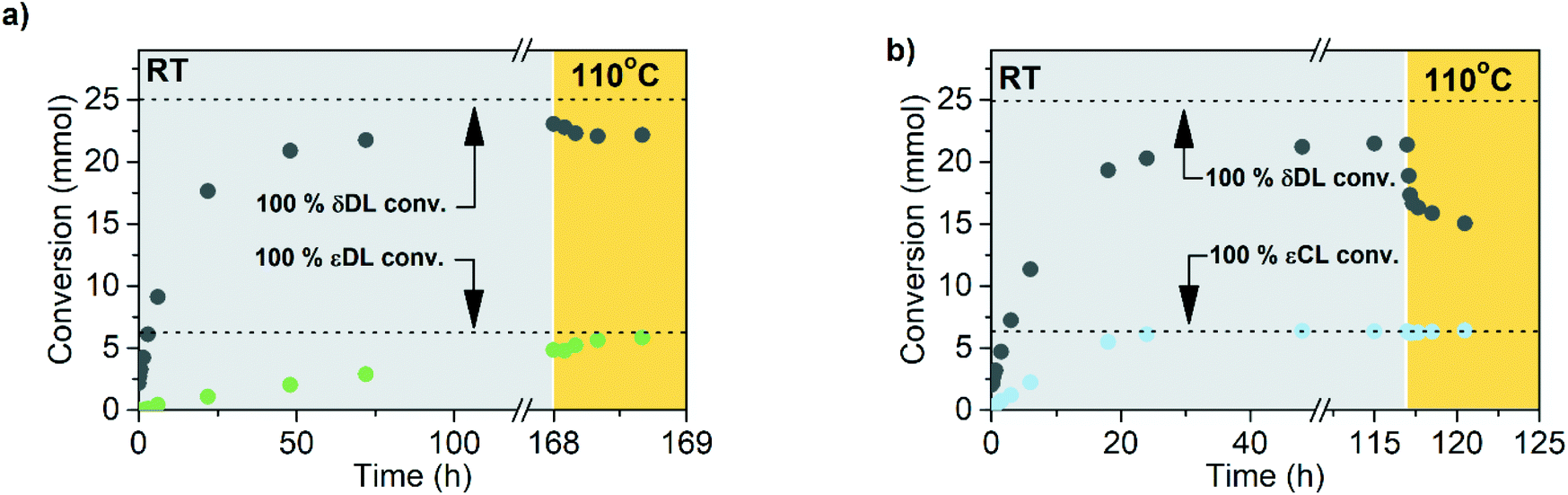 | ||
| Fig. 6 Kinetic and thermodynamic behavior with DPP (5 equiv.) as catalyst and 1,4-benzenedimethanol (1 equiv.) as initiator for ROcP of (a) PδDL-co-20εDL (100 equiv.) and (b) PδDL-co-20-εCL (100 equiv.). The conversion of respective monomer (grey = δDL, green = εDL, blue = εCL) is presented as a function of time, and was calculated from 1H NMR and 2D HSQC NMR. Lt grey and yellow areas represent data from the system at RT and 110 °C. | ||
As previously described, the depolymerization of PδDL took place by unzipping monomers from the chain end and reforming the cyclic monomer (Fig. 4). Since both εDL and εCL can be classed as thermodynamically favored monomers at 110 °C (Fig. 1), their thermodynamic equilibrium is still pushed towards polymer at this temperature. Hence, unzipping of the copolymer would only take place as long as a δDL unit was exposed at the chain end. How an average polymer chain would grow during the ROcP with 20 mol% commoner was schematically illustrated in Fig. 7. The illustration was based on the reaction kinetics of respectively monomer (determined by 2D HSQC NMR and 1H NMR, details on pages S4–S6 (Fig. S3–S5, eqn (S10)–(S13)) in ESI†) during ROcP and average DP of the polymer chain (under the assumption that no transesterification occurs in the system). To simplify the illustration, the figure shows the growth of an average chain from the initiator in only one direction, although the polymer was bifunctional. For PδDL-co-20εCL, the structure of the copolymer appeared rather random. This was also confirmed by 13C NMR (Fig. 8b) where four peaks in the carbonyl region were observed, two corresponding to homopolymer peaks and two dyad peaks from the transition between δDL and εCL segments. With a higher feed ratio of δDL compared to εCL (80 mol% δDL, 20 mol% εDL) in combination with the slightly faster conversion of εCL, the probability of the chain end being constituted by a δDL rather than εCL unit was substantially higher. Hence, upon heating, the δDL conversion dropped from 85% to 65% before εCL units started to appear at the ends. This was in large contrast to PδDL-co-20εDL, for which the δDL conversion decreased only slightly from 93% to 88%. Due to the slower conversion of εDL compared to δDL, the copolymer had a εDL gradient, with most εDL