
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of acrylamide-based block-copolymer brushes under flow: monitoring real-time growth and surface restructuring upon drying†
Joydeb
Mandal
 a,
Andrea
Arcifa
b and
Nicholas D.
Spencer
*a
a,
Andrea
Arcifa
b and
Nicholas D.
Spencer
*a
aLaboratory for Surface Science and Technology, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, CH-8093 Zurich, Switzerland. E-mail: nspencer@ethz.ch
bEMPA Dubendorf, CH-8600 Zurich, Switzerland
First published on 14th April 2020
Abstract
A series of block-copolymer brushes has been synthesized under continuous flow by surface-initiated, atom-transfer radical polymerization of the acrylamide derivatives: N-isopropylacrylamide (NIPAM), N,N-dimethylacrylamide (DMAM) and N-hydroxyethyl acrylamide (HEAM). The formation of individual blocks was monitored in real time by means of a quartz-crystal microbalance with dissipation monitoring (QCM-D). The chemical composition, as well as the surface properties of the block-copolymer brushes, were examined in their dry state with X-ray photoelectron spectroscopy (XPS) and by measuring the water contact angle following each stage of the polymerization. In most cases, the block-copolymer brushes exhibited surface compositions and properties that closely resembled those of a homopolymer brush of similar composition to the final block. This was not the case when the final block consisted of p(HEAM), where the surface composition and properties were found to resemble those of the penultimate block. XPS depth-profile analysis and dynamic-contact-angle (DCA) measurements indicated that p(HEAM)-terminated block-copolymer brushes undergo interlayer mixing upon drying, driven by surface-energy effects.
Introduction
Block-copolymers are a fascinating class of materials, which possess the ability to assemble into a wide variety of structures that are ordered on the nanoscale. Even the most basic A–B type diblocks can self-assemble into many interesting nanostructured morphologies.1 Theoretical studies of ABC type triblock-copolymers predict the existence of 30 different morphologies and the process becomes even more complicated upon increasing number of blocks further.2 Facilitated by recent progress in controlled-radical-polymerization techniques, self-assembly of block-copolymers provides immense opportunities in the field of materials science.3 However, this interesting aspect has not been significantly investigated in the case where the block-copolymer chains are tethered to a solid substrate, which is the situation for block-copolymer brushes. Such brushes are presumed to organize in a simple, layered manner due to their limited degrees of freedom resulting from one end being tethered to a solid substrate.4,5 Nevertheless, such layered assembly of block-copolymer brushes can provide a valuable approach to the design of novel functional materials with structural gradients resembling those observed in the interfacial region of many biological systems, including cartilage.6Layered architectures can also be achieved by selectively swelling one component while deswelling the other in a mixed polymer brush. Tsukruk and co-workers showed that a binary polymer brush containing two incompatible polymers, such as poly(methyl acrylate) (PMA) and poly(styrene-co-2,3,4,5,6-pentafluorostyrene) (PSF) can give rise to two very different surface morphologies when treated with toluene and THF, respectively, as a result of selective swelling of only one component in each solvent.7 Later, Ionov et al.8 and Vyas et al.9 exploited this solvent-selective swelling of either of the components in mixed polymer brushes to design bi-layered architectures and create responsive surfaces. However, achieving architectures such as tri or tetra-layered systems using mixed polymer brushes is far more complex, and requires the individual swelling properties of each component to be considered.
Layer-by-layer (LbL) assembly of dissimilar polymeric systems, on the other hand, has been extensively used to create materials with multilayered architectures bearing graded physical and chemical functionalities.10 However, the dynamic nature of the interlayer electrostatic, H-bonding or hydrophobic interactions11 makes them vulnerable to structural instability and mixing of the successive layers.12 Moreover, the self-limiting adsorption kinetics of the LbL process reduces the possibility of achieving precise control over the length-scale of the individual layers.10
Block-copolymer brushes, however, can provide optimal stability as a result of being covalently tethered to a substrate. Furthermore, if synthesised via surface-initiated controlled-radical-polymerization (SI-CRP) techniques, block-copolymer brushes can provide excellent control over the length of individual blocks and furnish sharp compositional transitions, thanks to the living nature of the polymerization mechanism.
Surface-initiated, atom-transfer radical polymerization (SI-ATRP) is considered to be the most versatile among all the SI-CRP techniques for growing polymers from surfaces and has been successfully implemented to construct block-copolymer brushes with precise block lengths.13–16 However, achieving such architectures with water-soluble monomers has remained elusive due to the uncontrolled nature of ATRP kinetics, when carried out in an aqueous medium. This is a result of the displacement of the halide ions from the ATRP deactivator (CuX2Ln; X = Br or Cl, L = ligand) by water molecules. However, the effective concentration of CuX2Ln can be elevated and hence the polymerization kinetics brought under control by the use of a halide salt such as KX or tEtAmX (X = Br or Cl, tEtAm = tetraethylammonium) as demonstrated by Tsarevsky and co-workers.17 Very recently, we demonstrated that even the SI-ATRP kinetics of water-soluble acrylamides can be greatly improved following similar reaction conditions.18 We also demonstrated that the growth of homopolymer brushes can be interrupted and reinitiated multiple times without any notable loss in polymerization efficiency, which is an essential prerequisite for the successful construction of block-copolymer brushes. A further condition for the growth of such brushes is successful chain-end reinitiation with a second monomer, rather than the formation of new chains from the unreacted initiators remaining on the substrate surface, as reported by Li et al.19
In the present study, we introduce a flow system coupled with a switching valve that can seamlessly switch the monomer solutions from one to another without interrupting the polymerization process. Previously, we have demonstrated that the dispersity of the tethered polymer chains in such reaction environment is fairly low which is possible only if there is no continuous reinitiation from unreacted initiators at the surface.18 This becomes possible because the growing chain ends are principally near the outer edge of the brush, and therefore there is a steep negative concentration gradient of monomer from there to the substrate surface. Therefore, if, in a flow system, the impinging monomer is changed, the polymerization will principally continue at the outer edge, leading to a block structure and also avoids any undesired growth of new chains from the remaining unreacted initiators on the substrate surface.
