DOI:
10.1039/D0PY00192A
(Paper)
Polym. Chem., 2020,
11, 2381-2389
Mono- and dimeric zinc(II) complexes for PLA production and degradation into methyl lactate – a chemical recycling method†
Received
5th February 2020
, Accepted 25th February 2020
First published on 27th February 2020
Introduction
Petroleum-based polymers (plastics) dominate modern-day society, with annual global production exceeding 300 Mtonnes, of which ca. 99% were derived from petrochemical feedstocks in 2015.1 However, it is clear polymer production based on diminishing fossil fuel reserves is unsustainable, with the oil demand for plastics anticipated to rise to 20% by 2050 from today's value of 6%.2–5 Moreover, plastic pollution, particularly in our oceans, has reached epidemic proportions in recent years, partly due to the intrinsic robustness and durability of plastics, but primarily through the irresponsible handling of plastic waste.6 Indeed, it is projected 250 Mtonnes of plastic waste will reside in our oceans by 2025 at current disposal rates.4,7 There is therefore a demand for renewable and sustainable alternatives that, ideally, possess facile degradation pathways, a prerequisite to mitigating such concerns. Consequently, renewable polymers have emerged as a promising alternative to traditional polymers since they can be produced from biomass, which remains a relatively untapped, abundant resource.3 Poly(lactic acid) (PLA), a sustainable and biocompatible aliphatic polyester, has emerged as one of the outstanding renewable alternatives within the last 20 years.8 PLA has attracted considerable commercial and academic interest, owing to its comparable physical properties with existing synthetic plastics.8,9 As such, PLA has found use in single-use, disposable applications in the packaging sector, but also the biomedical industry, owing to its inherent biocompatibility.3,5,8 However, the widespread use of PLA remains limited by a high production cost, which involves the fermentation of lactic acid, prior to subsequent pre-polymerisation steps to produce the cyclic diester monomer, lactide.9 Possible solutions include the use of shape selective catalysts and gas–phase reactions, both of which render the monomer preparation less energy and resource intensive.10,11 However, despite its green credentials, PLA is still a possible source of plastic pollution since biodegradation occurs exclusively under industrial composting conditions.12 Moreover, conventional waste management strategies, including landfill and incineration, align with a linear economic model, a primary contributor to the mounting plastic waste crisis. Indeed, as of 2015, ca. 6300 Mtonnes of plastic waste has been produced, and by 2050 it is estimated ca. 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mtonnes of plastic waste will reside in either landfill or the natural environment.6 Thus, there is a clear need to develop alternative targeted waste management technologies to facilitate this transition, namely through recycling.4,7 Mechanical recycling is one possible solution but is limited by eventual material downgrading due to thermomechanical degradation, resulting in material repurposing to applications that require polymer of lower quality. A promising alternative is chemical recycling, which has the potential to reduce both waste and operational costs through material regeneration, achieved either through the depolymerisation or degradation of PLA.13 A number of different methods have been reported in the literature, including hydrolytic and thermal degradation, as well as enzymatic processes.14–23 However, the former two rely on high temperature conditions (200–400 °C), resulting in high operational costs, whilst uncertainty surrounds the scalability of the latter. Alternatively, the transesterification of PLA with alcohols to generate lactate esters has gathered appreciable momentum in recent years (Scheme 1). Low-molecular-weight lactate esters have been cited as green substitutes to traditional hydrocarbon-based solvents, owing to their ease of handling, low toxicity and inherent biodegradability.24 As such, lactate esters lend themselves to a variety of different sectors, from polymer and pharmaceuticals manufacturing through to the paints and agricultural chemical industry, to name but a few.25 Indeed, it is envisaged that this process will be viable at an industrial scale, both from an economic and environmentally sustainable perspective.26 Furthermore, there is the potential to transform lactate esters into platform chemicals, such as lactic acid or lactide, further promoting a circular economy approach.27–29 A number of different PLA transesterification methods have been reported in the literature.30–44 DuPont possess a patent for PLA degradation in the presence of H2SO4, achieving high conversion (69–87%) to various lactate esters (R = Me, Et and nBu) within 2 h between 150–190 °C.30 Coszach et al.17 demonstrated the hydrolysis of PLA to lactic acid, observing enhanced PLA dissolution and polymer separation with lactate esters as the solvent of choice. Hydrolysis proceeded both with or without NaOH, requiring elevated temperatures between 80–180 °C and pressures of up to 10 bar. Catalysts based on rare metals, including iridium and ruthenium, have also been reported for hydrogenation and hydrosilylation processes, but are unattractive options to industry due to a high metal cost.36–38 Both Fliedel et al.40 and McKeown et al.41 have reported systems based on Zn(II)-complexes, with the latter achieving up to 100% Me-LA conversion within 1 h at 90 °C. Interestingly, McKeown et al.42 recently demonstrated shifting to an analogous propylenediamine system afforded superior activity, achieving 81% conversion within 30 minutes at 50 °C, highlighting the influence of metal–ligand relationships. Petrus et al.43 prepared a series of lactate esters from a variety of different alcohols, reporting reaction temperatures as low as 80 °C in the presence of earth abundant metal-based catalysts, including Mg(II) and Ca(II). However, in the absence of a catalyst, reaction temperatures increased to 260 °C, thus highlighting the benefits of metal-mediated degradation, particularly from an economic and environmental standpoint. However, despite the need for simple, controlled metal-mediated degradation pathways, literature examples remain limited. There is therefore a clear opportunity to develop robust, industrially viable catalysts based on both earth abundant metals and scalable ligand systems. Herein, we report the synthesis of a series of mono- and dimeric Zn(II)-Schiff-base complexes, and their application to the ROP of rac-lactide in both solution and under industrially preferred melt conditions is demonstrated. The metal-mediated degradation of PLA into Me-LA under mild conditions is also reported, in conjunction with a reaction kinetic study.
000 Mtonnes of plastic waste will reside in either landfill or the natural environment.6 Thus, there is a clear need to develop alternative targeted waste management technologies to facilitate this transition, namely through recycling.4,7 Mechanical recycling is one possible solution but is limited by eventual material downgrading due to thermomechanical degradation, resulting in material repurposing to applications that require polymer of lower quality. A promising alternative is chemical recycling, which has the potential to reduce both waste and operational costs through material regeneration, achieved either through the depolymerisation or degradation of PLA.13 A number of different methods have been reported in the literature, including hydrolytic and thermal degradation, as well as enzymatic processes.14–23 However, the former two rely on high temperature conditions (200–400 °C), resulting in high operational costs, whilst uncertainty surrounds the scalability of the latter. Alternatively, the transesterification of PLA with alcohols to generate lactate esters has gathered appreciable momentum in recent years (Scheme 1). Low-molecular-weight lactate esters have been cited as green substitutes to traditional hydrocarbon-based solvents, owing to their ease of handling, low toxicity and inherent biodegradability.24 As such, lactate esters lend themselves to a variety of different sectors, from polymer and pharmaceuticals manufacturing through to the paints and agricultural chemical industry, to name but a few.25 Indeed, it is envisaged that this process will be viable at an industrial scale, both from an economic and environmentally sustainable perspective.26 Furthermore, there is the potential to transform lactate esters into platform chemicals, such as lactic acid or lactide, further promoting a circular economy approach.27–29 A number of different PLA transesterification methods have been reported in the literature.30–44 DuPont possess a patent for PLA degradation in the presence of H2SO4, achieving high conversion (69–87%) to various lactate esters (R = Me, Et and nBu) within 2 h between 150–190 °C.30 Coszach et al.17 demonstrated the hydrolysis of PLA to lactic acid, observing enhanced PLA dissolution and polymer separation with lactate esters as the solvent of choice. Hydrolysis proceeded both with or without NaOH, requiring elevated temperatures between 80–180 °C and pressures of up to 10 bar. Catalysts based on rare metals, including iridium and ruthenium, have also been reported for hydrogenation and hydrosilylation processes, but are unattractive options to industry due to a high metal cost.36–38 Both Fliedel et al.40 and McKeown et al.41 have reported systems based on Zn(II)-complexes, with the latter achieving up to 100% Me-LA conversion within 1 h at 90 °C. Interestingly, McKeown et al.42 recently demonstrated shifting to an analogous propylenediamine system afforded superior activity, achieving 81% conversion within 30 minutes at 50 °C, highlighting the influence of metal–ligand relationships. Petrus et al.43 prepared a series of lactate esters from a variety of different alcohols, reporting reaction temperatures as low as 80 °C in the presence of earth abundant metal-based catalysts, including Mg(II) and Ca(II). However, in the absence of a catalyst, reaction temperatures increased to 260 °C, thus highlighting the benefits of metal-mediated degradation, particularly from an economic and environmental standpoint. However, despite the need for simple, controlled metal-mediated degradation pathways, literature examples remain limited. There is therefore a clear opportunity to develop robust, industrially viable catalysts based on both earth abundant metals and scalable ligand systems. Herein, we report the synthesis of a series of mono- and dimeric Zn(II)-Schiff-base complexes, and their application to the ROP of rac-lactide in both solution and under industrially preferred melt conditions is demonstrated. The metal-mediated degradation of PLA into Me-LA under mild conditions is also reported, in conjunction with a reaction kinetic study.
