
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrder–disorder transition in supramolecular polymer combs/brushes with polymeric side chains†
Milad
Golkaram
a,
Giuseppe
Portale
 a,
Pascal
Mulder
a,
Dina
Maniar
a,
Shirin
Faraji
b and
Katja
Loos
*a
a,
Pascal
Mulder
a,
Dina
Maniar
a,
Shirin
Faraji
b and
Katja
Loos
*a
aMacromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
bTheoretical Chemistry Group, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 3rd March 2020
Abstract
Supramolecular polymer combs/brushes can be used as smart materials which incorporate the properties of linear polymers with branch polymers depending on the temperature, grafting density, branch molecular weight and most importantly type of non-covalent interaction between the main chain and side chains. Three sets of polymers were synthesized and mixed based on a specific interaction between the main chain and the side chain polymers. The first two sets based on the combination of 2,4-diamino-1,3,5-triazine (DAT)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thymine (THY) and 2-ureido-4[1H]-pyrimidinone (UPy):(1-(6-isocyanatohexyl)-3-(7-oxo-7,8-dihydro-1,8-naphthyridin-2-yl)urea) (ODIN) were a blend and the third one using ODIN:ODIN interactions was used without a main chain. The polymer set using ODIN:ODIN interaction showed long-range ordering due to strong ODIN aggregation, whereas UPy:ODIN-based polymer combs showed comb-like clusters without any ordering. The phase separation in the THY:DAT system was more pronounced and was further improved after the addition of more equivalents of side chains. Moreover, using melt rheology, consistent with the SAXS data, it was concluded that long-range ordering is responsible for the elastic properties and the flow temperature (Tflow) is lower than the order–disorder transition temperature (TODT). This work can be considered as a toolkit for the design of bottlebrush and comb polymers as well as supersoft elastomers and stimuli-responsive materials.
:thymine (THY) and 2-ureido-4[1H]-pyrimidinone (UPy):(1-(6-isocyanatohexyl)-3-(7-oxo-7,8-dihydro-1,8-naphthyridin-2-yl)urea) (ODIN) were a blend and the third one using ODIN:ODIN interactions was used without a main chain. The polymer set using ODIN:ODIN interaction showed long-range ordering due to strong ODIN aggregation, whereas UPy:ODIN-based polymer combs showed comb-like clusters without any ordering. The phase separation in the THY:DAT system was more pronounced and was further improved after the addition of more equivalents of side chains. Moreover, using melt rheology, consistent with the SAXS data, it was concluded that long-range ordering is responsible for the elastic properties and the flow temperature (Tflow) is lower than the order–disorder transition temperature (TODT). This work can be considered as a toolkit for the design of bottlebrush and comb polymers as well as supersoft elastomers and stimuli-responsive materials.
Introduction
In the last few decades, there have been solid efforts to design complex supramolecular structures. In polymer science, more recently, a few researchers coined the terms supramolecular graft/comb/brush polymer.1–15 These reversible comb-shaped polymers can dissociate depending on the strength of the used supramolecular entity (sticker) at different temperatures.4 Moreover, the second parameter influencing the association is the steric hindrance of the side chains in highly grafted polymers, which can lead to significant chain stretching.16 Therefore, depending on the graft density a variety of polymers with different topologies can be obtained. At low grafting densities, the main chain of the polymer and the side chains behave as a random coil, whereas with increasing grafting density, both the side and main polymer chains are stretched, leading to loosely grafted combs (LC), dense combs (DC), loosely grafted bottlebrushes (LB) and dense bottlebrushes (DB).16The synthesis of supramolecular comb/brush polymers has been performed through a variety of synthetic methods and the stickers that were incorporated differed in polarity, association constant and difficulty of synthesis. They consist of pyridine/phenol, 2,4-diamino-1,3,5-triazine (DAT)/thymine (THY), adenine/THY, 2-ureido-4[1H]-pyrimidinone (UPy)/2,7-diamido-1,8-naphthyridine (Napy), bis/triurea and terpyridine ruthenium metal complex.1,2,4–8,17 Although, in these works no comparative investigation was carried out to check the effect of the sticker on the self-assembly and comb/brush formation, extensive studies were done on the simpler cases of supramolecular polymers with sticky side-groups.18–25
The side chain in all the above-mentioned supramolecular comb/brush polymers consists of either a small amphiphilic compound with a sticker at one end or in rare cases a polymer. Mono-functionalized polymers which form the side chains have been exclusively and widely studied in the framework of supramolecular telechelic polymers. For instance, for polymers based on THY or DAT end-functionalized poly(ethylene) (PE), a lamellar morphology could be obtained when THY is solely used as end-groups. THY could crystallize readily and govern the morphology whereas DAT functionalized PE or its mixture with PE-THY was only slightly ordered and needed crystallization of PE to form a lamellar morphology, as DAT is known to pack poorly.26–28 When a non-crystalline polymer such as poly(propylene oxide) (PPO) was used, long-range ordering is found for THY–PPO–THY but not for DAT–PPO–DAT.29,30 The authors attributed this to the crystallization of THY and a stronger interaction parameter for THY and PPO in comparison with DAT and PPO. For strongly non-polar polymers such as poly(isobutylene) (PIB) mono-functionalized with DAT, a body-centered cubic (BCC) morphology was observed, whereas mesophases were not observed for bifunctional PIB.31
In the case of stronger end-groups such as UPy, a lateral aggregation into high aspect nanofibers was observed when urea functionalities were used. Although, in this case no lamellar morphology was reported,32 in some other studies on UPy-functionalized polymers, a well-ordered lamellar morphology was observed at high UPy concentrations.33,34
Herein, we systematically study the interplay between aggregation strength and grafting density in supramolecular graft polymers with polymeric side chains of different sticker strengths and concentrations. Telechelic supramolecular polymers and comb polymers with short side-groups were discussed extensively in the past.4,9,12–14,26–30 Therefore, in our design, the focus is on the synthesis and self-assembly of novel supramolecular polymers with long (polymeric) side-branches. Dalcanale et al. recently introduced a new sticker, namely (1-(6-isocyanatohexyl)-3-(7-oxo-7,8-dihydro-1,8-naphthyridin-2-yl)urea) (ODIN), which can undergo sextuple hydrogen bonding.35 We have shown that ODIN possesses a high propensity for aggregation and stacking. Although the stacking strength is proven to be much stronger, the hydrogen bonding strength is weaker compared to UPy.36 Therefore, four stickers (UPy, ODIN, THY and DAT) were chosen to check the effect of stacking, polarity and hydrogen bonding in poly(n-butyl acrylate) (PnBA) as the polymer matrix. PnBA has been reported to be a good hydrogen bonding acceptor and also possesses medium polarity. It is, therefore, a good candidate to differentiate between the aggregation abilities of stickers (Scheme 1).37
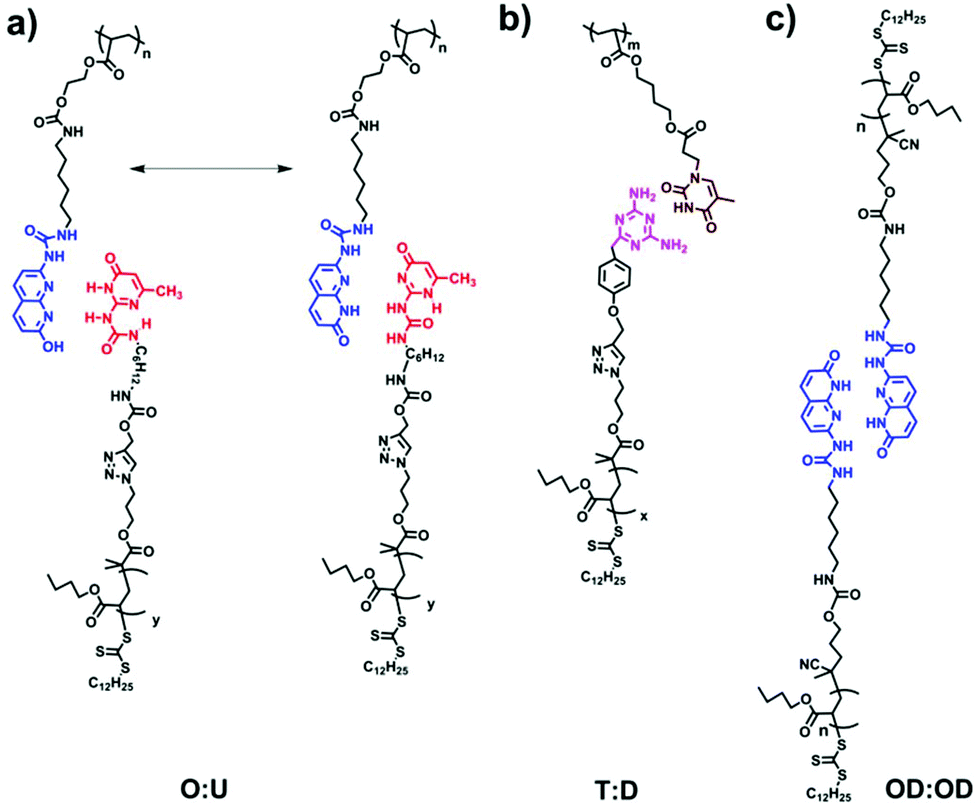 | ||
| Scheme 1 The design of supramolecular polymer combs/brushes based on (a) ODIN:UPy, (b) THY:DAT and (c) ODIN:ODIN interactions. | ||
Results and discussion
Synthesis of polymers
The aim of this work was to comprehensively study the self-assembly in supramolecular polymers when a polymer with stickers as side-groups is mixed with a polymer with stickers as end-groups, so that a hypothetical comb or brush (depending on the mixing ratio: LC, DC, LB or DB) can be formed. Therefore, two sets of polymers are synthesized with different stickers: (1) mono-functionalized (side chain) and (2) functionalized on each repeating unit (main chain). These were later mixed with suitable partners (O:U, T:D or OD:OD) in different ratios to check the effect of sticker type and grafting densities on self-assembly (Scheme 1). Moreover, the effect of the side chain molecular weight is addressed by changing the length of polymers D, U or OD (the main chain molecular weight is kept constant). Precursor polymers (main and side chains before mixing) are also separately investigated as they can readily phase separate in the pure form without the need for blending.
For a perfect comb formation, all the side chain end-groups should be modified and carry a sticker. For crystalline polymers like PE, it has been reported that unfunctionalized PE chains can be expelled from the lamellar domains and form a separate region.28 In this study, reversible addition–fragmentation chain transfer (RAFT) polymerization was employed, providing relatively good molecular weight distribution and end-group functionalization.38 Furthermore, using a chain transfer agent (CTA) readily carrying a sticker can be much more efficient than final post-polymerization functionalization, since the unmodified chains are usually hard to separate. Therefore, out of the three polymers, two were synthesized via a sticker containing a CTA (U and DScheme 2). In the case of polymer OD, although the formation of a CTA carrying sticker 10 was possible, the purification via column chromatography failed due to low solubility of the product and interaction of this CTA with the column. Therefore, post-polymerization modification was employed (Scheme 2, polymer OD).