In addition, a quartz-crystal microbalance with dissipation monitoring (QCM-D) was coupled to the flow system to allow in situ monitoring of both polymer growth and changes in polymer-brush viscoelastic properties, by measuring the change in the resonance frequency (ΔF) and the dissipation (ΔD) of the QCM-D sensor, respectively. The flow system is presented in Scheme 1. Although QCM-D has been successfully used by others, including ourselves, to study the growth kinetics of surface-initiated polymerization,18,20–25 in this study we have demonstrated that QCM-D can also be extremely useful in monitoring the real-time growth of block-copolymer brushes.
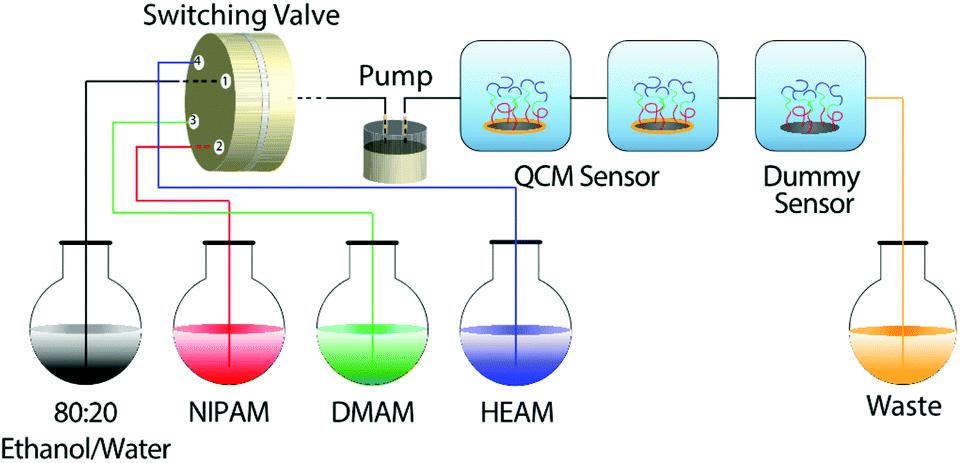 | ||
| Scheme 1 Illustration of the flow system used to synthesize the block-copolymer brushes and to monitor their growth in situ. | ||
Results and discussion
In situ monitoring of block-copolymer brush formation by means of QCM-D
Since our primary objective was to synthesize hydrophilic block-copolymer brushes and monitor their formation in situ, we chose to sequentially polymerize three water-soluble acrylamide monomers: N-isopropylacrylamide (NIPAM), N,N-dimethylacrylamide (DMAM) and N-hydroxyethyl acrylamide (HEAM) from a monolayer of SI-ATRP initiator, immobilized onto SiO2-coated QCM sensors. Similar experiments were also carried out on silicon wafers cut to the size and shape of QCM sensors, which will be referred to as “dummy sensors” in the following sections. The dummy sensors were used to carry out further ex situ characterizations, such as ellipsometry, X-ray photoelectron spectroscopy (XPS) and contact-angle measurements.In an exemplary case, a baseline was first established by flowing 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 ethanol/water through the QCM-D cells; the flow was then switched to the reaction mixture containing NIPAM (80:20 ethanol/water + NIPAM + CuBr + Me6TREN + CuBr2 + tEtAmBr) to initiate the polymerization and construct the first block of p(NIPAM). Subsequently the flow was switched to reaction mixtures containing DMAM and then to HEAM to construct the desired p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer brush and finally to 80:20 ethanol/water to stop the polymerization. The growth of the block-copolymer brushes was monitored in situ by following the decrease in the resonance frequency (ΔF) and the concomitant increase in the dissipation (ΔD) of the QCM-D sensors, as shown in Fig. 1a. Additionally, the dry thicknesses of the block-copolymer brushes were measured at each stage using ellipsometry and are presented in Table 1.
:20 ethanol/water through the QCM-D cells; the flow was then switched to the reaction mixture containing NIPAM (80:20 ethanol/water + NIPAM + CuBr + Me6TREN + CuBr2 + tEtAmBr) to initiate the polymerization and construct the first block of p(NIPAM). Subsequently the flow was switched to reaction mixtures containing DMAM and then to HEAM to construct the desired p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer brush and finally to 80:20 ethanol/water to stop the polymerization. The growth of the block-copolymer brushes was monitored in situ by following the decrease in the resonance frequency (ΔF) and the concomitant increase in the dissipation (ΔD) of the QCM-D sensors, as shown in Fig. 1a. Additionally, the dry thicknesses of the block-copolymer brushes were measured at each stage using ellipsometry and are presented in Table 1.