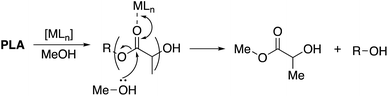 |
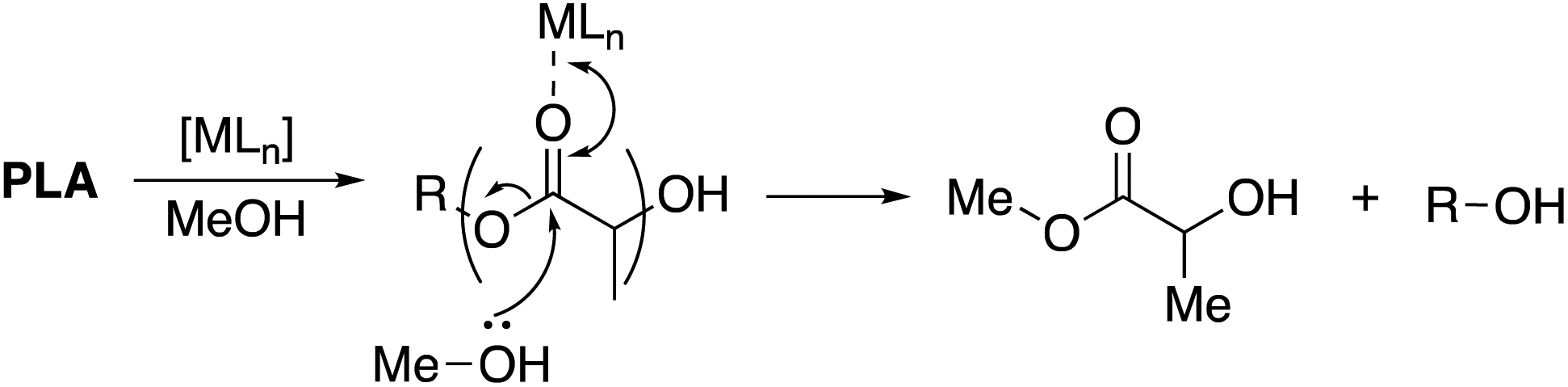
| | Scheme 1 Metal-mediated degradation mechanism of PLA into methyl lactate via transesterification with MeOH, where R denotes the growth polymer chain. | |
Results and discussion
Synthesis
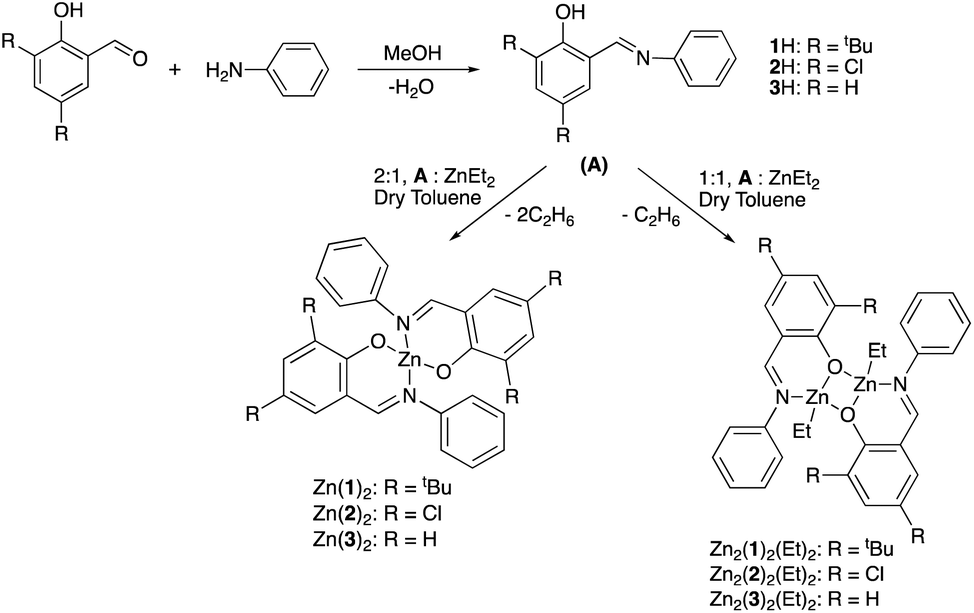
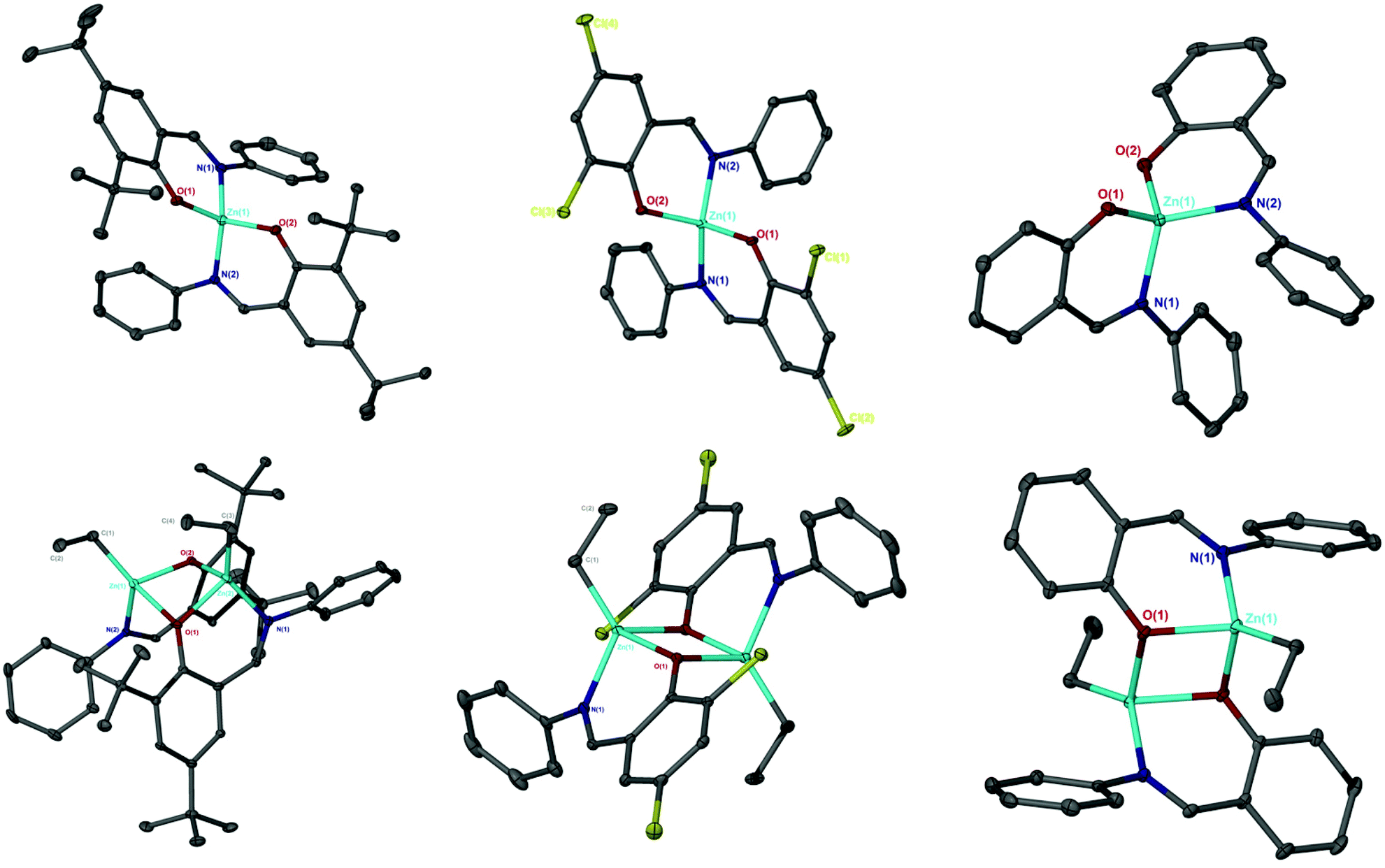
A range of simple Schiff-base ligands were prepared via an imine condensation reaction (Scheme 2) and characterised by 1H NMR spectroscopy and mass spectrometry.1H NMR singlets at ca. δ = 8 and 14 ppm were assigned to characteristic HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and O–H resonances respectively. Mono- and dimeric complexes of Zn(II) were then prepared in anhydrous toluene and purified by hexane recrystallisation or via washing (Scheme 2). To alleviate safety concerns associated with the pyrophoric alkyl precursor employed (ZnEt2), the synthesis of Zn(1)2 was attempted from Zn(OAc)2·2H2O in EtOH under reflux conditions but proved unsuccessful. In the solid state, Zn(1–3)2 and Zn2(1,3)2(Et)2 exhibited a distorted tetrahedral geometry around the metal centre, consistent with a τ4 value close to 1 (Table 1). XRD analysis revealed Zn2(1–3)2(Et)2 to adopt a phenoxy-bridged dimeric structure. In all instances, the Zn–O bond lengths increased between mono- and dimeric analogues, consistent with the observed shift from a 2c–2e− to 3c–2e− bonding system {Zn(2)2: Zn(1)–O(1) = 1.9297(11) Å, Zn2(2)2(Et)2: Zn(1)–O(1) = 2.0293(17) Å}. A Zn(1)–N(2) bond length of ca. 2 Å confirmed the retention of the imine functionality on coordination to Zn(II) for all complexes (Fig. 1), consistent with previously reported Zn(II)-complexes.45 This was reaffirmed by 1H NMR spectroscopic analysis, which exhibited a characteristic singlet peak at ca. 8 ppm corresponding to a HCN resonance. Additionally, the absence of a singlet resonance around 14 ppm, present in the isolated ligand, confirmed the loss of O–H functionality on coordination to the Zn(II) centre (Fig. 1). In all cases,1H NMR spectra suggested identical ligand coordination environments for each Zn(II)-complex in solution, with the exception of Zn2(1)2(Et)2. Four singlet peaks corresponding to tBu resonances were observed for Zn2(1)2(Et)2, suggesting multiple species present in solution. Diffusional ordered spectroscopy (DOSY) NMR analysis indicated the presence of two distinct species in solution, as a consequence of the Schlenk type equilibrium, corresponding to Zn(1)2 and Zn(1)Et with diffusion constants (D) of 0.6 × 10−9 and 0.8 × 10−9 m2 s−1 respectively (see ESI†). Zn(1)Et, instead of Zn2(1)2Et2, is proposed due to the large D observed.