units being located towards the ends of the polymers. Hence, PδDL-co-20εDL could be illustrated more by the structure of a triblock copolymer, supported by 13C NMR (Fig. 8a), where only two peaks corresponding to homopolymer peaks could be observed in the carbonyl region. In addition, as δDL had reached its equilibrium conversion, there were still residual εDL present in the reaction mixture, and at the moment the temperature was raised these residuals were quickly polymerized. Hence, opposite to PδDL-co-20εCL, it was more likely that an εDL unit would constitute the chain end, or would be localized very close to it. Consequently, only a few δDL units could be unzipped until a εDL unit was exposed at the chain end. Additionally, residual εDL might also endcap terminal δDL units as soon as the temperature was raised to 110 °C as this temperature favors εDL polymerization, why the depolymerization was hampered. Consequently, the results point out the importance of the copolymerization kinetics when tailoring the thermodynamic behavior of PδDL by ROcP. At higher temperature, the rate of transesterification increases, which was indicated by an increased dispersity for all the copolymers at 110 °C. This would lead to the formation of new chain ends composed of δDL from where the unzipping could proceed. From the kinetics (Fig. S6–S9†) it is however clear that the depolymerization rate decreased significant after 40 min at 110 °C, even though a slight decrease in conversion still is observed.
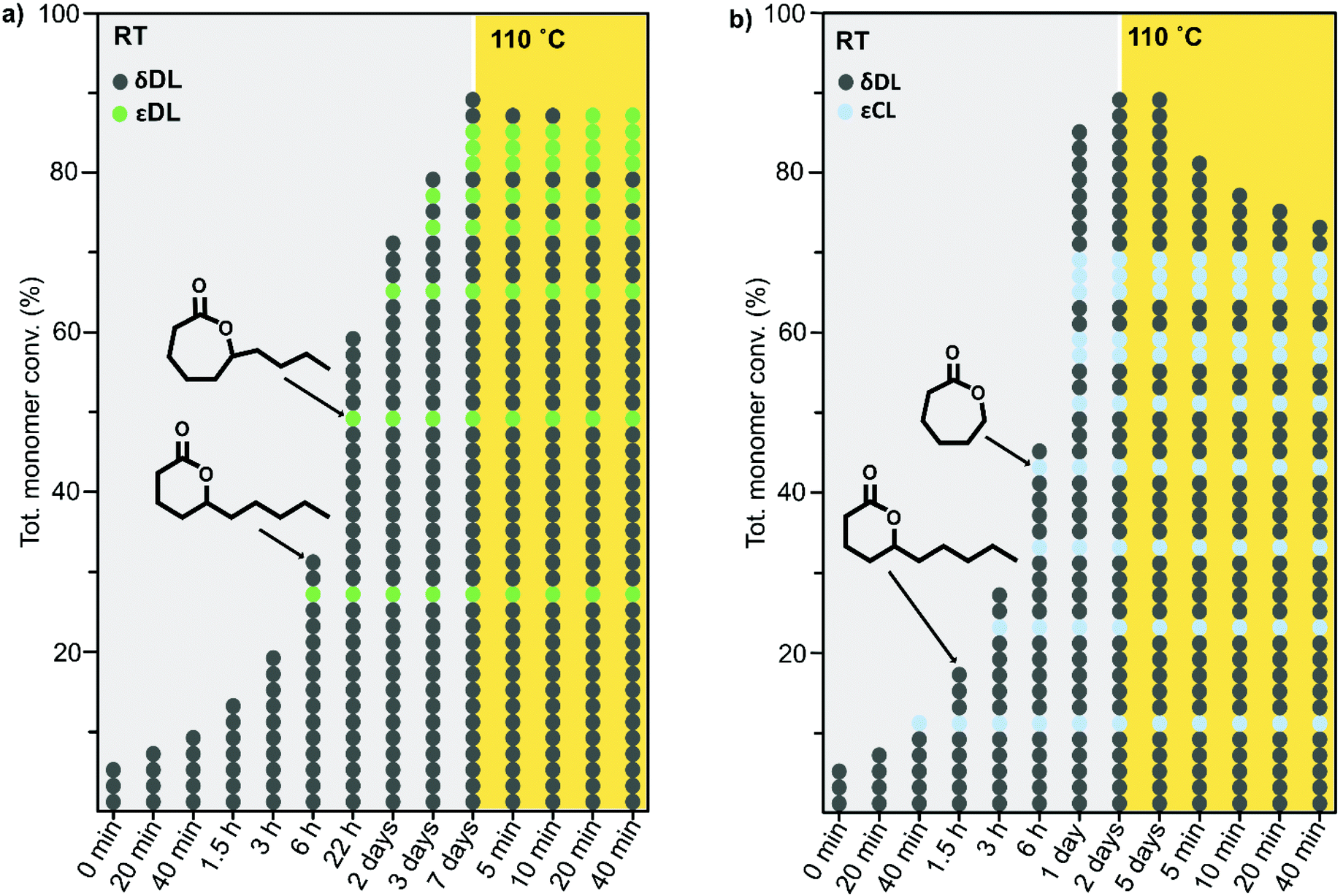 | ||
| Fig. 7 Schematic illustration of an average ROcP of (a) PδDL-co-20εDL and (b) PδDL-co-20εCL. For simplicity, the figure illustrates the growth of an average chain in one of the directions, although the polymer was bifunctional. Each circle represents one monomer unit (dk grey = δDL, green = εDL, blue = εCL) in the polymer chain. Lt grey and yellow areas represent data from the system at RT and 110 °C. The figure is based on kinetics of respectively monomer during ROcP and average DP of the polymer chain, with the assumption that no transesterification occurs in the system. The calculations were performed on 1H NMR and 2D HSQC NMR data. For more details, see pages S3–S5 in ESI.† | ||
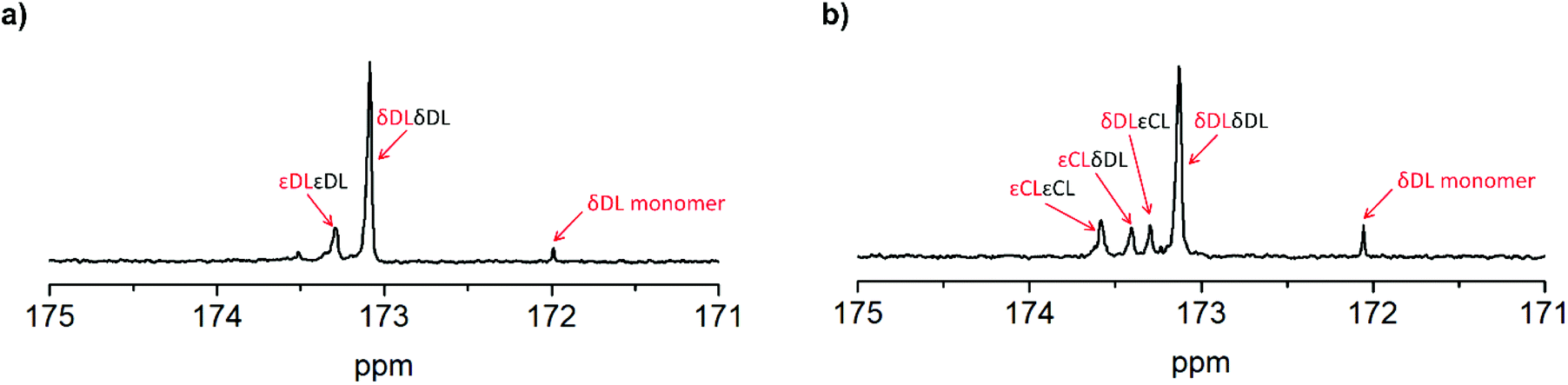 | ||