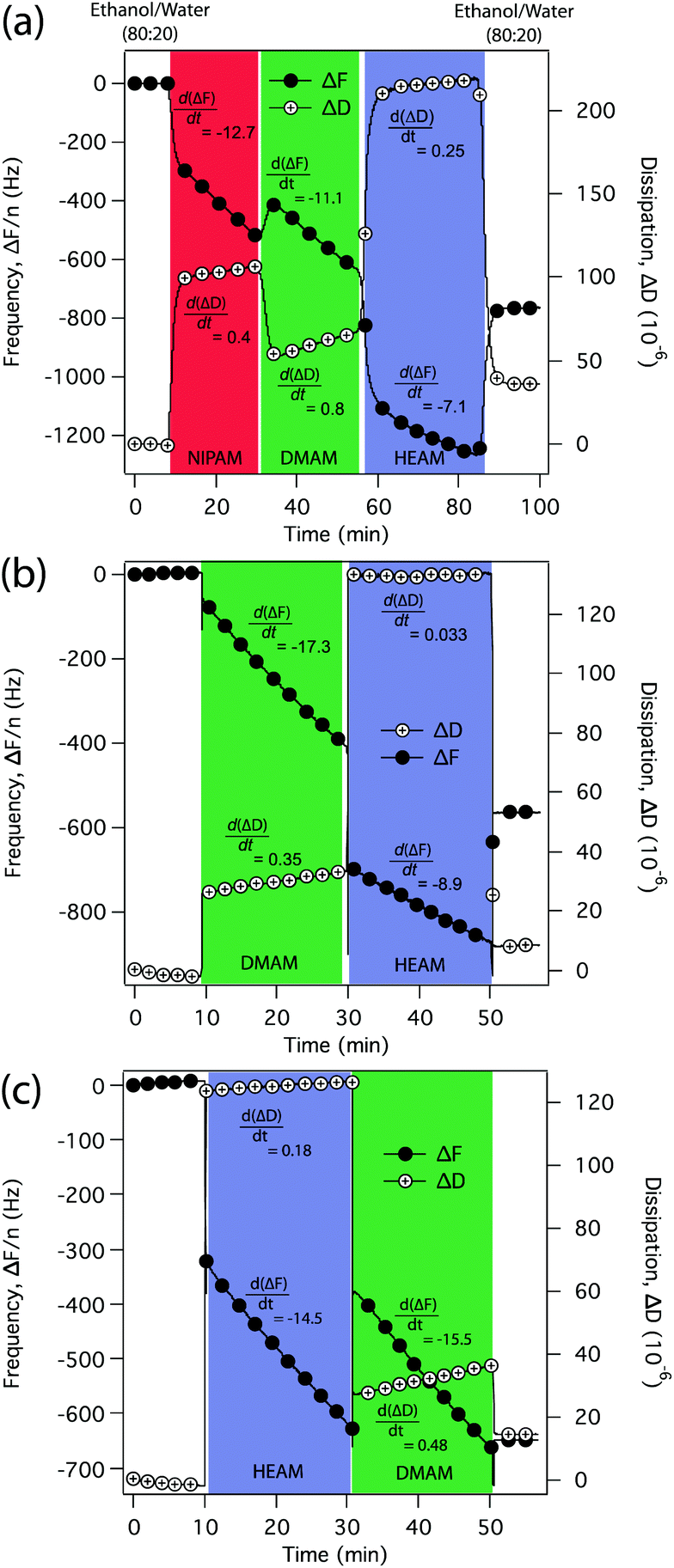 | ||
| Fig. 1 Formation of (a) p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer, (b) p(DMAM)-b-p(HEAM) diblock-copolymer and (c) p(HEAM)-b-p(DMAM) diblock-copolymer brushes, monitored in situ by QCM-D by measuring the variation in the resonance frequency (ΔF) and dissipation (ΔD) as a function of polymerization time (filled circles represent ΔF while the open circles with + signs represent ΔD). | ||
| Sample | Dry thickness (nm) | Static water contact angle (θs) |
|---|---|---|
| p(NIPAM) | 28.0 ± 0.4 | 58.0 ± 0.5° |
| p(NIPAM)-b-p(DMAM) | 50.0 ± 0.5 | 30.0 ± 0.5° |
| p(NIPAM)-b-p(DMAM)-p(HEAM) | 62.0 ± 1.0 | 29.0 ± 1.0° |
| p(DMAM) | 25.0 ± 0.2 | 29.0 ± 0.5° |
| p(DMAM)-p(HEAM) | 40.0 ± 1.0 | 26.0 ± 1.0 |
| p(HEAM) | 23.0 ± 0.5 | 9.0 ± 2.0° |
| p(HEAM)-b-p(DMAM) | 37.0 ± 1.0 | 30.0 ± 0.4° |
Although ΔF and ΔD values represent the total mass and viscoelasticity of the solvated polymer films, the slopes of the ΔF-t and ΔD-t profiles represent the instantaneous changes that occur as a result of the addition of each monomer to the growing films. Therefore, the linear ΔF-t profiles corresponding to the polymerization of NIPAM, DMAM and HEAM clearly demonstrate a steady increase in the brush thickness as a result of the living nature of the SI-ATRP process—as we have previously reported for the homopolymers.18,23 The sharp changes, however, while switching from one monomer to another, are caused by sudden changes in the viscosity of the medium. To rule out the possibility of the QCM-D profiles being affected by physisorption of the monomers, we also carried out a similar experiment with HEAM but in the absence of ATRP-catalyst and the result is presented in Fig. S7.† As expected, other than the viscosity-induced sharp changes upon introduction of the monomer, we did not observe any change in the ΔF-t or ΔD-t profiles.
The QCM-D study, however, revealed a few more interesting features: although NIPAM and DMAM exhibited very similar polymerization kinetics with ΔF-t profiles of comparable slopes of −12.7 and −11.1, polymerization of DMAM exhibited a significantly faster increase in the dissipation, showing a ΔD-t profile with a slope of 0.8 compared to 0.4 for the polymerization of NIPAM.
It has been widely reported in the literature that the degree of swelling of the tethered polymer chains dramatically influences the dissipation behavior of the polymer brush,26 also observed in our previous study of the urea-induced collapse of p(NIPAM) brush.27 Therefore, it is reasonable to assume that the higher rate of increase in dissipation seen while polymerizing DMAM is a result of the greater degree of swelling of the p(DMAM) block compared to the p(NIPAM) block, because of the much higher hydrophilicity of the p(DMAM) brush. This is demonstrated by its low water contact angle of 29° compared to 58° for the p(NIPAM) homopolymer brush (Fig. S1†).