N and O–H resonances respectively. Mono- and dimeric complexes of Zn(II) were then prepared in anhydrous toluene and purified by hexane recrystallisation or via washing (Scheme 2). To alleviate safety concerns associated with the pyrophoric alkyl precursor employed (ZnEt2), the synthesis of Zn(1)2 was attempted from Zn(OAc)2·2H2O in EtOH under reflux conditions but proved unsuccessful. In the solid state, Zn(1–3)2 and Zn2(1,3)2(Et)2 exhibited a distorted tetrahedral geometry around the metal centre, consistent with a τ4 value close to 1 (Table 1). XRD analysis revealed Zn2(1–3)2(Et)2 to adopt a phenoxy-bridged dimeric structure. In all instances, the Zn–O bond lengths increased between mono- and dimeric analogues, consistent with the observed shift from a 2c–2e− to 3c–2e− bonding system {Zn(2)2: Zn(1)–O(1) = 1.9297(11) Å, Zn2(2)2(Et)2: Zn(1)–O(1) = 2.0293(17) Å}. A Zn(1)–N(2) bond length of ca. 2 Å confirmed the retention of the imine functionality on coordination to Zn(II) for all complexes (Fig. 1), consistent with previously reported Zn(II)-complexes.45 This was reaffirmed by 1H NMR spectroscopic analysis, which exhibited a characteristic singlet peak at ca. 8 ppm corresponding to a HCN resonance. Additionally, the absence of a singlet resonance around 14 ppm, present in the isolated ligand, confirmed the loss of O–H functionality on coordination to the Zn(II) centre (Fig. 1). In all cases,1H NMR spectra suggested identical ligand coordination environments for each Zn(II)-complex in solution, with the exception of Zn2(1)2(Et)2. Four singlet peaks corresponding to tBu resonances were observed for Zn2(1)2(Et)2, suggesting multiple species present in solution. Diffusional ordered spectroscopy (DOSY) NMR analysis indicated the presence of two distinct species in solution, as a consequence of the Schlenk type equilibrium, corresponding to Zn(1)2 and Zn(1)Et with diffusion constants (D) of 0.6 × 10−9 and 0.8 × 10−9 m2 s−1 respectively (see ESI†). Zn(1)Et, instead of Zn2(1)2Et2, is proposed due to the large D observed.
