
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning polymer properties of non-covalent crosslinked PDMS by varying supramolecular interaction strength†
Brigitte A. G.
Lamers
a,
Marcin L.
Ślęczkowski
a,
Fabian
Wouters
a,
Tom A. P.
Engels
b,
E. W.
Meijer
 *a and
Anja R. A.
Palmans
*a
*a and
Anja R. A.
Palmans
*a
aInstitute for Complex Molecular Systems and Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl; a.palmans@tue.nl
bDepartment of Mechanical Engineering, Materials Technology Institute, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 25th March 2020
Abstract
Non-covalently crosslinked polymeric networks are promising materials towards sustainable and recyclable plastics. Here, we present the post-functionalization of poly(dimethyl siloxane) (PDMS) with supramolecular moieties, attached as grafts to the PDMS backbone, to obtain recyclable PDMS networks. We select three different supramolecular motifs that differ in interaction strength and investigate how these differences affect the dynamic behavior of the networks. The introduction of dinitrohydrazones (hydz), which afford weak supramolecular interactions by π-stacking, resulted in a viscous material at room temperature. Stronger self-association was achieved by the introduction of benzene-1,3,5-carboxamides (BTAs) and ureidopyrimidinones (UPys), which self-assemble via triple and quadruple hydrogen bonding, respectively. This resulted in a thermoplastic elastomeric material for BTA-based PDMS and brittle materials for Upy-based PDMS. Time- and temperature-dependent mechanical measurements reveal that the dynamic nature of the supramolecular bonds becomes slower upon increasing the interaction strength. The polymers are fully recyclable by solvation or compression molding without the loss of material properties. Thereby, by using one linear PDMS backbone, we demonstrate how fundamentally different material properties are obtained by changing the supramolecular interaction strength and type of non-covalent crosslinks. These molecular insights broaden the scope and application of PDMS-based sustainable materials.
Introduction
Polymer chemistry research has advanced the development of plastics that show a wide variety of tuneable material properties. More recently, focus has shifted to the reuse and reshaping of materials by developing re-organisable polymer networks using dynamic covalent bonds.1–6 Apart from dynamic covalent bonds, dynamic non-covalent bonds have also been applied in polymeric networks.7–9 Here, a linear polymer backbone is typically post-modified to incorporate supramolecular moieties along the backbone as grafts. Pioneering work performed by Stadler and coworkers showed that thermoplastic elastomers and viscoelastic materials could be accessed from functionalized polybutadienes.10–12 The facile synthesis of these materials permitted to easily vary the nature of the non-covalent crosslinks, and hereby polymer properties were readily tuned. Over the past decades, a variety of non-covalent interactions has been investigated, which are all amenable to introduction into polymer materials. Examples are hydrogen bonding,13 π-stacking,14 ionic paring,15 and metal–ligand association.16Polydimethylsiloxane (PDMS) elastomers are of great interest due to their high backbone flexibility, good thermal stability and biocompatibility,17 giving rise to applications ranging from biomaterials to soft robotics.18,19 The broad range of applications owing to the various chemistries available for functionalization of PDMS has resulted in a variety of sustainable solutions for crosslinked PDMS.20–24 However, there are only few examples in literature of post-modified PDMS and hereby introducing supramolecular moieties as grafts on the linear siloxane backbone. Bouteiller and coworkers grafted bis-ureas to PDMS, which resulted in a phase segregated system with good tensile properties due to the strong hydrogen bonding.25 Brook and coworkers attached coumarin groups, which afford a much weaker interaction, to the PDMS backbone. Also in this case, the self-association of coumarin groups by π-stacking resulted in a thermoplastic elastomer.26 These examples show that modifying PDMS, even with weakly associating groups, can dramatically affect the material's properties.
Recently, we studied the supramolecular interactions and morphologies of discrete length oligodimethylsiloxanes functionalized with dinitrohydrazones (hydz).27 Highly aligned and long-range ordered cylinders of hydrazone were obtained originating from weak self-association via π-stacking. In addition, the stacking and hydrogen bonding of benzene-1,3,5-carboxamides (BTA) has been extensively studied in solution as well as in the liquid crystalline phase.28 Furthermore, we showed the cooperative formation of multi-chain copolymer aggregates in solution when the BTAs are grafted to PDMS.29 Finally, very strong supramolecular interactions have been introduced by quadruple hydrogen bonding of ureidopyrimidinones (UPy).30 The donor–donor–acceptor–acceptor array results in a bond dissociation barrier of 70 ± 2 kJ mol−1 for the UPys in chloroform. Hence, the high dimerization constant gives rise to the use of UPys in many types of polymers among which are also siloxanes.31–33 However, post-functionalization of UPys as grafts onto linear PDMS have not been reported to date.