Interestingly, polymerization of HEAM, on the other hand, exhibited the slowest rate of increase in the dissipation signal, even though p(HEAM) is the most hydrophilic among the three polymers as demonstrated by its low water contact angle (θs) of ∼9° as shown in Fig. S1.† This anomalous behavior observed while constructing the p(HEAM) block could be a possible consequence of interchain hydrogen bonding between the secondary amides and hydroxyl groups that are present along the p(HEAM) block, which would tend to resist swelling and thereby have a stiffening effect.27
To gain a clearer understanding of interchain hydrogen bonding and its effect on the ΔD-t profile, we also examined the real-time growth of two more block-copolymer brushes: p(DMAM)-b-p(HEAM) and p(HEAM)-b-p(DMAM) using QCM-D as shown in Fig. 1b and c. In both cases, polymerization of HEAM exhibited a significantly slower increase in the dissipation signal compared to the polymerization of DMAM, again possibly due to interchain hydrogen bonding between the p(HEAM) segments. Such observations clearly demonstrate the potential of QCM-D in monitoring the growth of block-copolymer brush in real time.
Reorganization of block-copolymer brushes
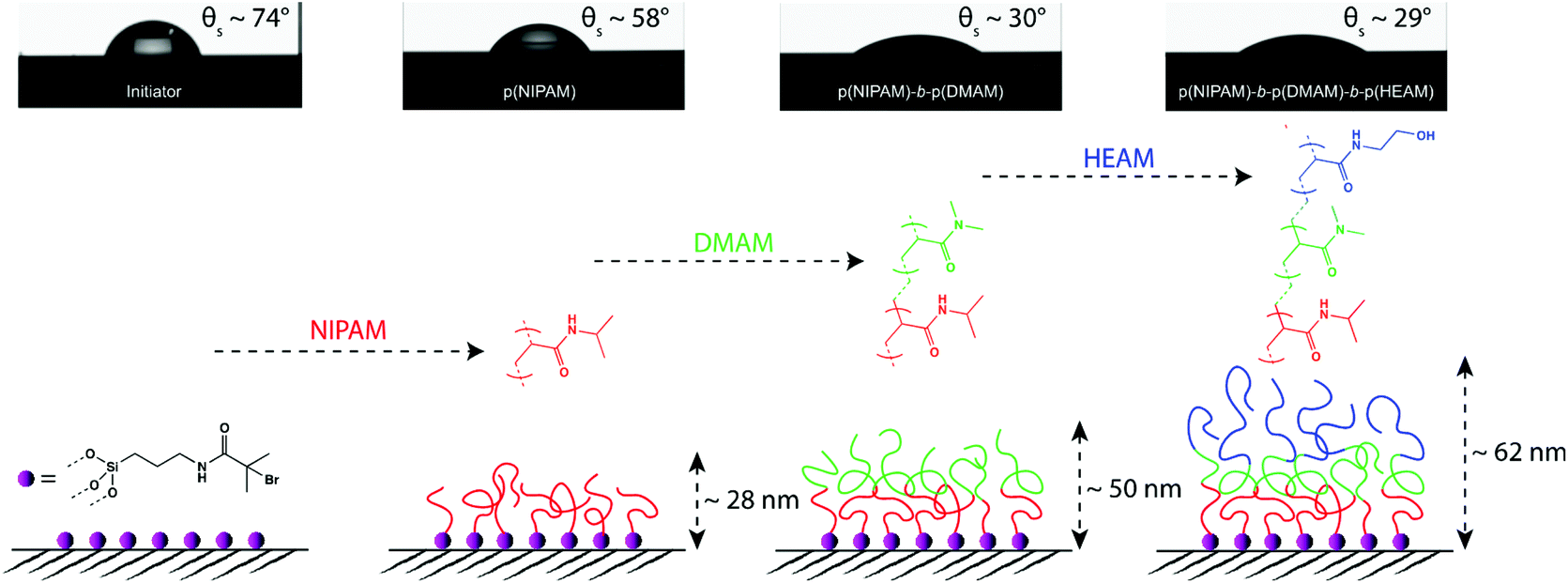 | ||
| Scheme 2 Schematic illustration of the construction of p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer brush along with the dry brush thicknesses and static water contact angles (θs) measured at each stage. Each block was constructed by polymerizing the respective monomer for 20 minutes. | ||
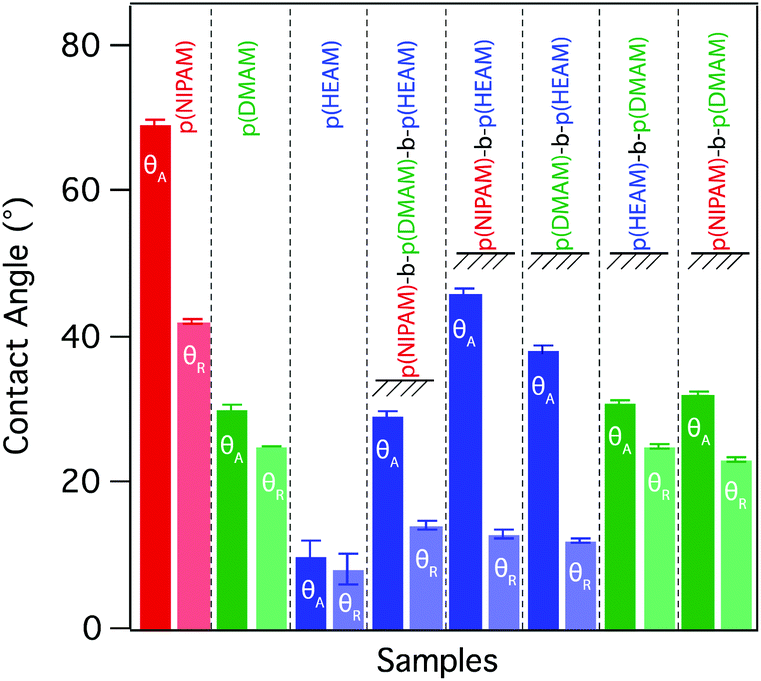 | ||
| Fig. 2 Advancing and receding contact angles of five block-copolymer brushes and the three homopolymer brushes, measured with Milli-Q water. | ||
The NIPAM monomer possesses both hydrophobic (isopropyl) and hydrophilic (secondary amide) moieties and their selective interactions towards the surrounding medium can significantly alter the wetting behaviour.30 In their pioneering work, Takei and co-workers showed that conformational freedom of grafted p(NIPAM) chains and hence their ability to reorganize in response to the interacting medium, dramatically influence the wetting behavior of p(NIPAM) grafted surfaces.31 Therefore, the contact-angle hysteresis observed for the p(NIPAM) brush is a result of reorganization of the grafted p(NIPAM) chains in response to the interacting medium, i.e. air or water.
