
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticles decorated with folate based on a site-selective αCD-rotaxanated PEG-b-PCL copolymer for targeted cancer therapy†
Giovanni
Dal Poggetto
 a,
Salvatore Simone
Troise
ab,
Claudia
Conte
c,
Roberta
Marchetti
b,
Francesca
Moret
d,
Alfonso
Iadonisi
b,
Alba
Silipo
b,
Rosa
Lanzetta
b,
Mario
Malinconico
a,
Fabiana
Quaglia
*c and
Paola
Laurienzo
*a
a,
Salvatore Simone
Troise
ab,
Claudia
Conte
c,
Roberta
Marchetti
b,
Francesca
Moret
d,
Alfonso
Iadonisi
b,
Alba
Silipo
b,
Rosa
Lanzetta
b,
Mario
Malinconico
a,
Fabiana
Quaglia
*c and
Paola
Laurienzo
*a
aInstitute for Polymers, Composites and Biomaterials, CNR, Via Campi Flegrei 34, 80078 Pozzuoli, Napoli, Italy. E-mail: paola.laurienzo@ipcb.cnr.it
bDepartment of Chemical Sciences, University of Naples Federico II, Via Cintia 4, I-80126 Napoli, Italy
cDrug Delivery Laboratory, Department of Pharmacy, University of Napoli Federico II, Via Domenico Montesano 49, 80131 Napoli, Italy
dDepartment of Biology, University of Padova, Via Ugo Bassi 58/B, 35121 Padova, Italy
First published on 18th May 2020
Abstract
Nanoparticles (NPs) made up of biodegradable block copolymers offer significant potential in the field of innovative anticancer therapies. However, the accumulation of NPs in tumour cells remains challenging. Surface decoration with targeting ligands recognizing specific receptors that are overexpressed on the cancer cell membrane represents the most common approach, but the effective exposure of the targeting molecule on the NP surface is often limited. An original method to favor the exposure of folate (Fol) as a targeting ligand on the NP surface is reported here. We focused on the introduction in NPs based on a PEG-b-PCL diblock copolymer of Fol-PEG-b-PCL in which the PEG chain is threaded with α-cyclodextrin (αCD). This selectively rotaxanated copolymer (Fol-PEG(αCD)-b-PCL) was prepared from an α,ω-azido-tosyl-PEG(αCD) pseudorotaxane, taking advantage of the different terminal groups of PEG. Fol and the PCL block, which act as caps to prevent αCD dethreading, were covalently linked to PEG with a “one-step” procedure, giving a rotaxanated copolymer with a high yield and a low host coverage (∼9%). The formation of the inclusion complex was confirmed by 2D-DOSY spectroscopy. Rotaxanated copolymers were fully characterized by elemental analysis, FTIR and 1H NMR spectroscopy, thermal and thermogravimetric analysis, wide angle X-ray diffraction and polarised optical microscopy. Finally, NPs fabricated from a mixture of PEG-b-PCL and Fol-PEG(αCD)-b-PCL showed, as preliminary results, promising internalisation in KB cells and a good active targeting effect.
Introduction
Nanotechnologies promise to drive a drug to tumours with an improvement of the activity profile, alleviation of side effects and increased patient survival. Core–shell nanoparticles (NPs) made up of biodegradable block copolymers able to self-assemble in solution have attracted enormous attention in the field of innovative anticancer therapies.1–3 The most common approach to improving their accumulation in tumour cells remains surface decoration with targeting ligands recognizing specific receptors that are overexpressed on the cell membrane of endothelial cells in the tumour capillary bed or cancer cells. Nevertheless, a significant challenge that remains is the adequate exposure of the targeting molecule on the NP surface4,5 that can greatly change depending on the mode of the copolymer assembly.When dealing with nanoprecipitation, which is the most common method to produce NPs, the polarity of the solvent/antisolvent system strictly controls the solubility of hydrophilic/hydrophobic segments in the copolymer. In aqueous solution, the insoluble hydrophobic block typically collapses to form discrete NPs where the hydrophilic blocks extend into the solution. In the case of PEG-modified diblock copolymers, the peculiar solubility profile of PEG, which is soluble in both aqueous and organic solvents, can generate NPs where PEG segments are segregated in the lipophilic core and thus not exposed completely on the NP surface.6,7 Moreover, when lipophilic targeting moieties (e.g., folate) are covalently linked to the –OH end of PEG chains, PEG segregation in the hydrophobic blocks can further increase, thus subtracting targeting elements from the NP surface.8
In the context of folate-decorated NPs able to deliver a drug cargo to cancer cells overexpressing the folate receptor (FR)α, we tried to tackle this drawback by encouraging folate location on the surface of biodegradable poly(ethylene glycol)-poly(ε-caprolactone) (PEG-b-PCL) NPs via non-covalent interactions of folate with the hydroxypropyl derivative of β-cyclodextrins (CDs).5 We interestingly found out that CDs not only promoted folate exposure but also were able to increase the number of PEG chains on the NP surface through non-covalent interactions of PEG-CDs.
Supramolecular structures consisting of macrocycles as CDs threaded on a linear polymer are emerging in biomaterial research. When CDs are blocked by bulky groups linked at the ends of a polymer, polyrotaxanes are formed,9,10 while in the absence of end stoppers, pseudo-polyrotaxanes originate. The specific nomenclature is based on the number of CDs per polymer chain.11 When the number of threaded macrocycles is higher than 5 or unknown, the formed structures are termed polyrotaxanes, otherwise they are referred to as rotaxanes. In a pioneering study by Harada and Kamachi, pseudo-polyrotaxanes could form spontaneously from αCDs and PEG mixtures in an aqueous environment.12 Since then, CD-based polyrotaxanes derived from PEG have been developed to encompass a broad range of medical applications, from erodible hydrogels to drug nanocarriers.13–15 NPs for drug delivery were fabricated from self-assembling amphiphilic polyrotaxanes, obtained by conjugation of hydrophobic bulky groups to PEG terminals.16 Targeting moieties were conjugated to the hydroxyl groups of CDs to enhance ligand–receptor recognition. It was reported that the flexible motion of CDs, which could slide along and rotate around the PEG chains, avoided the mismatch of ligand–receptor interaction.17,18 Herein, our approach is based on the hypothesis that αCDs locking on PEG chains impart a more extended conformation to the chains, promoting the exposure of Fol-targeted PEG brushes. We prepared a Fol-PEG-b-PCL copolymer in which the PEG segment is threaded with αCDs through a novel synthetic strategy. To date, αCD-threaded PEG polyrotaxanes (PEG(αCD)) have been prepared directly from PEG diol or by first converting the hydroxyl end groups to more reactive functional groups.10 Hence, PEG polyrotaxanes bearing identical bulky terminal groups were obtained. Here, we have developed a new strategy to convert an α,ω-hetero-bifunctional PEG(αCD) pseudo-rotaxane to a Fol-PEG(αCD)-b-PCL polyrotaxane, taking advantage of the different terminal functional groups of PEG. PCL (4 kDa molecular weight) and Fol, able to act as bulky stoppers, were bonded respectively via “click” chemistry on one side, and via a classical nucleophilic substitution on the other side.
Rotaxanated copolymers were analysed by solubility tests, Fourier Transform Infrared (FTIR), 1H NMR and 2D Diffusion Ordered (DOSY) spectroscopy, Gel Permeation Chromatography (GPC), Differential Scanning Calorimetry (DSC), Wide Angle X-ray Diffraction (WAXD), Thermogravimetric Analysis (TGA), and polarised optical microscopy. The composition was determined by elemental analysis. NPs were prepared with a mixture of mPEG1k-b-PCL4k and Fol-PEG1.5k(αCD)-b-PCL4k by nanoprecipitation. The different lengths of the PEG block in the Fol-modified copolymer and mPEG-b-PCL (respectively, 1.5 and 1 kDa) encourage accumulation in cancer cells overexpressing FRα, as we demonstrated in a previous work.6 For comparison, NPs were prepared from mPEG1k-b-PCL4k and from a mixture of mPEG1k-b-PCL4k and Fol-PEG1.5k-b-PCL4k (without αCD threading). NPs were characterised for size, polydispersity, surface charge and PEG shell thickness. The amount of PEG and αCD on the NP surface was calculated by 1H NMR. Finally, uptake studies on KB cancer cells overexpressing FRα were carried out to verify the occurrence of Fol-mediated endocytosis of the various NPs.