| Fig. 8 13C NMR spectrum of (a) PδDL-co-20εDL show two single peaks corresponding to the homopolymer peaks, (b) PδDL-co-20εCL show four peaks corresponding to the homopolymer peaks as well as two dyad peaks representing the transitions between δDL and εCL. | ||
Feedstock recycling of PδDL-co-εDL
We have now shown how a thermodynamically disfavored monomer, like δDL, can be ROcP together with a second more thermodynamically favored monomer, in order to increase the thermodynamic stability of the formed polymer. This could open up for a broader utilization of natural six-membered lactones within the polymer field. However, not only the feedstock origin is of importance for a material to be truly sustainable, its circularity is also vital. Chemical recyclability of polymers is a field of high current interest.64 Polymers synthesised by ROP have been successfully recycled by employing catalysts such as Sn(Oct)2,65 TBD,66 ZnCl2,8 LaCl367 and La[N(SiMe3)2]3.66
The monomer to polymer equilibrium of a polymer synthesised by ROP can be controlled by changing the thermodynamic features of the system. In order to favor cyclization, the system has to be pushed upwards the G-slope (Fig. 3) to the green area where ΔG ≥ 0, e.g. by increasing the temperature. The ceiling temperature for δDL has been calculated15 to 141 °C, from polymerization thermodynamic parameters (ΔHp = −17.1 kJ mol−1, ΔSp = −54 J mol−1 K−1)7 determined in bulk. Hence, above this temperature, the monomer to polymer equilibrium should be completely driven towards the monomeric from. However, due to the copolymerization with εDL, the depolymerization path by instant unzipping from the propagating end is hindered. In order to recover the cyclic δDL monomers from the PδDL-co-εDL copolymers, the polymer chain must therefore be cut to reveal δDL units at the chain ends. One way to initiate the depolymerization is by transesterifications within and between chains, which could lead to exposure of terminal δDL units. In order to investigate the effect of transesterifications in the presence of DPP, crude PδDL-co-20εDL was heated to 150 °C. After 4 h, a surprisingly large number of ring-closed monomers could be directly recovered (42%) (1, Table 2 and Table S1 in ESI†). But the 1H NMR also indicated formation of alkenes, which could originate from the elimination of the terminal OH-group. This was also observed when performing