The difference in hydrophilicity between the moieties (methyl and tertiary amide) residing on the DMAM monomer is significantly lower than that of NIPAM and hence the p(DMAM) brush exhibits much lower hysteresis compared to the p(NIPAM) brush. Similarly, in the case of p(HEAM) brush, both moieties (hydroxyl and secondary amide) are hydrophilic and as a result the brush exhibits very low advancing (θA) and receding contact angles (θR) that are close to the detection limit of the instrument.
While considering the block-copolymer brushes, it might be reasonable to assume that the wetting behaviour of these systems would resemble that of a homopolymer brush corresponding to the composition of the final block, as the contact angle commonly reflects the chemical composition of the very-near surface region (i.e. down to the molecular scale). This was indeed the case for the block-copolymer brushes terminating in p(DMAM), with advancing (θA) and receding contact angles (θR) that were nearly identical to that observed for the p(DMAM) homopolymer brush, as shown in Fig. 2 and S2.† However, the block-copolymer brushes bearing p(HEAM) as the final block exhibited θA values that were very similar to that of a homopolymer brush corresponding to the penultimate block, but θR values that closely resembled those of the p(HEAM) brush. Such behavior indicates that the block-copolymer brushes adopt the expected conformation with the final p(HEAM) block at the surface when wet, but reorganize upon drying, the p(HEAM) block being partially buried by the penultimate block, as depicted in Fig. 3a.
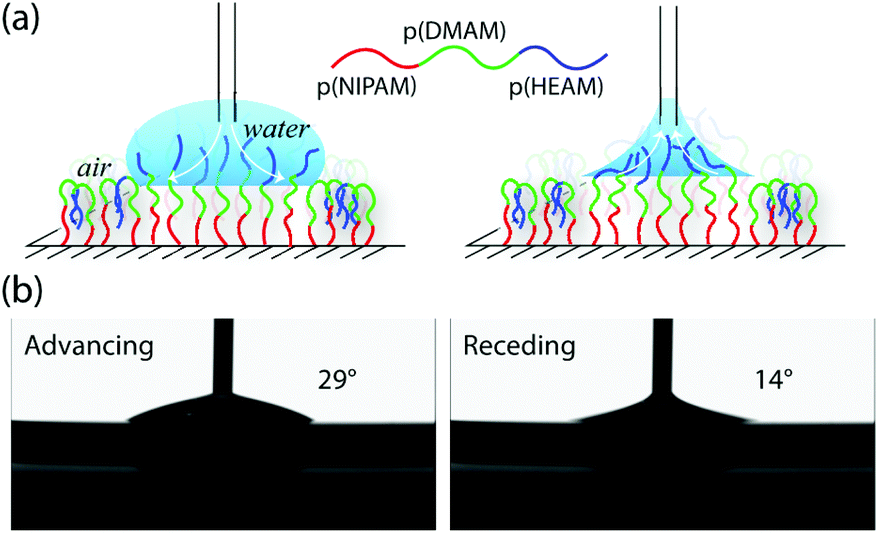 | ||
| Fig. 3 (a) Schematic illustration of the reorganization of the p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer brush in response to the surrounding medium; (b) advancing and receding contact angles of p(NIPAM)-b-p(DMAM)-b-p(HEAM) triblock-copolymer brush measured with Milli-Q water. | ||
Such reorganization of block-copolymer brushes has been observed by others as well. Zhao and co-workers while studying the effect of solvent treatment on different polystyrene-b-polyacrylate brushes observed that they undergo self-reorganization and exhibit very different surface properties.16 More recently Xu and co-workers observed that even the relative length of the blocks plays a critical role in the self-organization process of the tethered polymer chains.13
XPS analysis
The reorganization of the tethered polymer chains taking place at the surface of dry block-copolymer brushes with p(HEAM) as the final block, was further examined by angle-resolved X-ray photoelectron spectroscopy (ARXPS). The analysis of the O1s region of the spectra is particularly informative, as the HEAM monomer contains two different types of oxygen (carboxamide and hydroxyl) in a 1:1 ratio, while both DMAM and NIPAM monomers contain only one type of oxygen (carboxamide). Fig. 4 shows the O1s spectra of p(DMAM), p(HEAM) and p(DMAM)-b-p(HEAM) polymer brushes, all acquired in standard-lens mode.