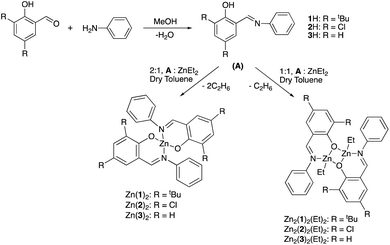 |
| | Scheme 2 Schiff-base ligand preparation and their complexation to Zn(II) to afford a series of mono-, Zn(1–3)2, and analogous dimeric, Zn2(1–3)2(Et)2, complexes. | |
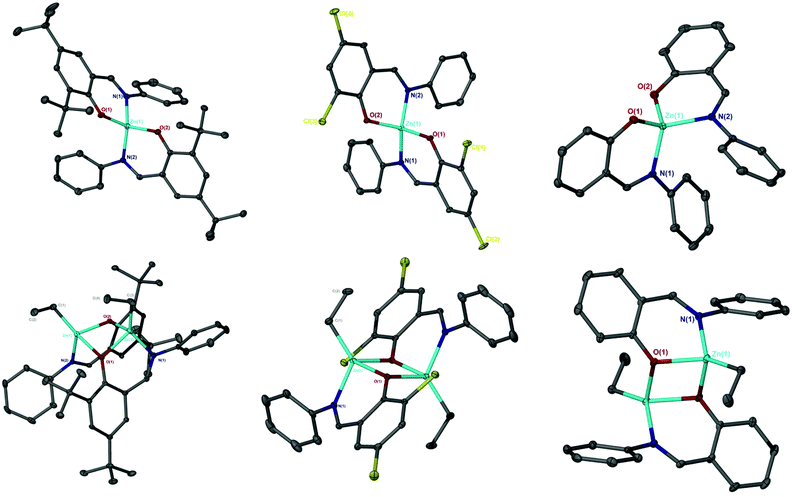 |
| | Fig. 1 Solid-state structures of Zn(1–3)2(top left to right) and Zn2(1–3)2(Et)2 (bottom left to right). Ellipsoids shown at 30% probability, with all hydrogen atoms, and one toluene molecule for Zn(1–2)2, omitted for clarity. | |
Table 1 Selected bond angles for Zn(1–3)2 and Zn2(1–3)2(Et)2 with calculated τ4 values. Ideal tetrahedral geometry corresponds to τ4 = 1
| Init. |
Bond |
Bond angle/° |
τ
4
|
| Zn(1)2 |
O(1)–Zn(1)–N(2), O(2)–Zn(1)–N(1) |
115.26(8), 155.48(8) |
0.92 |
| Zn(2)2 |
O(2)–Zn(1)–N(1), N(1)–Zn(1)–N(2) |
113.54(5), 126.19(5) |
0.85 |
| Zn(3)2 |
O(2)–Zn(1)–N(1), O(1)–Zn(1)–N(2) |
119.02(6), 121.68(6) |
0.85 |
| Zn2(1)2(Et)2 |
C(1)–Zn(1)–O(1), C(1)–Zn(1)–N(2) |
124.10(15), 132.40(16) |
0.73 |
| Zn2(2)2(Et)2 |
C(1)–Zn(1)–N(1), C(1)–Zn(1)–O(1) |
124.86(10), 129.61(10) |
0.75 |
| Zn2(3)2(Et)2 |
C(1)–Zn(1)–O(1), C(1)–Zn(1)–N(1) |
125.13(8), 134.65(8) |
0.71 |
A comparison between the 1H NMR spectra of Zn2(2–3)2Et2 with their respective monomeric counterparts indicates no presence of the homoleptic species, and thus are not susceptible to the Schlenk type equilibrium noted for Zn2(1)2(Et)2. It is proposed Zn(1)Et is stabilised by the steric bulk of 1H. For Zn2(1–3)2(Et)2, resonances in the region of ca. δ 0.5–1.5 ppm confirmed coordination of Et- to Zn(II) in solution, consistent with XRD analysis. Interestingly, trans-Et coordination was observed in Zn2(2–3)2(Et)2 compared to cis-Et in Zn2(1)2(Et)2. This is likely a consequence of reduced steric demand in Zn2(2–3)2(Et)2 compared to Zn2(1)2(Et)2.13C{1H} NMR analysis was consistent with XRD and 1H NMR data, although poor peak resolution was observed for both Zn(2)2 and Zn2(2)2(Et)2, indicative of fluxionality on the 13C {1H} NMR timescale. All Zn(II)-complexes were generally in good agreement with elemental analysis (EA) data obtained. The scalability of these complexes was also demonstrated, producing 14.6 g of Zn(2)2 in exceptional yield (87%).
Polymerisation
All Zn(II)-complexes were trialled for the polymerisation of rac-lactide (rac-LA) under both industrially preferred conditions in the absence of solvent at elevated temperatures (Tables 2 and 3) and in solution (Table 4). Catalysts that operate under industrially viable conditions are desirable since they remove the need for solvents, which are typically a significant source of waste.45–49 The lactide monomer, rac-LA, was recrystallised from toluene once prior to use, and benzyl alcohol was employed as a co-initiator. Conversion was determined via analysis of the methine region (ca. δ 4.9–5.2 ppm) by means of 1H NMR spectroscopy. All initiators demonstrated rapid polymerisation at 130 °C, achieving high conversion within minutes {[rac-LA]:[Init]:[BnOH] = 300:1:1} (Table 2). The reduction in activity of Zn(1)2 compared to Zn(2–3)2 could likely be attributed to reduced steric bulk of the substituted phenoxy-fragment. In all instances, Zn2(1–3)2(Et)2 exhibited superior activity compared to their respective monomeric counterparts. It is proposed Zn(1–3)2 operate via an activated monomer mechanism, whilst Zn2(1–3)2(Et)2 operate via a coordination- insertion mechanism once the labile Et- group is displaced by –OBn. To investigate this the stability of Zn(1)2 and Zn2(3)2(Et)2 in BnOH was studied. 1H NMR analysis revealed the former to be stable, whilst the latter converted to the corresponding BnO– analogue via elimination of the terminal –Et to afford dissolved ethane (see ESI†), consistent with the emergence of a single CH3 resonance at ca. δ = 0.80 ppm, as expected. These results are compatible with the respective suggested mechanisms. Zn(3)2 and Zn2(3)2(Et)2 achieved the highest conversions of 88% and 96% respectively within 2 minutes, suggesting reduced sterics dominates electron withdrawing inductive effects in both subseries. All initiators offered moderate number average molecular weight (Mn) control, with broad dispersities (Đ = 1.67–2.20) observed under these conditions. Reduced Mn control may also be due to rapid polymerisation (<1 min) inhibiting homogeneity, thus preventing optimal initiation. Zn(1)2 was repeated at {[rac-LA]:[Init]:[BnOH] = 300:1:6} to afford PLA of sufficiently low Mn weight for end group characterisation. MALDI-ToF analysis confirmed the polymer (Table 2, entry 2) to be –OBn and –H end-capped with multiple series present suggesting poor polymerisation control, consistent with GPC data. For all Zn(II)-initiators, polymerisation of rac-LA afforded atactic PLA (Pr ≈ 0.5). Whilst maintaining a constant rac-LA : co-initiator ratio at reduced catalyst loading {[rac-LA]:[Init]:[BnOH] = 3000:1:10} (Table 3), increasing the temperature to 180 °C resulted in a shift to very slight heterotactic PLA (Pr = 0.58). Generally, accelerated polymerisation (<1 min) was observed at higher temperatures, with the exception of Zn(1,3)2. Zn2(1–3)2(Et)2 exhibited superior activity compared to their monomeric analogues, consistent with observations at 130 °C. At 3000:1:10, moderate Mn control and broad dispersities (Đ = 1.83–2.04) were observed, consistent with observations at 130 °C. However, GPC analysis revealed both Zn(2–3)2 produced PLA of significantly higher Mn than predicted. Superior Mn control was achieved at lower polymerisation temperatures (80 °C) in solution {[rac-LA]:[Init]:[BnOH] = 100:1:1} (Table 4), producing atactic PLA (Pr ≈ 0.5) with broad dispersities (Đ = 1.21–1.83). All Zn(II)-initiators achieved high conversion (≥92%) within 10 to 30 minutes, with the exception of Zn(1)2, which achieved 70% conversion after 2 h, consistent with observations at 130 and 180 °C. MALDI-ToF analysis revealed the polymer (Table 4, entry 1) to be BnO– and –H end-capped, with molecular weight consistent with GPC and observations in the melt (see ESI†). A second series of lower intensity was observed corresponding to transesterified polymer, an initial indication of the catalysts propensity to facilitate PLA degradation.