ROP of εDL at elevated temperatures with DPP as catalyst (Fig. S10 in ESI†). However, since this side reaction results in a “dead end”, terminating the unzipping reaction, it could be an explanation to why higher monomer yield was not obtained. In order to essay another track, we took inspiration from previous studies on recycling of poly((glycolic acid)-co-(γ-butyrolactone)).8 PδDL-co-20εDL was, hence, treated at 220 °C, in a nitrogen atmosphere after purification and without catalyst, or at 120 °C with ZnCl2 as catalyst. However, only 5% monomer yield was obtained after 8 h at 120 °C, with ZnCl2 as catalyst (3, Table 2 and Table S2 in ESI†). Slightly higher monomer yield (12%) was obtained after 6 h at 220 °C (2, Table 2 and Table S3 in ESI†), and alkene formation was observed in both cases.
| # | Starting material | Catalyst | BnOH (equiv.) | Temperature (°C) | Time (h) | Cyclic monomera (%) | Polymera (%) | Terminal OHa (%) | Alkenea (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated from 1H NMR peaks assigned in Fig. 9. | |||||||||
| 1 | PδDL-co-20εDL | DPP | — | 150 | 4 | 42 | 50 | 2 | 6 |
| 2 | PδDL-co-20εDL | — | — | 220 | 6 | 12 | 67 | <1 | 21 |
| 3 | PδDL-co-20εDL | ZnCl2 | — | 120 | 8 | 5 | 91 | 2 | 1 |
| 4 | PδDL-co-20εDL | TBD | 0.2 | 150 | 6 | 17 | 73 | 9 | — |
| 5 | PδDL-co-20εDL | DPP | 0.2 | 150 | 6 | 43 | 42 | 7 | 9 |
| 6 | PδDL-co-20εDL | DPP | 1 | 150 | 6 | 37 | 25 | 30 | 8 |
| 7 | PδDL-co-20εDL | DPP | 5 | 150 | 6 | 35 | 14 | 48 | 2 |
| 8 | δDL | DPP | 0.2 | 150 | 4 | 71 | 15 | 6 | 7 |
| 9 | δDL | DPP | 1 | 150 | 4 | 54 | 14 | 25 | 7 |
| 10 | δDL | DPP | 5 | 150 | 4 | 32 | 4 | 60 | 5 |
One way to avoid the alkene formation could be by enhancing the rate and amount of transesterification by the addition of an external nucleophile. Alcohols, like ethanol or ethylene glycol, have earlier been used for chemical recycling of polyesters like PLA and PET,68,69 with the hydroxyl group acting as an external nucleophile, and the polymer chains were degraded through transesterification into e.g. diethyl terephthalate, bis(2-hydroxyethyl)terephthalate or ethyl lactate. Such an approach could be applicable here as well. However, in order to obtain cyclic δDL monomers, the reaction should be performed above the ceiling temperature of δDL (141 °C), why a high boiling point alcohol is needed. For that