Experimental
Materials
Poly(ethylene glycol), HO–PEG–OH, with Mn 1.5 kDa (PEG1.5k Sigma-Aldrich, Milan, Italy) and monomethoxy-poly(ethylene glycol), CH3O–PEG–OH, with Mn 1.0 kDa (mPEG1.0k, Nanocs Inc., New York, USA) were dehydrated by azeotropic distillation with dry toluene in a Dean–Stark trap. α-Cyclodextrin (αCD) and all reagents were purchased from Sigma-Aldrich (Milan, Italy). ε-Caprolactone (CL) was distilled over CaH2 under vacuum. Tin(II) bis(2-ethylhexanoate) (Sn(Oct)2), tosyl chloride (TsCl), folic acid (Fol), sodium hydride and sodium azide were used without further purification. 3-Butyn-1-ol was anhydrified through silica gel. Copper wires (Carlo Erba, Milan, Italy) were treated with H2SO4 for 3 minutes just before the reaction, repeatedly washed with water and methanol, and finally dried under vacuum in an oven for 30 minutes at 60 °C. All solvents (Sigma-Aldrich, Milan, Italy) were dried before use according to standard procedures. Milli-Q H2O was used throughout the work.Synthesis of mPEG1k-b-PCL4k
The linear diblock copolymer was prepared by ring-opening polymerization (ROP) of CL at 120 °C for 24 h using mPEG1.0k as the initiator and Sn(Oct)2 as the catalyst (20% mol). CL/initiator molar ratio = 36. 1H NMR (CDCD3, δ in ppm), ε-PCL block: 1.38 (72 H, m), 1.65 (144 H, m), 2.30 (72 H, t), 4.10 (72 H, t), 4.31 (2 H, t); PEG block: 3.64 (88 H, s), 3.38 (3 H, t). Mn determined by 1H NMR = 5160 Da.Synthesis of butynyl-PCL4k
Butynyl-PCL4k was synthesized by ROP using 3-butyn-1-ol as the initiator and Sn(Oct)2 as the catalyst (20% mol). Yield 99.6%. 1H NMR (CDCD3, δ in ppm), 1.37 (72 H, m), 1.64 (144 H, m), 2.29 (72 H, t), 3.63 (2 H, t), 4.1 (72 H, t). Mn determined by 1H NMR = 4168 Da.Synthesis of α-tosyl-ω-azido PEG1.5k (TsO–PEG1.5k–N3)
Monoazido-PEG1.5k (N3–PEG1.5k–OH) was prepared as described in a previous paper.6 In a round flask under an argon stream, 1.2 g (0.8 mmol) of N3–PEG1.5k–OH were dissolved under stirring in dry THF (15 mL). 274 μL (2.0 mmol) of N,N-diethylethanamine (NEt3) and 375 mg (2.0 mmol) of TsCl were added. The reaction was carried out at 25 °C under vigorous stirring for 24 h. The solution was first filtered on paper, concentrated using a rotovapor and then precipitated in cold n-hexane. After hexane removal, the product was dried under vacuum overnight. Yield: 87.5%. 1H NMR (d6-DMSO, δ in ppm): 7.79 (2 H, d), 7.49 (2 H, d), 4.12 (2 H, t), 3.51 (126 H, s), 3.40 (2 H, m), 2.41 (3 H, s).Synthesis of the Fol-targeted PEG-b-PCL copolymer (Fol-PEG1.5k-b-PCL4k)
Fol was treated with NaH before the reaction. 97.6 mg (0.22 mmol) of Fol and 8.0 mg (0.33 mmol) of dry NaH were dissolved in DMSO (3 mL) under an Ar stream at T = 45 °C. The reaction was allowed to proceed until complete H2 evolution. In a second flask, 40 mg of TsO–PEG1.5k–N3 were dissolved in DMSO (3 mL) under an Ar stream at T = 45 °C. 92 mg of Cu and 121.6 mg (0.03 mmol) of butynyl-PCL were then added under stirring. Fol solution was added dropwise. The reaction was carried out at 45 °C under vigorous stirring for 48 h. The solution was poured in 60 mL of acetone. After filtration, the solvent was removed under vacuum to recover the product. Yield: 94%. Fol functionalisation degree: 93%. 1H NMR (CDCl3, δ in ppm): ε-PCL block: 1.38 (72 H, m), 1.65 (144 H, m), 2.30 (t, 72 H), 4.1 (72 H, t); PEG block: 3.4 (2H, t), 3.5 (127H, s, PEG backbone), 3.6 (2H, t), Mn of PCL evaluated by 1H NMR = 4.6 kDa. Elemental analysis. Theoretical: C% 59.99; H% 8.55; (O + N)% 31.44. Found: C%56.8; H%8.8; (O + N)% 34.4.Preparation of the TsO–PEG1.5k(αCD)–N3 pseudo-rotaxane
In a round flask, 10.2 g (10.5 mmol) of αCD were dissolved in H2O (80 mL) at room temperature under a N2 atmosphere with vigorous magnetic stirring for 20 min. The αCD solution was poured in a second flask containing a solution of 1.0 g (0.618 mmol) of TsO–PEG–N3 in H2O (40 mL) and then stirred overnight at room temperature (αCD concentration in the final solution: 8% w/v). A white precipitate (inclusion complex, IC, of PEG and αCD) appeared after around 30 min. Water was removed at 40 °C under an air stream for 4 h, and the recovered product was dried in a vacuum oven at 40 °C until constant weight.Synthesis of the Fol-PEG1.5k(αCD)-b-PCL4k rotaxane
Two different protocols are reported. In the first case, the end-capping reagents were added simultaneously to the pseudo-rotaxane; in the second case, the end-capping reactions were performed in two separate steps.Copolymer characterisation
Elemental analysis was performed with a LECO Italy Mod 840 analyser. Fourier Transform Infrared (FTIR) analysis was performed with a PerkinElmer spectrometer (Paragon 500) equipped with a ZnSe attenuated total reflectance (ATR) crystal accessory. Samples were placed in direct contact with the ATR crystal and pressed with a pressure clamp positioned over the crystal/sample area to allow intimate contact between the material and the crystal. Spectra were acquired in the 4000–400 cm−1 range, at a resolution of 2 cm−1 (average of 20 scans). Gel Permeation Chromatography (GPC) analyses were performed in THF at 35 °C and a flow rate of 0.8 mL min−1 with a Malvern-Viscotek GPC MAX/TDA 305 quadruple detector array, using a precolumn and two Phenogel columns (Phenomenex, USA) with exclusion limits of 103 and 500 Da, respectively. Samples (100 μL) were filtered through a 0.22 μm PTFE membrane filter before injection. Triple detector calibration was based on a standard of polystyrene with a molecular weight of 104![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 959 Da. 1H NMR spectra were obtained on a Bruker DRX-400 spectrometer using 5 mm tubes. Sample concentrations were about 0.7% (w/v). All spectra were measured with 16 accumulations and a 10 s recycle delay. Diffusion-ordered NMR spectroscopy (DOSY) experiments were carried out at 298 K for R2 and 313 K for R1 on a Bruker 600 DRX equipped with a cryo-probe. The concentration of the samples was about 0.7% (w/v) in d6-DMSO. The ledbpgp2s pulse sequence from the Bruker library, incorporating a bipolar gradient pulse pair and 2 spoil gradients, with a longitudinal eddy current delay, was run with a linear gradient (5.35 G cm−1) stepped between 2.0% and 95.0%, incremented in 32 steps. The gradient duration (δ) was held constant at 2 ms and the echo delay (Δ) at 200 ms. After Fourier transformation and baseline correction of the 1D 1H spectra (F2 dimension), the diffusion dimension (F1) of the 2-dimensional DOSY was processed using TopSpin software (version 2.1). Differential Scanning Calorimetry (DSC) analysis (Q2000 TA Instruments) was performed under a nitrogen flow with a 2 °C min−1 scanning rate. The sample (about 4 mg) sealed in an aluminum crucible was heated from −20 to 90 °C, cooled at −20 °C and finally heated again to 90 °C. X-ray powder diffraction profiles were obtained at room temperature with Ni filtered CuKa radiation using an X-Pert automatic diffractometer from PANalytical. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer Pyris Diamond TG-DTA apparatus from 25 to 500 °C under a nitrogen flow (50 ml min−1) at 10 °C min−1 heating rates. Polarised optical microscopy was performed on an Axioscop-Zeiss instrument equipped with a THMS 600 hot stage and a Linkam TMS 91 temperature programmer. Samples were squeezed between two slides, heated to 80 °C, then cooled down to 48 °C and allowed to crystallise. Dry nitrogen was purged throughout the hot stage during the experiments. The amount of Fol linked to the copolymer was quantified by UV–vis spectroscopy on DMSO polymer solutions (0.2–2 mg mL−1), using Fol standard solutions in DMSO to construct calibration curves. The absorbance of the sample was evaluated at 360 nm on a Shimadzu 1800 spectrophotometer.