Table 2 Polymerisation of rac-LA at 130 °C using Zn(II)-complexes
| Init. |
[rac-LA]:[I]:[BnOH] |
Time/min |
Conv.a/% |
M
n/theo
|
M
n
|
Đ
|
Prd |
| Conditions: rac-LA, solvent free (130 °C). Determined via1H NMR spectroscopy. Theoretical average number molecular weight (Mn) dependent on conversion and co-initiator added {(Mr,LA × 3 × %conv) + Mn,BnOH)}. Determined via GPC analysis (in THF). Determined via homonuclear decoupled NMR spectroscopy. Note {[I]:[BnOH] = 1:1} corresponds to 1 equivalent of BnOH per Zn centre. |
| Zn(1)2 |
300:1:1 |
9 |
69 |
29900 |
16650 |
1.86 |
0.56 |
| 300:1:6 |
10 |
90 |
6600 |
13000 |
1.46 |
0.54 |
| Zn(2)2 |
300:1:1 |
2 |
78 |
33800 |
26600 |
1.75 |
0.54 |
| Zn(3)2 |
300:1:1 |
2 |
88 |
38100 |
43700 |
1.98 |
0.54 |
| Zn2(1)2Et2 |
300:1:1 |
2 |
88 |
38100 |
25350 |
1.99 |
0.55 |
| Zn2(2)2Et2 |
300:1:1 |
<1 |
72 |
31200 |
17000 |
1.67 |
0.54 |
| Zn2(3)2Et2 |
300:1:1 |
2 |
96 |
41600 |
46600 |
2.20 |
0.53 |
Table 3 Polymerisation of rac-LA at 180 °C using Zn(II)-complexes
| Init. |
[rac-LA]:[I]:[BnOH] |
Time/min |
Conv.a/% |
M
n/theo
|
Mnc |
Đ
|
P
r
|
| Conditions: rac-LA, solvent free (180 °C). Determined via1H NMR spectroscopy. Theoretical average number molecular weight (Mn) dependent on conversion and co-initiator added {(Mr,LA × 3 × %conv) + Mn,BnOH)}. Determined via GPC analysis (in THF). Determined via homonuclear decoupled NMR spectroscopy. Note {[I]:[BnOH] = 1:1} corresponds to 1 equivalent of BnOH per Zn centre. |
| Zn(1)2 |
3000:1:10 |
18 |
47 |
20400 |
63450 |
1.96 |
0.58 |
| Zn(2)2 |
3000:1:10 |
<1 |
55 |
23850 |
124400 |
1.95 |
0.58 |
| Zn(3)2 |
3000:1:10 |
10 |
72 |
31200 |
116800 |
2.04 |
0.58 |
| Zn2(1)2Et2 |
3000:1:10 |
<1 |
92 |
39850 |
61200 |
2.04 |
0.58 |
| Zn2(2)2Et2 |
3000:1:10 |
<1 |
94 |
40700 |
54550 |
1.91 |
0.56 |
| Zn2(3)2Et2 |
3000:1:10 |
<1 |
88 |
38100 |
40000 |
1.83 |
0.58 |
Table 4 Polymerisation of rac-LA at 80 °C using Zn(II)-complexes
| Init. |
[rac-LA]:[I]:[BnOH] |
Time/h |
Conv.a/% |
M
n/theo
|
M
nc |
Đ
|
P
r
|
| Conditions: rac-LA, solvent (toluene, 80 °C). Determined via1H NMR spectroscopy. Theoretical average number molecular weight (Mn) dependent on conversion and co-initiator added {(Mr,LA × %conv) + Mn,BnOH)}. Determined via GPC analysis (in THF). Determined via homonuclear decoupled NMR spectroscopy. RT. Note {[I]:[BnOH] = 1:1} corresponds to 1 equivalent of BnOH per Zn centre. |
| Zn(1)2 |
100:1:1 |
2 |
70 |
10200 |
8800 |
1.27 |
0.46 |
| Zn(2)2 |
100:1:1 |
0.50 |
95 |
13800 |
12600 |
1.83 |
0.51 |
| Zn(3)2 |
100:1:1 |
0.50 |
96 |
13950 |
16200 |
1.73 |
0.50 |
| Zn2(1)2Et2 |
100:1:1 |
0.17 |
92 |
13350 |
12600 |
1.21 |
0.53 |
| Zn2(2)2Et2 |
100:1:1 |
0.17 |
96 |
13900 |
12650 |
1.42 |
0.55 |
| 100:1:1 |
30e |
83 |
12050 |
11900 |
1.22 |
0.59 |
| Zn2(3)2Et2 |
100:1:1 |
0.17 |
96 |
13900 |
12000 |
1.24 |
0.47 |
| 100:1:1 |
24e |
99 |
14350 |
10850 |
1.44 |
0.58 |
Interestingly, all dimers exhibited exceptional Mn control, which is considerably poorer under extended periods of polymerisation (Đ = 1.63–2.21 after 2 h), presumably due to detrimental transesterification. It was envisaged the trans-Et-configuration in Zn2(2–3)2(Et)2 would facilitate coordination of initiated polymer chains above and below the plane of the Zn(II)–O–Zn(II) framework, resulting in superior monomer selectivity. Therefore, the solution polymerisations were repeated at RT. Both Zn2(2–3)2(Et)2 exhibited a shift to very slight heterotactic PLA (Pr = 0.58–0.59) and narrower dispersities (Đ = 1.22–1.44) compared to 80 °C, suggesting enhanced control at the expense of high catalyst activity, consistent with the literature.45 Zn2(2)2(Et)2 displayed exceptional Mn control under these conditions (Mn, = 11900; Mn,theo = 12050). Reactivity trends were consistent with those observed in the melt at 130 °C, with Zn2(3)2(Et)2 achieving 99% conversion within 24 h. Under solution conditions, it is tentatively suggested the original dimeric framework of Zn2(2–3)2Et2, is retained, although there is existing literature precedent for benzoxy bridged dinuclear Zn(II)-initiators.50–53 In light of DOSY NMR analysis, presence of the Schlenk type equilibrium for Zn2(1)2Et2 inhibits the rationale for the proposed species. In summary, all Zn(II)-initiators exhibited high catalyst activity in the polymerisation of rac-LA, consistent with previous examples in the literature.41,42,45,46,50–63
Polymerisation kinetics
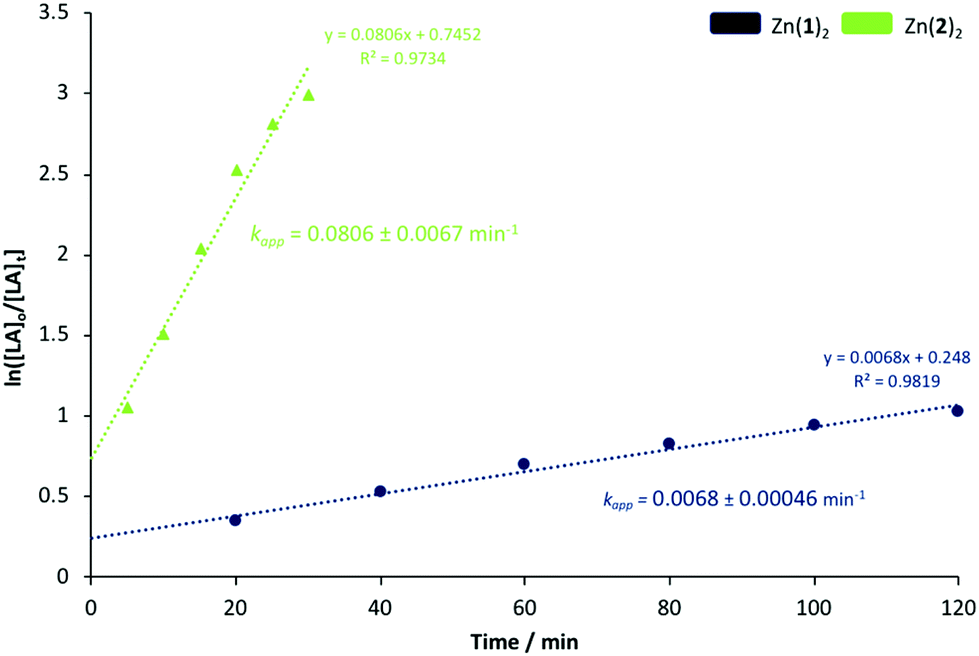
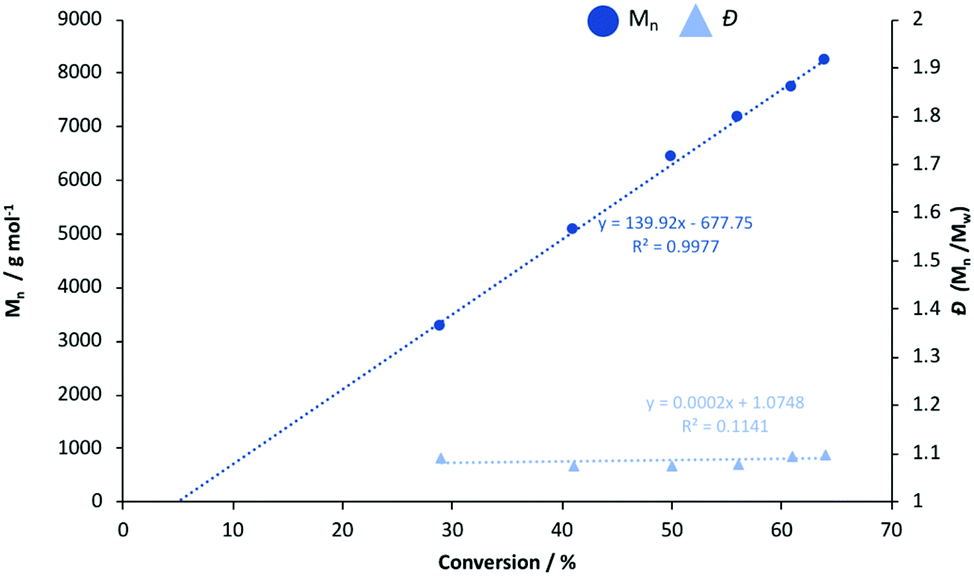
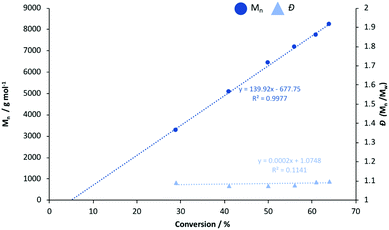
Generally, Zn2(1–3)2Et2 outperformed their monomeric counterparts under both melt and solution conditions. A mono- vs. dimeric system kinetic study was envisaged but not possible due to the extremely high activity of the latter. To this end, Zn(1)2 and Zn(2)2 were chosen to better understand the impact of steric and electronic effects on polymerisation activity in solution (Fig. 2 and 3). The consumption of rac-LA was shown to adopt first-order kinetics as demonstrated by the linear relationship of ln([LA]0/[LA]t) against time (Fig. 2). Zn(1)2 and Zn(2)2 exhibited apparent rate constants (kapp) of 0.0068 and 0.081 min−1 respectively, indicating reduced sterics, coupled with a more electron withdrawing ligand, affords enhanced polymerisation activity with statistical significance. GPC analysis of the retained aliquots for Zn(1)2 confirmed the polymerisation to be well controlled and living, demonstrated by a linear increase in Mn with conversion, whilst retaining a narrow Đ range (Fig. 3).