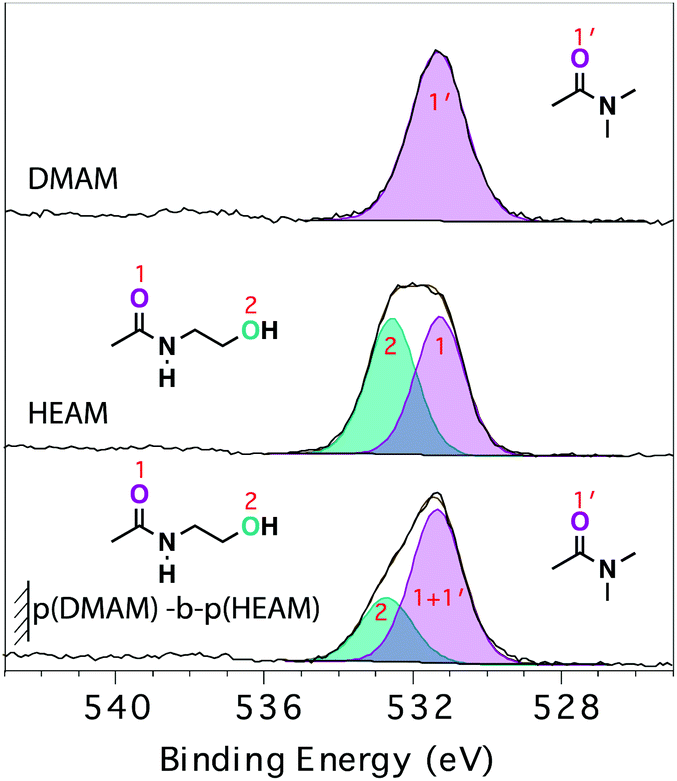 | ||
| Fig. 4 O1s XP-spectra of p(DMAM), p(HEAM) homopolymer brushes and p(DMAM)-b-p(HEAM) diblock-copolymer brush. All the spectra were acquired in standard-lens mode (θe = 53°). | ||
The O1s spectrum of p(DMAM) was fitted to a single peak, which is consistent with the chemical composition of p(DMAM), which bears only carboxamide oxygen. One additional component, representing the hydroxyl oxygen, was added to fit the spectrum of p(HEAM). In agreement with the stoichiometry of the carboxamide and hydroxyl oxygens present in p(HEAM), the ratio of the areas under the two peaks was found to be close to 1. The same curve-fitting model was applied to the O1s spectra of p(DMAM)-b-p(HEAM) (Fig. 4, bottom) and p(NIPAM)-b-p(DMAM)-b-p(HEAM) brushes as well (Fig. S6†). In both cases, the ratio of the areas corresponding to carboxamide and hydroxyl components was found to be significantly higher than 1, indicating that the near-surface region probed by the technique is not composed entirely of p(HEAM), but also has a significant contribution from the penultimate block.
The analysis of the relative intensity of the two components as a function of θe (Fig. 5) provided further details on the self-organization of the block-copolymer brushes in their dry state. The depth from which 99.7% of the signal originates—information depth (ID)—is estimated to be 7.2 and 1.5 nm for θe values of 26.75° and 79.25°, respectively (section S E4†). The higher contribution of the carboxamide component to the O1s signal acquired at higher θe points to an enrichment of the near-surface region by the relatively less hydrophilic p(DMAM) block of the p(DMAM)-b-p(HEAM) diblock-copolymer brush. This picture is in line with the DCA measurements on the block-copolymer brushes with a final p(HEAM) block, which exhibited advancing contact angles representative of the penultimate block.
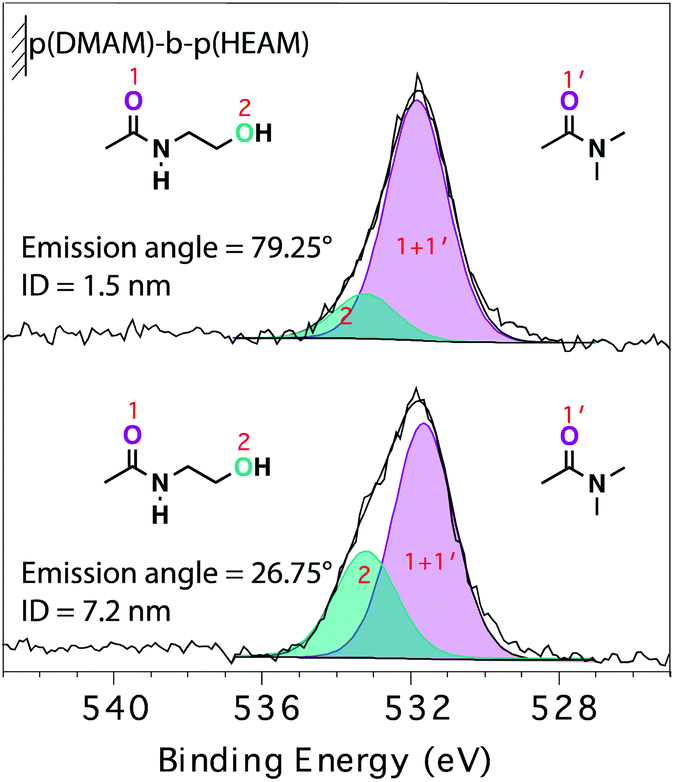 | ||
| Fig. 5 O1s angle-resolved XP-spectra of p(DMAM)-b-p(HEAM) diblock-copolymer brush, collected at emission angles of 79.25° (top) and 26.75° (bottom). | ||
To further illustrate the effect of the final block on the reorganization, and hence the wetting behaviour of the block-copolymer brushes, we also carried out XPS analysis on the p(NIPAM)-b-p(DMAM) diblock-copolymer brush, as shown in Fig. S5.† In this case, no interlayer mixing between the two layers was evident from either chemical state or quantitative analysis. The results were in excellent agreement with the outcomes of DCA measurements as all the copolymers with p(DMAM) as the final block exhibited a wetting behaviour closely resembling that of the p(DMAM) homopolymer brush.
Further information obtained by XPS is presented in the ESI.† In particular, the quantitative elemental analysis of the homopolymer brushes was found to be in excellent agreement with the expected stoichiometry (Table S2†), indicating high chemical purity and integrity of the polymers obtained with the method described in this work. In addition, the survey spectrum presented in Fig. S3† did not show any detectable signal attributable to the silicon substrate, indicating that the dry brushes were uniformly thicker than the ID of the method.