959 Da. 1H NMR spectra were obtained on a Bruker DRX-400 spectrometer using 5 mm tubes. Sample concentrations were about 0.7% (w/v). All spectra were measured with 16 accumulations and a 10 s recycle delay. Diffusion-ordered NMR spectroscopy (DOSY) experiments were carried out at 298 K for R2 and 313 K for R1 on a Bruker 600 DRX equipped with a cryo-probe. The concentration of the samples was about 0.7% (w/v) in d6-DMSO. The ledbpgp2s pulse sequence from the Bruker library, incorporating a bipolar gradient pulse pair and 2 spoil gradients, with a longitudinal eddy current delay, was run with a linear gradient (5.35 G cm−1) stepped between 2.0% and 95.0%, incremented in 32 steps. The gradient duration (δ) was held constant at 2 ms and the echo delay (Δ) at 200 ms. After Fourier transformation and baseline correction of the 1D 1H spectra (F2 dimension), the diffusion dimension (F1) of the 2-dimensional DOSY was processed using TopSpin software (version 2.1). Differential Scanning Calorimetry (DSC) analysis (Q2000 TA Instruments) was performed under a nitrogen flow with a 2 °C min−1 scanning rate. The sample (about 4 mg) sealed in an aluminum crucible was heated from −20 to 90 °C, cooled at −20 °C and finally heated again to 90 °C. X-ray powder diffraction profiles were obtained at room temperature with Ni filtered CuKa radiation using an X-Pert automatic diffractometer from PANalytical. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer Pyris Diamond TG-DTA apparatus from 25 to 500 °C under a nitrogen flow (50 ml min−1) at 10 °C min−1 heating rates. Polarised optical microscopy was performed on an Axioscop-Zeiss instrument equipped with a THMS 600 hot stage and a Linkam TMS 91 temperature programmer. Samples were squeezed between two slides, heated to 80 °C, then cooled down to 48 °C and allowed to crystallise. Dry nitrogen was purged throughout the hot stage during the experiments. The amount of Fol linked to the copolymer was quantified by UV–vis spectroscopy on DMSO polymer solutions (0.2–2 mg mL−1), using Fol standard solutions in DMSO to construct calibration curves. The absorbance of the sample was evaluated at 360 nm on a Shimadzu 1800 spectrophotometer.
Preparation and characterisation of nanoparticles
Non-targeted NPs (ntNPs) were prepared from mPEG1k-b-PCL4k whereas folate-decorated NPs were prepared from a mixture of mPEG1k-b-PCL4k and either Fol-PEG1.5k-b-PCL4k or R1 (respectively, Fol-NPs and R1-NPs, 10 and 20% by wt) by the solvent diffusion method. Briefly, an organic phase (1 mL) containing 5 mg of copolymer or copolymer mixture (acetone for ntNPs and Fol-NPs; THF/DMSO 1/1 v/v for R1-NPs) was added dropwise in water (4 mL) under magnetic stirring (300 rpm). Acetone was removed by rotary evaporation, whereas in the case of R1-NPs, THF was evaporated and DMSO was removed by dialysis against water. After solvent evaporation, NPs were filtered through 0.45 μm Phenex® filters (Phenomenex, USA). Fluorescent NPs for uptake studies were prepared with Nile Red (0.1% theoretical content). Nile Red was co-dissolved with the copolymers in the organic phase. ntNPs and Fol-NPs could be freeze-dried (Christ Alpha 2–4 LSCplus, Martin Christ Freeze Dryers, Germany, sample frozen in liquid N2) after the addition of HPβCD as a cryoprotectant (polymer/HPβCD 1/10 wt ratio), whereas R1-NPs could be freeze-dried without the help of any cryoprotectant. The recovery yield of the production process was evaluated by weighing the solid residue after freeze-drying (yield >99% for all formulations).The hydrodynamic diameter (DH), polydispersity index (PI) and zeta potential (ζ) of NPs were determined on a Zetasizer Nano Z (Malvern Instruments Ltd). 1H NMR was carried out on NP aqueous dispersions to evaluate the amount of PEG or PEG/αCD on the surface. Spectra were recorded for either NPs dispersed in D2O or NPs dissolved in the organic phase (5 mg mL−1). Fixed aqueous layer thickness (FALT) was measured by monitoring the influence of ionic strength on ζ. Different amounts of NaCl stock solutions were added to NPs dispersed in water and the zeta potential of the samples was measured. A plot of lnζ against 3.33 × [NaCl]0.5 gives a straight line where the slope represents the thickness of the shell in nm.19 TEM analysis was performed with a FEI Tecnai G12 (LAB6 source) equipped with a FEI Eagle 4 K CCD camera (USA) operating with an acceleration voltage of 120 kV. Samples were prepared by air-drying a droplet of NP suspension directly onto a copper TEM grid (3 mm).
Cell cultures and NP uptake studies
KB carcinoma cells (American Type Culture Collection, ATCC, Rockville, USA) overexpressing FRα were grown in Eagle's medium (MEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin G and 100 μg mL−1 streptomycin and maintained at 37 °C under a humidified atmosphere containing 5% CO2. Flow cytometry (FACS) experiments were performed in order to study the uptake of NR-loaded NP formulations. For these experiments, cells were seeded in 24 wells per plate (50000 cells per well) in folate-deficient RPMI medium (Life Technologies) supplemented with 10% FBS. After 24 h of growth at 37 °C, the cells were incubated for 1 h with 100 μg mL−1 of NPs diluted in RPMI medium. After NP incubation, the cells were washed, detached from the plates with 0.25% trypsin (Life Technologies), centrifuged and resuspended in Versene™ before measuring NR fluorescence using a BD FACSCanto™ II instrument (Becton Dickinson). A blue laser at 488 nm was used as an excitation source and the PE channel (585/42 nm) was selected for the detection of NR fluorescence. 104 events per sample were acquired and analysed using the FACSDiva Software. The competition experiments were carried out by incubating the cells with 1 mM free Fol 1 h prior to the addition of NPs in order to saturate the FR binding sites on the cell surface.
Results and discussion
Synthesis of copolymers and rotaxanes
The mPEG1k-b-PCL4k copolymer was synthesized by typical ROP polymerization of CL using mono-hydroxyl PEG1k as the initiator and stannous octoate as the catalyst.20 The molecular weight of the PCL block was controlled by the CL/initiator molar ratio in the feed, and calculated by the ratio between the intensities of the resonance associated with CH3–O– protons of the mPEG terminal at 3.38 ppm and –CH2–O–CO– units in the PCL chain at 4.10 ppm of the 1H NMR spectrum.The Fol-PEG1.5k-b-PCL4k copolymer and its rotaxane derivatives were prepared following a multi-step procedure. First, tailoring of PEG was performed through a method that allows the selective introduction of different α,ω-moieties,21,22 as described in Scheme 1. The key step of the reaction is the desymmetrisation of PEG diol, achieved using the method of Bouzide and Sauve.23 Tosylate and azide groups were introduced for successive reactions with Fol and butinyl-PCL, respectively.