purpose, benzyl alcohol (BnOH) (bp = 205 °C) was chosen. The crude PδDL-co-20εDL (Mn = 12500 Da) was heated to 150 °C with varying amounts of BnOH (0.2, 1 and 5 equiv. to repeating units) (5–7, Table 2 and Tables S4–S6 in ESI†). The reactions were monitored by 1H NMR, and four different structures/structural units were distinguished: cyclic monomer (δDL and εDL), unaffected polymer chain segments, end-groups with a terminal OH or end-groups with an alkene (Fig. 9). However, due to the overlapping peaks, the type of monomer, εDL or δDL, could not be distinguished within the structures.
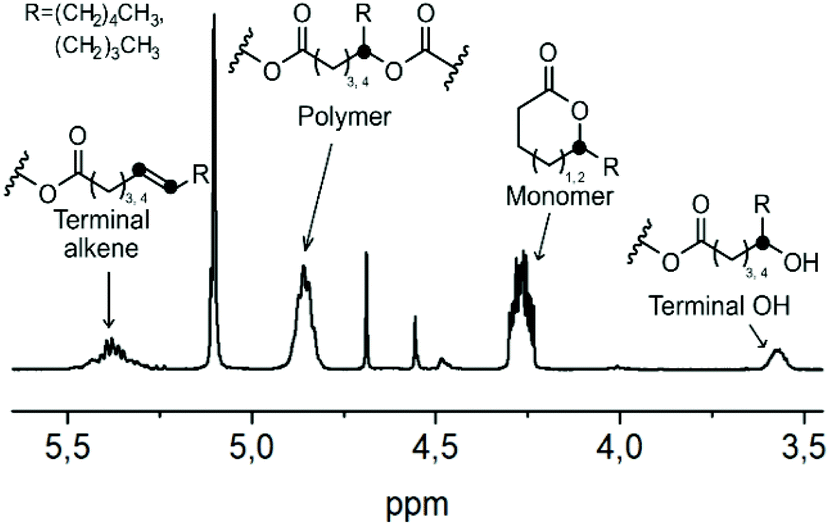 | ||
| Fig. 9 Structure assignment of 1H NMR peaks used for quantification of recycling products. | ||
When 0.2 and 1 equiv. BnOH was used, the amount of terminal OH decreased with time as the amount alkenes increased. However, with increased amount of BnOH the amount of alkene formation decreased drastically (5–7, Table 2). This could possibly be explained by the significantly lower DPP concentration in the reaction vessel, when large amount of BnOH was added and the copolymer became more diluted. Interestingly, no alkene formation was observed with 0.2 equiv. BnOH when the catalyst was TBD (4, Table 2 and Table S7 in ESI†). As expected, the amount of terminal OH-groups increased with increasing amount of BnOH (5–7, Table 2). It could also be noted that after 3 h, the concentration of ring-closed monomers reached a plateau and were thereafter more or less unchanged (Tables S4–S6 in ESI†). Surprisingly, the ratio of end-groups with a terminal OH exceeded the ratio of εDL units in the polymer, which must mean that not all terminal δDL units underwent cyclization. The same trend was also observed when δDL monomer was treated with BnOH (0.2, 1, and 5 equiv.) at 150 °C in the presence of the same concentration of DPP, where an increased amount of BnOH resulted in an increased concentration of end-groups with terminal OH (8–10, Table 2 and Tables 8–10 in ESI†). The high temperature and the dilution with BnOH are both thermodynamic features that would favor ring-closing. However, it is possible that at this temperature and with a large excess of nucleophile, the kinetic driving force for ring-opening is competing with the thermodynamic driving force for ring-closing, hindering complete depolymerization.