Conclusion
We have introduced a flow system coupled with a QCM-D that enables the uninterrupted construction of block-copolymer brushes and monitoring of their growth in real time. The ability of QCM-D to measure any changes in the dissipation behavior of the growing polymer brushes provides an opportunity to study the formation of individual blocks, since the dissipation is heavily dependent on the degree of swelling of individual blocks. The block-copolymer brushes were further characterized at each stage by measuring both water contact angle and the chemical composition of the surfaces using XPS. Interestingly, dried block-copolymer brushes with p(HEAM) as the final block always exhibited surface properties and chemical compositions that were representative of the penultimate block. Further experiments with angle-resolved XPS and dynamic contact angle (DCA) measurements demonstrated that the block-copolymer brushes with p(HEAM) as the final block undergo a reorganization upon drying. This is likely to be the result of surface-energy minimization, which leads to the less hydrophilic, penultimate block being exposed at the surface, as shown in Fig. 3a.Even though surface reorganization can have an unexpected impact on this surface-tailoring methodology, the resulting high contact-angle hysteresis could be useful, having potential applications in areas where droplet positioning is of importance, such as in diagnostic devices.
Experimental section
Materials
α-Bromoisobutyryl bromide (98%), (3-aminopropyl) triethoxysilane (99.8%, extra dry), triethylamine (99.5%), copper(II) bromide (99%), copper(II) chloride (99%), tris[2-(dimethylamino)ethyl]amine (97%), tetraethylammonium bromide (98%) and tetraethylammonium chloride (98%) were purchased from Sigma Aldrich (Germany) and used as received. N-Isopropylacrylamide (97%) and N,N-dimethylacrylamide (99%) were purchased from Sigma Aldrich (Germany), whereas N-hydroxyethylacrylamide (>97%) was purchased from Tokyo Chemical Industries (Germany). N-Isopropylacrylamide was purified by crystallization from a 60:40 (v/v) toluene/hexane mixture and was dried in vacuum prior to use, while other monomers were used without any purification. Cu(I) chloride (99.99%) and Cu(I) bromide (99.99%) were purchased from Sigma Aldrich (Germany) and purified by stirring overnight in glacial acetic acid, filtering, and washing several times with acetone, diethyl ether and finally drying under vacuum. Silicon wafers (P/B h100i) were purchased from Si-Mat Silicon Wafers (Germany) while SiO2-coated QCM sensors were purchased from Q-Sense (Sweden).
Immobilization of SI-ATRP initiator
SiO2-coated sensors and the Si-wafers were first cleaned by sonicating them in toluene and isopropanol followed by UV/ozone (ProCleanerTM and ProCleanerTM Plus, BioForce, (USA)) treatment for 35 min. The substrates were then functionalized with (3-aminopropyl)triethoxysilane (APTES) by vapor deposition and subsequently washed with toluene and dried under a stream of nitrogen. The APTES-modified substrates were then reacted with α-bromoisobutyryl bromide (BIBB) by immersing them in a 10 mg mL−1 solution of BIBB in dry dichloromethane (DCM) containing triethylamine and kept under an argon atmosphere. The reaction was continued for three hours and finally the substrates were cleaned with DCM, dried under nitrogen gas and used to carry out SI-ATRP.Preparation of the reaction mixtures
In order to achieve maximum control over the SI-ATRP kinetics, which is necessary for the construction of block-copolymer brushes, the polymerizations were performed in the presence of additional CuX2 and tEtAmX (X = Cl or Br). The reaction mixtures were prepared following our previously reported procedure18 and this is reported in detail in the ESI.†In an exemplary case, a mixture of NIPAM (4.57 g, 40.35 mmol), Me6TREN (43 μL, 0.16 mmol), CuBr2 (3.6 mg, 0.016 mmol), tEtAmBr (0.84 g, 4.0 mmol) and 80:20 ethanol–water mixture (10 mL) was degassed by bubbling nitrogen gas for 45 min. The degassed mixture was then transferred using a syringe to a flask containing CuBr (11.6 mg, 0.08 mmol), kept under nitrogen. The syringe was flushed 3–4 times with nitrogen gas before drawing the reaction mixture. The solution was stirred for 10–15 min for the complete dissolution of the catalyst.
Flow synthesis of block-copolymer brush
The block-copolymer brushes were grown inside QCM-D flow cells from initiator-modified, SiO2-coated QCM-D sensors and Si-wafers cut into the shape and size of the sensors. Also, the growth of the block-copolymer brushes was monitored in situ using a QCM-D instrument (Q-Sense E4, Västra Frölunda, Sweden).In an exemplary procedure, a stable baseline was first established by flowing an 80:20 ethanol/water mixture at a constant flow rate of 2.5 μL s−1. The polymerization was then initiated by switching the flow to the NIPAM reaction mixture and then to DMAM and HEAM and finally to 80:20 ethanol/water mixture to stop the reaction, using a multi-position HPLC valve (Vici, model no.: EUHA, Serial no.: EUA08048, Valco Instruments, Houston, Texas, USA). Polymerizations were carried out for ∼20 minutes with each of the three monomers. The flow rate was maintained at 2.5 μL s−1 using a piston pump (milliGAT Low Flow pump, Model CP-DSM-GF, Valco Instrument Co. Inc., Houston, Texas, USA). Finally, the sensors and the Si-wafers (dummy sensors) were removed from the cells, washed several times with water and ethanol and finally dried under a nitrogen stream.
To prevent gathering information from the penultimate layers while performing XPS measurements, the samples used for the XPS study were prepared by polymerizing each block for longer times. For example, the p(DMAM)-b-p(HEAM) diblock-copolymer brush used for the XPS measurements was prepared by polymerizing both DMAM and HEAM for 30 min each and the dry thicknesses at the two stages were measured to be 50 and 80 nm.
Characterization of the films
The dry thicknesses of the polymer films were measured using variable-angle spectroscopic ellipsometry, (VASE) M-2000F (LOT Oriel GmbH, Darmstadt, Germany). The brush-supporting Si wafers were in all cases assumed to be covered with a 2 nm-thick silicon dioxide film. Determination of ψ and Δ as a function of wavelength (275–827 nm) was carried out by employing the WVASE32 software package (LOT Oriel GmbH, Darmstadt, Germany). The analysis of the brush layers was performed based on a Cauchy model: n = A + Bλ − 2 where n is the refractive index, λ is the wavelength and A and B were assumed to be 1.45 and 0.01, respectively, as values for transparent organic films.Contact-angle measurements were carried out with Milli-Q water, using a Krüss DSA100 (Germany) instrument, equipped with a high-resolution camera in sessile-drop mode. A 5 μL drop was placed on the surface and the advancing (θA) and receding (θR) contact angles were measured by addition to and withdrawal from the drop at a rate of 4 μL min−1.