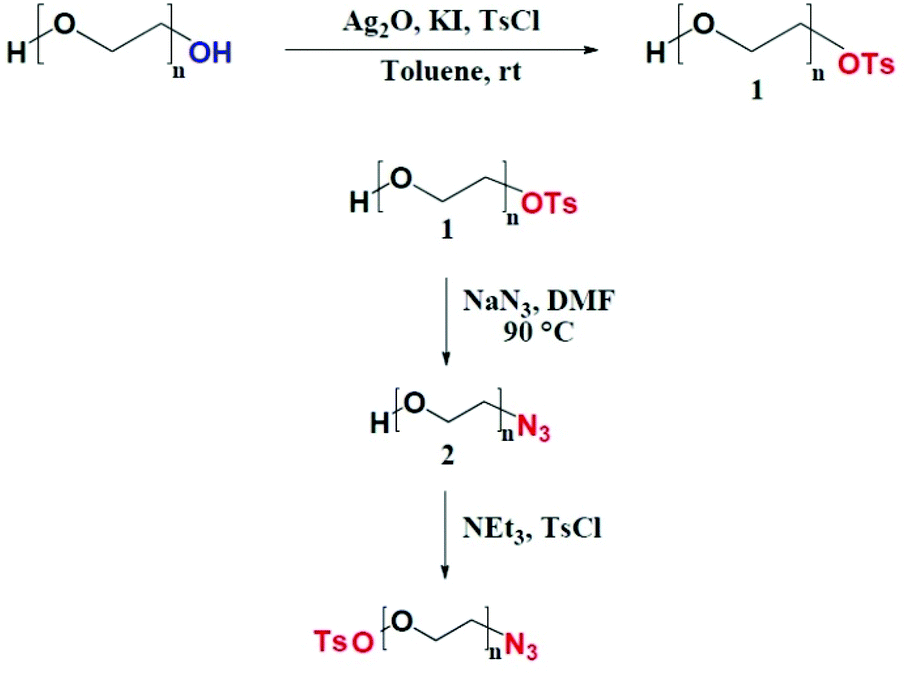 | ||
| Scheme 1 Synthesis pathway to the TsO–PEG–N3 derivative (adapted from ref. 18). | ||
The molecular structure of all intermediates was confirmed by the presence of the corresponding peaks and peak integration in the 1H NMR spectrum. The azide group was identified by the characteristic band at 2105 cm−1 in the FTIR spectrum.
The Fol-PEG1.5k-b-PCL4k copolymer and rotaxane derivatives were then prepared following the procedure described hereafter (Scheme 2).
 | ||
| Scheme 2 General scheme for end-capping reactions of the TsO–PEG1.5k–N3 pseudo-rotaxane. | ||
The TsO–PEG1.5k(αCD)–N3 pseudo-rotaxane was formed by simply mixing components in water. The pseudo-rotaxane was first isolated and successively re-dissolved in DMSO for end-capping reactions (“two-pot” procedure). Purification steps were performed directly on the final rotaxane since the removal of unthreaded αCDs from a pseudo-rotaxane is troublesome.
Different capping reactions are reported in the literature.24,25 All of them use α,ω-homo-bifunctional pseudo-polyrotaxane PEG as the precursor. Then, identical bulky groups, acting as physical stoppers, are coupled to the functional end groups of PEG through a variety of reactions, such as nucleophilic substitution or condensation.26–28 In the present work, end-capping was realised for the first time using two different stoppers, starting from an α,ω-hetero-bifunctional PEG pseudo-rotaxane. Fol was coupled to the α-tosyl terminal group of PEG via nucleophilic substitution, after activating the amine functionality of Fol with sodium hydride whereas butynyl-PCL4k was coupled to the ω-azido terminal group via Cu(I)-catalyzed Huisgen [2 + 3] dipolar cycloaddition (“click” reaction), described in the literature to be an ideal and versatile solution to the problem of efficiently end-capping αCD-based polyrotaxanes.29 Since the PCL chain is thin enough to fit within αCD, the possible involvement of PCL in complex formation could be envisaged. Nevertheless, PCL is reported to form ICs only for molecular weights up to 3 kDa under similar preparation conditions.30 Furthermore, for extremely hydrophobic polymers such as PCL, slow CD threading kinetics makes complexation unlikely. Hence, a PCL block with Mn of 4 kDa was expected to be efficient enough as a stopper. With regard to folate, it was already described in the literature as a fitted cap to prevent unthreading of CD from PEG.31 Besides, Fol conjugation through the amine functionality was preferred, since other possible reactions involving the γ-carboxylic group of Fol may also concern α-carboxyl, negatively affecting the folate receptor-mediated endocytosis.32
A selectively threaded copolymer was finally obtained (so-called “site-selective complexation”).33 Selective complexation of block copolymers is usually based on the different binding affinity of the two blocks towards a specific ring size. In the case of triblock copolymers of PCL and PEG, it was found that both blocks were complexed by αCD because there is no preference for one block.34 Hence, the strategy described here allows a selective αCD-complexation of the PEG block in PEG-b-PCL copolymers not achievable otherwise.
Two slightly different pathways were pursued and compared. The difference just consisted of adjusting the timeline. Capping was carried out simultaneously on both ends in the “one-step” strategy, whereas the two reactions were carried out in sequence with the “two-step” strategy. Particularly noteworthy is the excellent yield obtained for both rotaxanated copolymers (>50%), significantly higher as compared to that reported in the literature for most PEG polyrotaxanes (<10%).35
The FTIR analysis of Fol-PEG1.5k-b-PCL4k and rotaxanated copolymers evidenced the complete disappearance of the peak at 2105 cm−1, corresponding to the specific vibration of the azide group, suggesting that all PEG-N3 groups were reacted (Fig. S1, ESI†). Peaks attributable to αCD (3350 and 1640 cm−1) were visible in the FTIR spectra, although their low intensity suggests a low rotaxanation degree.
The functionalisation degree of Fol was determined by UV-vis spectroscopy, a validated technique for Fol quantification,36 and was found to be very different between R1 and R2 (respectively, 94 and 19%). The difference can be ascribed to the different reactivity of the tosyl-end group on PEG. When the capping reactions were carried out in sequence, the nucleophilic substitution of tosyl with folate reduced the efficiency, since the tosyl group of the TsO–PEG-b-PCL copolymer is likely less accessible. In contrast, a high functionalisation degree was obtained with one-step capping since substitution of the tosyl group with folate involves mainly free TsO–PEG–N3.
The Fol content has a high impact on solubility properties. R1 is insoluble in THF and CHCl3 and barely soluble in DMSO at room temperature, whereas it easily dissolves in DMSO at T > 40 °C and in a THF/DMSO 1/1 v/v mixture at room temperature. In contrast, R2 retains the solubility of the precursor copolymer.