Even though only 35–43% cyclic monomers could be recovered (5–7, Table 2) SEC analysis of the products after 22 h reaction showed complete disappearance of the polymer peak at 14.1 mL retention volume (Ret. Vol.) (Mn = 12500 Da, Đ = 1.2) (Fig. 10). Instead peaks between 16–19 mL Ret. Vol. appeared, which were all outside the calibration range (1200–400000 Da). Nevertheless, it was clear that a higher number of BnOH equiv. resulted in a more homogenous low molecular weight product, and for 7 (PδDL-co-20εDL, 5 equiv. BnOH) mainly two narrow peaks could be observed. The large peak corresponding to the lowest molecular weight, at 18.9 mL Ret. Vol. could, hence, be a combination of cyclic monomers (170 Da) and BnOH (205 Da). Comparing the number of terminal OH (48%) to units in polymer (14%) indicates that the majority of ring-opened monomer units occurred as BnOH-DL dimers, which could correspond to the peak at 18.3 mL Ret. Vol. The low intensity peaks, 17.0–17.9 mL Ret. Vol., could thereby be trimers (BnOH-DL-DL) and higher.
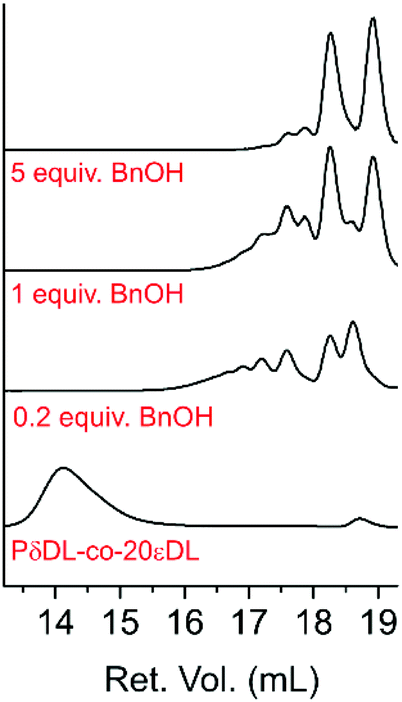 | ||
| Fig. 10 CHCl3 SEC elution diagram of the starting material PδDL-co-20εDL and the recycling product after 22 h at 150 °C with BnOH as external nucleophile and DPP as catalyst. | ||
Conclusions
A method to address the polymerization thermodynamics of naturally occurring δ-lactones, by controlling their equilibrium behavior was developed. This widens the possibility to utilize δ-lactones within the polymer field. δDL was ROcP with ε-lactones with more favorable polymerization thermodynamics, where these comonomers acted as internal capping agents that prevent the copolymer from depolymerization through unzipping. The comparison between εDL and εCL as comonomers illustrated how the copolymerization kinetics influenced the structure of the copolymer and subsequently the final thermodynamic properties of the copolymer. ROcP of δDL with εDL resulted in a “block-like” copolymer structure, with a higher concentration of εDL towards the chain ends, attributed to the slower polymerization rate of εDL compared to δDL. Thanks to the terminal εDL units, depolymerization of the copolymer was hindered, and the polymer was stable also at 110 °C. In contrast, due to the faster polymerization rate of εCL compared to δDL, εCL was consumed before δDL when copolymerized, resulting in a long δDL homosequence at the chain end. Hence, ROcP of δDL with εCL did not result in the same increase in thermodynamic stability compared to ROcP of δDL with εDL. When the copolymer was subjected to an increase in temperature (110 °C), it started to depolymerize by unzipping from the chain end until an εCL unit was exposed at the chain end. This highlights the importance of considering the copolymerization kinetics to circumvent the poor equilibrium behavior of δ-lactones.This knowledge was further utilized to recycle a copolymer of PδDL-co-εDL with an external nucleophile (BnOH) at 150 °C. Even though the reaction temperature was above the ceiling temperature of δDL, a large amount of δDL units were ring-opened by BnOH. In order to understand the driving force for ring-opening and cyclization of δDL with an external nucleophile the effect of different concentration of nucleophiles was evaluated. A large excess of BnOH (5 equiv. to repeating units), enabled high yield recovery of the monomers either as ring-closed monomers or as BnOH-DL dimers, both products being valuable as monomeric precursors.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge funding from the Knut and Alice Wallenberg foundation through Wallenberg Wood Science Center.References
- D. Sengupta and R. W. Pike, Handbook of Climate change Mitigation and Adaption, 2017, 1723, DOI:10.1007/978-3-319-14409-2.