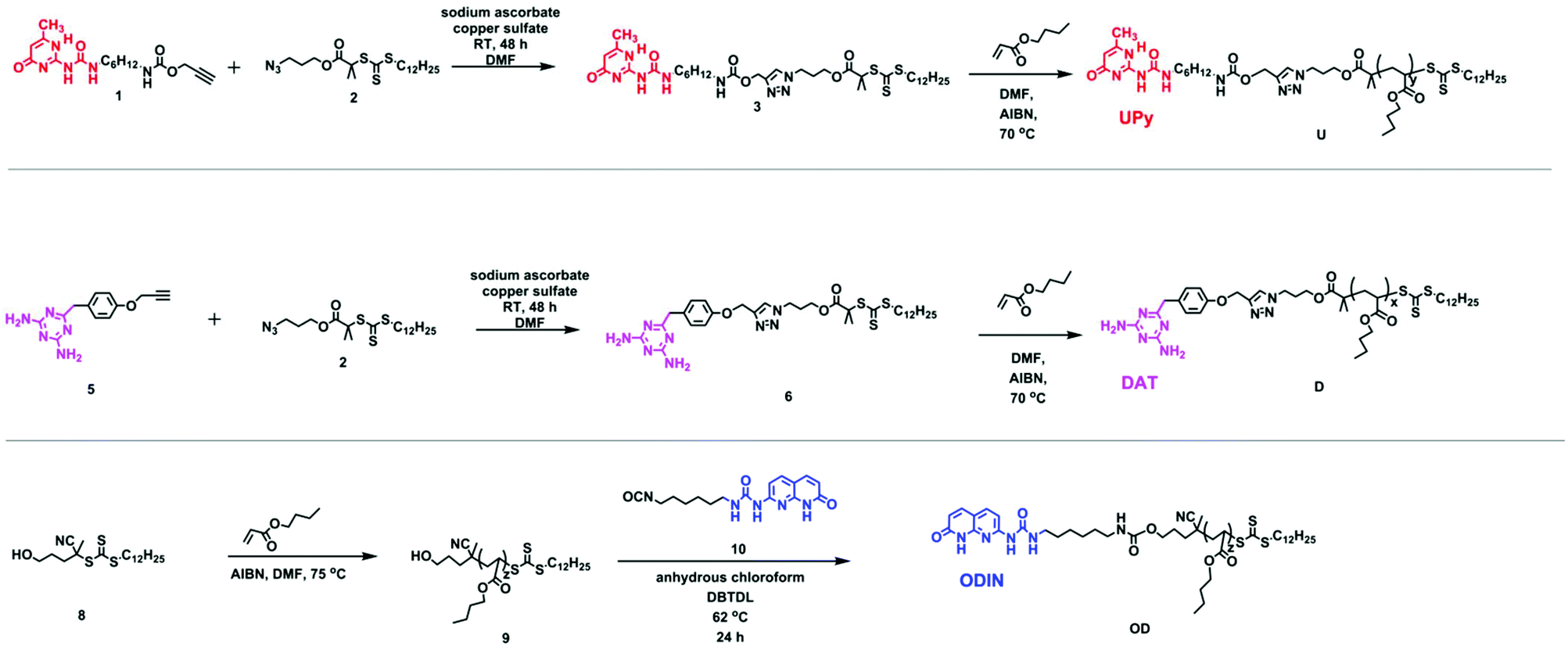 | ||
| Scheme 2 Synthesis of chain transfer agents 3 and 6 and polymers U, D and ODvia reversible addition–fragmentation chain transfer (RAFT) polymerization. | ||
Synthesis of 1 and 5 was performed following the literature.37,39 Afterwards, these two alkyne-terminated stickers were attached to the azide-terminated CTA using the protocol of Sharpless et al.40 The singlets at 7.63 ppm (Fig. 1a) and 8.20 ppm (Fig. 1b) prove the formation of the triazole ring in the 1H NMR of 3 and 6, respectively. Furthermore, the presence of the peaks assigned 1–5 substantiates the coupling of 1 with 2 (Fig. 1a) and 2 with 5 (Fig. 1b). On the other hand, 13C NMR substantiated the coupling (Fig. S8 and S10†). Subsequently, polymerization of n-butyl acrylate (nBa) was performed to yield three different molecular weights of U and D (UX and DX, with X being the molecular weight of the polymers in g mol−1), carrying 3 and 6 as end-groups, respectively (Table 1 and Fig. S1–S4†). Good control over the molecular weight was achieved (Đ < 1.3).
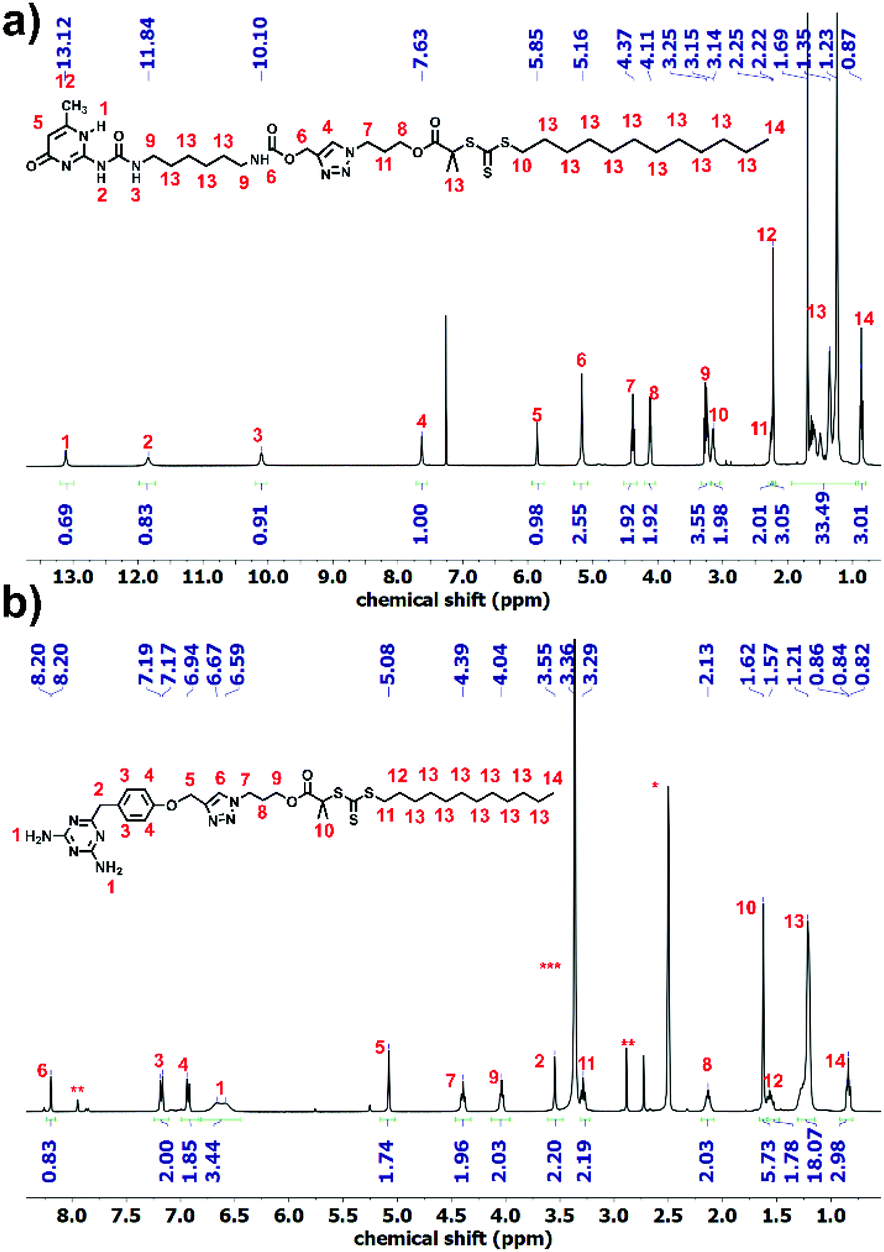 | ||
| Fig. 1 1H NMR spectra of (a) CTA 3 and (b) CTA 6. (*: DMSO-d6, **: DMF, ***: water). | ||
| Entry | Sample | M n theory (kg mol−1) |
M
na (kg mol−1) |
Đ |
|---|---|---|---|---|
| a Calculated via GPC measurements. b Calculated via1H NMR. | ||||
| 1 | U4500 | 5.1 | 4.5 | 1.30 |
| 2 | U12500 | 15.6 | 12.5 | 1.13 |
| 3 | U26000 | 33.0 | 26.0 | 1.29 |
| 4 | D3400 | 4.0 | 3.4 | 1.21 |
| 5 | D4100 | 5.7 | 4.1 | 1.24 |
| 6 | D15600 | 22.5 | 15.6 | 1.31 |
| 7 | OD2700 | 3.5 | 2.7 | 1.12 |
| 8 | OD8300 | 11.1 | 8.3 | 1.13 |
| 9 | OD9500 | 14.3 | 9.5 | 1.12 |
| 10 | O | 55.6 | 30.3b | — |
| 11 | T | 133.0 | 90.0b | — |
Polymer 9 was synthesized using a CTA carrying a carboxyl group (8) to yield PnBA with three different molecular weights (Fig. S5†). For the synthesis of polymer OD (ODX, with X being the molecular weight of the polymers in g mol−1), the previously synthesized sticker 10 was coupled to polymer 9 by the formation of a urethane bond. The detailed study of this sticker is published elsewhere.35,36Fig. 2 shows the 1H NMR of OD. The urethane proton at 4.79 ppm and the protons assigned 1–7 (which correspond to sticker 10) prove the coupling. For the synthesis of polymers O and T, different approaches were used (Scheme 3). HEA was coupled to sticker 10 using a similar reaction performed for OD. The formation of monomer 12 was proved using 1H NMR (Fig. 3a). The urethane peak at 4.86 ppm shows that the coupling occurred successfully. Furthermore, 13C NMR proved the coupling, although the low solubility of 10 and 12 hindered the analysis and some peaks showed a low signal to noise ratio (Fig. S9 and S11†). This monomer was then polymerized using S,S-dibenzyl trithiocarbonate (DBTTC) as the CTA. The phenyl protons corresponding to the end-groups are visible in Fig. 3b (assigned 16–18). Therefore, the average number of stickers per polymer in polymer O can be estimated to be around 70.