GPC analysis
GPC analysis can definitively distinguish between a polyrotaxane and a pseudo-polyrotaxane (which unthreads in solution giving signals at the retention times of the polymer and CD).37 The results for Fol-PEG1.5k-b-PCL4k and R2 are presented in Table 1. The poor solubility of R1 prevented a reliable analysis.| Sample | α | M w (Da) | M n (Da) | PI |
|---|---|---|---|---|
| Fol-PEG1.5k-b-PCL4k | 0.55 | 6740 | 5990 | 1.12 |
| R2 | 0.66 | 8690 | 6350 | 1.36 |
GPC confirmed that αCDs are threaded on PEG in R2, since only one peak was detected in correspondence of the copolymer (Fig. S2, ESI†). Moreover, the increase of the Mark–Houwink constant α accounted for the modification of PEG chain conformation in the IC. An increase of Mw, in line with the presence of CDs on the PEG chain, was observed. Nevertheless, it must be kept in mind that, according to the literature, the exact molecular weight of a polyrotaxane cannot be determined by GPC since polyrotaxanes behave unlike any commercial standard, due to their rod-like morphology.11
1H NMR analysis and 2D-DOSY spectroscopy
1H NMR allowed the evaluation of the PEG/αCD ratio in the copolymers, although it does not allow the establishment of whether or not PEG chains are included in the macrocycle. Signals of the copolymer and αCD were clearly visible in both R1 and R2 spectra. Peak assignment of the copolymer and αCD protons is shown in Fig. S3, ESI.† From the ratio between the integrals of the signal of protons of the –O–CH2– unit of the PEG chain at 3.4 ppm and the signal of the anomeric proton H1 of αCD at 4.8 ppm, the number of αCDs associated or threaded per PEG chain was calculated. Coverage percentage was estimated by assuming that 100% complexation corresponds to a αCD/PEGunits ratio = 1/2, as previously reported in the literature.12 A value of around 2 αCDs per PEG chain was obtained, corresponding to around 9% coverage. This value is far below that reported with traditional stoppers, such as 3,5-dimethylphenol (∼70%).35 The threading level in a polyrotaxane is limited by competition between the end-capping reaction and unthreading of the pseudo-rotaxane precursor, since threading complexation is generally a reversible process.38 The formation of a rotaxane in DMSO solution is a quite difficult task to carry out, and a lower inclusion ratio or even failure to trap CD on a polymeric backbone in DMSO has been described.39 Furthermore, limited coverage is often reported in the case of a polymer with a molecular weight higher than 1 kDa, since threaded segments aggregate and precipitate before full coverage has been achieved. For these reasons, it is supposed that unthreading prevails, finally obtaining a low coverage ratio. It is worth underlining, however, that the formation of polyrotaxanes with low coverage is a desired goal for applications as “slide-ring” materials.40 To this purpose, some authors have resorted to hydroxypropyl-CD instead of the native CD to hamper hydrogen bonds between adjacent CD and minimise the CD number on polymer chains.41DOSY is a very sensitive method for discriminating threaded from unthreaded macrocycles in rotaxanated polymers.35,42 In a pseudo-2D DOSY spectrum, the chemical shifts are displayed along the horizontal axis, while the diffusion coefficients are reported along the F1 dimension. Thus, it was observed from the analysis of R1 that the diffusion coefficient of the PEG axis was coincident with that of the αCD rings, providing strong evidence of rotaxanation (Fig. 1a). Instead, the two horizontal lines in the R2 spectrum (Fig. 1b) marking the diffusion coefficients for αCD and the PEG-b-PCL copolymer indicated that the material exists as two separate components and not as a threaded structure. This result is apparently in contrast with GPC analysis. A possible interpretation is that the R2 is actually a pseudo-rotaxane, due to the low % of Fol capping, and rapidly unthreads upon dissolution in DMSO. On the other hand, the poor solubility of free αCDs in THF might be responsible for the tendency of αCDs to be retained on the PEG chain, giving rise to a single peak in the GPC chromatogram.
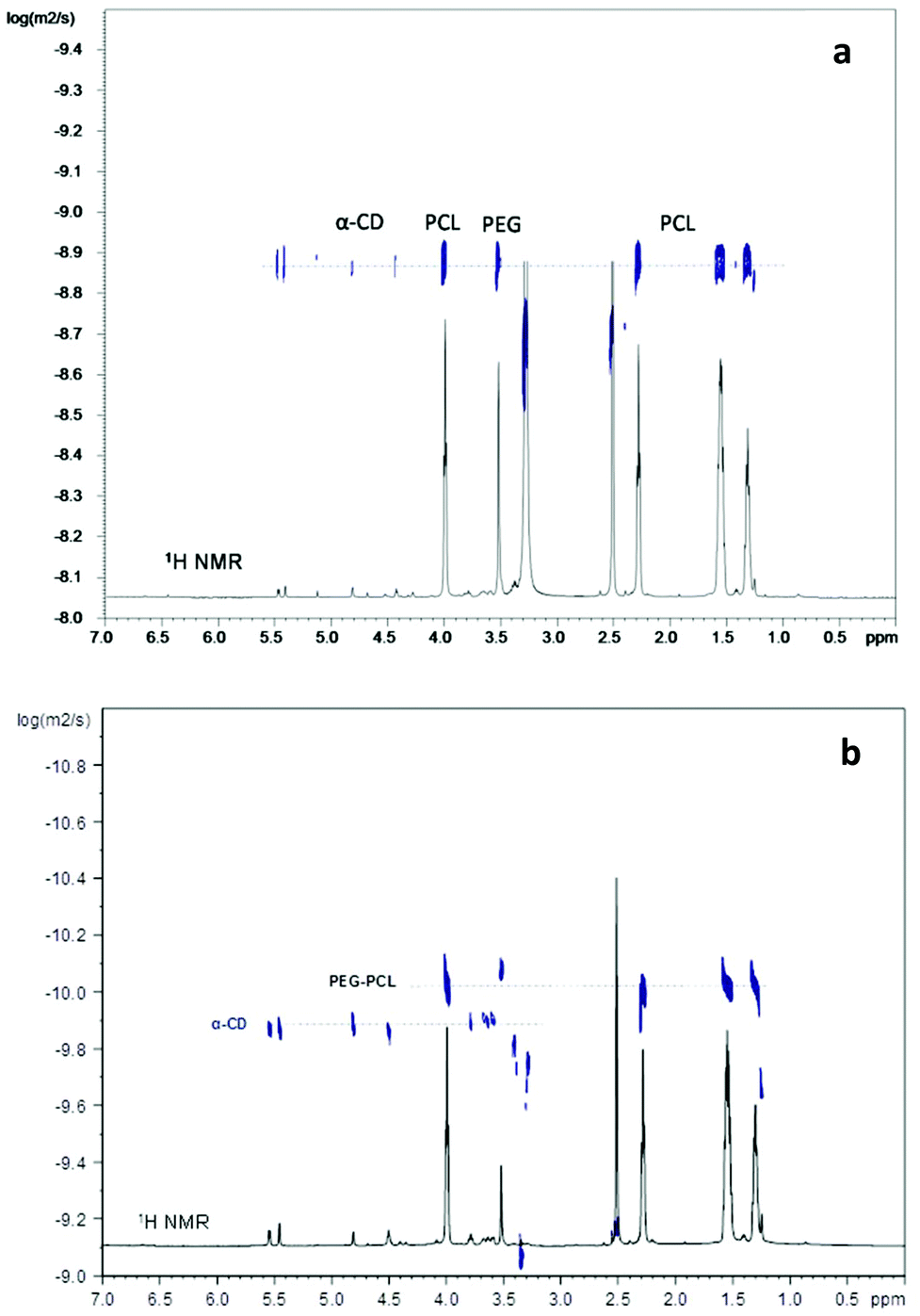 | ||
| Fig. 1 Superimposition of the 1H NMR and 2D DOSY spectra in d6-DMSO of R1 at 313 K (a) and R2 at 298 K (b). | ||
Thermal analysis
DSC analysis was performed in order to study the influence of the PEG/αCD complex on the crystallinity of PEG and PCL blocks. A low scanning rate was chosen to promote the formation of well-separated exo/endotherms for the two different blocks. The thermal properties of the Fol-PEG-b-PCL copolymer, rotaxanes and the precursors (butynyl-PCl4k, PEG1.5k and azido-PEG-PCL) are presented in Table 2. Melting peaks of PEG and PCL are distinguished in the N3-PEG1.5k-b-PCL4k copolymer, indicating phase segregation of blocks. Since the PEG block is shorter, the crystallisation of PCL is dominant, leading to imperfect crystallisation of PEG.43 In fact, both Tm and ΔHm decrease. The melting endotherm of PEG was fully absent in the thermogram of Fol-PEG1.5k-b-PCL4k. This result can be ascribed to the effect originating from folate, a bulky terminal group, which significantly affects the crystallinity and melting temperature of PEG.43 Similarly to unthreaded Fol-PEG-b-PCL, rotaxanes did not show a separate PEG melting endotherm; therefore, the influence of the αCD ring on PEG crystallinity cannot be deduced by DSC analysis.| Sample | T m1 | ΔHm1 | T m1 | ΔHm1 | T m2 | ΔHm2 | T m2 | ΔHm2 |
|---|---|---|---|---|---|---|---|---|
| PEG | PEG | PCL | PCL | PEG | PEG | PCL | PCL | |
| Fol-PEG1.5k-b-PCL4k | — | — | 54 | 121 | — | — | 51 | 103 |
| R1 | — | — | 57 | 196 | — | — | 53 | 135 |
| R2 | — | — | 59 | 227 | — | — | 56 | 160 |
| Butynyl-PCL4k | 58 | 105 | 54 | 75 | ||||
| PEG1.5k | 43 | 187 | 43 | 173 | ||||
| N3-PEG1.5k-b-PCL4k | 39 | 41 | 55 | 93 | 32 | 61 | 50 | 70 |
The presence of the PCL melting endotherm around 55 °C in rotaxanes confirms that PCL chains are not involved in inclusion complexation. Interestingly, the high value of ΔHm observed for Fol-PEG1.5k-b-PCL4k and rotaxanated copolymers, both in the first and second runs, suggests that the endotherm cannot be ascribed to PCL melting alone (whose  value corresponds to 139.5 J g−1), but there is a contribution from PEG. This hypothesis is in line with the literature reports on αCD/PEG ICs, which show the complete absence of PEG crystallinity only for a coverage ratio above 70%. It is believed that crystallisation and melting temperatures of the two blocks approach until they overlap, due to the reduced mobility of the PEG chain in the Fol terminated copolymers which causes an increase of the PEG crystallisation temperature.