- S. J. Bennett and H. A. Page, Handbook of Climate Change Mitigation, 2017, 383, DOI:10.1007/978-3-319-14409-2.
- E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305 CrossRef CAS.
- M. J.-I. Tschan, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836 RSC.
- R. A. Gross and B. Kalara, Science, 2002, 297, 803 CrossRef CAS PubMed.
- V. Arias, P. Olsén, K. Odelius, A. Höglund and A. C. Albertsson, Polym. Chem., 2015, 6, 3271 RSC.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2012, 1, 131 CrossRef CAS.
- X. Liu, M. Hong, L. Falivene, L. Cavallo and E. Y. X. Chen, Macromolecules, 2019, 52, 4570 CrossRef CAS.
- H. Quilter, M. Hutchby, M. Davidson and M. Jones, Polym. Chem., 2017, 8, 833 RSC.
- D. Tang, C. Macosko and M. Hillmyer, Polym. Chem., 2014, 5, 3231 RSC.
- W. Farhat, A. Biundo, A. Stamm, E. Malmström and P.-O. Syrén, J. Appl. Polym. Sci., 2020, 137, 48949 CrossRef CAS.
- A. E. Neitzel, M. A. Petersen, E. Kokkoli and M. Hillmyer, ACS Macro Lett., 2014, 3, 1156 CrossRef CAS.
- O. Haba and H. Itabashi, Polym. J., 2014, 46, 89 CrossRef CAS.
- J. Zhao and N. Hadjichristidis, Polym. Chem., 2015, 6, 2659 RSC.
- P. Olsén, K. Odelius and A. C. Albertsson, Biomacromolecules, 2016, 17, 699 CrossRef PubMed.
- M. Hong and E. Y.-X. Chen, Angew. Chem., 2016, 128, 4260 CrossRef.
- D. Zhang, M. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091 CrossRef CAS PubMed.
- R. Szymanski, Makromol. Chem., 1991, 192, 2943 CrossRef CAS.
- M. Bednarek, T. Biedron, P. Kubisa and S. Penczek, Makromol. Chem., Makromol. Symp., 1991, 42–43, 475 CrossRef.
- M. Nishiura, Z. Hou, T. Koizumi, T. Imamoto and Y. Wakatsuki, Macromolecules, 1999, 32, 8245 CrossRef CAS.
- M. Hong, X. Tang, B. Newell and E. Y.-X. Chen, Macromolecules, 2017, 50, 8469 CrossRef CAS.
- S. Agarwal and X. Xie, Macromolecules, 2003, 36, 3545 CrossRef CAS.
- K. Tada, Y. Numata, T. Saegusa and J. Furukawa, Makromol. Chem., 1964, 77, 220 CrossRef CAS.
- P. Olsén, J. Undin, K. Odelius and A. C. Albertsson, Polym. Chem., 2014, 5, 3847 RSC.
- J. Undin, P. Olsén, J. Godfrey, K. Odelius and A. C. Albertsson, Polymer, 2016, 87, 17 CrossRef CAS.
- C. E. Schweitzer, R. N. Macdonald and J. O. Punderson, J. Appl. Polym. Sci., 1959, 1, 158 CrossRef CAS.
- C. T. Walling, B. Frank and B. K. William, investors; Celanese Corp., assignee. Copolymers, US3027352, 1962.
- R. Ferrari, A. Agostini, L. Brunel, L. Morosi and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3788 CrossRef CAS.
- K. K. Bansal, D. Kakade, L. Purdie, D. J. Irvine, S. M. Howdle, G. Mantovani and C. Alexander, Polym. Chem., 2015, 6, 7196 RSC.
- G. S. Lee, B. R. Moon, H. Jeong, J. Shin and J. G. Kim, Polym. Chem., 2019, 10, 539 RSC.
- D. Bandelli, C. Helbing, C. Weber, M. Seifert, I. Muljajew, K. D. Jandt and U. S. Schubert, Macromolecules, 2018, 51, 5567 CrossRef CAS.