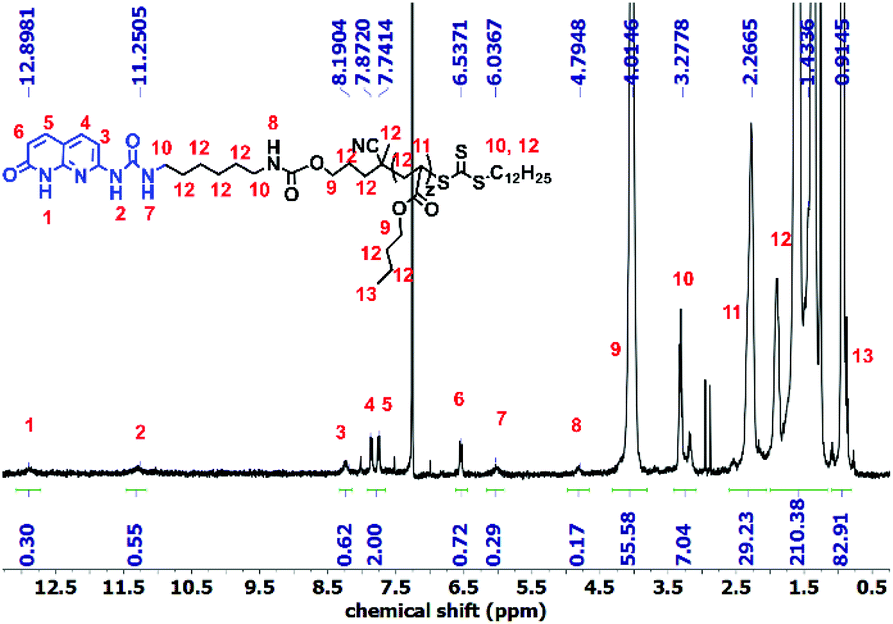 | ||
| Fig. 2 1H NMR spectra of polymer OD. | ||
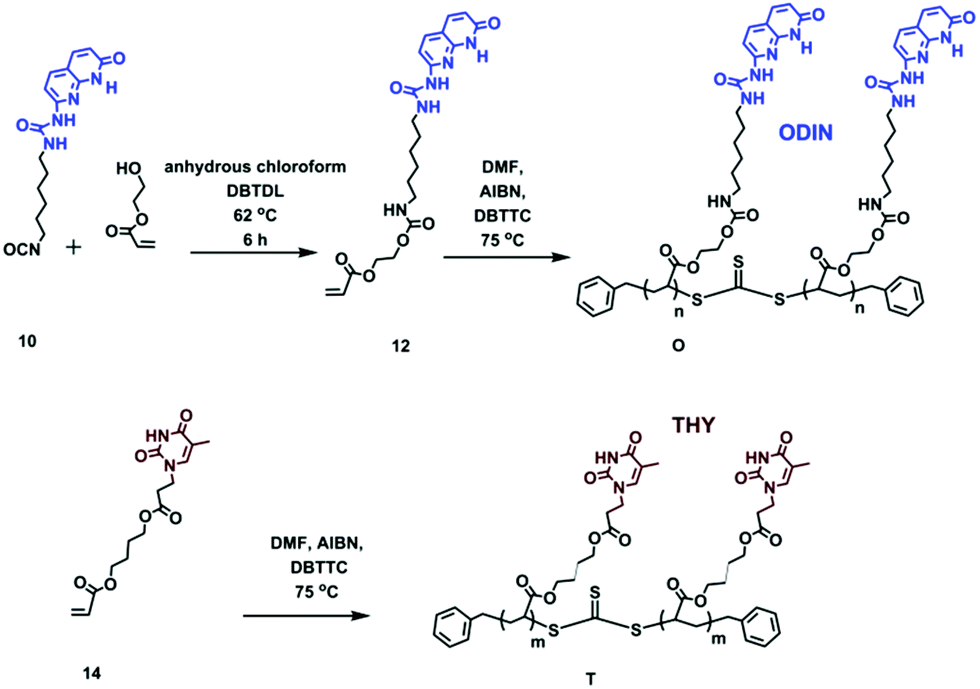 | ||
| Scheme 3 Synthesis of monomer 12 and polymers O and T. | ||
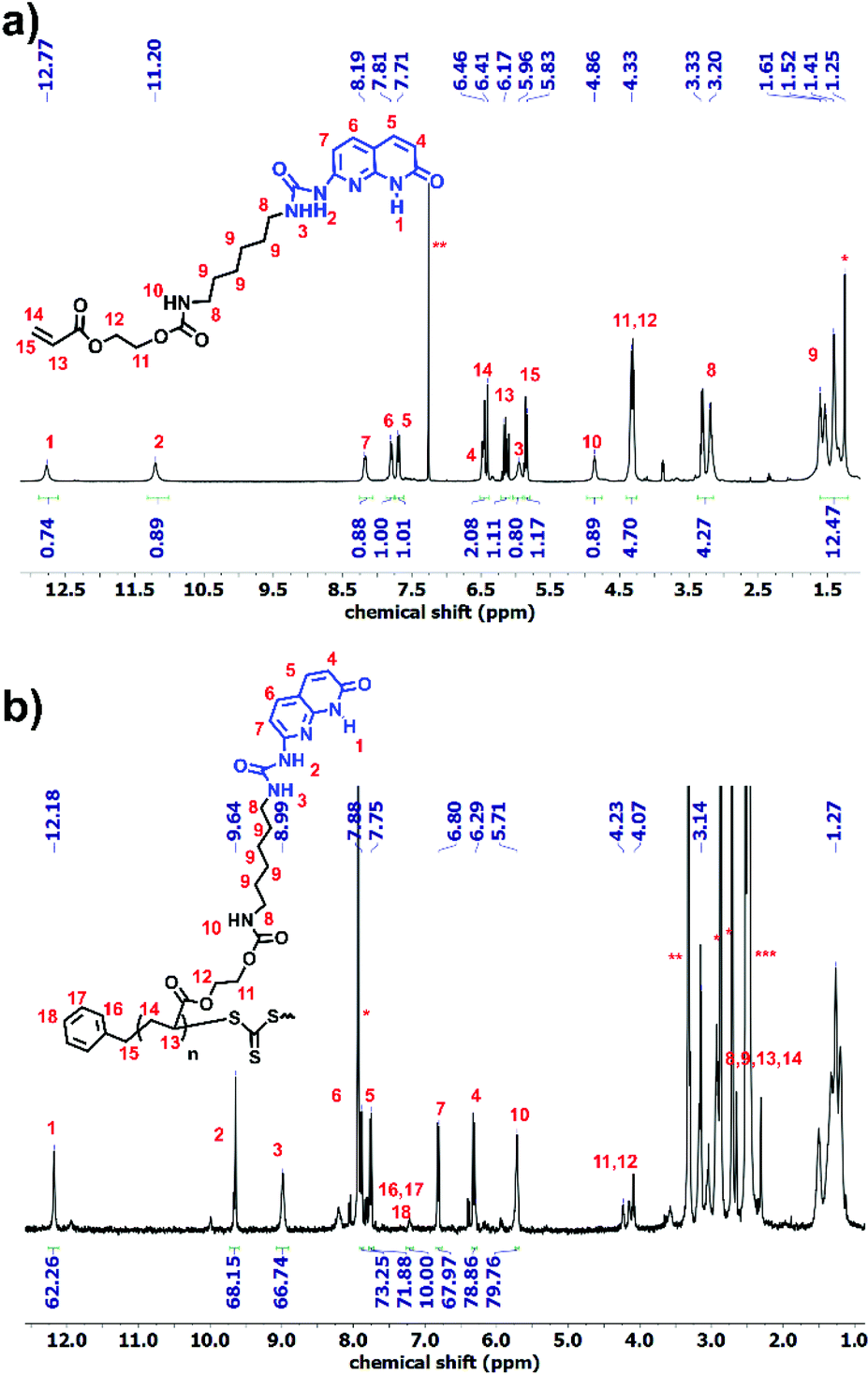 | ||
| Fig. 3 1H NMR spectra of (a) monomer 12 and (b) polymer O. | ||
The polymer carrying thymine (T) was synthesized using a previously published method (Fig. S6†).25 Since GPC was not possible for polymers O and T due to their interaction with the column, 1H NMR was used for the molecular weight calculations (Fig. 3b and S6,†Table 1).
In order to form a supramolecular polymer comb or brush, the polymers constructing the side chains, namely entries 1–6 in Table 1, should be mixed with the polymers carrying stickers along the chain (main chain), entries 10 and 11. It has to be noted that entries 1–3 (polymers carrying UPy, U) have the potential to form multiple H-bondings with polymer O (entry 10) as depicted in Scheme 1 and Fig. 4. However, we rule out this association as follows. Fig. 4 shows the possible dimerization and association of the two stickers. Electronic structure calculations at the density functional level of theory, DFT (omega B97X-D functional/cc-PVDZ basis set), were used to evaluate the molecular structures and dimerization energies (Edim). Calculations were conducted both in a vacuum and with chloroform using the polarizable continuum model (PCM). All calculations were performed using the Q-Chem electronic structure program.41 The keto tautomer of sticker 10 shows significant dimerization using 6 H-bondings. This is also the case for UPy consistent with the previous reports.42 However, hetero-association of the two stickers is not favourable. This conclusion was supported by a 1H NMR titration experiment, showing no significant change in the chemical shifts when they were mixed in different ratios (Fig. S7†).
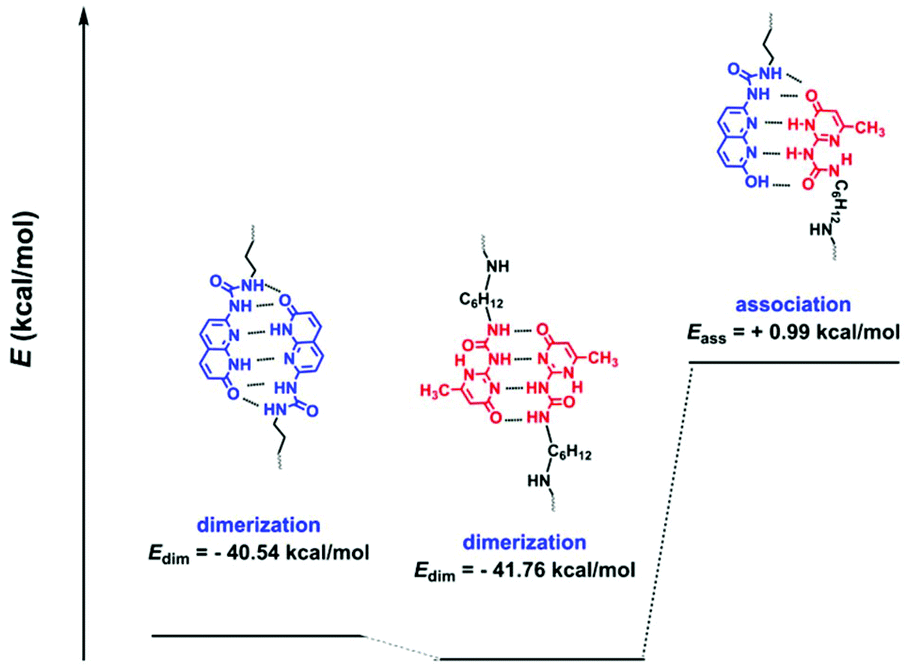 | ||
| Fig. 4 Energy diagram of UPy and ODIN and their possible dimerization and association in solution (polarizable continuum model (PCM), chloroform), showing unfavourable interaction between UPy and ODIN. The values for E are in reference with the monomeric units without association. | ||
Therefore, three different molecular weights of U were mixed with O to check whether they can form any comb-shaped morphology due to solely π–π stacking and phase separation of the sticker from the polymer matrix as was reported for the polymer with two polar end-groups.26,43 Exclusively for the shortest polymer U4500 two different main chain: side chain experimental molar ratios, namely 1:10 and 1:30, were used (called O:U4500-1:10 and O:U4500-1:30) to check the effect of the side-chain density on the morphology. Moreover, in order to check the effect of the molecular weight on the morphology, three different molecular weights of U were mixed with O in the fixed ratio of 1:10 (O:U4500-1:10, O:U12500-1:10 and O:U26000-1:10) (Table 2).
| Entry | Sample | Side chain Mn (kg mol−1) | Molar ratio [main chain]/[side chain] |
|---|---|---|---|
| 1 |
O:U4500-1:30
|
4.5 | 1:30 |
| 2 |
O:U4500-1:10
|
4.5 | 1:10 |
| 3 |
O:U12500-1:10
|
12.5 | 1:10 |
| 4 |
O:U26000-1:10
|
26.0 | 1:10 |
| 8 |
T:D3400-1:50
|
3.4 | 1:50 |
| 5 |
T:D4100-1:200
|
4.1 | 1:200 |
| 6 |
T:D4100-1:100
|
4.1 | 1:100 |
| 7 |
T:D4100-1:50
|
4.1 | 1:50 |
| 9 |
T:D15600-1:50
|
15.6 | 1:50 |
For polymers D carrying DAT and T carrying THY also a similar approach was implemented. Polymer D4100 was mixed with polymer T in three different molar ratios (1:50, 1:100 and 1:200) and called T:D4100-1:50, T:D4100-1:100 and T:D4100-1:200 to investigate the side-chain density effect, whereas to check the effect of the molecular weight on the morphology, the polymers were mixed in a 1:50 molar ratio with different side-chain molecular weights (T:D3400-1:50, T:D4100-1:50 and T:D15600-1:50) (Table 2). The DAT:THY hetero-association is well known and is not discussed here as it is reported elsewhere.44
The third class of comb polymers (polymer OD:OD) consists of sticker 10 at the end-groups. This means that they have the potential to self-associate and form a brush architecture. The brush backbone is comprised of stacked ODIN moieties and the brush polymers are called OD:OD2700, OD:OD8300, OD:OD9700, depending on the OD molecular weight.
Before analysing self-assembly and phase separation (using small angle X-ray scattering (SAXS)), the thermal properties of one sample from each of the three supramolecular comb/brush polymers (OD:OD9700, O:U12500-1:10 and T:D15600-1:50) are compared with the precursors in Fig. S12 and S13.† Differential scanning calorimetry (DSC) did not show any Tg for O which implies that the Tg is beyond the experimentally investigated range. This is expected considering the bulky associating groups in each repeating unit, similar to the effect of thymine on polymer T. Therefore, the presence of more thymine or ODIN on the polymer leads to higher Tg.45 Interestingly, their thermal degradation followed a similar trend where both T and O showed the least thermal stability. Comparing their backbone chemistry, probably the associating groups are responsible for this behaviour and are cleaved off first. Moreover, the DSC thermogram of T:D15600-1:50 shows no change in the glass transition temperature of the poly(n-butyl acrylate) phase (Tg = −53 °C). However, a weak but visible transition can be seen at T = Tg of polymer T (Tg = 44 °C). This shows that the addition of T to D15600 leads to the association of end/side groups and no T is dispersed in the poly(n-butyl acrylate) phase, proving the formation of the comb-like architecture. For O:U12500-1:10 only one unaltered Tg (corresponding to the poly(n-butyl acrylate) phase) was observed, since the Tg for the O phase was beyond the experimentally studied temperature.
Variable-temperature infrared (VT-IR) spectroscopy was also employed to further investigate the effect of temperature on sticker dissociation (Fig. S14–S18†). In Fig. S16,† the peaks at 1654 and 3400 cm−1 correspond to the hydrogen bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –N–H, respectively. The increase in the temperature up to 200 °C did not influence the hydrogen bonding significantly. This is consistent with the SAXS data discussed later in this manuscript. On the other hand, for sample O the peak corresponding to the hydrogen bonded CO (at 1654 cm−1) disappears and the peak for free CO at 1690 cm−1 intensifies (Fig. S14†), proving that the hydrogen-bonding tends to dissociate at elevated temperature. For T the peaks at 1672 and 3176 cm−1 corresponding to the hydrogen bonded CO and –N–H shift to higher wavenumbers which implies the disruption of hydrogen bonding with increasing temperature (Fig. S15†). For the two mixtures T:D15600-1:50 and O:U12500-1:10 the presence of hydrogen bonding was harder to be studied due to the very low concentration of the stickers and weak signals (similar to the DSC studies). Nevertheless, the disappearance of the hydrogen bonded CO (at 1659 cm−1) with increasing temperature for O:U12500-1:10 was clearly visible (Fig. S17†).
O and –N–H, respectively. The increase in the temperature up to 200 °C did not influence the hydrogen bonding significantly. This is consistent with the SAXS data discussed later in this manuscript. On the other hand, for sample O the peak corresponding to the hydrogen bonded CO (at 1654 cm−1) disappears and the peak for free CO at 1690 cm−1 intensifies (Fig. S14†), proving that the hydrogen-bonding tends to dissociate at elevated temperature. For T the peaks at 1672 and 3176 cm−1 corresponding to the hydrogen bonded CO and –N–H shift to higher wavenumbers which implies the disruption of hydrogen bonding with increasing temperature (Fig. S15†). For the two mixtures T:D15600-1:50 and O:U12500-1:10 the presence of hydrogen bonding was harder to be studied due to the very low concentration of the stickers and weak signals (similar to the DSC studies). Nevertheless, the disappearance of the hydrogen bonded CO (at 1659 cm−1) with increasing temperature for O:U12500-1:10 was clearly visible (Fig. S17†).
Small angle X-ray scattering (SAXS)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :U comb polymers.
The self-assembly of the comb polymers was investigated using small angle X-ray scattering (SAXS). Fig. 5a shows the SAXS profiles of polymers O and U4500 and the mixture of O and U4500 in two different polymer molar ratios of 1:30 (O:U4500-1:30) and 1:10 (O:U4500-1:10) (1 mole of O was added to 10 or 30 moles of U4500). Therefore, considering 70 stickers per polymer chain of O, the mixture O:U4500-1:30 contains approximately two to three ODIN per UPy, whereas this value is seven to one in O:U4500-1:10. Therefore, O:U4500-1:30 has a higher grafting density than O:U4500-1:10. The high-angle peaks located at q = 3.6, 4.5, 7.6, and 8.9 nm−1 belong to the unit cell of the phase separated ODIN moieties.36 This observation suggests that, in the pure polymer O, the functional groups can aggregate and form domains consisting of stacked ODIN molecules. The presence of a small-angle broad peak centered at q* = 1.26 nm−1 indicates that the interdomain distance between the ODIN aggregates is about 5 nm. By the addition of PnBA carrying UPy end-groups at one end (polymer U4500), the characteristic peaks of the unit cell disappear. Since UPy and ODIN were shown to be incapable of hydrogen bond formation, probably due to stacking between UPy and ODIN, the ODIN stacking is perturbed. This can be a sign of comb formation in the two mixtures. In fact, a similar phenomenon has been reported by Soulié-Ziakovic et al. when THY end-functionalized PE was mixed with DAT end-functionalized PE. Therefore, after mixing DAT and THY, THY crystals were not formed and phase separation occurred due to disparity between the aggregates of end-groups and non-polar PE.28 The lack of significant ordering or phase separation in the two mixtures (no sharp peak in Fig. 5a) can be a sign of gel phase formation, whereby the stickers are more or less randomly dispersed in a comb-like polymeric matrix (see Fig. 6 for clarity). However, the rheological experiments show no gel formation which means that the samples are below the gel-point and only a partial gel phase is formed as will be discussed at the end of this manuscript (Fig. 14a). Another explanation is that the polarity difference between the stickers and the polymer matrix (PnBA) is not sufficient to induce phase separation.27Fig. 5b compares the effect of the side chain molecular weight on phase separation and comb formation in the mixtures with a molar ratio of 1:10 (entries 2–4 in Table 2). The morphology in all samples seems to be the same. Similarly, in the pure side chains (U4500, U12500 and U26000), no phase separation is observed (Fig. 7b). Here, the formation of hydrogen bonding does not play a significant role and although UPy can dimerize strongly, it does not necessarily imply better phase separation. It has been shown that moieties incapable of hydrogen bond formation such as methylated thymine26,39 or cytosine and adenine derivatives can show mesoscopic organization.43 This implies that for the stickers, crystallization and hydrogen bonding with the matrix are two opposing effects that determine the phase separation.26–28
:U comb polymers.
The self-assembly of the comb polymers was investigated using small angle X-ray scattering (SAXS). Fig. 5a shows the SAXS profiles of polymers O and U4500 and the mixture of O and U4500 in two different polymer molar ratios of 1:30 (O:U4500-1:30) and 1:10 (O:U4500-1:10) (1 mole of O was added to 10 or 30 moles of U4500). Therefore, considering 70 stickers per polymer chain of O, the mixture O:U4500-1:30 contains approximately two to three ODIN per UPy, whereas this value is seven to one in O:U4500-1:10. Therefore, O:U4500-1:30 has a higher grafting density than O:U4500-1:10. The high-angle peaks located at q = 3.6, 4.5, 7.6, and 8.9 nm−1 belong to the unit cell of the phase separated ODIN moieties.36 This observation suggests that, in the pure polymer O, the functional groups can aggregate and form domains consisting of stacked ODIN molecules. The presence of a small-angle broad peak centered at q* = 1.26 nm−1 indicates that the interdomain distance between the ODIN aggregates is about 5 nm. By the addition of PnBA carrying UPy end-groups at one end (polymer U4500), the characteristic peaks of the unit cell disappear. Since UPy and ODIN were shown to be incapable of hydrogen bond formation, probably due to stacking between UPy and ODIN, the ODIN stacking is perturbed. This can be a sign of comb formation in the two mixtures. In fact, a similar phenomenon has been reported by Soulié-Ziakovic et al. when THY end-functionalized PE was mixed with DAT end-functionalized PE. Therefore, after mixing DAT and THY, THY crystals were not formed and phase separation occurred due to disparity between the aggregates of end-groups and non-polar PE.28 The lack of significant ordering or phase separation in the two mixtures (no sharp peak in Fig. 5a) can be a sign of gel phase formation, whereby the stickers are more or less randomly dispersed in a comb-like polymeric matrix (see Fig. 6 for clarity). However, the rheological experiments show no gel formation which means that the samples are below the gel-point and only a partial gel phase is formed as will be discussed at the end of this manuscript (Fig. 14a). Another explanation is that the polarity difference between the stickers and the polymer matrix (PnBA) is not sufficient to induce phase separation.27Fig. 5b compares the effect of the side chain molecular weight on phase separation and comb formation in the mixtures with a molar ratio of 1:10 (entries 2–4 in Table 2). The morphology in all samples seems to be the same. Similarly, in the pure side chains (U4500, U12500 and U26000), no phase separation is observed (Fig. 7b). Here, the formation of hydrogen bonding does not play a significant role and although UPy can dimerize strongly, it does not necessarily imply better phase separation. It has been shown that moieties incapable of hydrogen bond formation such as methylated thymine26,39 or cytosine and adenine derivatives can show mesoscopic organization.43 This implies that for the stickers, crystallization and hydrogen bonding with the matrix are two opposing effects that determine the phase separation.26–28
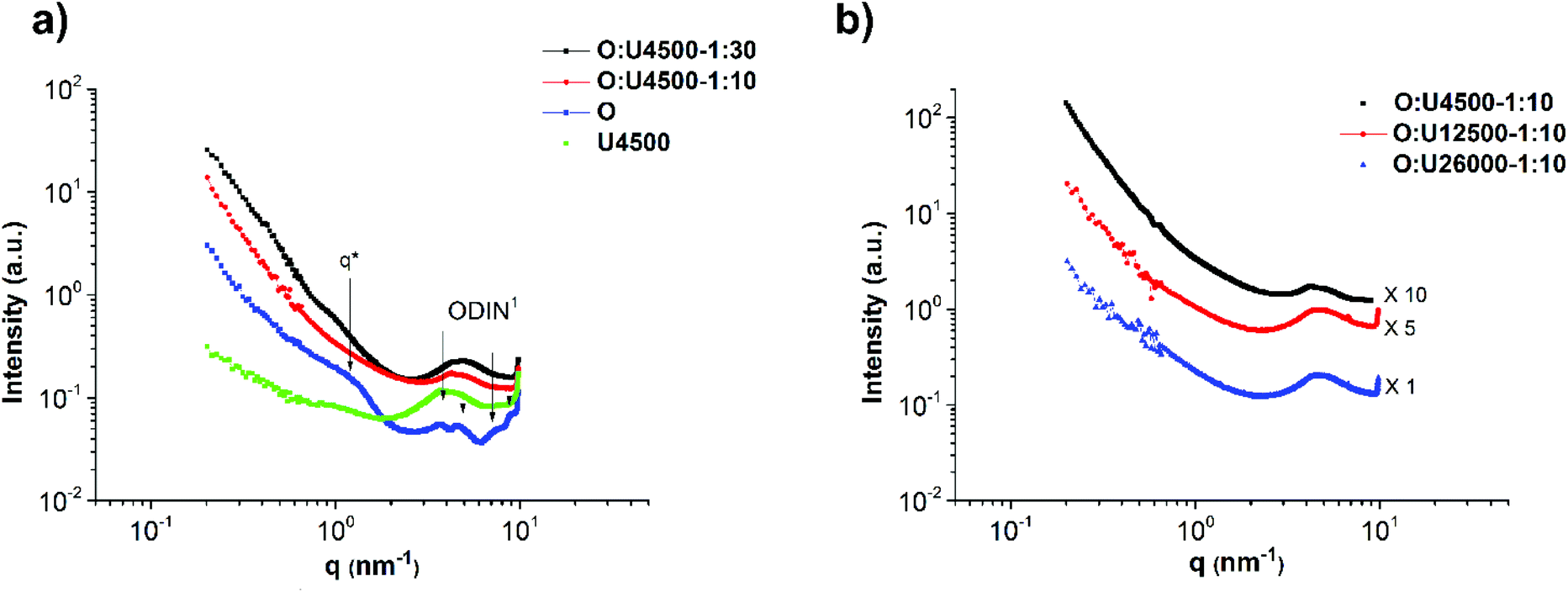 | ||
| Fig. 5 SAXS profiles showing (a) the comparison of the morphology and phase separation in the main chain polymer O, side chain polymer U4500 and the mixture of O and U4500 with two different polymer molar ratios of 1:30 (O:U4500-1:30) and 1:10 (O:U4500-1:10) and (b) the effect of the side chain molecular weight in a fixed ODIN:UPy ratio (O:U4500-1:10, O:U12500-1:10 and O:U26000-1:10). (1: ODIN characteristic peaks.) | ||
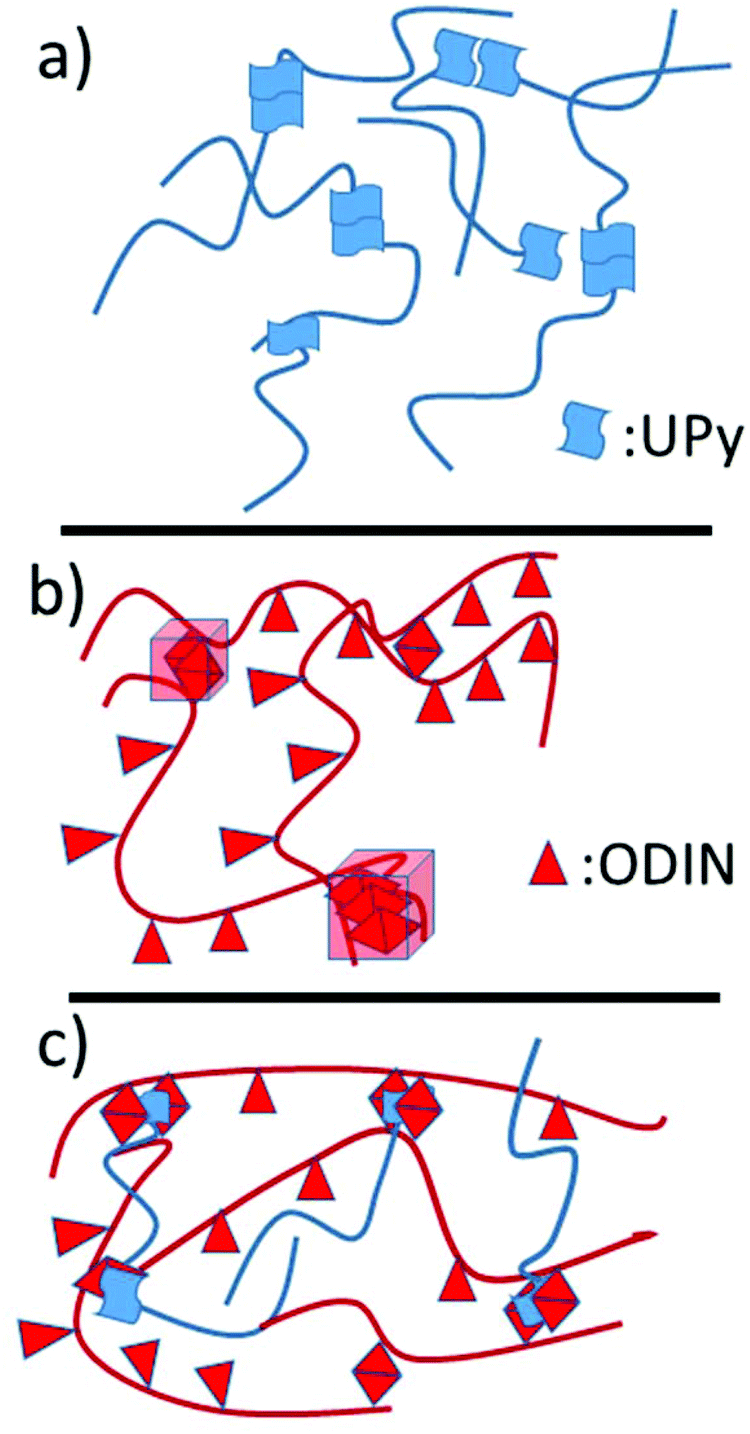 | ||
| Fig. 6 Schematic representation of (a) side chain polymer U4500, (b) main chain polymer O and (c) the corresponding comb polymer based on UPy:ODIN interactions. | ||
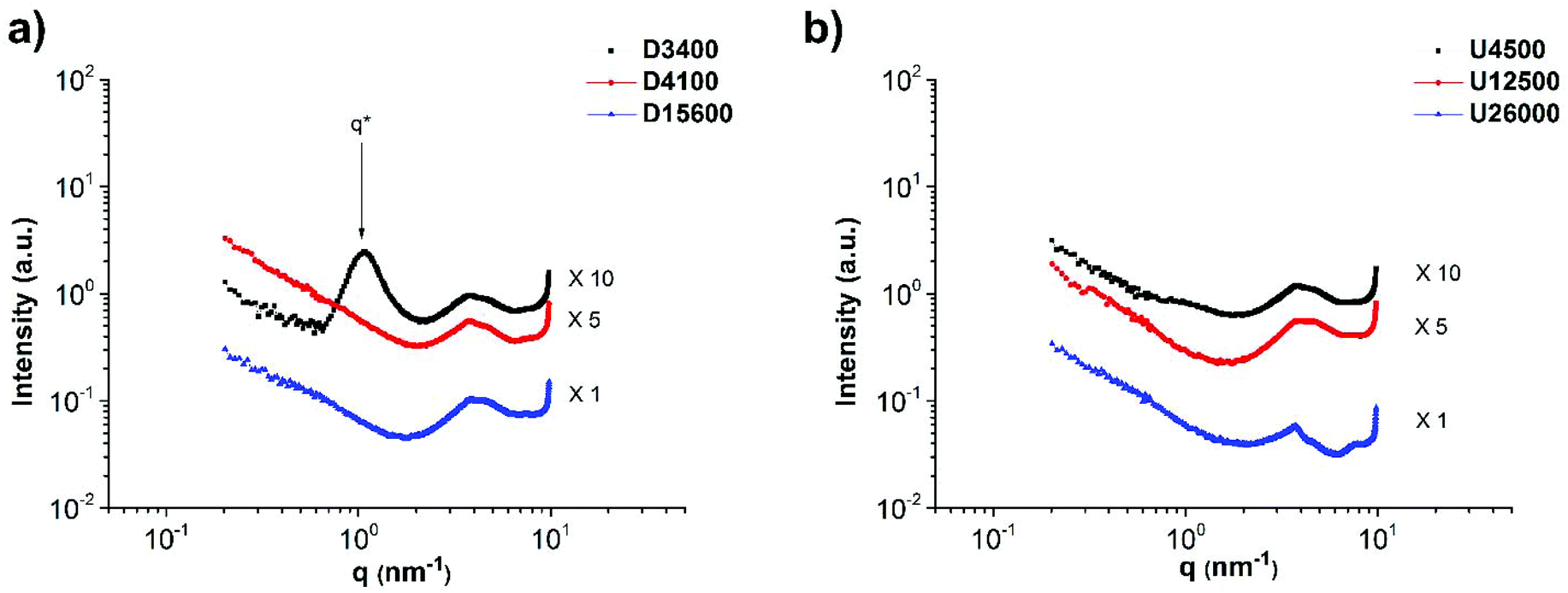 | ||
| Fig. 7 SAXS profiles of side chain polymers (a) D3400, D4100 and D15600 and (b) U4500, U12500 and U26000 showing the effect of the molecular weight on phase separation and morphology. | ||
:T comb polymers.
The main chain polymer T has been shown to be homogeneous with negligible phase separation.45Fig. 7a shows the SAXS profiles of polymers D3400, D4100 and D15600. In particular, in the case of sample D3400 short-range phase separation is observed with a domain size of 5.7 nm (d* = 2π/q* and q* = 1.1 nm−1). The formation of micelle-like aggregates has been observed in DAT/THY/barbituric acid functionalized poly(isobutylene) (PIB)s too.31,46,47 With increasing molecular weight, the material becomes more homogeneous as the concentration of the stickers decreases and they are too dispersed to diffuse and form aggregates.27 The same phenomenon (weaker phase separation in higher molecular weights) occurs in the mixtures T:D3400-1:50, T:D4100-1:50 and T:D15600-1:50 (Fig. 8a). In these mixtures, the ratio of THY:DAT was kept constant, namely 6:1. Moreover, for comparison, the data for polymer D3400 are added to Fig. 8a. The characteristic peak exhibits a shift from q* = 1.1 to 0.67 nm−1 (domain size d* from 5.7 nm to 9.4 nm). This change in d* shows that after the addition of the main chain (T) to polymer D3400, the polymer chains forming the side chains are stretched, leading to a higher feature size. Therefore, it can be implied that a comb-like polymer mixture is formed. The sharpening of the characteristic peak after the addition of the main chain polymer T also indicates that upon comb formation a material with more ordering (in comparison with the micelle-like aggregates in pure D3400) is obtained, although this ordering seems to be short-ranged. Fig. 9 reports the proposed schemes for the structures and clarifies this behaviour.
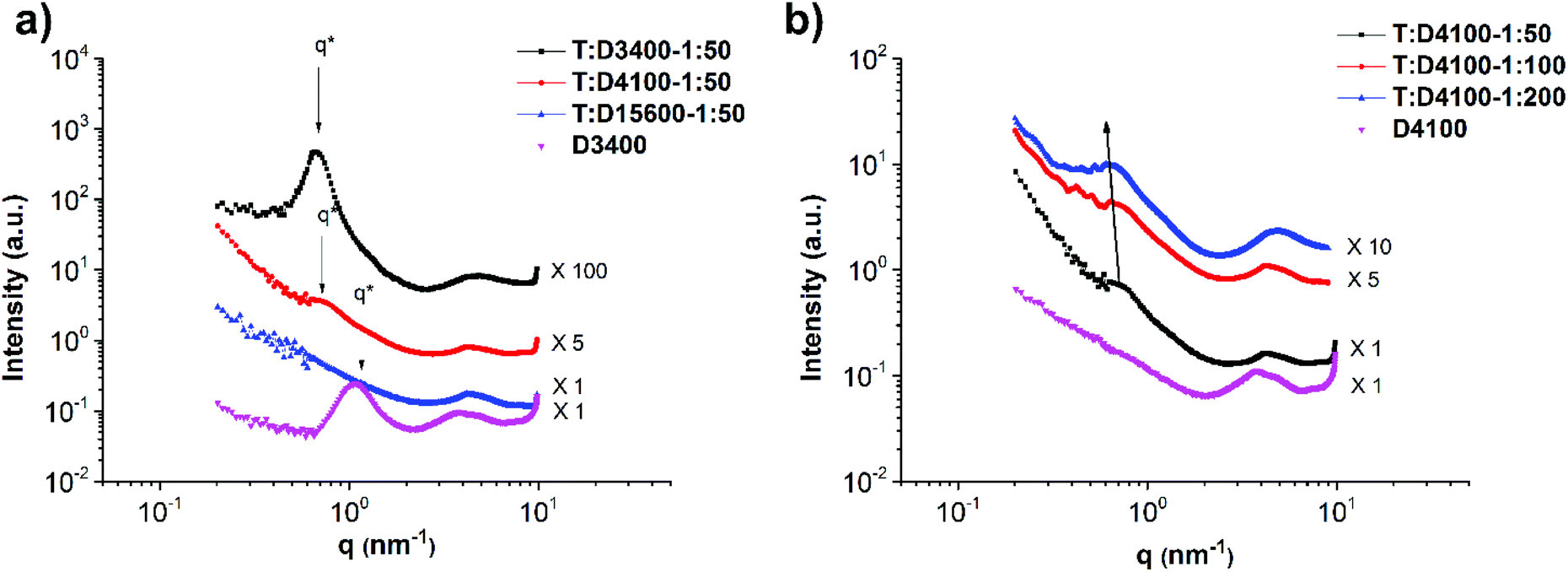 | ||
| Fig. 8 SAXS profiles of (a) mixtures T:D3400-1:50, T:D4100-1:50 and T:D15600-1:50 and pure side chain D3400 showing the effect of the branch molecular weight on phase separation and (b) mixtures T:D4100-1:50, T:D4100-1:100 and T:D4100-1:200 and pure D4100 indicating the effect of the branch density on phase separation. | ||
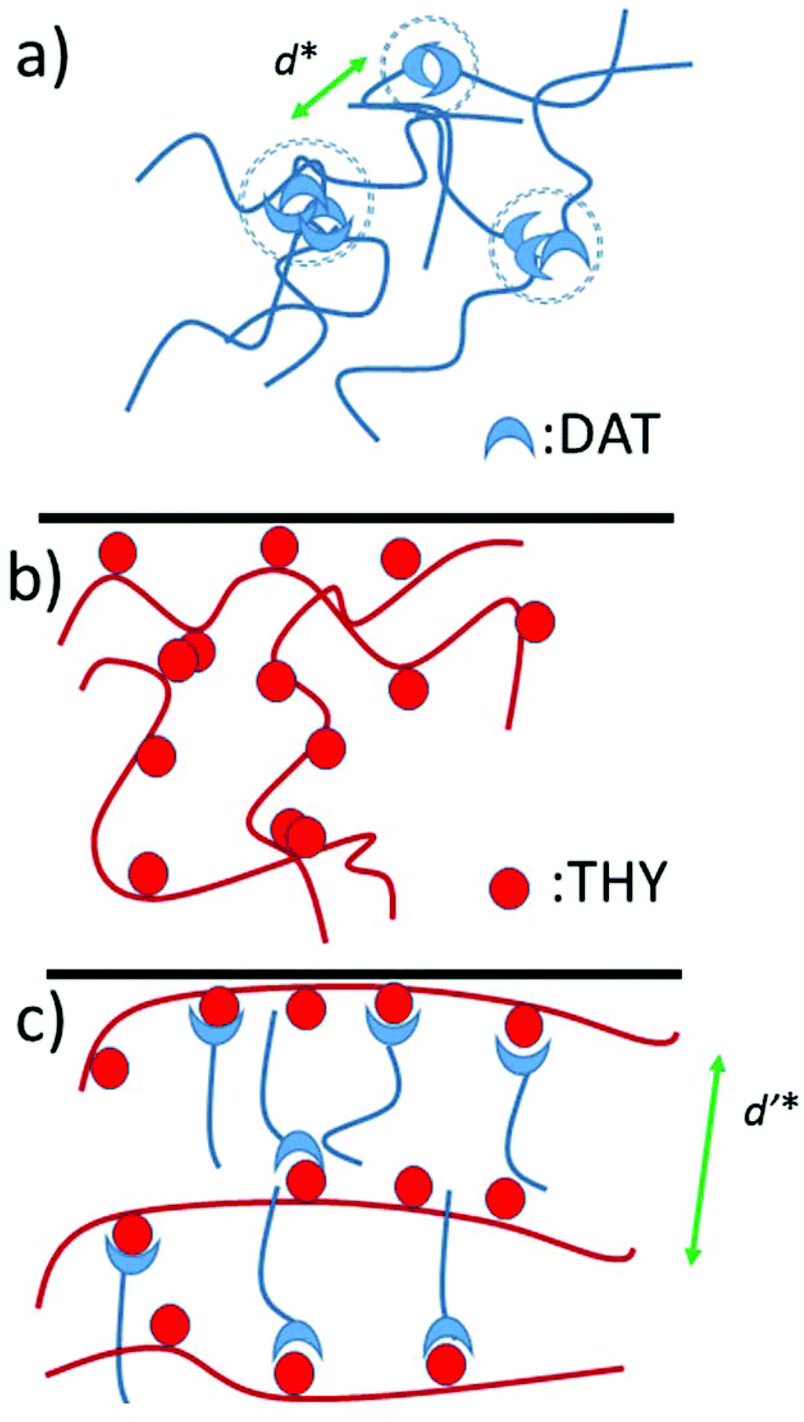 | ||
| Fig. 9 Schematic representation of (a) side chain polymer D4100, (b) main chain polymer T and (c) the corresponding comb polymer based on THY:DAT interactions. | ||
In mixture T:D4100-1:50 (with a side chain molecular weight of 4100 g mol−1), it was shown that with a ratio of 1:6 for DAT:thymine, a weak phase separation is obtained. In order to check whether the addition of more side chains can promote phase separation, two more mixtures called T:D4100-1:100 and T:D4100-1:200 were prepared. The former contains a ratio of approximately 1 to 3 and the latter 1 to 1 for DAT:thymine. It is clearly visible that the addition of more side chains can improve the phase separation (stronger characteristic peak). Although this improvement is not significant, compared to the pure side chain polymer D4100 the effect stands out (Fig. 8b). Moreover, the characteristic peak tends to shift slightly to lower q values as more side chains are added. This can be a sign of chain stretching although the effect is insignificant, probably due to random self-association of DAT end-groups outside the main chain domains. Similarly, with increasing molecular weight of the side chain, the domain size does not change significantly (Fig. 8a) which is because of random self-association of DAT end-groups outside the main chain domains. In other words, the disordered side chains aggregate between the ordered comb-shaped domains and compensate for the domain distance, and the feature size remains more or less the same with increasing side chain molecular weight.
:O and D:T comb polymers.
In order to compare the combs formed in the UPy and DAT systems, O:U4500-1:10 and T:D4100-1:50 are compared in Fig. 10. O:U4500-1:10 contains seven ODIN per UPy (with a side chain molecular weight of 4500 g mol−1) whereas T:D4100-1:50 has six THY per DAT (with a molecular weight of 4100 g mol−1). Therefore, within the experimental error, the two mixtures are good candidates for comparison. The SAXS profile in Fig. 10 shows that the two curves almost overlap, except that there is a small characteristic peak in T:D4100-1:50. The absence of this peak in the UPy system (O:U4500-1:10) may seem counterintuitive as UPy and ODIN tend to aggregate better and have both higher dimerization constant and electron density difference in comparison with the DAT system (T:D4100-1:50).31,33,36,42,48 However, we support this behaviour with the following two reasons:
First, we have recently investigated the same polymer with different amounts of thymine stickers (10, 20, 30 and 100% stickers per chain) and concluded that although small aggregates are present, no continuous network is generated. Nevertheless, clusters below the gel point are formed.25 For polymer O considering the SAXS data in Fig. 5a, one may speculate that the formation of aggregates (and a network) is more probable. This also makes sense considering the stronger stacking and dimerization constant of ODIN in comparison with thymine.25,31,35,36 Therefore, the addition of side chains in the DAT system can disturb the clusters easily in comparison with the UPy system wherein strong ODIN aggregates tend to form a gel. This means in T:D4100-1:50 the cluster is almost completely dissociated and a comb-shaped polymer with a main chain–main chain distance of ca. 9 nm is formed (Fig. 9).
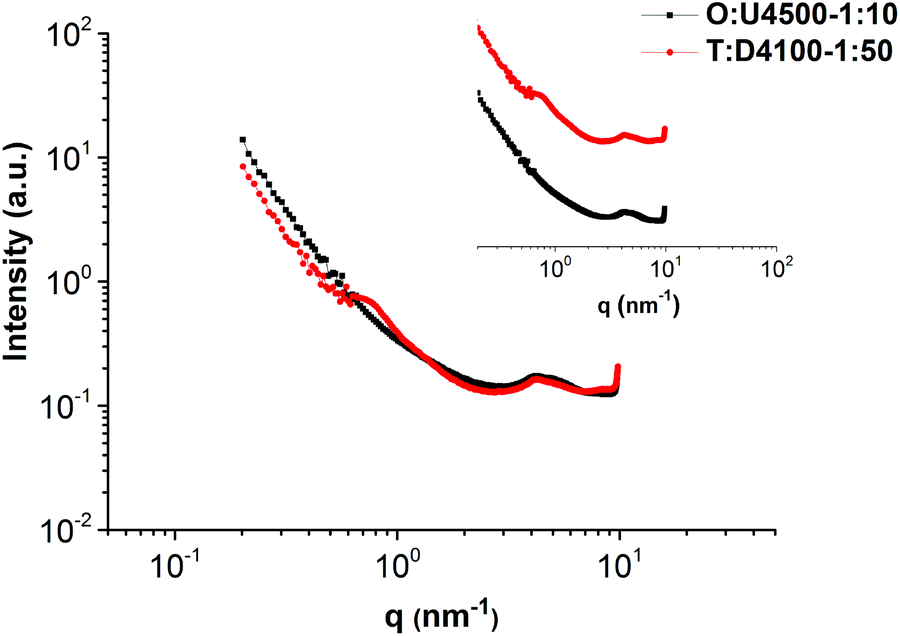 | ||
| Fig. 10 SAXS profiles of O:U4500-1:10 and T:D4100-1:50 comparing the phase separation in DAT and UPy blends (in the inset, the data are shifted vertically for clarity). | ||
The second explanation is that UPy is prone to hydrogen bond formation with the carboxyl group of PnBA which means that a fraction of UPy end groups does not aggregate in the ODIN domain but rather in the PnBA domain.37
:OD brush polymers.
In order to check the order–disorder transition in PnBA with an ODIN end-group (polymer OD), three different molecular weights were investigated using SAXS. In principle this system can be considered as the third method to prepare a supramolecular graft polymer (in addition to D:T and O:U systems), with two differences. (1) Polymer OD can only prepare a polymer brush (rather than a comb polymer). According to the widely accepted definition, when one or more side chains are located on each main chain repeating unit, leading to strong chain stretching, a polymer can be called a polymer brush.16,17 In other words, in DAT and UPy systems the grafting density can be tuned but in polymer brush OD:OD this value is constant (two branches per main chain repeating unit). (2) In OD:OD polymer brushes, the ODIN end-groups are stacked to form a non-covalent backbone without the need for mixing an additional polymer.
Fig. 11a shows the SAXS profile of OD:OD with three different molecular weights (OD:OD2700, OD:OD8300 and OD:OD9700). The peak at q = 5.8 nm−1 reveals a characteristic size of d = 1.1 nm for the ODIN domains, the size between binary associations. Moreover, for all samples the following characteristic peaks were observed: q*, 2q*, 3q*, 4q*, 5q*, 6q*, 7q*, 8q*, and 9q* implying that a lamellar morphology with long-range ordering is obtained. The lamellar spacing d* also scales linearly with the molecular weight with the slope approximately equal to 1.0 (Fig. 11b). This means the chains are significantly stretched similar to our observations in poly(tetrahydrofuran)-based polymer brushes, although a better ordering is achieved in PnBA-based polymers.36 The fact that for all molecular weights (∼2–10 kg mol−1) mesoscopic organization was observed means that considering the strong stacking ability of ODIN36 the volume fraction of the sticker is high enough to induce long-range ordering. While for THY-functionalized PEG, with THY volume fractions below 5%, no organization could be obtained;27 this value was shown to be 8% for UPy-functionalized PDMS.49 This threshold was significantly lower for tris-urea PnBA (1.5%),17 and for PnBA-ODIN containing 3.4% stickers, the mesoscopic organization is still present. This value has to be much higher for UPy and DAT-functionalized PnBA as no ordering was observed even at the lowest studied polymer molecular weight (highest sticker concentration). This is consistent with the conclusion by Soulié-Ziakovic et al. that the sticker concentration below which the organization is lost depends on the sticker properties.27 In order to compare the polymer brush OD:OD with the DAT system, polymers D3400 and T:3400-1:50 (red and blue points in Fig. 11b, respectively) were chosen as they showed the most significant phase separation between the two studied systems (O:U and T:D). Fig. 11b shows that the addition of T leads to the stretching of the polymer D3400 (vertical shift of the domain size) and the domain size gets closer to the one in ODIN-based polymers (OD:OD). Fig. 12 depicts the self-assembly in ODIN:ODIN-based polymer brushes.
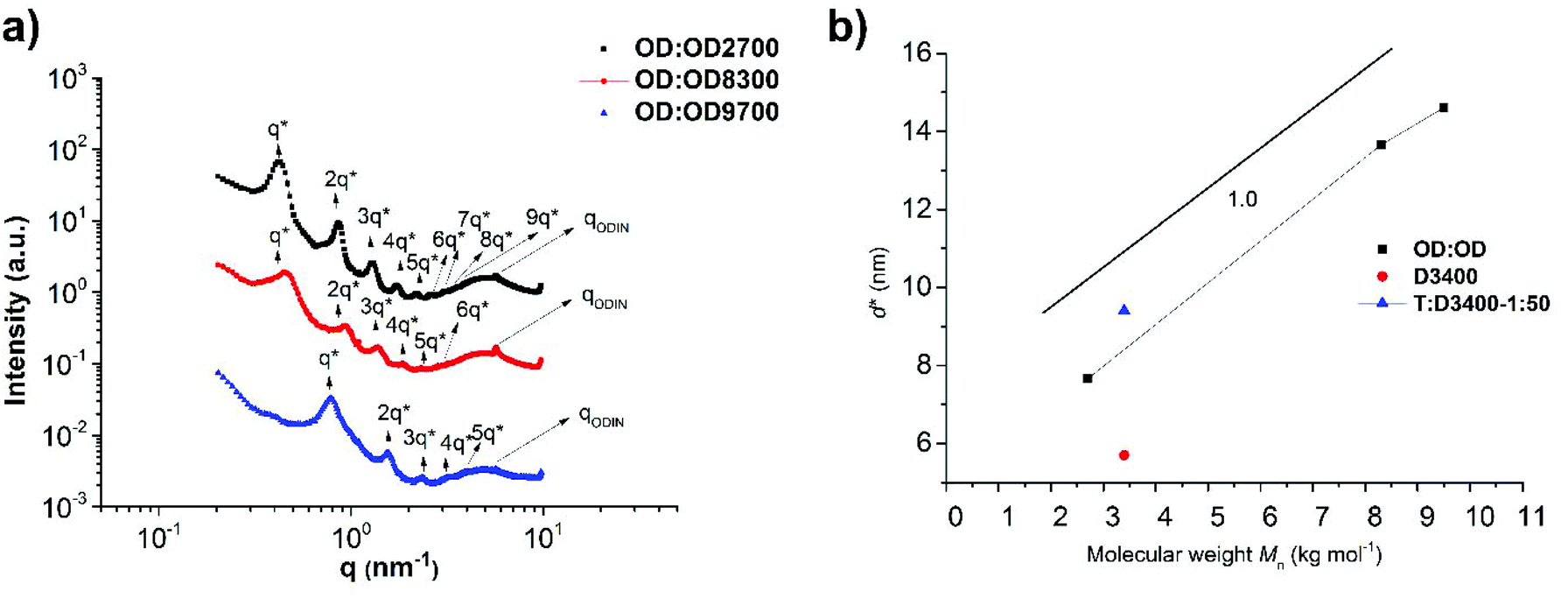 | ||
| Fig. 11 (a) SAXS profiles of OD:OD2700, OD:OD8300 and OD:OD9700, showing a long-range ordered lamellar morphology and (b) molecular dependency of the feature sizes. | ||
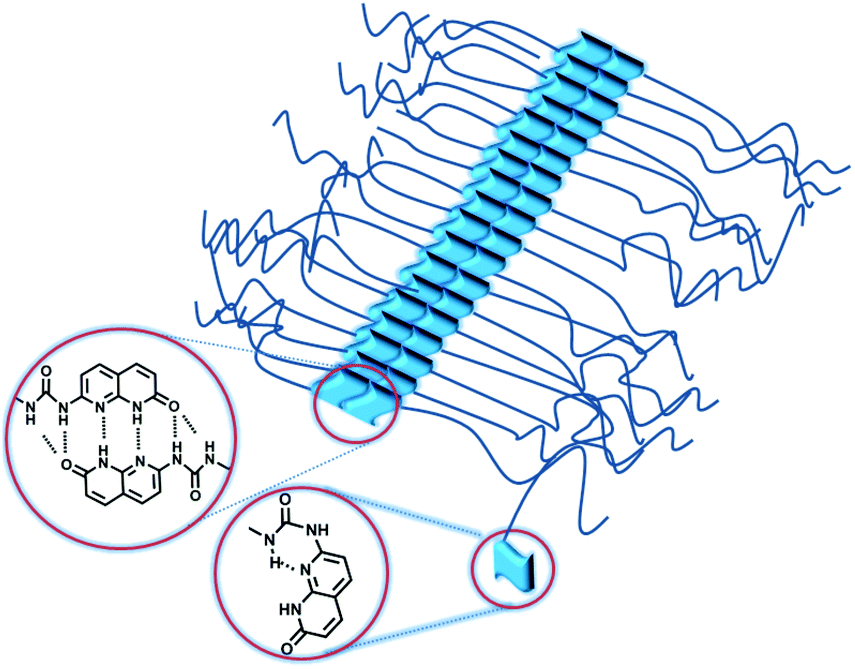 | ||
| Fig. 12 Schematic representation of polymer brush OD:OD based on ODIN:ODIN interactions. | ||
To investigate the order–disorder transition temperature (TODT), sample OD:OD2700 was heated and studied using temperature resolved SAXS (Fig. 13). The lamellar morphology disappears at elevated temperatures around 180 °C and an order–disorder transition is observed at 250 °C. At 180 °C also the domain size changes significantly which means probably the portion of the dissociated chains (no more π–π stacked) moves to the polymer domains and this leads to an increase in the domain size. However, even at 180 °C the peak at q = 0.24 nm−1 indicates that hydrogen bonding is still present between stickers and they aggregate within the polymer matrix. This is consistent with the VT-IR data in which hydrogen bonding was present up to 200 °C.
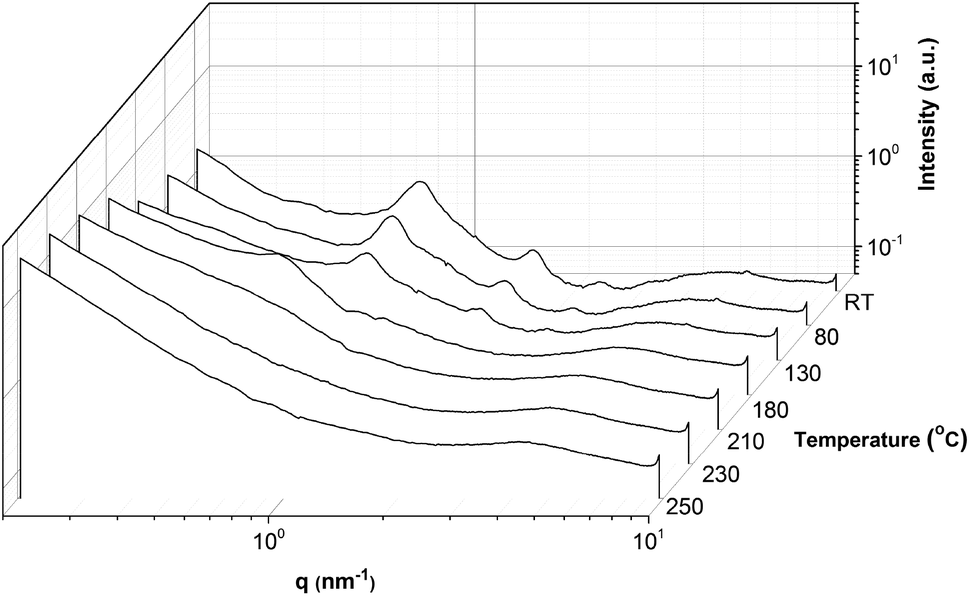 | ||
| Fig. 13 Variable temperature SAXS (VT-SAXS) of polymer brush OD:OD3400. | ||
These results show that in contrast to the previously reported works on telechelic and comb polymers,2,7,8,31,33,37,46 in order to achieve better phase separation and comb/brush formation, hydrogen bonding can even have detrimental effects (for instance, UPy with carboxylic groups of the matrix) and the important factors for long-range ordering and phase separation are π–π stacking and the aggregation ability (or crystallization). Comparing OD:OD and O:U (or T:D), the stronger stacking and packing abilities of ODIN36 in comparison with DAT and UPy led to a much better ordering. Phase separation although improved by the addition of the main chain polymers U and T is still negligible in comparison with polymer OD. This conclusion was made also for telechelic polymers with THY end-groups, as THY can crystallize and lead to long-range ordering without the need for hydrogen bonding.26–28,43
Melt rheology
The melt dynamics in one sample from each of the three supramolecular comb/brush polymers (OD:OD9700, O:U12500-1:10 and T:D15600-1:50) were analysed using melt rheology. Fig. 14a shows that in the case of DAT and UPy systems (O:U12500-1:10 and T:D15600-1:50), a Maxwell-like relaxation (with G′ ∝ ω2 and G′′′ ∝ ω1) is observed at room temperature. For O:U12500-1:10 however, a reliable G′ was not observed due to the low viscosity of the polymer. On the other hand, polymer OD:OD9700 shows a plateau with a modulus value close to 5 kPa. With increasing temperature (Fig. 14b) this value decreases and at 180 °C the plateau diminishes. Again, at this temperature for OD:OD9700 (similar to O:U12500-1:10), the viscosity was too low to show an accurate G′; however, from the G′′ frequency dependency, an obvious terminal relaxation is observed. Interestingly, the flow temperature (180 °C) was similar to the temperature at which the lamellar ordering disappears in SAXS experiments. This means that rather than aggregations, long-range ordering is necessary for the elastic behaviour of the polymer (TODT ≠ Tflow). The only report on the rheology of supramolecular comb polymers is the work done by Staropoli et al. where a polybutylene oxide (PBO)-based main chain randomly functionalized with THY groups was mixed with mono-DAT-functionalized PBO. Using SANS and melt rheology, it was concluded that a supramolecular comb shows dynamics between a permanent comb and a polymer mixture without any sticker. Moreover, the mechanism of the polymer relaxation was shown to be associated with arm retraction and reptation.2,7,8 Due to the high main chain molecular weight and low grafting density, a rubbery plateau was observed showing the reptation of the entire polymer in a dilated tube. In our case, we speculate that the main chain polymers T and O are below the critical molecular weight Mc and also they are extremely diluted in the matrix of side chains which are also below the Mc of PnBA (=20–30 kg mol−1).17 Therefore, no plateau was observed for O:U12500-1:10 and T:D15600-1:50. The situation for OD:OD9700 is rather different as the observed plateau does not correspond to the entanglement of the polymer main chain or side chain or network formation as was reported by Hayes et al.50,51 It is rather due to either colloidal properties36,52 or entanglement of the entire polymer brush as was seen in our previous study.36 The value of the plateau modulus (5 kPa) is also consistent with this conclusion.
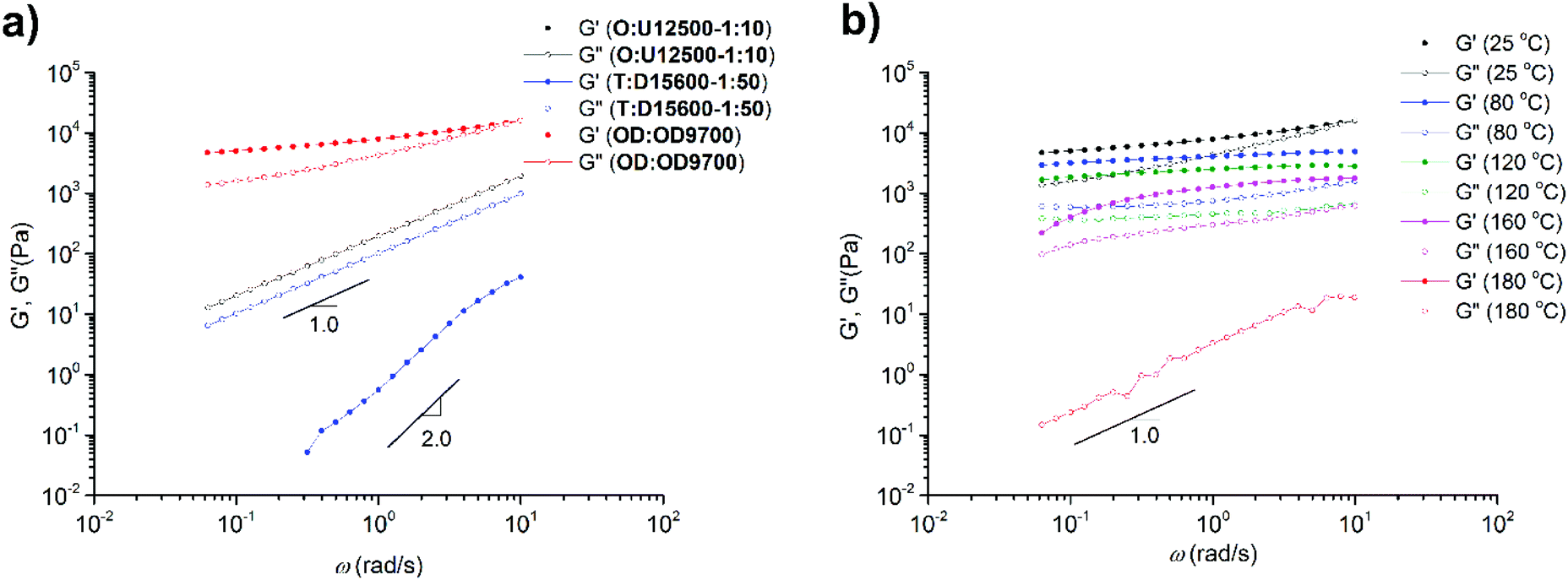 | ||
| Fig. 14 Melt (linear shear) rheology of (a) O:U12500-1:10, T:T15600-1:50 and OD:OD9700 at room temperature, and (b) OD:OD9700 at different temperatures (25, 80, 120, 160, 180 °C). | ||
Therefore, the results of melt rheology are consistent with SAXS investigations; long-range ordering and chain stretching lead to elastic properties. The absence of long-range ordering in the DAT and UPy systems leads to liquid-like behaviour. Although polymer T carrying THY groups has been shown to relax as a cluster,25 after the addition of side chain polymer D the cluster dissociates and a comb-like polymer is obtained which due to tube dilation relaxes as a Newtonian liquid.53
Conclusions
The synthesis and self-assembly of supramolecular polymer combs with short amphiphilic side groups have been investigated widely in the past. The goal in this work was to study the role of the stickers (hydrogen bonding moieties) in the association and phase separation in comb/brush polymers with polymeric side chains. Therefore, using RAFT polymerization, three different sets of polymer combs/brushes were synthesized each carrying a specific sticker: 2,4-diamino-1,3,5-triazine (DAT):thymine (THY), 2-ureido-4[1H]-pyrimidinone (UPy):(1-(6-isocyanatohexyl)-3-(7-oxo-7,8-dihydro-1,8-naphthyridin-2-yl)urea) (ODIN) and ODIN:ODIN, so that complementary interactions may be used to form comb polymers. For two sets of polymers (based on DAT:THY and UPy:ODIN interactions), a main chain (polymer with stickers on each repeating unit) and a side chain (mono-functionalized polymer) were mixed with different ratios and molecular weights. For the third class of comb polymers (based on ODIN:ODIN interactions), only one type of polymer was used (no need for mixing with a second component).
The mixtures and the precursors were studied using small angle X-ray scattering (SAXS) and it was shown that the aggregation ability and crystallization of the end-groups are important parameters for long-range ordering. On the other hand, it was proved that hydrogen bond formation is not a requirement for phase separation. In the case of ODIN:UPy interactions, without complementary hydrogen bonding between the two stickers, solely based on disruption of ODIN:ODIN stacks, a homogeneous comb-like cluster was formed. The ordering was better for the THY:DAT system as breaking of the THY:THY interaction of the main chain was much easier compared to the strong interactions of ODIN:ODIN and therefore, a comb-like polymer with short-range phase separation was formed. The addition of the main chain polymer to pure side chain polymers always showed an increase in phase separation, whereas the molecular weight showed an inverse relation with the ordering (a decrease in the sticker volume fraction deteriorates the ordering and phase separation).
The results were substantiated using melt rheology and it was shown that long-range ordering is necessary for elastic properties. Therefore, the solid-to-liquid transition (Tflow) occurs at lower temperatures than the order–disorder transition (TODT), where no long-range ordering is present.
This work can be used as a general toolkit for the formation of supramolecular comb polymers with side polymeric groups and is a promising approach for applications in supersoft elastomers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to A.J.J. Woortman (RUG) for GPC measurements. The research was supported by a NWO-VICI innovational research grant.Notes and references
- Y.-C. Wu, B. Bastakoti, P. Malay, Y. Yamauchi and S.-W. Kuo, Polym. Chem., 2015, 6, 5110–5124 RSC.
- M. Staropoli, A. Raba, C. Hövelmann, M.-S. Appavou, J. Allgaier, M. Kruteva, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, J. Rheol., 2017, 61, 1185–1196 CrossRef CAS.
- W.-T. Kuo, H.-L. Chen, R. Goseki, A. Hirao and W.-C. Chen, Macromolecules, 2013, 46, 9333–9340 CrossRef CAS.
- A. H. Hofman, Y. Chen, G. ten Brinke and K. Loos, Macromolecules, 2015, 48, 1554–1562 CrossRef CAS.
- H. Ohkawa, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2007, 40, 1453–1459 CrossRef CAS.
- U. S. Schubert and H. Hofmeier, Macromol. Rapid Commun., 2002, 23, 561–566 CrossRef CAS.
- M. Staropoli, A. Raba, C. H. Hövelmann, M. Krutyeva, J. Allgaier, M.-S. Appavou, U. Keiderling, F. J. Stadler, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, Macromolecules, 2016, 49, 5692–5703 CrossRef CAS.
- J. Allgaier, C. H. Hövelmann, Z. Wei, M. Staropoli, W. Pyckhout-Hintzen, N. Lühmann and S. Willbold, RSC Adv., 2016, 6, 6093–6106 RSC.
- E. Polushkin, G. O. R. Alberda van Ekenstein, M. Knaapila, J. Ruokolainen, M. Torkkeli, R. Serimaa, W. Bras, I. Dolbnya, O. Ikkala and G. ten Brinke, Macromolecules, 2001, 34, 4917–4922 CrossRef CAS.
- S. Pensec, N. Nouvel, A. Guilleman, C. Creton, F. Boué and L. Bouteiller, Macromolecules, 2010, 43, 2529–2534 CrossRef CAS.
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193–207 RSC.
- A. H. Hofman, M. Reza, J. Ruokolainen, G. ten Brinke and K. Loos, Angew. Chem., Int. Ed, 2016, 55, 13081–13085 CrossRef CAS PubMed.
- A. H. Hofman, I. Terzic, M. C. A. Stuart, G. ten Brinke and K. Loos, ACS Macro Lett., 2018, 7, 1168–1173 CrossRef CAS PubMed.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- I. Davidi, D. Hermida-Merino, K. Keinan-Adamsky, G. Portale, G. Goobes and R. Shenhar, Chem. – Eur. J., 2014, 20, 6951–6959 CrossRef CAS PubMed.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Nat. Mater., 2015, 15, 183 CrossRef PubMed.
- X. Callies, C. Véchambre, C. Fonteneau, S. Pensec, J.-M. Chenal, L. Chazeau, L. Bouteiller, G. Ducouret and C. Creton, Macromolecules, 2015, 48, 7320–7326 CrossRef CAS.
- M. Chen, D. L. Inglefield, K. Zhang, A. G. Hudson, S. J. Talley, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1844–1852 CrossRef CAS.
- X. Chen, K. Zhang, S. J. Talley, C. M. Orsino, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 13–23 CrossRef CAS.
- K. Zhang, M. Chen, K. J. Drummey, S. J. Talley, L. J. Anderson, R. B. Moore and T. E. Long, Polym. Chem., 2016, 7, 6671–6681 RSC.
- K. Zhang, S. J. Talley, Y. P. Yu, R. B. Moore, M. Murayama and T. E. Long, Chem. Commun., 2016, 52, 7564–7567 RSC.
- K. Zhang, M. Aiba, G. B. Fahs, A. G. Hudson, W. D. Chiang, R. B. Moore, M. Ueda and T. E. Long, Polym. Chem., 2015, 6, 2434–2444 RSC.
- K. Zhang, G. B. Fahs, M. Aiba, R. B. Moore and T. E. Long, Chem. Commun., 2014, 50, 9145–9148 RSC.
- M. Golkaram and K. Loos, Macromolecules, 2019, 52, 9427–9444 CrossRef CAS PubMed.
- M. Golkaram, C. Fodor, E. van Ruymbeke and K. Loos, Macromolecules, 2018, 51, 4910–4916 CrossRef CAS PubMed.
- J. Lacombe and C. Soulié-Ziakovic, Polym. Chem., 2017, 8, 5954–5961 RSC.
- J. Lacombe, S. Pearson, F. Pirolt, S. Norsic, F. D'Agosto, C. Boisson and C. Soulié-Ziakovic, Macromolecules, 2018, 51, 2630–2640 CrossRef CAS.
- I. German, F. D'Agosto, C. Boisson, S. Tencé-Girault and C. Soulié-Ziakovic, Macromolecules, 2015, 48, 3257–3268 CrossRef CAS.
- J. Cortese, C. Soulié-Ziakovic, M. Cloitre, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2011, 133, 19672–19675 CrossRef CAS PubMed.
- J. Cortese, C. Soulié-Ziakovic, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2012, 134, 3671–3674 CrossRef CAS PubMed.
- F. Herbst, K. Schröter, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006–10016 CrossRef CAS.
- W. P. J. Appel, G. Portale, E. Wisse, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- C.-C. Cheng, J.-H. Wang, W.-T. Chuang, Z.-S. Liao, J.-J. Huang, S.-Y. Huang, W.-L. Fan and D.-J. Lee, Polym. Chem., 2017, 8, 3294–3299 RSC.
- C.-C. Cheng, F.-C. Chang, J.-H. Wang, Y.-L. Chu, Y.-S. Wang, D.-J. Lee, W.-T. Chuang and Z. Xin, RSC Adv., 2015, 5, 76451–76457 RSC.
- J. Tellers, S. Canossa, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Macromolecules, 2018, 51, 7680–7691 CrossRef CAS.
- M. Golkaram, L. Boetje, J. Dong, L. E. A. Suarez, C. Fodor, D. Maniar, E. van Ruymbeke, S. Faraji, G. Portale and K. Loos, ACS Omega, 2019, 4, 16481–16492 CrossRef CAS PubMed.
- S. Bobade, Y. Wang, J. Mays and D. Baskaran, Macromolecules, 2014, 47, 5040–5050 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- F. Herbst and W. H. Binder, Polym. Chem., 2013, 4, 3602–3609 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed, 2002, 41, 2596–2599 CrossRef CAS PubMed.
- Y. Shao, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- D. Guo, R. P. Sijbesma and H. Zuilhof, Org. Lett., 2004, 6, 3667–3670 CrossRef CAS PubMed.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS PubMed.
- J. Cortese, C. Soulié-Ziakovic and L. Leibler, Polym. Chem., 2014, 5, 116–125 RSC.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS.
- T. Yan, K. Schröter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Macromolecules, 2014, 47, 2122–2130 CrossRef CAS.
- T. Yan, K. Schröter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Macromolecules, 2017, 50, 2973–2985 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- R. H. Zha, B. F. M. de Waal, M. Lutz, A. J. P. Teunissen and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 5693–5698 CrossRef CAS PubMed.
- P. J. Woodward, D. Hermida Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Macromolecules, 2010, 43, 2512–2517 CrossRef CAS.
- D. Hermida-Merino, B. O'Driscoll, L. R. Hart, P. J. Harris, H. M. Colquhoun, A. T. Slark, C. Prisacariu, I. W. Hamley and W. Hayes, Polym. Chem., 2018, 9, 3406–3414 RSC.
- L. Gury, M. Gauthier, M. Cloitre and D. Vlassopoulos, Macromolecules, 2019, 52, 4617–4623 CrossRef CAS.
- J. L. Viovy, M. Rubinstein and R. H. Colby, Macromolecules, 1991, 24, 3587–3596 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01915d |
| This journal is © The Royal Society of Chemistry 2020 |