value corresponds to 139.5 J g−1), but there is a contribution from PEG. This hypothesis is in line with the literature reports on αCD/PEG ICs, which show the complete absence of PEG crystallinity only for a coverage ratio above 70%. It is believed that crystallisation and melting temperatures of the two blocks approach until they overlap, due to the reduced mobility of the PEG chain in the Fol terminated copolymers which causes an increase of the PEG crystallisation temperature.
T m of PCL was not influenced by its covalent connection with PEG in rotaxanated copolymers, whereas a decrease was noted in the case of N3-PEG1.5k-b-PCL4k and Fol-PEG1.5k-b-PCL4k. As known, Tm is related to the crystallite size and structure. Since the semicrystalline nature of PCL and its higher hydrophobicity can likely give NPs with high kinetic stability and low rates of disassembly,45 the finding that the PCL block retains its crystalline structure unchanged in the rotaxanated copolymers might be a benefit in view of the stability of related NPs.
Rotaxanes show unique thermoresistant properties, due to the supramolecular structure. Thermogravimetric analysis was also performed, and the results are reported in the ESI.† Thermogravimetric curves of Fol-PEG1.5k-b-PCL4k, R1 and R2 clearly show an increase of the thermal stability of rotaxanes (Fig. S4†).
X-ray diffraction analysis
X-ray powder diffraction patterns for N3-PEG1.5k-b-PCL4k, Fol-PEG1.5k-b-PCL4k and the two rotaxanes are shown in Fig. 2. The peak characteristics of αCD (Fig. S5(a), ESI†) were not detected in the spectrum of rotaxanes, likely due to its low amount. All samples display the reflection peaks of PCL at 2θ 21.4° and 23.7° (Fig. S5(b), ESI†), whereas the characteristic reflection of PEG at 2θ 19.3° was detected only in the diffractogram of the N3-PEG1.5k-b-PCL4k copolymer. The formation of the IC could cause a change in the crystalline arrangement of PEG chains with a shift of the reflection peak, which would overlap with the one of PCL at 2θ 21.4°. A similar behavior was described by Ye et al. in the case of the PEG/urea inclusion complex.44 The authors suggested that PEG chains adopt a more extended conformation when included in urea channels compared to the conformation found in the bulk. As a matter of fact, there are no reports on the crystal structure of PEG in αCD ICs. The exact conformation of PEG chains in the complex has not yet been determined.46 | ||
| Fig. 2 X-ray diffraction patterns for N3-PEG1.5k-b-PCL4k, Fol-PEG1.5k-b-PCL4k, R1 and R2. | ||
Interestingly, a new weak reflection at 2θ 20° was present in the R1 pattern. According to the literature,47 the 2θ 20° peak is associated with the αCD crystalline organisation in the channel-like structure, characteristic of polymer/CD inclusion complexes. Hence, short αCD sequences organised in the channel-like structure are present in R1, despite the low coverage.
Optical microscopy
Optical photos of Fol-PEG1.5k-b-PCL4k, R1 and R2 after isothermal crystallization of PCL chains at 48 °C are shown in Fig. 3. It is evident that the formation of large spherulites was inhibited in the R1 copolymer. This behaviour is analogous to what is reported in the case of a βCD/PCL IC.48 Nevertheless, in our case, DSC and RX analyses did not provide evidence of rotaxanation of PCL blocks. Therefore, the presence in R1 of the αCD/PEG IC organised in a channel-like structure influences the morphology of PCL. It is supposed that the crystalline ICs embedded in the PCL matrix disturb the growth of spherulites and, at the same time, act as nucleation agents, giving rise to the growth of a higher number of spherulites of smaller dimension. | ||
| Fig. 3 Photographs of Fol-PEG1.5k-b-PCL4k (a), R1 (b) and R2 (c) obtained by polarised light optical microscopy. Samples are crystallised at 48 °C. | ||
Preparation and characterisation of NPs
In order to demonstrate the ability of the novel synthesized polyrotaxane to form NPs targeted to cancer cells overexpressing the FRα receptor and delivering lipophilic drugs, we performed a preliminary formulation study. In particular, we prepared polyrotaxane-based NPs by the solvent diffusion method (currently indicated as nanoprecipitation). Nanoprecipitation is a simple and versatile method for preparing polymeric NPs. Compared to micellization, a widely employed technique to form nanocarriers with well-defined structures, nanoprecipitation allows for an up-scalable and reproducible preparative process. Indeed, the solvent-displacement principle has been exploited to develop different microfluidic devices proposed as promising tools to accelerate the industrial preparation and the clinical translation of nanoparticles.49 NPs were prepared from a mixture of mPEG1k-b-PCL4k and R1 at different percentages (10 and 20% by wt) (R1-NPs 10% and R1-NPs 20%). R2 was not considered due to its low folate content which caused unthreading of CD in polar solvents. As a control, folate targeted NPs were prepared also from a mixture of mPEG1k-b-PCL4k and Fol-PEG1.5k-b-PCL4k at the same percentages (Fol-NPs 10% and Fol-NPs 20%), with the aim to understand the role of αCDs in folate exposure. Finally, the corresponding non-targeted NPs (ntNPs) from mPEG1k-b-PCL4.0k were prepared too. The hydrodynamic diameter (DH), polydispersity index (PI) and zeta potential (ζ) of NPs are presented in Table 3. The amount of PEG and αCD on the NP surface was evaluated by 1H NMR according to the literature.7| Formulation code | D H (nm) ± SD | PI | ζ (mV) ± SD | Surface PEG (wt%) | Surface αCD (wt%) |
|---|---|---|---|---|---|
| ntNPs | 85.7 ± 1.0 | 0.108 | −25.1 ± 0.3 | 7.1 | — |
| Fol-NPs 10% | 92.5 ± 3.3 | 0.215 | −22.4 ± 2.5 | 5.4 | — |
| Fol-NPs 20% | 102.5 ± 4.0 | 0.195 | −28.1 ± 1.3 | 4.2 | — |
| R1-NPs 10% | 171.0 ± 10 | 0.220 | −42.3 ± 1.0 | 5.5 | 84.4 |
| R1-NPs 20% | 167.2 ± 10 | 0.254 | −38.8 ± 0.5 | 7.1 | 88.2 |
All formulations gave NPs in an excellent yield (above 90%). Nanoprecipitation of mPEG1k-b-PCL4k gave NPs of a size below 100 nm with very low polydispersity and negative surface charge, in line with the previous studies.6 Upon R1 addition, NPs of higher dimension and much more negative ζ were formed, whereas Fol-NPs did not show such a size increase. The higher value of ζ is suggestive of the presence of Fol moieties on the surface. NPs exhibit spherical morphology and absence of aggregation phenomena, as evidenced by TEM analysis (Fig. S6, ESI†). The thickness of the external hydrophilic PEG shell of NPs was also evaluated by FALT, measuring the ζ values of NPs in NaCl solutions at different concentrations. The shell thickness results were lower for ntNPs (∼2 nm) and NPs decorated with folate without rotaxanated copolymers (2.2 and 2.7 nm for Fol-NPs 10% and Fol-NPs 20% respectively), compared to R1-NPs 10% (∼3.8 nm) and R1-NPs 20% (∼3.3 nm) thus demonstrating that the presence of αCD can modify the conformation of the PEG shell and the exposure of targeting moieties on the NP surface (Fig. 4).