- T. Isono, B. J. Ree, K. Tajima, R. Borsali and T. Satoh, Macromolecules, 2018, 51, 438 CrossRef.
- H. Tamura, M. Appel, E. Richling and P. Schreier, J. Agric. Food Chem., 2005, 53, 5397 CrossRef CAS PubMed.
- Y. Karagül-Yüceer, M. Drake and K. R. Cadwallader, J. Agric. Food Chem., 2001, 49, 2948 CrossRef PubMed.
- C. Romero-Guido, I. Belo, T. M. N. Ta, L. Cao-Hoang, M. Alchihab, N. Gomes, P. Thonart, J. A. Teixeira, J. Destain and Y. Waché, Appl. Microbiol. Biotechnol., 2011, 89, 535 CrossRef CAS PubMed.
- C. Fuganti, G. Zucchi, G. Allegrone, M. Barbeni and P. Cabella, Biotechnol. Lett., 1994, 16, 935 CrossRef CAS.
- M. I. Alam, T. S. Khan and M. A. Haider, ACS Sustainable Chem. Eng., 2019, 7, 2894 CrossRef CAS.
- H. Nishida, M. Yamashita, T. Endo and Y. Tokiwa, Macromolecules, 2000, 33, 6982 CrossRef CAS.
- J.-M. Raquez, P. Degée, R. Narayan and P. Dubois, Macromolecules, 2001, 34, 8419 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64 CrossRef CAS.
- L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687 RSC.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999 CrossRef CAS.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 3814 CrossRef CAS.
- T. Saito, Y. Aizawa, K. Tajima, T. Isono and T. Satoh, Polym. Chem., 2015, 6, 4374 RSC.
- P. Olsén, K. Odelius, H. Keul and A. C. Albertsson, Macromolecules, 2015, 48, 1703 CrossRef PubMed.
- E. J. Shin, H. A. Brown, S. Gonzalez, W. Jeong, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2011, 50, 6388 CrossRef CAS PubMed.
- F. S. Dainton and K. J. Ivin, Nature, 1948, 162, 705 CrossRef CAS.
- F. S. Dainton and K. J. Ivin, Q. Rev., Chem. Soc., 1958, 12, 61 RSC.
- A. V. Tobolsky, J. Polym. Sci., 1957, 25, 220 CrossRef CAS.
- A. V. Tobolsky and A. Eisenberg, J. Am. Chem. Soc., 1959, 81, 2302 CrossRef CAS.
- A. V. Tobolsky and A. Eisenberg, J. Am. Chem. Soc., 1960, 82, 289 CrossRef.
- A. Duda, A. Kowalski, J. Libiszowski and S. Penczek, Macromol. Symp., 2005, 224, 71 CrossRef CAS.
- A. Duda and A. Kowalski, Handbook of Ring-Opening Polymerization, 2009, p. 1 Search PubMed.
- D. K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641 RSC.
- P. Olsén, T. Borke, K. Odelius and A. C. Albertsson, Biomacromolecules, 2013, 14, 2883 CrossRef PubMed.
- M. T. Martello, D. K. Schneiderman and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2014, 2, 2519 CrossRef CAS.
- J. Bai, J. Wang, Y. Wang and L. Zhang, Polym. Chem., 2018, 9, 4875 RSC.
- H. R. Kricheldorf, C. Boettcher and K.-U. Tönnes, Polymer, 1992, 33, 2817 CrossRef CAS.
- O. H. Wheeler and E. L. G. De Rodriguez, J. Org. Chem., 1964, 29, 1227 CrossRef CAS.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692 RSC.
- J. P. Brutman, G. X. De Hoe, D. K. Schneiderman, T. N. Le and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11097 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42 CrossRef CAS PubMed.
- X. Tang, M. Hong, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2016, 138, 14326 CrossRef CAS PubMed.
- K. Fukushima, O. Coulembier, J. Lecuyer, H. Almegren, A. Alabdulrahman, F. Alsewilem, M. Mcneil, P. Dubois, R. Waymouth, H. Horn, J. Rice and J. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273 CrossRef CAS.
- A. Carné-Sánchez and S. R. Collinson, Eur. Polym. J., 2011, 47, 1970 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00270d |
| This journal is © The Royal Society of Chemistry 2020 |