XP-spectra were acquired with a VG Theta probe spectrometer (Thermo Fisher Scientific, East Grinstead, UK) operating in the constant-analyzer-energy (CAE) mode, using a monochromatic source with a beam diameter of 300 μm and a power of 60 W. High resolution spectra were acquired with a pass energy of 100 eV, a step site of 0.1 eV (full width at half-maximum (FWHM) of the peak height for Ag 3d5/2 = 0.91 eV) and with lenses operating in either standard or angle-resolved mode. In the standard-lens mode, a single spectrum was acquired with an average emission angle of 53° and an acceptance angle of 60°. In the angle-resolved mode, multiple spectra were acquired simultaneously by dividing the acceptance angle into channels. In the present study, eight channels were used; only the spectra corresponding to the two extreme angles are shown (26.75° and 79.75°).
All the Survey spectra were acquired in standard lens modes with a pass energy of 300 eV and step size of 1 eV. The linearity of the binding-energy scale was periodically checked using sputter-cleaned gold, silver, copper. Charging was corrected by referencing to aliphatic carbon at 285.0 eV. The high-resolution spectra were processed using CasaXPS software (v2.3.15, Casa Software Ltd, Wilmslow, Cheshire, UK). A Tougaard background subtraction was applied before peak fitting using a linear least-squares algorithm. Details on the curve fitting model are reported in the ESI.†
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors gratefully acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 669562).References
- F. S. Bates and G. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- W. Zheng and Z.-G. Wang, Macromolecules, 1995, 28, 7215–7223 CrossRef CAS.
- I. W. Hamley, Developments in block copolymer science and technology, John Wiley & Sons, 2004 Search PubMed.
- S. Kumar, Y. L. Dory, M. Lepage and Y. Zhao, Macromolecules, 2011, 44, 7385–7393 CrossRef CAS.
- S. N. Ramakrishna, M. Cirelli, E. S. Kooij, M. Klein Gunnewiek and E. M. Benetti, Macromolecules, 2015, 48, 7106–7116 CrossRef CAS.
- S. Lee and N. D. Spencer, Science, 2008, 319, 575 CrossRef CAS PubMed.
- M. Lemieux, D. Usov, S. Minko, M. Stamm, H. Shulha and V. V. Tsukruk, Macromolecules, 2003, 36, 7244–7255 CrossRef CAS.
- L. Ionov, M. Stamm, S. Minko, F. Hoffmann and T. Wolff, Macromol. Symp., 2004, 210, 229–235 CrossRef CAS.
- M. K. Vyas, K. Schneider, B. Nandan and M. Stamm, Soft Matter, 2008, 4, 1024–1032 RSC.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- C. Porcel, P. Lavalle, G. Decher, B. Senger, J.-C. Voegel and P. Schaaf, Langmuir, 2007, 23, 1898–1904 CrossRef CAS PubMed.
- C. Xu, T. Wu, C. M. Drain, J. D. Batteas, M. J. Fasolka and K. L. Beers, Macromolecules, 2006, 39, 3359–3364 CrossRef CAS.
- F. Zhou and W. T. Huck, Phys. Chem. Chem. Phys., 2006, 8, 3815–3823 RSC.
- J.-B. Kim, W. Huang, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 5410–5416 CrossRef CAS.
- B. Zhao and W. J. Brittain, Macromolecules, 2000, 33, 8813–8820 CrossRef CAS.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- J. Mandal, R. Simic and N. D. Spencer, Polym. Chem., 2019, 10(29), 3933–3942 RSC.
- A. Li, S. N. Ramakrishna, P. C. Nalam, E. M. Benetti and N. D. Spencer, Adv. Mater. Interfaces, 2014, 1, 1300007 CrossRef.
- S. E. Moya, A. A. Brown, O. Azzaroni and W. T. Huck, Macromol. Rapid Commun., 2005, 26, 1117–1121 CrossRef CAS.
- N. Cheng, O. Azzaroni, S. Moya and W. T. Huck, Macromol. Rapid Commun., 2006, 27, 1632–1636 CrossRef CAS.
- J. J. I. Ramos and S. E. Moya, Macromol. Rapid Commun., 2011, 32, 1972–1978 CrossRef CAS PubMed.
- J. Mandal, R. Varunprasaath, W. Yan, M. Divandari, N. D. Spencer and M. Dübner, RSC Adv., 2018, 8, 20048–20055 RSC.
- R. Heeb, R. M. Bielecki, S. Lee and N. D. Spencer, Macromolecules, 2009, 42, 9124–9132 CrossRef CAS.
- E. M. Benetti, E. Reimhult, J. de Bruin, S. Zapotoczny, M. Textor and G. J. Vancso, Macromolecules, 2009, 42, 1640–1647 CrossRef CAS.
- E. S. Dehghani, N. D. Spencer, S. N. Ramakrishna and E. M. Benetti, Langmuir, 2016, 32, 10317–10327 CrossRef CAS PubMed.
- J. Mandal, R. Simic and N. D. Spencer, Adv. Mater. Interfaces, 2019, 1900321 CrossRef.
- R. H. Dettre and R. E. Johnson Jr., J. Phys. Chem., 1965, 69, 1507–1515 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- H. Yim, M. Kent, D. Huber, S. Satija, J. Majewski and G. Smith, Macromolecules, 2003, 36, 5244–5251 CrossRef CAS.
- Y. G. Takei, T. Aoki, K. Sanui, N. Ogata, Y. Sakurai and T. Okano, Macromolecules, 1994, 27, 6163–6166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00219d |
| This journal is © The Royal Society of Chemistry 2020 |