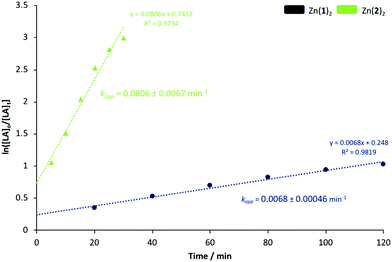 |
| | Fig. 2 First-order logarithmic plot for the polymerisation of rac-LA at 80 °C in toluene {[rac-LA]:[I]:[BnOH] = 100:1:1} using Zn(1)2 and Zn(2)2. Note [LA]o = 0.69 mol dm−3. | |
 |
| | Fig. 3
M
n and Đ against conversion for the solution polymerisation of rac-LA in toluene at 80 °C using Zn(1)2. | |
Degradation
All Zn(II)-complexes were investigated in the metal-mediated degradation of PLA into methyl lactate (Me-LA) in solution between 50–80 °C (Table 5). In addition to Me-LA being a green alternative to traditional petrochemical based solvents, it can also be directly converted to lactide and is therefore a potentially valuable chemical to the PLA supply chain.24,25,64 Commercially available polymer (0.25 g, Vegware™, PLA cup, Mn = 45510 g mol−1) and catalyst were dissolved in THF under Ar, with heat and stirring assisting dissolution. MeOH was then added, and the conversion to Me-LA was determined via1H NMR analysis of the methine region (ca. δ 4.2–5.2 ppm). We have previously demonstrated the production of Me-LA proceeds via a two-step process through the intermediate formation of chain-end groups (see ESI†).41 As such, the methine groups can be categorised as internal (int), chain end (CE) and those corresponding directly to the alkyl lactate (Me-LA), as denoted in Table 5. In general, reasonably good catalyst activity was observed under milder reaction conditions compared to previously reported recycling systems.14,15,18,33,37,38,43,65–69 In all instances, an increase in temperature coincided with higher Me-LA yield (YMe-LA) and shorter reaction times, consistent with the literature.35,41,43 Zn(2)2 exhibited superior activity compared to Zn(1,3)2 at 50 °C, suggesting inductive effects dominate, whilst Zn(1)2 exhibited the lowest degradation activity, both consistent with polymerisation results (Table 3). As noted previously, a shift to the corresponding dimeric analogue, Zn2(1)2(Et)2, coincided with superior activity at both 50 and 80 °C, with the exception of Zn(2–3)2. This was likely due to reduced sterics in the latter two rendering the dimeric counterparts more susceptible to decomposition at 50 °C, resulting in deactivation. It is suggested this effect was remediated by an increase in rate of reaction at 80 °C. Indeed, both Zn(2–3)2 and Zn2(3)2(Et)2 achieved 100% conversion to Me-LA within 8 h at 80 °C. Overall, mass transfer limitations due to polymer particle size and stirring speed were considered to be negligible based on previously reported work by McKeown et al.41 on an analogous Zn(II)-imino monophenolate system.
Table 5 Degradation of PLA into Me-LA using Zn(II)-complexesa,b
| Init. |
Time/h |
T/°C |
Y
Me-LA/% |
Y
CE/% |
X
int/% |
|
Reaction conditions: 0.25 g of PLA, VTHF:VMeOH = 4:1, [Zn(1–3)2/Zn2(1–3)2(Et)2] = 8 wt% cat. loading, nMeOH:nester = 7:1.
Me-LA and oligomer yield (YMe-LA and YCE respectively) determined by 1H NMR upon solvent (THF) removal.