 | ||
| Fig. 4 FALT measurements. The slope of the straight line represents the thickness of the outer hydrophilic shell expressed in nm. | ||
To evaluate the PEG amount on the NP surface, the 1H NMR spectrum of NPs in D2O was acquired, and the amount of surface PEG was determined by comparing the intensity of the peak relative to –CH2–O– units of PEG at 3.65 ppm in D2O with the corresponding peak in the spectrum of freeze-dried NPs dissolved in the organic phase (CDCl3). The 1H NMR spectra for R1-NPs are shown in Fig. 5. The experimental amount of PEG on the surface is much lower than the theoretical value, as already shown in a previous work.6 No variation of the surface PEG amount was detected upon R1 addition. The spectrum of R1-NPs in D2O also clearly shows the resonances relative to αCD and Fol units,50 attesting their good exposure on the surface. It is worth noting that Fol resonances were not visible at all in the spectrum of Fol-NPs (not shown), attesting a better exposure of Fol to the water pool in R1-NP formulations. Similarly to PEG, the αCD amount on the NP surface (defined as the percentage fraction of αCD located in the water pool with respect to total αCD) was also estimated by the ratio between the intensity of the H1 signal of αCD at 4.8 ppm in D2O and the intensity of the corresponding signal in CHCl3. As shown in Table 3, a percentage of αCD close to 90% was found. This result is significant, since the almost full surface exposure of αCD implies a corresponding good surface exposure of Fol-targeting molecules bonded to the R1 PEG end. Moreover, since αCD is threaded around PEG chains, we can reasonably assume that PEG from Fol-modified rotaxane copolymers is mostly present on the NP surface while the PEG fraction from mPEG1k-b-PCL4k mainly locates in the lipophilic core of NPs, as can be deduced by the low total amount of surface PEG presented in Table 3 (only 5–7%), which is similar for all formulations.
 | ||
| Fig. 5 1H-NMR spectra of R1-NPs 20% in D2O and CDCl3. | ||
Cell uptake studies
Following the full characterization of NP surface properties in terms of Fol and CD exposure and PEG density and thickness, we investigated whether these NPs could bind and be internalized in KB cancer cells, in which FRα is overexpressed. Uptake studies were performed with fluorescent NR-loaded NPs which showed no significant variation in terms of colloidal properties and NR entrapment as compared with their unloaded counterpart (Table S1 and Fig. S6, ESI†). To highlight the effect of the αCD/PEG rotaxane on Fol exposure, Fol-NPs were also tested for comparison. Fig. 6 shows the uptake of the different NPs in KB cells, pre-incubated or not with 1 mM free Fol before NP addition. Free Fol was used as a competitor to saturate FRα, which is overexpressed on the cell surface. As can be seen in the figure, all Fol-decorated NPs showed higher internalization compared to ntNPs, thus demonstrating the active involvement of the targeting agent in NP uptake. Importantly, the uptake of both formulations prepared with rotaxane-PEG (R1-NPs) was higher with respect to the corresponding formulation prepared without αCD threading, confirming a better exposure of Fol on the NP surface. Competition experiments showed significantly reduced uptake for all Fol-decorated NPs, confirming the involvement of FR-mediated endocytosis on the NP internalization process. However, since Fol-decorated NPs showed higher internalization compared to ntNPs also in the presence of free folate, this may suggest the involvement of additional uptake mechanisms besides FR-mediated endocytosis.6,51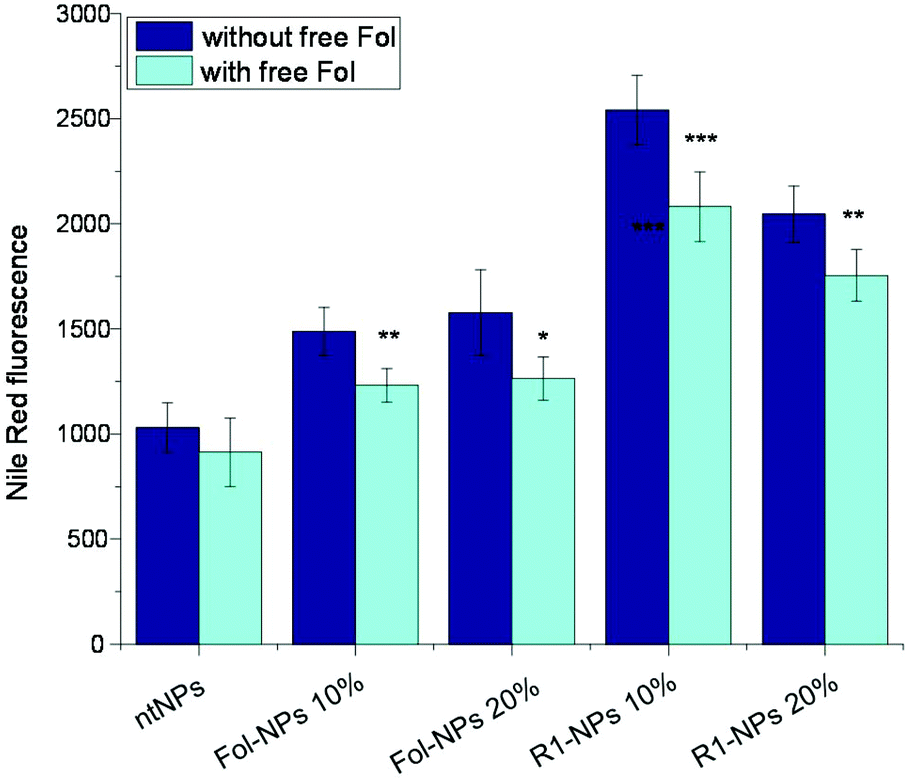 | ||
| Fig. 6 Flow cytometry measurement of NP uptake in KB cells incubated for 1 h with 100 μg mL−1 of NR-loaded NPs, in the absence or in the presence (competition experiment) of 1 mM free Fol. Data are the means of at least 2 independent experiments (mean ± SD), carried out in triplicate. Significant difference for *p < 0.05; **p < 0.01; ***p < 0.001, t test, Student's t-test. | ||
Conclusions
Selective complexation of PEG-b-PCL copolymers with αCD was successfully achieved from an α,ω-hetero-bifunctional PEG pseudo-rotaxane with an original methodology, involving two different end-capping reactions to prevent αCD unthreading. Folate and butynyl-PCL (Mn 4 kDa) were bonded on PEG ends as stoppers through, respectively, nucleophilic substitution and click reaction, finally realising a selectively rotaxanated Fol-PEG(αCD)-b-PCL copolymer. To the best of our knowledge, this is the first example of end-capping of a PEG pseudo-rotaxane with two different stoppers. We found that when the two end-capping reactions were carried out simultaneously (“one-step”, R1), the inclusion complex was stable in solution, as demonstrated by DOSY spectroscopy, whereas rapid unthreading was detected in the case of the rotaxane obtained by carrying out the reactions in sequence (“two-steps”, R2). This finding was ascribed to the low folate capping efficiency achieved with the two-step methodology. Overall, solid-state characterisation showed a modification of the PEG crystalline structure with a shift of the melting temperature upon Fol functionalisation and αCD complexation, the existence of the typical channel-like crystalline structure of αCDs in the inclusion complex, an improvement of the thermal stability of rotaxanated copolymers and an influence of rotaxanation of PEG on the morphology of PCL spherulites. NPs prepared from a mixture of mPEG-b-PCL and R1 exposed folate on the surface in the aqueous environment and entered cancer cells overexpressing the folate receptor much better than non-targeted NPs but also better than the corresponding non-rotaxanated targeted NPs.In conclusion, our findings suggest that NPs based on selectively rotaxanated PEG-b-PCL copolymers provide an effective niche capable of augmenting the potential of NPs in cancer therapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Italian Association for Cancer Research (IG2014 #15764). The authors are indebted to Prof. Elena Reddi (Department of Biology, University of Padova, Italy) for the support in the interpretation of the biological results. The authors are indebted to Prof. M. Trifuoggi and Dr M. Toscanesi (ACE Laboratory, Department of Chemical Sciences, University of Naples Federico II, Italy) for the elemental analysis. The authors wish to thank Dr M. Milazzo (Department of Chemical Sciences, University of Naples Federico II, Italy) for the WAXD analysis.References
- Y. Y. Yang, Y. Wang, R. Powell and P. Chan, Clin. Exp. Pharmacol. Physiol., 2006, 33, 557 CrossRef CAS PubMed.
- G. Chen, Y. Wang, R. Xie and S. Gong, Adv. Drug Delivery Rev., 2018, 130, 58 CrossRef CAS PubMed.
- H. Lee, A. Ponta and Y. Bae, Ther. Delivery, 2010, 1, 803 CrossRef CAS PubMed.
- A. Cheung, H. J. Bax, D. H. Josephs, K. M. Ilieva, G. Pellizzari, J. Opzoomer, J. Bloomfield, M. Fittall, A. Grigoriadis, M. Figini, S. Canevari, J. F. Spicer, A. N. Tutt and S. N. Karagiannis, Oncotarget, 2016, 7, 52553 CrossRef PubMed.
- C. Conte, I. Fotticchia, P. Tirino, F. Moret, B. Pagano, R. Gref, F. Ungaro, E. Reddi, C. Giancola and F. Quaglia, Colloids Surf., B, 2016, 141, 148 CrossRef CAS PubMed.
- A. Venuta, F. Moret, G. Dal Poggetto, D. Esposito, A. Fraix, C. Avitabile, F. Ungaro, M. Malinconico, S. Sortino, A. Romanelli, P. Laurienzo, E. Reddi and F. Quaglia, Eur. J. Pharm. Sci., 2018, 111, 177 CrossRef CAS.
- Q. Xu, L. M. Ensign, N. J. Boylan, A. Schon, X. Gong, J. C. Yang, N. W. Lamb, S. Cai, T. Yu, E. Freire and J. Hanes, ACS Nano, 2015, 9, 9217 CrossRef CAS PubMed.
- P. M. Valencia, M. H. Hanewich-Hollatz, W. Gao, F. Karim, R. Langer, R. Karnik and O. C. Farokhzad, Biomaterials, 2011, 32, 6226 CrossRef CAS PubMed.
- A. Hashidzume, H. Yamaguchi and A. Harada, Eur. J. Org. Chem., 2019, 3344 CrossRef CAS.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974 CrossRef CAS PubMed.
- S. Loethen, J.-M. Kim and D. H. Thompson, Polym. Rev., 2007, 47, 383–418 CrossRef CAS.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821 CrossRef CAS.
- T. Ooya, A. Yamashita, M. Kurisawa, Y. Sugaya, A. Maruyama and N. Yui, Sci. Technol. Adv. Mater., 2004, 5, 363 CrossRef CAS.
- W. K. Lee, J. Kobayashi, T. Ooya, K. D. Park and N. Yui, J. Biomater. Sci., Polym. Ed., 2002, 13, 1153 CrossRef CAS PubMed.
- L. Zhang, T. Su, B. He and Z. Gu, Nano-Micro Lett., 2014, 6, 108 CrossRef CAS.
- H. Harisaka and N. Yui, J. Mater. Chem. B, 2019, 7, 2123 RSC.
- N. Yui and T. Ooya, Chem. – Eur. J., 2006, 12, 6730 CrossRef CAS PubMed.
- N. Yui, R. Katoono and A. Yamashita, Adv. Polym. Sci., 2009, 222, 115 CrossRef.
- T. K. Endres, M. Beck-Broichsitter, O. Samsonova, T. Renette and T. H. Kissel, Biomaterials, 2011, 32, 7721 CrossRef CAS PubMed.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1996, 29, 8689 CrossRef CAS.
- R. Mahou and C. Wandrey, Polymers, 2012, 4, 561 CrossRef.
- D. Shenoy, W. Fu, J. Li, G. Jones, C. Dimarzio, S. Sridhar and M. Amiji, Int. J. Nanomed., 2006, 1, 51 CrossRef CAS PubMed.
- A. Bouzide and G. Sauvé, Org. Lett., 2002, 4, 2329 CrossRef CAS PubMed.
- F. H. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, J. Am. Chem. Soc., 1994, 116, 3192 CrossRef CAS.
- S. M. N. Simões, A. Rey-Rico, A. Concheiro and C. Alvarez-Lorenzo, Chem. Commun., 2015, 51, 6275 RSC.
- T. Ichi, J. Watanabe, T. Ooya and N. Yui, Biomacromolecules, 2001, 2, 204 CrossRef CAS PubMed.
- T. Ooya, K. Arizono and N. Yui, Polym. Adv. Technol., 2000, 11, 642 CrossRef CAS.
- S. Loethen, T. Ooya, H. S. Choi, N. Yui and D. H. Thompson, Biomacromolecules, 2006, 7, 2501 CrossRef CAS PubMed.
- A. Harada, Y. Kawaguchi and M. Kamachi, Macromol. Rapid Commun., 1997, 18, 535 CrossRef CAS.
- Y. Zhou, H. Wang, C. Wang, Y. Li, W. Lu, S. Chen, J. Luo, Y. Jiang and J. Chen, Mol. Pharmaceutics, 2012, 9, 1067 CrossRef CAS PubMed.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782 CrossRef CAS PubMed.
- J. Lu, I. D. Shin, S. Nojima and A. E. Tonelli, Polymer, 2000, 41, 5871 CrossRef CAS.
- T. Zhao, W. Haskell and H. W. Beckham, Macromolecules, 2003, 36, 9859 CrossRef CAS.
- R. Mathias, P. R. S. Ribeiro, M. C. Sarraguça and J. A. Lopes, Anal. Methods, 2014, 6, 3065 RSC.
- A. Harada, Coord. Chem. Rev., 1996, 148, 115 CrossRef CAS.
- Y. K. Agarwal and C. R. Sharma, Rev. Anal. Chem., 2005, 24, 35 Search PubMed.
- J. Araki and K. Ito, Soft Matter, 2007, 3, 1456 RSC.
- K. Ito, Curr. Opin. Solid State Mater. Sci., 2010, 14, 28 CrossRef CAS.
- L. Jiang, C. Liu, K. Mayumi, K. Kato, H. Yokoyama and K. Ito, Chem. Mater., 2018, 30, 5013 CrossRef CAS.
- T. Zhao, H. W. Beckham and H. W. Gibson, Macromolecules, 2003, 36, 4833 CrossRef CAS.
- B. Bogdanov, A. Vidts, A. Van Den Bulcke, R. Verbeeck and E. Schacht, Polymer, 1998, 39, 1631 CrossRef CAS.
- H.-M. Ye, Y.-Y. Song, J. Xu, B.-H. Guo and Q. Zhou, Polymer, 2013, 54, 3385 CrossRef CAS.
- J. Liu, F. Zeng and C. Allen, Eur. J. Pharm. Biopharm., 2007, 65, 309 CrossRef CAS PubMed.
- S. Takahashi, N. L. Yamada, K. Ito and H. Yokoyama, Macromolecules, 2016, 49, 6947 CrossRef CAS.
- L. Huang, E. Allen and A. E. Tonelli, Polymer, 1998, 39, 4857 CrossRef CAS.
- O. Jazkewitsch and H. Ritter, Macromolecules, 2011, 44, 375 CrossRef CAS.
- C. I. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66 CrossRef PubMed.
- X. Yang, S. Pilla, J. J. Grailer, D. A. Steeber, S. Gong, Y. Chend and G. Chen, J. Mater. Chem., 2009, 19, 5812 RSC.
- F. Moret, D. Scheglmann and E. Reddi, Photochem. Photobiol. Sci., 2013, 12, 823 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00158a |
| This journal is © The Royal Society of Chemistry 2020 |