In this work, we post-functionalize a PDMS backbone with hydz, BTA or UPy moieties to systematically vary the interaction strength of the non-covalent crosslinks in the polymeric material (Scheme 1). This gives rise to three distinct polymers with mechanical properties varying from viscous to brittle. A molecular explanation gives more insight into designing polymer properties depending on the supramolecular interaction strength and dynamics.
 | ||
| Scheme 1 Molecular structures of PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy. | ||
Results and discussion
Synthesis and characterization
Three functional PDMS polymers were synthesized by reacting alkene-functionalized supramolecular moieties to commercial poly(dimethylsiloxane-co-methylhydrosiloxane) (PDMS-co-PHMS) with an average molecular weight of 25 kg mol−1 and PHMS content of 5% (Schemes S1 and S2†). The synthesis of the dinitrohydrazone and BTA moieties has been described previously.27,29 The UPy moiety was synthesized from isocyanatodecene and methylisocytosine in good yield (64%) (Scheme S2†). Direct hydrosilylation of the hydrazone, BTA and UPy onto the PDMS-co-PHMS using Karstedt's catalyst resulted in PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy, respectively (Scheme 1).29 The molecular structure of PDMS-g-UPy is slightly different from the hydrazone and BTA functionalized polymers. Here, only part of the hydrides was reacted with the UPy moiety, and the rest with hexene resulting in a UPy graft density of 0.5 mol%. PDMS with a higher UPy density (4 mol%) yielded a material which was too brittle, precluding analysis of its mechanical properties. Therefore, the number of UPy grafts was lowered by including hexyl groups to the polymer backbone. These groups are necessary to prevent the hydride-containing PDMS from crosslinking covalently in atmospheric humidity via intermolecular dehydration as was shown by Opris et al.34Attaching the supramolecular moieties to the PDMS backbone resulted in dramatic changes of the material properties such as viscosity, elasticity and brittleness of the materials. Whereas PDMS-co-PHMS is a liquid, all functionalized silicones appear as solids. Hence, the supramolecular moieties act as non-covalent, intermolecular crosslinks between the linear PDMS chains. Films of the polymers could be dissolved in THF, demonstrating the reversibility of the network (Fig. 1).
 | ||
| Fig. 1 (A) PDMS-g-hydz polymer film in THF and (B) dissolved in THF after 1 minute at room temperature. (C) PDMS-g-BTA polymer film in THF and (D) dissolved in THF after 1 minute at 40 °C. (E) PDMS-g-UPy polymer film in THF and (F) dissolved in THF after 10 minutes at 65 °C. | ||
The polymers were fully characterized by size exclusion chromatography (SEC) and 1H and 29Si NMR spectroscopy (Fig. S1–S6†). The results are summarized in Table 1. The number average molecular weight (Mn) and dispersity (Đ) of the polymers were obtained by SEC (Fig. S1†). The Mn of PDMS-g-hydz and PDMS-g-UPy, 27.9 and 29.5 kg mol−1, respectively, were similar to the molecular weight of the PDMS-co-PHMS given by the supplier. In contrast, the Mn of PDMS-g-BTA was much higher (42 kg mol−1). Most likely the higher apparent Mn is caused by interactions of the BTAs with the SEC column. Therefore, multiple polymer chains cluster together resulting in a higher average molecular weight measured. The absence of covalent crosslinks was revealed by 29Si NMR where only one peak was observed at δ = 22 ppm (Fig. S4–6†). This signal is indicative for the presence of only double oxygen-bonded silicon atoms since triple and quadrupole oxygen-bonded silicon atoms appear at δ ≥ −55 ppm. The graft density was calculated by 1H NMR and was 5, 3 and 0.5 mol% for PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy, respectively (Table 1, see ESI for details†). For the latter, only the UPy grafts were included in the graft density, the hexyl groups are included in the dimethylsiloxane content.
| Polymer | M n [kg mol−1] | Đ | Mol% graftb | x | y | T g [°C] | T f [°C] |
|---|---|---|---|---|---|---|---|
| a Determined by SEC in THF, calibrated with polystyrene standards. b Determined by 1H NMR, see ESI† for calculation. c x is the number of siloxane units, y is the number of siloxane units with a supramolecular moiety attached. Calculated using the Mn, mol% supramolecular crosslink and average molecular weight of a monomer. d Determined by DMTA. e Includes both the number of dimethylsiloxane as well as methyl-hexyl-siloxane (x and x2 in Scheme 1). | |||||||
| PDMS-g-hydz | 29.5 | 2.2 | 5 | 320 | 17 | −105 | −4 |
| PDMS-g-BTA | 42.1 | 2.1 | 3 | 440 | 14 | −104 | 27 |
| PDMS-g-Upy | 27.9 | 1.5 | 0.5 | 380e | 2 | −113 | 100 |
Non-covalent interactions and phase segregation between hard and soft segments
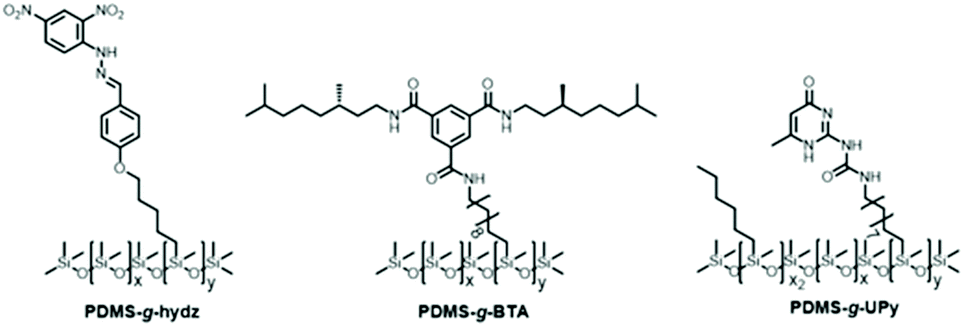
Fourier transform infrared (FT-IR) spectroscopy was applied to investigate the presence of hydrogen bonds in the polymers (Fig. 2A). First, the PDMS-g-hydz shows an absorption band at 3280 cm−1, representing an intramolecular hydrogen bond between the nitro and amine group according to previous studies.27,35 The presence of intermolecular hydrogen bonding between the amides of the BTAs in PDMS into helical aggregates was confirmed by an NH stretch band at 3238 cm−1 and CO stretch and amide II band at 1640 and 1560 cm−1, respectively.29 Finally, dimerization of the UPy units was corroborated by the presence of hydrogen bonded carbonyl groups at 1707 and 1672 cm−1.36 Due to the low concentration of UPys in the material, a weak stretching band for the hydrogen bonding NH groups was observed in the region at 3212 to 3147 cm−1.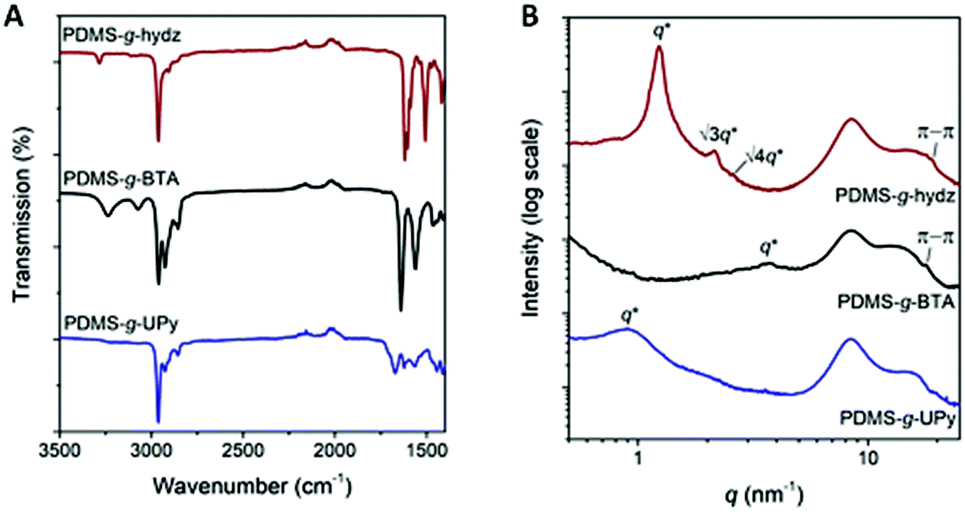 | ||
| Fig. 2 (A) FT-IR spectra and (B) 1-D transmission scattering profiles of PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy. Data is shifted for clarity. | ||
In addition, medium and wide angle X-ray scattering experiments showed the presence of a microphase segregated state for all grafted polymers (Fig. 2B). The phase segregation of PDMS-g-hydz is most pronounced and can be assigned to a hexagonally packed cylindrical phase by the presence of q* followed by √3q* and √4q*. The BTA- and UPy-grafted PDMS polymers are only weakly segregated as indicated by the weak principal scattering peak (q*). The wide angle region (q > 6 nm−1) shows the presence of π-stacking for both PDMS-g-hydz and PDMS-g-BTA by the appearance of a scattering peak at a spacing of 0.3 nm.
Mechanical properties
The (thermo)mechanical properties of the polymers were examined by dynamic mechanical thermal analysis (DMTA) and tensile tests. Films of PDMS-g-hydz and PDMS-g-BTA were produced by drop casting a concentrated solution of the copolymers in THF (∼1 g mL−1) into a Teflon mold. The THF slowly evaporated and the films were annealed in the oven at 40 °C overnight. In contrast, we could not obtain uniform films of PDMS-g-UPy by drop casting, thus uniform films were obtained by compression molding at 130 °C. The films were annealed overnight in the oven at 80 °C to ensure equilibrium in the polymeric system and that no stresses remain from compression.DMTA results show a glass transition (Tg) for the three polymers at −104 to −113 °C, which is close to the Tg of conventional PDMS (Fig. 3A).17 Therefore, we can conclude that the Tg is mainly governed by the PDMS backbone (Table 1). Interestingly, the tan(δ) peak of PDMS-g-hydz has a shoulder at −90 °C which we assign to the mobility of the hydrazone groups. Likewise, the gain in mobility of the BTAs is observed by the second maximum in tan(δ) at −67 °C for PDMS-g-BTA. The sharp peak in tan(δ) below the Tg (−118 °C) for PDMS-g-UPy is most likely attributed to a gamma transition, which may also involve associations with water. The flow temperature (Tf), defined as the change from elastic to a viscous response, is strongly influenced by the supramolecular bond strength of the grafts (Table 1). Fig. 3B clearly shows that the hydrazone grafted PDMS flows at the lowest temperature (−4 °C) while PDMS-g-UPy only flows at 100 °C. Hence, the interaction strength of the supramolecular moieties increases from hydrazone via BTA to UPy. Interestingly, the Tf of the BTA grafted PDMS is low compared to liquid crystalline BTAs (120 °C), measured by DSC.37 Most likely, this is due to the presence of small BTA stacks in the PDMS, i.e., dimers, trimers, tetramers, whereas liquid crystalline BTAs can form infinitely long stacks. Hence, dissociation of the small stacks in the PDMS costs less energy yielding a relatively low flow temperature. The storage moduli at the rubber plateau determined at −40 °C for PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy are 26, 13 and 4 MPa, respectively. This corresponds well to the number of non-covalent crosslinks incorporated on the PDMS (Table 1).
 | ||
| Fig. 3 (A) tan(δ) and (B) storage modulus of PDMS-g-hydz (red), PDMS-g-BTA (black) and PDMS-g-UPy (blue) as a function of temperature. | ||
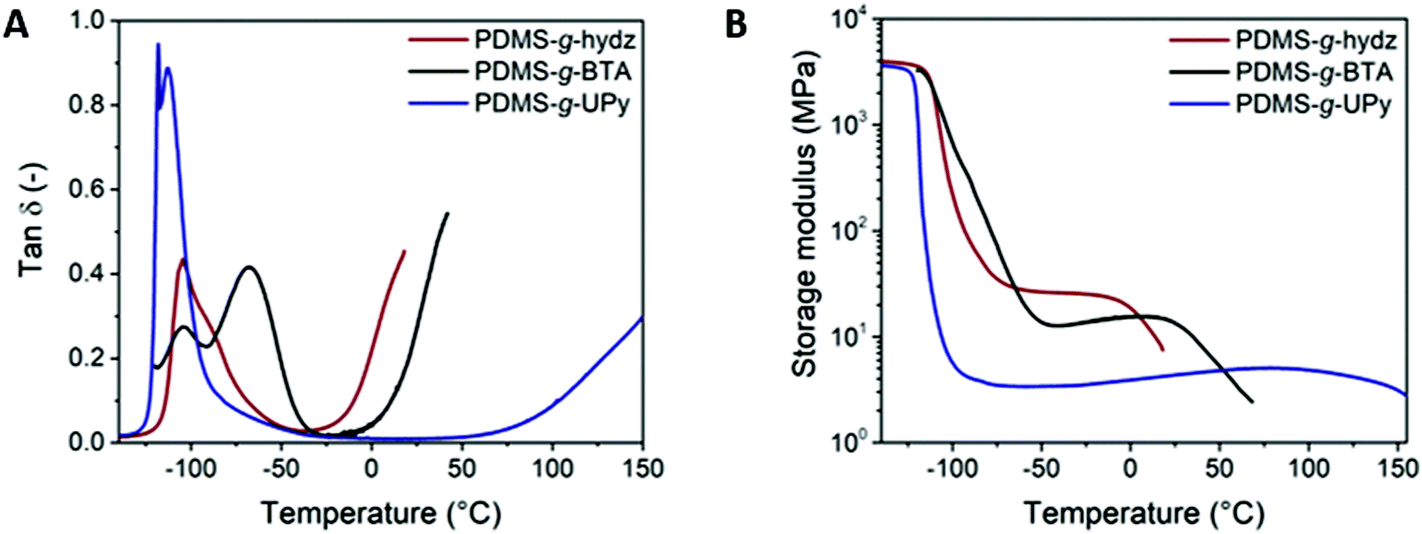
Tensile tests of the copolymers were performed at room temperature and the results are listed in Table 2 and Fig. 4. In addition, X-ray diffraction measurements were performed on samples stretched at different strains to attain information on the alignment of the supramolecular grafts (Fig. 5 and S7–9†). PDMS-g-hydz exhibits only viscous flow as the polymer chains are only associated by weak π–π stacking. Upon stretching the polymer sample beyond the yield point, the phase segregated cylinders are aligned in the direction of the force (Fig. S7†). Herein, the direction of the π-stacking originating from the hydrazones are found perpendicular to the cylindrical axis. The alignment of the microphase after stretching is also observed by polarized optical microscopy (POM) of the polymer film (Fig. S8†). Before stretching, the birefringence is non-directional while after elongation, the birefringence is more pronounced in the direction of the force indicating planar alignment of the cylinders. The stress–strain curve of PDMS-g-UPy shows a low maximum stress and strain at break combined with no plastic deformation which is indicative for a brittle material. PDMS-g-BTA has a yield point of 106% and elongation at break up to 518%. The order of the Young's modulus of the three polymers follow the modulus at room temperature from the DMTA (vide supra). Hence, the highest modulus is observed for PDMS-g-BTA, followed by PDMS-g-UPy and then PDMS-g-hydz (Table 2). In further agreement with the DMTA results, it follows that the PDMS-g-UPy has a rate independent modulus at room temperature while the Young's modulus of PDMS-g-BTA and PDMS-g-hydz are highly dependent on the rate (Fig. S10†).
 | ||
| Fig. 4 Engineering stress–strain curve of PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy, measured at 0.01 s−1. Inset shows the low strain region. | ||
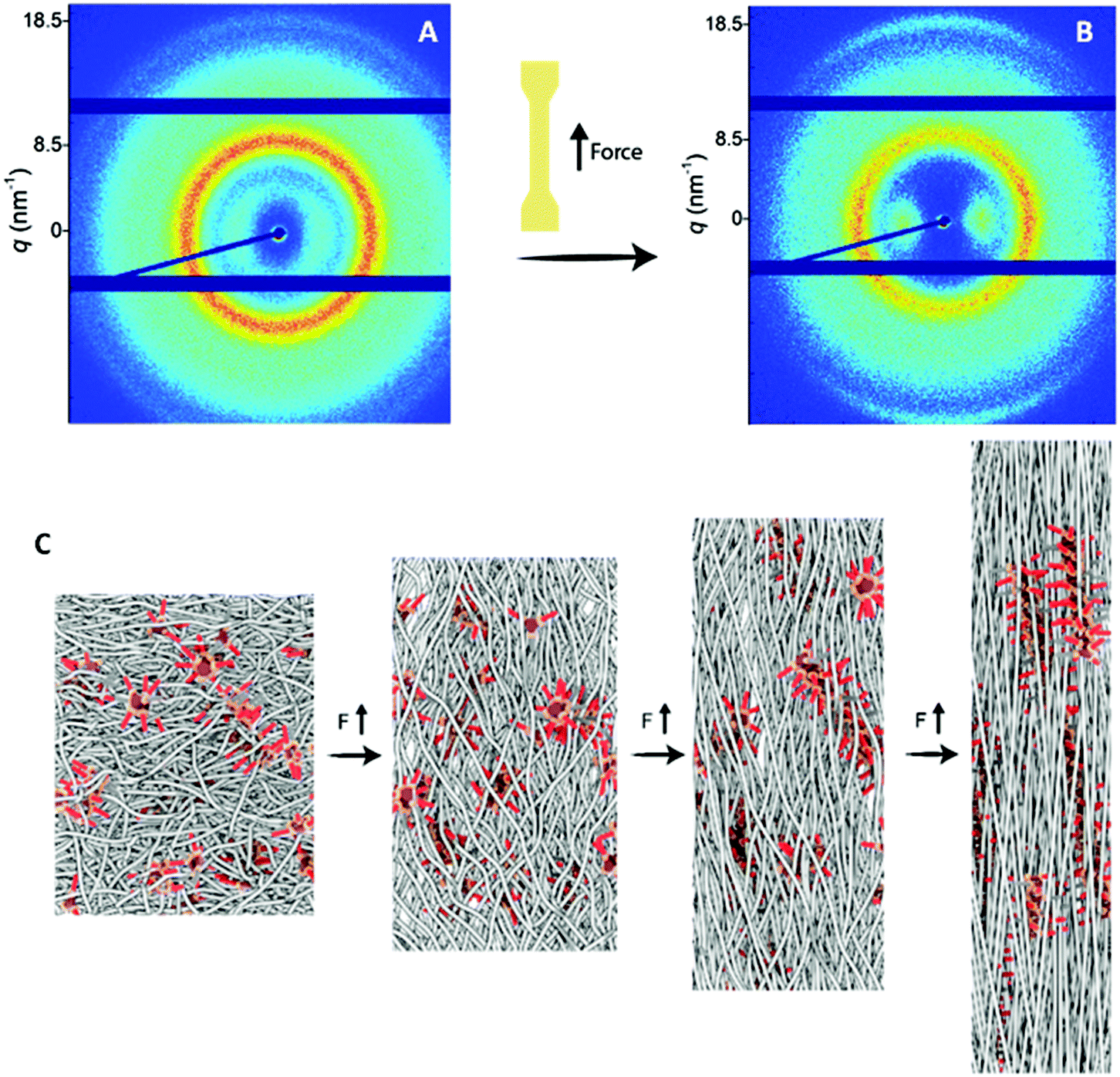 | ||
| Fig. 5 Two-dimensional transmission scattering profile of PDMS-g-BTA after (A) 80% and (B) 400% strain. (C) Schematic representation of phases during elongation of PDMS-g-BTA. The PDMS chains are represented in gray and the BTAs are represented in red. Initially, the BTAs are randomly distributed through the material (left) and upon elongation by force, the BTAs move from stack to stack and come closer together forming aligned helices within the stretched PDMS chains (right). | ||
| Polymer | E [MPa] | σ yield [kPa] | ε yield [%] | σ max [kPa] | ε break [%] |
|---|---|---|---|---|---|
| Values were averaged over four tensile tests; n.o.: not observed. | |||||
| PDMS-g-hydz | 2.4 ± 0.04 | 162 ± 27 | 25 ± 4 | 162 ± 27 | 467 ± 82 |
| PDMS-g-BTA | 5.3 ± 0.06 | 1014 ± 39 | 106 ± 7 | 1420 ± 109 | 518 ± 11 |
| PDMS-g-Upy | 3.9 ± 0.01 | n.o. | n.o. | 273 ± 36 | 7 ± 1 |
PDMS-g-BTA shows a typical rubber-like, elastomeric response below the yield point. Hence, when tested under quasi-static conditions at moderate to high strain rates, the mechanical response of the material resembles that of a thermoplastic elastomer. Beyond the yield point (strain >100%), plastic deformation occurs in which the BTA stacks dynamically break and reform to release stress. The yield point represents the dynamics of the BTA in the polymer whereas the yield point of conventional covalent polymers is associated with the segmental mobility of the main chain. Finally, strain hardening takes place in which the BTAs align in the direction of the force (Fig. 5A and B, ring at 18.5 nm−1). Herein, a large enhancement of the π-stacking is observed (Fig. S9†), indicative for larger domains of BTA stacks and hence longer BTA fibers in the PDMS. POM images of the stretched film confirmed the alignment of the BTAs, showing birefringence under crossed polarizers throughout the whole film after stretching while before stretching only small birefringent domains are observed (Fig. S8†). Additionally, the PDMS chains align slightly in the direction of the force observed by brightening of spots on the ring representing the PDMS (Fig. 5B, ring at 8.5 nm−1). To clarify the tensile tests at the molecular level, a schematic representation of the polymer with BTA stacks before alignment, below 80% and after 400% strain is shown (Fig. 5C). The polymer starts as random coil with small crystallites of the BTA as confirmed by the low Tf (vide supra). Then the polymer backbone start to elongate slightly after which the BTAs start to move in and out of the stacks and finally align into larger stacks in the direction of the force.
Dynamic properties of supramolecular crosslinks in PDMS
Dynamic properties of the three different supramolecular interactions in the PDMS networks were examined by stress relaxation experiments through DMTA. A constant strain of 1% was applied and the relaxation modulus was followed over time. The normalized stress relaxation moduli (E/E0) are shown in Fig. 6 at different temperatures. At room temperature, full stress relaxation was achieved in 160 and 210 s for PDMS-g-hydz and PDMS-g-BTA, respectively (Fig. 6A and B). The UPy grafted PDMS required more than 8 hours at room temperature to obtain full stress relaxation (Fig. S11†). The efficient stress relaxation is facilitated by the reversibility of the supramolecular networks as the bonds can exchange to relax the polymeric chains that are stressed. Furthermore, the faster decay of the modulus with increasing temperature for all polymers indicates an increased dynamics of non-covalent crosslinks at higher temperatures. The characteristic relaxation time (τ*) was determined from the stress relaxation curves at E/E0 equals 1/e, assuming the Maxwell model for stress relaxation. The temperature dependence of τ* is in good agreement with the Arrhenius equation arising from dynamic exchange. The relationship between ln(τ*) and 1000/T is plotted and fitted to the Arrhenius law (Fig. 6D) and approximate activation energies (Ea) are obtained (see ESI† for details). The resulting activation energies are 22, 39 and 69 kJ mol−1 for PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy, respectively. As expected, the Ea for PDMS-g-hydz is relatively low as the material is only crosslinked via weak physical interactions. The high Ea of PDMS-g-UPy originates from the strong hydrogen bonding of the UPys in the polymer and is in agreement with the dissociation energy of UPy dimers in chloroform.38 Hence, the solid state bond exchange at room temperature is slow in comparison to other non-covalent crosslinked polymers, yet, comparable to some dynamic covalent bonds in polymer networks.39–43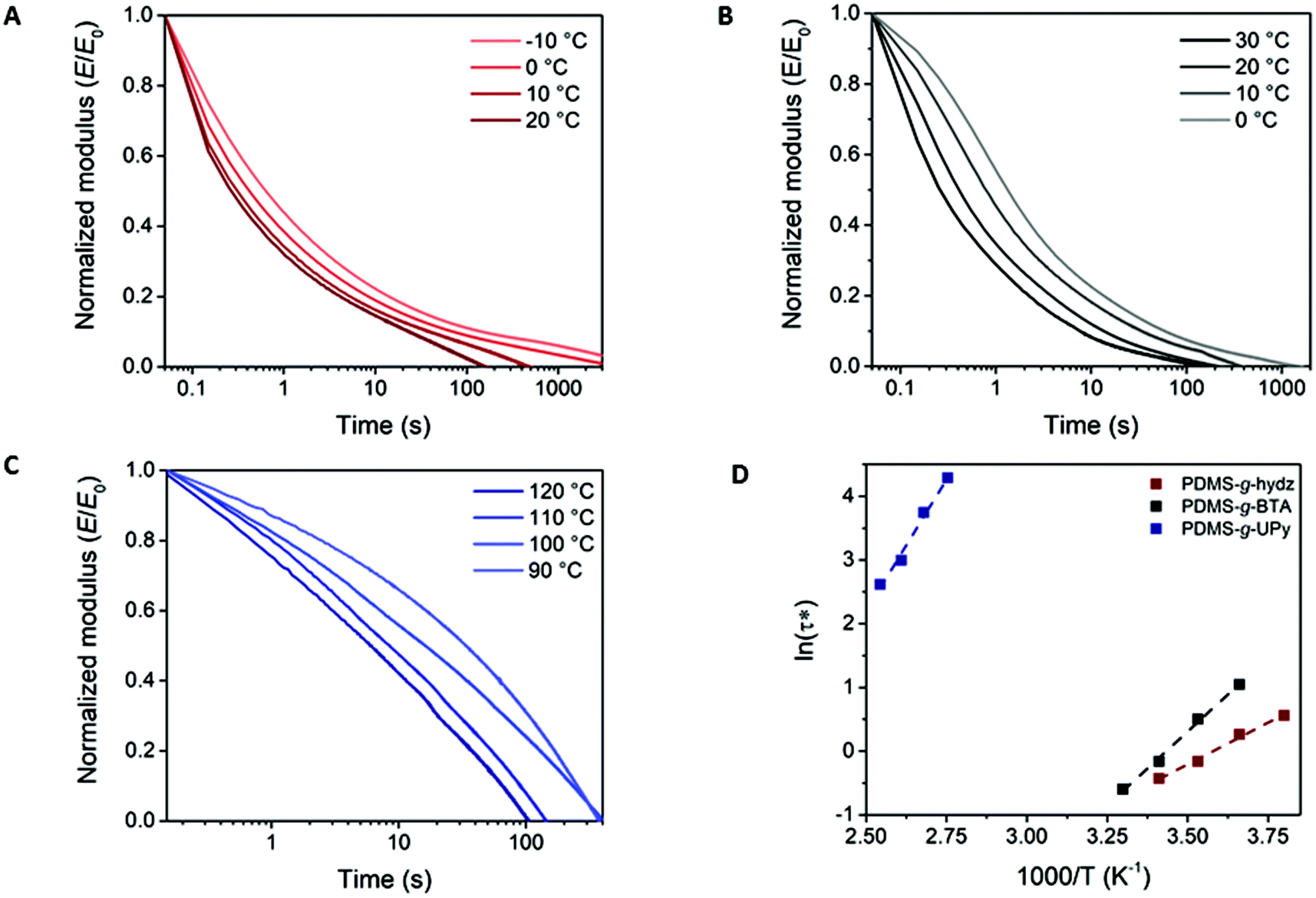 | ||
| Fig. 6 Stress relaxation curves of (A) PDMS-g-hydz, (B) PDMS-g-BTA and (C) PDMS-g-UPy, measured at 1% strain. (D) Arrhenius analysis of the characteristic relaxation time τ* obtained from the stress relaxation values at E/E0 = 1/e for PDMS-g-hydz (red), PDMS-g-BTA (black) and PDMS-g-UPy (blue). | ||
The dynamics of the networks were further verified by creep experiments in which the polymers were subjected to a constant stress of 2 kPa at room temperature and followed over time. After 20 minutes, a strain of 0.5% was measured for PDMS-g-UPy, whereas BTA and hydrazone grafted PDMS increased to 2 and 8% strain, respectively (Fig. S12†). Consequently, PDMS-g-hydz is the best energy absorbing material which is in accordance with its viscoelastic properties at room temperature. In contrast, the energy absorption of PDMS-g-UPy is negligible, as expected for a brittle material. The origin of the creep in PDMS-g-hydz and PDMS-g-BTA is the exchange of supramolecular moieties at room temperature on the 2–4 minutes time scale, shown by the stress relaxation experiments.
Recyclability of the non-covalent crosslinked PDMS networks
The supramolecular origin of the three PDMS networks enables recycling of the polymers, both thermally and via solvation. Polymeric films can be obtained by dropcasting from THF solution (vide supra), thus the films can be redissolved in THF and dropcasted to make new films. Although this method yielded poor films for PDMS-g-UPy, the material can still dissolve in THF, showing the reversibility and recyclability of the network. Alternatively, recycling of the polymers is accomplished by compression molding of the material, giving homogeneous films (Fig. 7A). In this way, the polymers can be reprocessed in any shape by either molding or cutting the films into the desired shape. The recycled polymers still have the same properties as is shown by the tensile tests (Fig. 7B).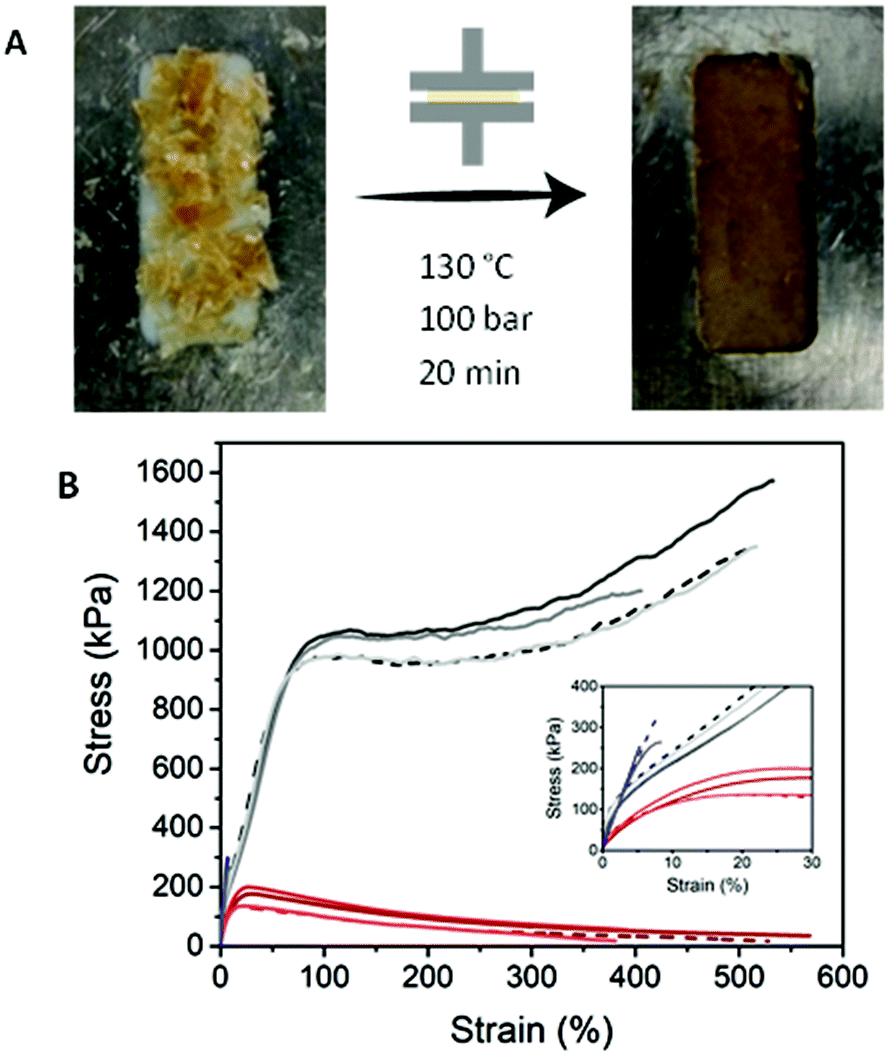 | ||
| Fig. 7 (A) Photo showing the thermal recycling of PDMS-g-UPy into a rectangular shape. (B) Tensile tests of PDMS-g-hydz, PDMS-g-BTA and PDMS-g-UPy after a three times recycling process. Dashed line represents the original sample. Inset shows the low strain region. | ||
Conclusions
In conclusion, we utilized supramolecular grafts to successfully crosslink linear PDMS and obtain strikingly different mechanical properties by tuning the strength of the non-covalent interactions. Hereby, we developed a way to finely tune the PDMS polymer properties only by changing the nature of the supramolecular interactions. Association of the hydrazones resulted in a viscous material, which is weakly crosslinked at room temperature. Hydrogen bonding by BTAs gives a thermoplastic elastomer, while a brittle material was obtained by quadruple hydrogen bonding in UPys. Stress relaxation experiments revealed that the dynamics of the supramolecular moieties was slowest for the UPy, followed by the BTA and the hydrazone. The slow dynamics of the UPy due to strong association results in a lack of elasticity and therefore a brittle material is obtained. The BTA dynamics in the PDMS provides an equilibrium of the BTAs reorganizing their superstructure. Thereby, the stress is reduced due to the dynamic nature of the crosslinks originating from reversible dissociation and association of the BTAs. Finally, all polymers are fully recyclable without any loss of the material properties. These molecular insights into supramolecular crosslinking of PDMS broadens the scope of applications for PDMS in e.g. the field of biomaterials. We are confident that this, in combination with the facile post-functionalization synthesis, will lead to the further development of recyclable plastics with novel polymer properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Eveline Maassen for the fruitful discussions.We acknowledge funding from the Dutch Ministry of Education, Culture and Science (gravity program 024.001.035) and European Union's Horizon 2020 research and innovation program under the grant agreement no. 642083.
Notes and references
- C. Jehanno and H. Sardon, Nature, 2019, 568, 467–468 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. DuPrez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- W. G. Skene and J. M. P. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS PubMed.
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 CrossRef.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef PubMed.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef PubMed.
- C. Hilger and R. Stadler, Makromol. Chem., 1990, 191, 1347–1361 CrossRef.
- C. Hilger and R. Stadler, Makromol. Chem., 1991, 192, 805–817 CrossRef.
- M. Müller, U. Seidel and R. Stadler, Polymer, 1995, 36, 3143–3150 CrossRef.
- L. Bouteiller, Hydrogen Bonded Polymers, Advances in Polymer Science, 2007, vol. 207, pp. 79–112 Search PubMed.
- L. R. Hart, N. A. Nguyen, J. L. Harries, M. E. Mackay, H. M. Colquhoun and W. Hayes, Polymer, 2015, 69, 293–300 CrossRef.
- M. A. Rahman, M. Penco, I. Peroni, G. Ramorino, A. M. Grande and L. Di Landro, ACS Appl. Mater. Interfaces, 2011, 3, 4865–4874 CrossRef PubMed.
- J. C. Lai, X. Y. Jia, D. P. Wang, Y. B. Deng, P. Zheng, C. H. Li, J. L. Zuo and Z. Bao, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- E. Yilgör and I. Yilgör, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- Y. Mi, Y. Chan, D. Trau, P. Huang and E. Chen, Polymer, 2006, 47, 5124–5130 CrossRef.
- G. M. Whitesides, Angew. Chem., Int. Ed., 2018, 2–18 Search PubMed.
- L. Dodge, Y. Chen and M. A. Brook, Chem. – Eur. J., 2014, 20, 9349–9356 CrossRef PubMed.
- T. Rambarran, A. Bertrand, F. Gonzaga, F. Boisson, J. Bernard, E. Fleury, F. Ganachaud and M. A. Brook, Chem. Commun., 2016, 52, 6681–6684 RSC.
- T. Stukenbroeker, W. Wang, J. M. Winne, F. E. Du Prez, R. Nicolaÿ and L. Leibler, Polym. Chem., 2017, 8, 6590–6593 RSC.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef PubMed.
- M. Kathan, C. Jurissek, P. Kovaříček and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2378–3282 CrossRef.
- O. Colombani, C. Barioz, L. Bouteiller, C. Chanéac, L. Fompérie, F. Lortie and H. Montés, Macromolecules, 2005, 38, 1752–1759 CrossRef.
- A. S. Fawcett and M. A. Brook, Macromolecules, 2014, 47, 1656–1663 CrossRef.
- B. A. G. Lamers, R. Graf, B. F. M. de Waal, G. Vantomme, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 15456–15463 CrossRef.
- S. Cantekin, T. F. A. De Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- M. L. Ślęczkowski, E. W. Meijer and A. R. A. Palmans, Macromol. Rapid Commun., 2017, 38, 1–5 CrossRef.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef.
- R. H. Zha, B. F. M. De Waal, M. Lutz, A. J. P. Teunissen and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 5693–5698 CrossRef.
- N. E. Botterhuis, D. J. M. van Beek, G. M. L. van Gemert, A. W. Bosman and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3877–3885 CrossRef.
- Y. Meng, W. Xu, M. R. Newman, D. S. W. Benoit and M. Anthamatten, Adv. Funct. Mater., 2019, 1903721, 1903721 Search PubMed.
- C. Racles, M. Alexandru, A. Bele, V. E. Musteata, M. Cazacu and D. M. Opris, RSC Adv., 2014, 4, 37620–37628 RSC.
- W. Paulus, H. Ringsdorf, S. Diele and G. Pelzl, Liq. Cryst., 1991, 9, 807–819 CrossRef.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Chem. – Eur. J., 2009, 15, 2071–2080 CrossRef.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. Van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- A. Chao and D. Zhang, Macromolecules, 2019, 52, 495–503 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, Schemes S1, 2 and Fig. S1–12. See DOI: 10.1039/d0py00139b |
| This journal is © The Royal Society of Chemistry 2020 |