|
| Zn(1)2 |
18 |
50 |
41 |
22 |
37 |
| 8 |
80 |
54 |
18 |
28 |
| Zn(2)2 |
18 |
50 |
88 |
6 |
6 |
| 8 |
80 |
100 |
0 |
0 |
| Zn(3)2 |
18 |
50 |
77 |
13 |
10 |
| 8 |
80 |
100 |
0 |
0 |
| Zn2(1)2(Et)2 |
18 |
50 |
57 |
17 |
26 |
| 8 |
80 |
81 |
11 |
8 |
| Zn2(2)2(Et)2 |
18 |
50 |
71 |
13 |
16 |
| 8 |
80 |
88 |
7 |
5 |
| Zn2(3)2(Et)2 |
18 |
50 |
52 |
21 |
27 |
| 8 |
80 |
100 |
0 |
0 |
Degradation kinetics
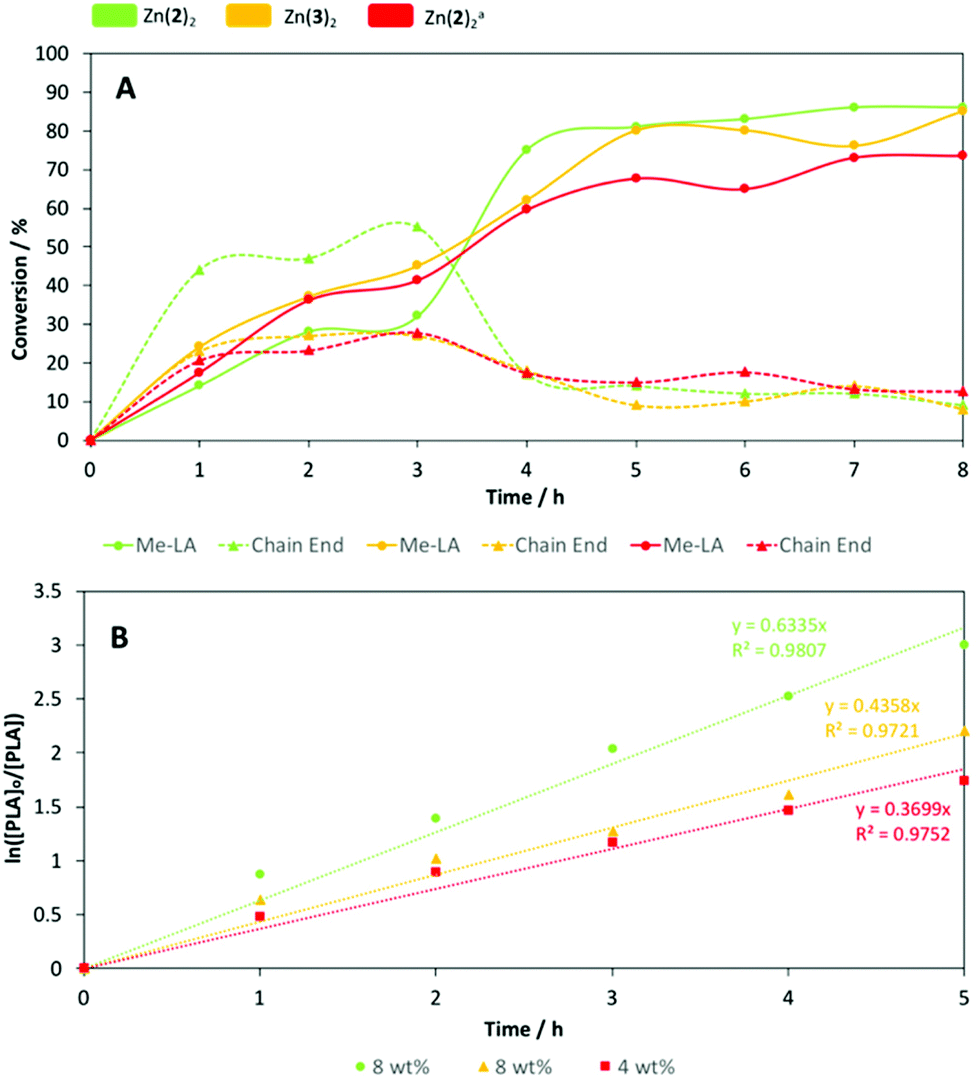
Both Zn(2)2 and Zn(3)3 were chosen for further analysis owing to their high degradation activity, stability and potential scalability. To ensure sufficiently short reaction times, degradation reactions were repeated under identical conditions at 80 °C (Table 5). Reaction progress was monitored hourly by taking aliquots for 1H NMR (CDCl3) analysis of the methine region (Fig. 4). Zn(2–3)2 achieved comparable yields of Me-LA after 8 h, although 100% conversion to Me-LA was not achieved as previously noted (Table 5). PLA degradation using Zn(2)2 was also tested at 4 wt%. Interestingly, at a lower catalyst loading, a higher Me-LA conversion was achieved within 3 h compared to 8 wt%, suggesting preferential transesterification of oligomer compared to polymer. However, the general degradation profile was retained, albeit with a less severe rapid conversion event, likely due to a shift in transesterification selectivity. As expected, a lower overall Me-LA conversion (78%) was observed compared to Zn(2)2 at 8 wt%. In all instances, fluctuations in Me-LA and oligomer yield (YMe-LA and YCE respectively) can likely be attributed to an equilibrium process.70 In accordance to kinetic work by Mckeown et al.,41 PLA consumption was presumed to adopt pseudo-first-order kinetics (Fig. 2b). Thus, the gradient of the logarithmic plot is equivalent to the apparent rate constant, kapp (Table 6). Zn(2–3)2 exhibited kapp values of 0.63 ± 0.051 and 0.44 ± 0.029 h−1 respectively at 8 wt%, indicating reduced sterics and electronics imparts a statistically significant reduction in activity. For Zn(2)2, a shift to 4 wt% coincided with a statistically significant reduction in kapp (0.37 ± 0.021 h−1) as expected.
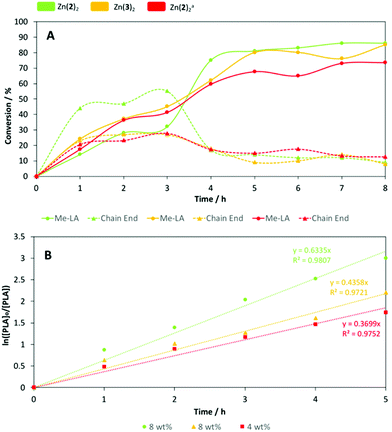 |
| | Fig. 4 (A) Vegware PLA cup degradation plot of conversion vs. time with Zn(2–3)2 at 8 wt% in THF at 80 °C. a [Zn(2)2] = 4 wt%. (B) Pseudo-first-order logarithmic plots for the degradation of Vegware PLA cup with Zn(2–3)2 in THF at 80 °C. Line of best fit constrained to pass through the origin. | |
Table 6 PLA cup degradation with Zn(2–3)2 in THF at 80 °Ca,b,c
| Init. |
Y
Me-LAb/% |
k
app/h−1 |
|
Reaction conditions: 0.25 g of PLA, VTHF:VMeOH = 4:1, [Zn(2–3)2] = 8 wt% cat. loading, nMeOH:nester = 7:1.
Maximum Me-LA conversion (YMe-LA) determined via1H NMR (CDCl3) analysis after 8 h upon solvent (THF) removal.
Error associated with kapp using linear regression.
[Zn(2)2] = 4 wt% cat. loading.
|
| Zn(2)2 |
87 |
0.63 ± 0.051 |
| Zn(3)2 |
82 |
0.44 ± 0.029 |
| Zn(2)2d |
78 |
0.37 ± 0.021 |
Conclusion
A series of novel mono- and dimeric Zn(II)-Schiff-base complexes were successfully synthesised and fully characterised. Their application in the ROP of rac-LA to produce biocompatible atactic PLA was demonstrated in both solution and under industrially preferred melt conditions, albeit with generally poor Mn control and broad dispersities (Đ). Additionally, their capacity to facilitate PLA degradation into Me-LA under mild conditions was shown. Zn(2)2 and Zn(3)2 emerged as the outstanding candidates, with both achieving 100% conversion to Me-LA within 8 h at 80 °C in THF. Further degradation kinetic analysis revealed Zn(2–3)2 to possess kapp values of 0.63 ± 0.051 and 0.44 ± 0.029 h−1 respectively at 8 wt%, indicating steric and electronic effects have a statistically significant impact on degradation activity. A statistically significant reduction in kapp was observed for Zn(2)2 between 8 and 4 wt%.
Experimental
An exemplar synthesis is provided below, see ESI† for full details.
Ligand
1H: To a solution of 3,5-di-tert-butyl-2-hydroxybenzaldehyde (4.68 g. 20 mmol) dissolved in MeOH (50 mL), aniline (1.86 g, 1.82 mL, 20 mmol) was added with stirring. The reaction mixture was then left to stir at RT for 18 h, affording a yellow solid product. The product was then separated by filtration, washed with MeOH (5 × 5 mL) and dried in vacuo. Yield = 4.74 g, 77%.
Zn(II)-Complex
Zn(1)2: To a solution of 1H (0.62 g, 2 mmol) dissolved in anhydrous toluene (10 mL), ZnEt2 (1 M in hexane, 1 mmol) was added dropwise with stirring. After complete addition, the solution was stirred at RT for 15 minutes before being left to stand for 1 h. The solvent was then removed via cannula filtration and the desired Zn(II) complex was recrystallised from hexane as yellow crystals. Yield = 0.50 g, 73%.
Polymerisation
Polymerisations were conducted in a Youngs ampoule under argon. rac-LA was recrystallised once from toluene prior to use. All melt polymerisations were performed in the absence of solvent. The reaction commenced on melting of the monomer and deemed finished once a polymer melt of sufficient viscosity stopped the stirrer bar. The reaction was then quenched in air and the product dissolved in DCM (20 mL) with stirring. The solvent was then removed in vacuo and a crude 1H NMR spectrum of the polymer was obtained. The polymer was then washed with copious amounts of MeOH (100 mL) to remove initiator and any unreacted monomer, dried in vacuo and retained for materials characterisation. Solution polymerisations were conducted in anhydrous toluene (10 mL) using the purification method described for the melt.
Degradation
All reactions were performed in a Youngs ampoule under argon. The flask was loaded with Zn(II) catalyst (8 wt% – 1 mol% relative to PLA ester linkages, 0.02 g) in a Glovebox to which PLA (0.25 g, Vegware™, PLA cup, Mn = 45510 g mol−1) was added under a flow of argon. The polymer was then dissolved in THF (4 mL), with heating and stirring assisting dissolution. The flask was then submerged in a preheated oil bath (50 or 80 °C) to which MeOH (1 mL) was added. Aliquots were taken for 1H NMR (CDCl3) analysis of the methine region. After the reaction, the solvent was removed in vacuo and the residual methyl lactate (Me-La) was analysed further.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We wish to thank the EPSRC for funding and the University of Bath and MC2 for use of their analysis facilities. We would like to thank the EPSRC for funding (EP/L016354/1) for a PhD studentship to JP and (EP/P016405/1) for PM.
References
-
European Bioplastics, Facts and Fig. 2016, http://docs.europeanbioplastics.org/2016/publications/EUBP_Facts_and_Figures_2017.pdf, (Accessed: 30th October 2018) Search PubMed.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
-
Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the future of plastics, 2016, https://www.ellenmacarthurfoundation.org/publications/the-new-plastics-economy-rethinking-the-future-of-plastics, (Accessed: 7th January 2019) Search PubMed.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 1–5 Search PubMed.
-
Ellen MacArthur Foundation, The New Plastics Economy: Catalysing Action, 2016, https://www.ellenmacarthurfoundation.org/assets/downloads/New-Plastics-Economy_Catalysing-Action_13–1–17.pdf, (Accessed: 9th January 2019) Search PubMed.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner, B. Springs, R. P. O'Connor, J. Kolstad and P. R. Gruber, Macromol. Biosci., 2004, 4, 551–564 CrossRef CAS PubMed.
- E. T. H. Vink, D. A. Glassner, J. Kolstad, R. J. Wooley and R. P. O'Connor, Biotechnology, 2007, 3, 58–81 CAS.
- M. Dusselier, P. V. Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- R. De Clercq, M. Dusselier, C. Poleunis, D. P. Debecker, L. Giebeler, S. Oswald, E. Makshina and B. F. Sels, ACS Catal., 2018, 8, 8130–8139 CrossRef CAS.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- H. Tsuji, H. Daimon and K. Fujie, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS PubMed.
- Y. Aoyagi, K. Yamashita and Y. Doi, Polym. Degrad. Stab., 2002, 76, 53–59 CrossRef CAS.
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS PubMed.
-
P. Coszach, J.-C. Bogaert and J. Willocq, US Pat, 8431683B2, 2013 Search PubMed.
- H. Tsuji, T. Saeki, T. Tsukegi, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2008, 93, 1956–1963 CrossRef CAS.
- V. Piemonte and F. Gironi, J. Polym. Environ., 2013, 21, 313–318 CrossRef CAS.
- C. F. Van Nostrum, T. F. J. Veldhuis, G. W. Bos and W. E. Hennink, Polymer, 2004, 45, 6779–6787 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460–2466 CrossRef CAS.
- S. Lazzari, F. Codari, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2014, 110, 80–90 CrossRef CAS.
- K. Odelius, A. Höglund, S. Kumar, M. Hakkarainen, A. K. Ghosh, N. Bhatnagar and A. C. Albertsson, Biomacromolecules, 2011, 12, 1250–1258 CrossRef CAS PubMed.
- C. T. Bowmer, R. N. Hooftman, A. O. Hanstveit, P. W. M. Venderbosch and N. van der Hoeven, Chemosphere, 1998, 37, 1317–1333 CrossRef CAS PubMed.
- C. S. M. Pereira, V. M. T. M. Silva and A. E. Rodrigues, Green Chem., 2011, 13, 2658–2671 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Y. Fan, C. Zhou and X. Zhu, Catal. Rev.: Sci. Eng., 2009, 51, 293–324 CrossRef CAS.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
-
L. D. Brake, US Pat, 5264617, 1993 Search PubMed.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- X. Song, Z. Bian, Y. Hui, H. Wang, F. Liu and S. Yu, Polym. Degrad. Stab., 2019, 168, 108937–108944 CrossRef CAS.
- X. Song, H. Wang, X. Zheng, F. Liu and S. Yu, J. Appl. Polym. Sci., 2014, 131, 40817–40823 CrossRef.
- F. Nederberg, E. F. Connor, T. Glausser and J. L. Hedrick, Chem. Commun., 2001, 2066–2067 RSC.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, 1–8 Search PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. McIlrath, A. A. Fischer and M. J. Carney,
et al.
, Chem. Commun., 2014, 50, 4884–4887 RSC.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- P. McKeown, L. A. Román-Ramírez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 12, 5233–5238 CrossRef CAS PubMed.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- H. Liu, X. Song, F. Liu, S. Liu and S. Yu, J. Polym. Res., 2015, 22, 135–141 CrossRef.
- P. McKeown, S. N. McCormick, M. F. Mahon and M. D. Jones, Polym. Chem., 2018, 9, 5339–5347 RSC.
- M. Fuchs, S. Schmitz, P. M. Schäfer, T. Secker, A. Metz, A. N. Ksiazkiewicz, A. Pich, P. Kögerler, K. Y. Monakhov and S. Herres-Pawlis, Eur. Polym. J., 2020, 122, 109302–109308 CrossRef.
- P. M. Schäfer, M. Fuchs, A. Ohligschläger, R. Rittinghaus, P. McKeown, E. Akin, M. Schmidt, A. Hoffmann, M. A. Liauw, M. D. Jones and S. Herres-Pawlis, ChemSusChem, 2017, 10, 3547–3556 CrossRef PubMed.
- J. Börner, I. Dos Santos Vieira, A. Pawlis, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2011, 17, 4507–4512 CrossRef PubMed.
- J. Börner, U. Florke, K. Huber, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362–2376 CrossRef PubMed.
- H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS.
- H.-L. Chen, H.-J. Chuang, B.-H. Huang and C.-C. Lin, Inorg. Chem. Commun., 2013, 35, 247–251 CrossRef CAS.
- W.-C. Hung, Y. Huang and C.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6466–6476 CrossRef CAS.
- D. Jędrzkiewicz, G. Adamus, M. Kwiecień, Ł. John and J. Ejfler, Inorg. Chem., 2017, 56, 1349–1365 CrossRef PubMed.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 222–224 RSC.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS PubMed.
- M. H. Chisholm and K. Phomphrai, Inorg. Chim. Acta, 2003, 350, 121–125 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS.
- S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS PubMed.
- D. E. Stasiw, A. M. Luke, T. Rosen, A. B. League, M. Mandal, B. D. Neisen, C. J. Cramer, M. Kol and W. B. Tolman, Inorg. Chem., 2017, 56, 14366–14372 CrossRef CAS PubMed.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. A. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- P. P. Upare, Y. K. Hwang, J.-S. Chang and D. W. Hwang, Ind. Eng. Chem. Res., 2012, 51, 4837–4842 CrossRef CAS.
- A. C. Sanchéz and S. R. Collinson, Eur. Polym. J., 2011, 47, 1970–1976 CrossRef.
- C. Alberti, N. Damps, R. R. R. Meißner, M. Hofmann, D. Rijono and S. Enthaler, Adv. Sustainable Syst., 2020, 4, 1900081 CrossRef CAS.
- C. Alberti, N. Damps, R. R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845–6848 CrossRef CAS.
- A. Plichta, P. Lisowska, A. Kundys, A. Zychewicz, M. Dębowski and F. Florjańczyk, Polym. Degrad. Stab., 2014, 108, 288–296 CrossRef CAS.
- K. Hirao and H. Ohara, Polym. Rev., 2011, 51, 1–22 CrossRef CAS.
- D. Bykowski, A. Grala and P. Sobota, Tetrahedron Lett., 2014, 55, 5286–5289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the experimental protocols with selected spectra and raw data. CCDC 1972259–1972264. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py00192a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab