
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh Tm poly(L-lactide)s via REP or ROPPOC of L-lactide†
Hans R.
Kricheldorf
*a,
Steffen M.
Weidner
 b and
Andreas
Meyer
c
b and
Andreas
Meyer
c
aInstitut für Technische und Makromolekulare Chemie, Universität Hamburg, Bundestr. 45, D-20146 Hamburg, Germany. E-mail: hrkricheldorf@aol.de
bBAM, Federal Institute for Materials Research and Testing, Richard-Willstätter-Straße 11, D-12489 Berlin, Germany
cInstitut für Physikalische Chemie, Universität Hamburg, Grindelallee 117, D-20146 Hamburg, Germany
First published on 19th February 2020
Abstract
A new kind of high melting (HTm) pol(L-lactide) was discovered when cyclic poly(L-lactide)s were prepared by ring-expansion polymerization with cyclic tin catalysts at 130–160 °C in bulk. By DSC measurements with 10 K min−1 melting temperatures (Tm) in the range of 190–196 °C were found. The WAXS and SAXS measurements evidenced that not a new crystal lattice but a well-ordered morphology and a higher perfection of the crystallites are responsible for the high Tm values and high crystallinities. Under identical reaction conditions SnOct2-catalyzed and alcohol-initiated ROPs do not yield these crystallites. Furthermore, it was found that the standard crystallites are kinetically favored upon rapid crystallization, whereas the high melting form of poly(L-lactide) is thermodynamically more stable.
Introduction
Over the past 40 years, studies of syntheses, properties and applications of polylactides have rapidly increased. It was found, that stereo-copolymers of D- and L-lactic acid are amorphous with glass transitions in the range of 50–55 °C (depending of molar mass and stereo sequence), whereas poly(L-lactide) and its mirror image are crystalline materials having a glass transition temperature (Tg) around 60–63 °C and melting temperatures (Tm) of 176–180 °C after annealing.1–4 All these properties were determined from samples exclusively or predominantly consisting of linear chains and prepared by alcohol-initiated ROPs catalyzed by Sn(II) 2-ethylhexanoate (SnOct2).About four years ago the authors have launched a research project dealing with synthesis and characterization of optically pure, high molecular weight cyclic polylactides. In the first paper ring-expansion polymerizations catalyzed by 2,2-dibutyl-2-stanna-1,3-dithiolane (DSTL) were described.5 Weight average molecular weights (Mw's) in the range of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–150000 g mol−1 were obtained and a cyclic topology was confirmed. Furthermore, it was found that, when the time was extended from 4 h (standard) to 20 h, complete crystallization of the initially amorphous poly(L-lactide) occurred in several experiments. This phenomenon has never been observed bevor by the authors in alcohol-initiated polymerizations catalyzed by SnOct2. It was speculated, that the cyclic poly(L-lactide)s possess a higher crystallization tendency than their linear counterparts.
000–150000 g mol−1 were obtained and a cyclic topology was confirmed. Furthermore, it was found that, when the time was extended from 4 h (standard) to 20 h, complete crystallization of the initially amorphous poly(L-lactide) occurred in several experiments. This phenomenon has never been observed bevor by the authors in alcohol-initiated polymerizations catalyzed by SnOct2. It was speculated, that the cyclic poly(L-lactide)s possess a higher crystallization tendency than their linear counterparts.
During the past fifteen years, several synthetic methods were elaborated allowing for the preparation of cyclic polylactides.6–11 All these methods have in common, that the resulting polylactides are partially racemized and had Mw values < 40000 g mol−1. An optically pure cyclic poly(L-lactide) having a Mw of 30000 g mol−1 was reported by Piedra-Arroni et al.12 without information about the polymerization mechanism. Furthermore, Sugai et al.13,14 described the preparation of guest–host copoly(L-lactide)s with low melting temperatures (125 or 128 °C) indicating a strong influence of the guest monomer.
More recently, optically pure cyclic poly(L-lactide)s with Mw values up to 300000 g mol−1 were prepared either by ring-expansion polymerization (REP) with cyclic tin catalysts15–17 or by the so-called ROPPOC approach,18–20 which means simultaneous ring-opening polymerization and polycondensation. This mechanism initially generates linear chains having highly reactive end groups, so that low and high molecular weight cyclics are formed by back-biting and by end-to-end cyclization (Scheme 3). When the reaction time in those studies was extended to 22–44 h, again spontaneous crystallization was observed at 160 °C and the DSC measurements revealed unusually high melting endotherms (Tm values) around 189–192 °C (not described in those previous publications). These unexpected findings prompted a more detailed study of this phenomenon, the results of which are presented below. In the course of this study it turned out, that the polymerization mechanism plays an important role and thus, the following text is subdivided according to the three mechanisms studied in this work:
(1) Ring-expansion polymerization (REP, Scheme 1).
 | ||
| Scheme 1 Simplified polymerization mechanism of the DSTL-catalyzed REP of L-lactide. | ||
(2) Alcohol-initiated ring-opening polymerizations (ROPs, Scheme 2) and
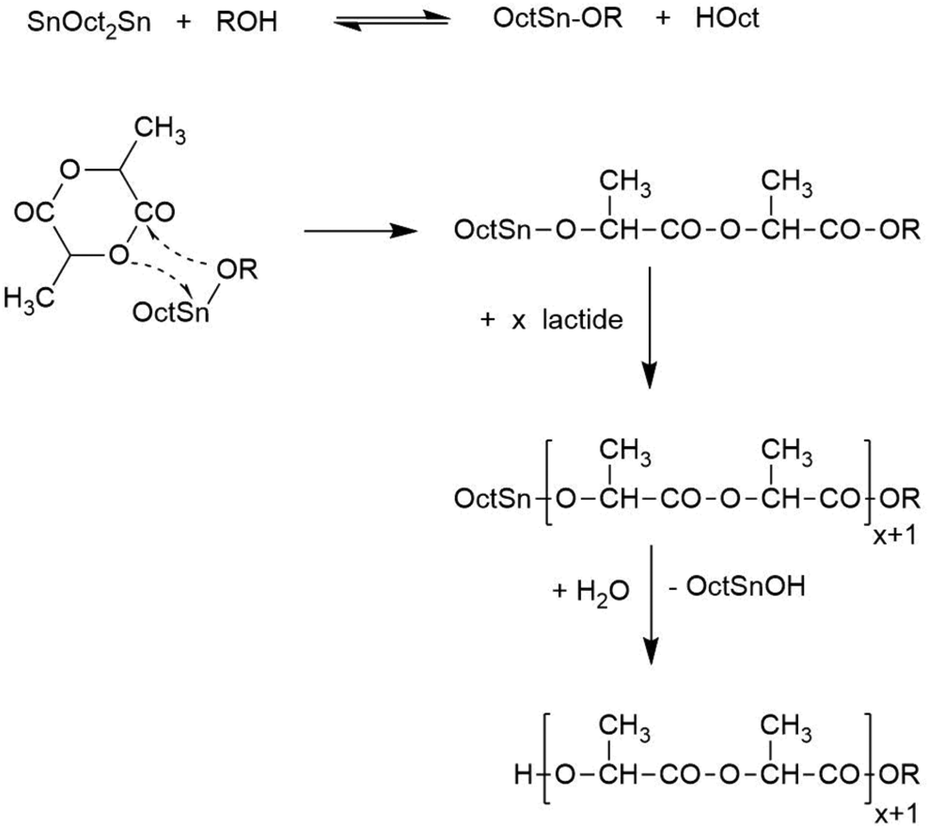 | ||
| Scheme 2 Simplified polymerization mechanism of alcohol-initiated and SnOct2-catalyzed ROPs of L-lactide. | ||
(3) Syntheses of polylactides via the ROPPOC method (ring-opening polymerization combined with simultaneous polycondensation, Scheme 3).
 | ||
| Scheme 3 Simplified mechanism of a ROPPOC synthesis of cyclic polylactides. | ||
The present study served two purposes. First, it should be explored how the formation of the high melting (HTm) polylactides depends on experimental parameters such as temperature, time, catalyst concentration and nature of the catalyst. The virgin reaction products should be characterized without any post-treatment. Second, it should be explained, why this phase has not been detected earlier despite 50 years of intensive research on synthesis and properties of polylactides. WAXS measurements of the first HTm specimens (no. 3–5, Table 1) proved, that they consist of the normal α-modification based on antiparallel 10/3 helices (first described by DeSantis and Kovacs21) and not of a new crystal modification. Therefore, the HTm phase discussed in this work will be termed HTm crystallites or HTm polylactides in contrast to the relatively low melting (Tm < 181 °C) “normal” (LTm) crystallites.
| Exp. no | Lac/Cat | t [h] | M n [g mol−1] | M w [g mol−1] | T g [°C] | T m [°C] | ΔHm [−J g−1] | Cryst. [%] |
|---|---|---|---|---|---|---|---|---|
| a After precipitation into ligroin and annealing at 160 °C for 1 d. b Based on five measurements with repeated calibration. c Repetition with another batch of L-lactide one year later gave a Tm of 195.5 °C. d Only part of the reaction mixture had crystallized. | ||||||||
| 1A | 100/1 | 4 | 27500 |
138000 |
59.8 | (175.0)a | (59.0)a | — |
| 1B | 100/1 | 24 | 24000 |
127000 |
58.5 | No crystall. | — | — |
| 2A | 200/1 | 4 | 27000 |
142000 |
57.5 | (176.0) | (41.0)a | — |
| 2B | 200/1 | 24 | 24000 |
131000 |
— | 192.5–195.50b | 90.5–91.5b | 85–86 |
| 3A | 500/1 | 4 | 26500 |
137000 |
55.5 | (174.5) | (47.8) | — |
| 3B | 500/1 | 24 | 23000 |
123000 |
60.3 | 190.0–192.5c | 83.5–85.0b | 78–80 |
| 4A | 1000/1 | 4 | 27000 |
140000 |
56.5 | (175.5) | (49.0) | — |
| 4B | 1000/1 | 24 | 23000 |
119000 |
— | 194.5–196.5b | 90–92b | 84–86 |
| 5 | 1500d | 48 | 19000 |
105000 |
— | 193.5d | 97.0 | 91.5 |
| 6 | 2000/1 | 48 | 14000 |
58000 |
58.0 | No crystall. | — | — |
Experimental
Materials
L-Lactide produced by PURAC was kindly supplied by Uhde Inventa-Fischer (Berlin, Germany). It was recrystallized from “Toluene 99.89% extra dry” (ACROS Organics, Geel Belgium) prior to polymerization. Tin(II) 2-ethylhexanoate (SnOct2), n-butanol, 10-undeceneol, ethyl L-lactate and salicylic acid were purchased from Aldrich Co (Steinheim, Germany) and used as received. Dichloromethane and toluene were distilled over P4O10. A linear, isopropanol-initiated poly(L-lactide), Purapol L105, was purchased from Carbion Purac for comparison of intrinsic viscosities with the polylactides synthesized in this work or in previous publications.5,15,22,23The catalysts used in this work (Scheme 4) were those prepared and used previously for syntheses of cyclic pol(L-lactide)s: DSTL, BuSnBuca,24 BuSnBiph,15 BuSnNaph,15 SnBiph,17 SnNaph,17 BuSnOPF,18 BuSnSBn19 and BuSnSPF.19
 | ||
| Scheme 4 Structures and labels of the catalysts discussed in this work. | ||
Polymerizations
All reaction mixtures were prepared under a blanket of argon. Table 1: L-Lactide (50 mmol) and DSTL (0.125 mmol) were weighed into a 50 mL Erlenmeyer flask (dried with hot air >300 °C). The reaction vessel was immersed into an oil bath preheated to 161 °C. After 10 min the temperature was lowered to 160 °C. After 4 h part of the viscous melt was removed with a spatula. In a parallel experiment the reaction vessel was destroyed after 20 h to isolate the completely crystallized reaction product. Polymerizations with other Lac/Cat ratios were conducted analogously.Table 2: L-Lactide (50 mmol) was polymerized with DSTL as described above.
| Exp. no. | T [°C] | t [h] | M n [g mol−1] | M w [g mol−1] | T m [°C] | ΔHm [−J g−1] | Cryst. [%] |
|---|---|---|---|---|---|---|---|
| 1 | 180 | 22 | Dark tar | — | — | — | — |
| 2 | 170 | 44 | No crystallization | — | |||
| 3 | 165 | 44 | No crystallization | — | |||
| 4 | 160 | 22 | 31000 |
146000 |
195.0 | 92.5 | 88 |
| 5 | 150 | 22 | 45000 |
156000 |
192.5 | 79.0 | 74 |
| 6 | 140 | 24 | 57000 |
167000 |
191.0 | 80.0 | 75 |
| 7A | 130 | 24 | 65000 |
187000 |
189.0 | 78.0 | 74 |
| 7B | 130 | 44 | 27000 |
69000 |
192.5 | 78.0 | 74 |
| 8A | 120 | 24 | 69000 |
170000 |
176.5 | 57.5 | 54 |
| 8B | 120 | 44 | 15000 |
61000 |
175.5 | 66.5 | 62 |
Table 3: L-Lactide (50 mmol) and SnBiph (0.25 mmol) were weighed under argon into a 50 mL Erlenmeyer flask and polymerized in a preheated thermostated oil bath. Polymerizations with other catalysts were performed analogously.
| Exp. no. | Catalyst | Lac/Cat | T [°C] | t [h] | M n [g mol−1] | M w [g mol−1] | T m [°C] | ΔHm [−J g−1] | Cryst. [%] |
|---|---|---|---|---|---|---|---|---|---|
| a Repetition with another batch of recrystallized L-lactide gave a slower crystallization, but a Tm of 191.5 after 48 h. b Beginning crystallization observed after 44 h. | |||||||||
| 1A | BuSnBuCa | 600/1 | 160 | 24 | 14000 |
73000 |
189.0 | 82.5 | 78 |
| 1Bb | BuSnBuCaa | 1000/1 | 160 | 24 | 16000 |
76000 |
191.0 | 89.5 | 84 |
| 2 | BuSnBiph | 1000/1 | 160 | 22 | 38000 |
105000 |
189.0 | 75.0 | 71 |
| 3 | BuSnNaph | 1000/1 | 160 | 22 | 41000 |
120000 |
191.5 | 74.0 | 70 |
| 4 | SnNaph | 1000/1 | 160 | 24 | 70000 |
159000 |
193.5 | 88.5 | 83 |
| 5 | SnBiph | 1000/1 | 120 | 24 | 56000 |
167000 |
191.2 | 73.0 | 70 |
| 6 | SnBiph | 1 0001 | 130 | 24 | 75000 |
156000 |
190.5 | 70.5 | 67 |
| 7 | SnBiph | 1000/1 | 140 | 24 | 72000 |
170000 |
189.0 | 63.5 | 60 |
| 8 | SnBiph | 1000/1 | 150 | 44 | 69000 |
203000 |
189.0 | 72.5 | 69 |
| 9 | SnBiph | 1000/1 | 160 | 24 | 78000 |
221000 |
192.5 | 76.0 | 72 |
| 10 | SnBiph | 1000/1 | 170 | 24 | No crystal. | — | — | ||
| 11 | SnBiph | 600/1 | 160 | 24 | 61000 |
163000 |
194.5 | 83.5 | 79 |
| 12 | SnBiph | 1500/1 | 160 | 44 | 83000 |
232000 |
191.5 | 83.0 | 78 |
| 13 | SnBiph | 2000/1 | 160 | 72b | 55000 |
124000 |
194.8 | 87.0 | 82 |
Table 4: L-Lactide (50 mmol) was weighed under argon into a 50 mL Erlenmeyer flask and 0.1 mL of a 5 M solution of an alcohol in toluene and 0.1 mL of a 0.5 m solution of SnOct2 in xylene were injected. The polymerizations were performed as described for Table 1.
| Exp. no. | Catalyst | Cocatalyst | t [h] | M n [g mol−1] | M w [g mol−1] | T m [°C] | ΔHm [−J g−1] | Cryst. [%] |
|---|---|---|---|---|---|---|---|---|
| a After 48 h beginning crystallization was observed. b 24 h annealed at 120 °C. c After annealing at 120 °C for 2 h the Tm's were in the range of 170.0–171.5 °C (see Table 6). d Lac/CoCat = 1000/1. | ||||||||
| 1 | SnOct2 | n-Butanola | 72a | 17500 |
29000 |
178.5 | 77.5 | 73 |
| 2A | SnOct2 | Ethyl lact. | 48 | 15000 |
25500 |
No cryst. | — | — |
| 2Bc | +24b | 173.5 | 76.5 | 72 | ||||
| 3A | SnOct2 | 11-Undecenol | 48 | 16000 |
28000 |
No cryst. | — | — |
| 3B | +24b | 178.0 | 78.5 | 74 | ||||
| 4 | SnOct2 | Salicylic acidd | 48 | 40000 |
65500 |
192.5 | 86.5 | 82 |
| 5A | DSTL | n-Butanol | 48 | 20000 |
31000 |
No cryst. | — | — |
| 5Bc | +24b | 179.0 | 82.0 | 77 | ||||
| 6A | DSTL | Ethyl lact. | 48 | 21000 |
35000 |
No cryst. | — | — |
| 6Bc | +24b | 179.0 | 87.5 | 83 | ||||
| 7A | BuSnBuCa | n-Butanol | 48 | 19000 |
33000 |
No cryst. | — | — |
| 7B | +24b,c | 171.0 | 69.5 | 65 | ||||
| 8A | BuSnBuCa | Ethyl lact. | 48 | 18000 |
31000 |
No cryst. | — | — |
| 8B | +24b,c | 170.5 | 70.0 | 65 | ||||
| 9 | BuSnBi | n-Butanol | 48 | 17000 |
31000 |
No cryst. | — | — |
Measurements
The 150 MHz 13C NMR spectra were recorded on a Bruker Avance 600 FT spectrometer in 5 mm sample tubes using CDCl3 as solvent and TMS as internal shift reference. DSC measurements of all samples were performed with a Mettler-Toledo DSC-1 equipped with the Stare Software version 11.0 at a heating and cooling rate of 10 K min−1 (indium and zinc were used for calibration). Indium and zinc were used for calibration. The crystallinities were calculated with ΔHmmax (for 100% crystallinity) of −106 J g−1.The SEC measurements were performed using a modular HPLC system consisting of a pump (Chromophor C 1130) running at 1 mL min−1 flow rate. The system was kept at 40 °C. 100 μL of each sample with a concentration of 2–4 mg mL−1 was manually injected. A triple detector combination consisting of a refractive index detector RI-501 (Shodex), a multi angle laser light scattering detector DAWN and a viscometer Viscostar (both Wyatt Germany, Dernbach) was used to determine Mark–Houwink–Sakurada (MHS) parameter. Clarity software (GPC extension, DataApex) was used for instrument control and molecular weight calculation, whereas ASTRA 6 (Wyatt) was used to perform the MHS calculations. SEC calibration was performed using commercially available polystyrene standard sets (Polymer Standards Service – PSS, Mainz, Germany). Using triple detection, the intrinsic viscosities of narrow fractions were compared with those of the commercial, linear poly(L-lactide) Purapol L105.
The MALDI-TOF mass spectra were recorded in the linear mode with a mass spectrometer AutoflexMax (Bruker Daltonik, Bremen, Germany) equipped with a laser running at 355 nm. The sample targets were prepared from pre-mixed solutions of sample (CHCl3, 4 mg ml−1) with matrix (20/50 v/v), which consisted of a mixture of (50/1 v/v) trans-2-[4-tert-butylphenyl-2-methyl-2-propylidene] malonitrile (DCTB) (THF, 10 mg mL−1) and potassium trifluoroacetate (THF, 2 mg mL−1) as dopant. For one spectrum 8000 laser shots were recorded at 4 different positions of the sample spot.
The wide-angle X-ray scattering (WAXS) measurements were performed using our in-house SAXS/WAXS apparatus equipped with an Incoatec™ X ray source IμS and Quazar Montel optics. The wavelength of the X ray beam was 0.154 nm and the focal spot size at the sample position was 0.6 mm2. The samples were measured in transmission geometry. The sample-detector distance was 0.1 m, allowing us to detect an angular range of 2Θ = 5°–33°. The patterns were recorded with a Rayonix™ SX165 CCD-Detector and the accumulation time per WAXS measurement was 600 s. DPDAK25 a customizable software for reduction and analysis of X-ray scattering data sets was used for gathering 1D scattering curves.
The small angle X-ray scattering (SAXS) measurements were performed with a self-built SAXS apparatus, equipped with a high brilliance Incoatec IμS X-ray source and Quazar Montel optics. The wavelength was 0.154 nm and the focal spot size at the sample had a diameter of 700 μm. The distance between the sample and the CCD detector (Rayonix 165 SX) was 1.6 m, allowing us to detect a q-range from 0.08 to 2.5 nm−1. The recording time per sample was 1200 s. After subtracting the background, the 2D scattering patterns were radially integrated with the software DPDAK25 and transformed into Kratky-plots (q vs. Iq2). These plots enabled observation and analysis of positions of the lamellar reflections, which can easily be transformed into real space dimensions using the equation L = 2 π/q.
Optical rotation was measured with a Krüss P 8000 T polarimeter using the Na-D line as light source. Chloroform solutions thermostated at 25 °C were measured in a glass tube with 10 cm length.
Results and discussion
REPs catalyzed by DSTL
As reported recently, heating of L-lactide with the cyclic catalyst DSTL yields cyclic poly(L-lactide)s via ring-expansion polymerization (REP) the schematic illustration of which is presented in Scheme 1.5 Characteristic for this special kind of (ROP) is a slow initiation step which generates a tin alkoxide bond which is several orders of magnitude more reactive towards cyclic esters than the thermodynamically more stable Sn–S bond. Therefore, the slow initiation is followed by rapid chain growth (i.e. ring expansion). However, from time to time, the thermodynamically more stable Sn–S bond is restored with extrusion of the catalyst, so that a tin-free cyclic polylactide is formed. The rapid equilibration of the initially formed even-numbered polylactide cycles with odd-numbered cycles also proves that an intracircular “back-biting” reactions must exist (quite analogous to the “back-biting” equilibration of linear polyester chains).22 The existence of a rapid equilibration process is also in agreement with the relatively high dispersities (D-values in the range of 3–4) found for the polylactides listed in Table 1. Such a rapid odd-even equilibration was also observed for the other cyclic tin catalysts used in this work.5,15,17,24 The existence of a rapid ring-ring equilibration is, in turn, essential for the proper understanding of the results discussed below.When polymerizations of lactide were conducted in bulk at 160 °C with reaction times of 4 h (A-experiments in Table 1), highly viscous transparent melts were obtained which partially crystallized upon slow cooling. The typical DSC trace of such a poly(L-lactide) isolated with rapid cooling (and thus, with suppression of crystallization) is presented in Fig. 1. The glass transition is followed by crystallization exotherm which is the followed by the melting endotherm. The maximum of this endotherm transition appears around 174–176 °C regardless of the Lac/Cat ratio, as demonstrated by the data listed in Table 1. Hence, the normal crystallization of cyclic poly(L-lactide)s yields the same crystal modification and crystalline phase as crystallization of linear poly(L-lactide)s.
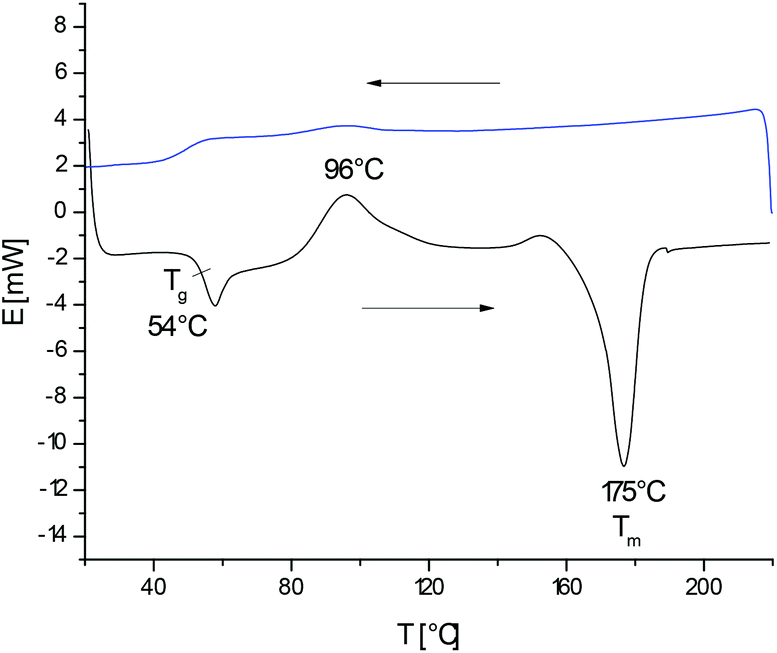 | ||
| Fig. 1 DSC measurements of no. 4A Table 1 rapidly cooled from the melt. | ||
When an A-sample of Table 1 was precipitated into ligroin so that most of the catalyst was removed, annealing at 160 °C had consequences that were typical for annealing of almost all crystalline polymers. The degree of crystallinity increased (as indicated by the disappearance of the glass-transition step) and size plus perfection of the crystallites increased, so that the maximum of the melting endotherm shifts to somewhat higher values and the melting enthalpy increased. This effect is exemplarily illustrated by the DSC trace of Fig. 2(A). The maximum Tm achieved in this way was 178–179 °C regardless of the sample and the maximum melting enthalpy was around −87/−89 J g−1. These values are in perfect agreement with literature data concerning linear poly(L-lactide)s.1–4
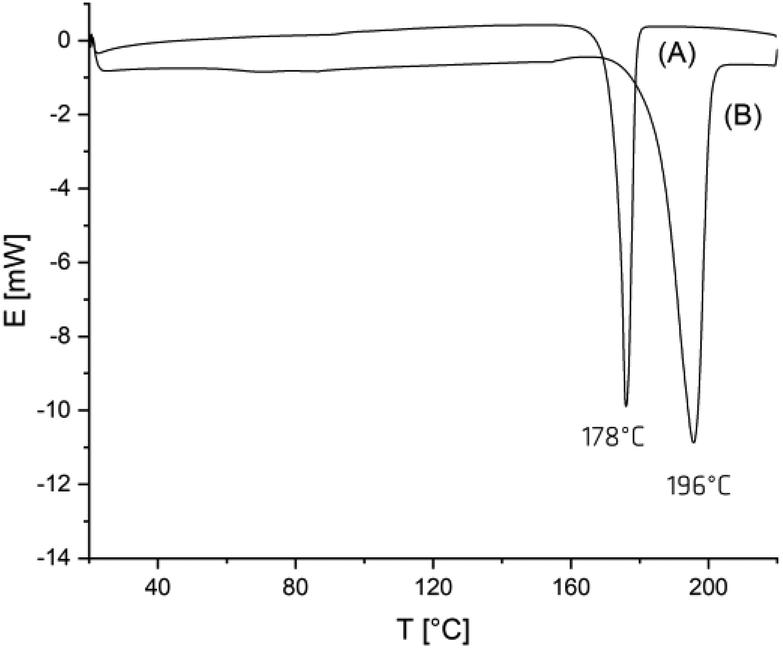 | ||
| Fig. 2 DSC measurement of (A) no. 4A, Table 1 after precipitation into ligroin followed by annealing at 160 °C for 1 day, (B) no. 4B, Table 1. | ||
However, when the polymerization time was extended beyond 4 h (not reported in ref. 5), the cyclic poly(L-lactide)s began to crystallize and after 24 h the entire product looked crystalline for the naked eye (B-experiments in Table 1). When the samples of these B-experiments were subjected to DSC measurements, no glass transition was detectable and Tm's in the range of 190–196.5 °C were found (Fig. 2(B)). These high temperatures are far outside the range of melting temperatures which can be realized by normal annealing of crystalline poly(L-lactide)s.
For a better understanding of the correlation between reaction conditions and HTm crystallites the following observations are important. First, when the B-samples were precipitated into ligroin to remove most of the catalyst, and annealed at 160 °C for 1 d, Tm's around 178 °C corresponding to the normal LTm crystallites (Fig. 2).1–4 In other words, the formation of HTm-crystallites requires the presence of the catalyst. Second, when the first heating of HTm samples up to 220 °C is followed by rapid cooling and a 2nd heating, the resulting heating trace looked exactly like that of Fig. 1 showing a distinct glass transition and Tm's in the range of 173–175 °C characteristic for normal LTm-crystallites. This result indicates that upon rapid crystallization the LTm-crystallites are the kinetically favored phase, and the HTm-crystallites are the thermodynamically more stable alternative. This interpretation is also supported by the results discussed in the next section.
In order to get information on the influence of the polymerization temperature on the formation of HTm polylactides, several DSTL-catalyzed REPs were conducted at 180, 170, 165, 150, 140, 130 and 120 °C (Table 2) with the favorable Lac/Cat ratio of 1000/1. At 180 °C rapid crystallization was not observed and annealing times beyond 4 h finally turned the reaction mixture into a brown tar. Again, no crystallization was observed at 170 and 165 °C.
The experiments performed at temperatures between 165 and 120 °C may be subdivided into two groups. At 160–130 °C crystalline samples having Tm values around 190 °C were obtained indicating formation of HTm poly(L-lactide), whereas at 120 only crystallites with Tm's < 180 °C were found. In other words, the relatively rapid crystallization at 140 or 130 °C did not hinder formation of HTm crystallites, when an efficient transesterification catalyst favoring formation of cyclic polylactides was present. In summary, it may be said that formation of the HTm phase is confined to certain limits with regard to Lac/Cat ratio and temperature. For DSTL the effective Lac/Cat ratio is in the range of 200–1500 and the temperature range is as narrow as 130–160 °C.
REPs with various cyclic tin catalysts (Table 3)
To find out, if the results obtained with DSTL are representative for other cyclic tin catalysts, polymerizations with five other catalysts were performed and compiled in Table 3. All five catalysts yielded the HTm form of poly(L-lactide) under the standard conditions (160 °C/22 h, Lac/Cat = 1000/1). With the most reactive catalyst, SnBiph, both, Lac/Cat ratio and temperature were varied (no. 5–12, Table 3). In contrast to DSTL, it was found that the HTm crystallites can be obtained even at a Lac/Cat ratio of 2000/1, but a longer time (72 h) was necessary to achieve an almost complete crystallization of the reaction mixture. Furthermore, the minimum temperature needed for the HTm form was as low as 120 °C whereas 130 °C was the minimum for DSTL. Hence, these results evidence that a higher transesterification activity of the catalyst is favorable for formation of the HTm form. In summary, the experiments listed in Table 3, together with those summarized in Tables 1 and 2, clearly demonstrate that ring-expansion polymerization with covalent cyclic tin catalysts is particularly favorable for the formation of HTm poly(L-lactide)s regardless of the structure of the catalyst, provided its transesterification activity is high enough.Finally, structure and topology of the polylactides listed in Tables 1–3 need a short comment. As reported previously, the poly(L-lactide)s prepared by REP with cyclic tin catalysts at short reaction times almost completely consist of cycles.5,15,26,27 The long reaction times studied in this work have the consequence that thermal degradation comes into play, so that cycles are slowly transformed into linear chains as evidenced by MALDI TOF mass spectrometry. This change is favored by higher temperatures and longer times and it depends on the nature of the catalyst. Fig. 3A, presents an example with a mass spectrum bare of “linear peaks” obtained with DSTL as catalyst at 160 °C after 4 h. As demonstrated by Fig. 3B, more than 50% of the cycles were transformed into linear species by thermal degradation after 24 h. The lower Mn and Mw values obtained after 1 d ((B) experiments in Table 1) also indicated beginning thermal degradation at 160 °C. Yet, when the temperature was lowered to 140 or 130 °C, this rapid degradation was avoided as shown by the mass spectra of Fig. 4A or Fig. S1 (ESI†). The MHS measurement presented in Fig. 4B proves that not only the mass fraction below 10000 g mol−1, but also the mass fraction above 50000 g mol−1 almost completely consists of cycles. This information is based on the long-known fact that the intrinsic viscosity of cyclic polymers is approx. 33% lower than that of a linear counterpart, due to the lower hydrodynamic volume. When other catalysts, such as SnNaph or SnBiph are used (Table 3), the degradation is much slower and even after 1 d at 160 °C the cycles are the predominant species as evidenced by Fig. 5 and Fig. S2A–4 in the ESI.† Yet, when the time is extended to 3d considerable transformation of cycles into linear species was also observable for SnBiph-catalyzed samples (Fig. 2B, ESI†).
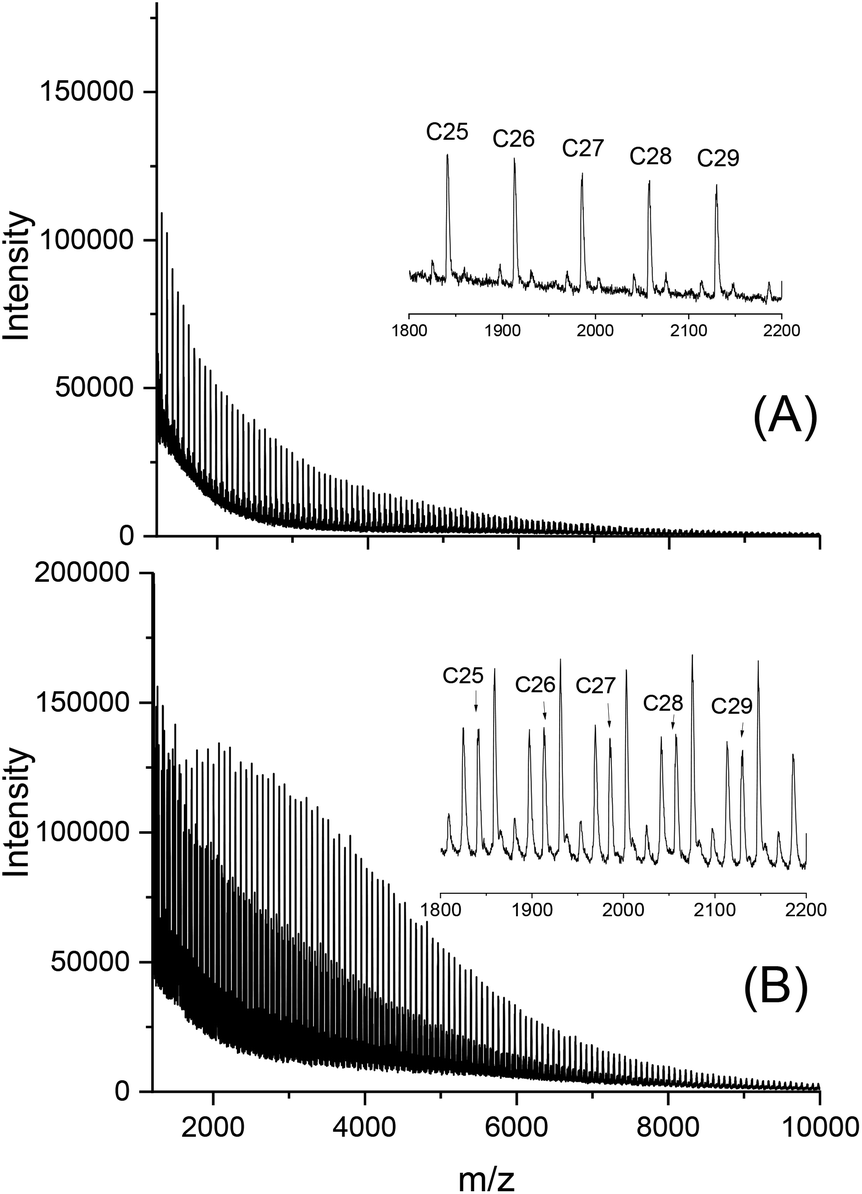 | ||
| Fig. 3 MALDI-TOF mass spectra of polylactides polymerized with DSTL at 160 °C: (A) after 4 h, no. 4A, Table 1, (B) after 1 d no. 4B, Table 1. | ||
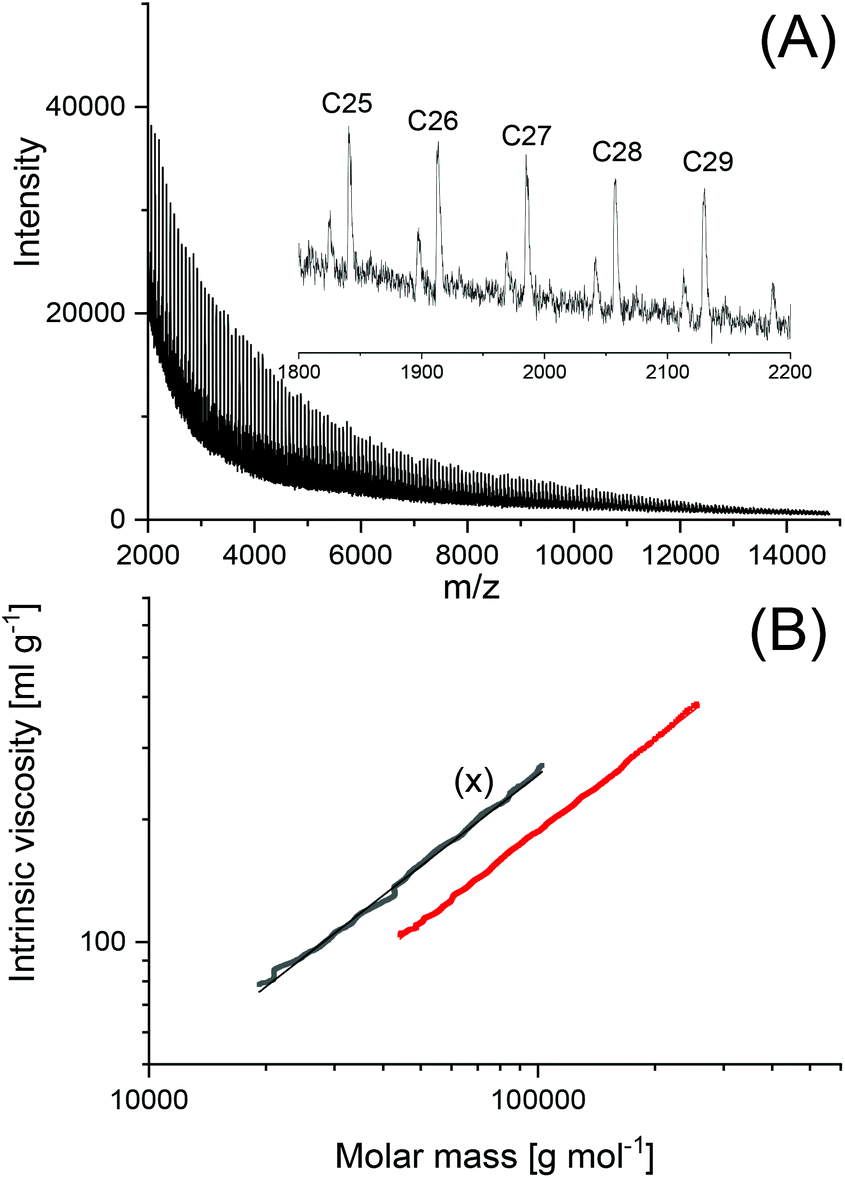 | ||
| Fig. 4 Polylactide polymerized with DSTL at 140 °C/1 d (no. 6, Table 2): (A) MALDI-TOF mass spectrum, (B) MHS measurements (X = linear Purapol L105). | ||
 | ||
| Fig. 5 Polylactide polymerized with SnNaph at 160 °C/1 d: (no. 4, Table 3): (A) MALDI-TOF mass pectrum, (B) MHS measure, ent (X = linear Purapol L105). | ||
These observations allow for the conclusion that formation and presence of linear polylactides do not hinder the formation of HTm crystallites. A further discussion of this point is given below in connection with the experiments of Table 5.
| Exp. no. | Catalyst: dibutyltin | Lac/Cat | t [h] | M n [g mol−1] | M w [g mol−1] | T m [°C] | ΔHm [−J g−1] | Cryst. [%] |
|---|---|---|---|---|---|---|---|---|
| a With another batch of L-lactide a Tm of 192.0 °C and a ΔHm of −80.0 J g−1 were obtained. | ||||||||
| 1 | Bisbenzylmercaptide | 200/1 | 24 | 4 000 | 16000 |
184.5 | 76 | 72 |
| 2 | Bisbenzylmercaptide | 400/1 | 24 | 4 000 | 15000 |
189.5 | 79 | 75 |
| 3 | Bisbenzylmercaptide | 1000/1 | 24 | 3 500 | 21000 |
189.5 | 74 | 70 |
| 4 | Bisbenzylmercaptide | 2000/1 | 24 | 3 500 | 22000 |
No crystallization | — | |
| 5 | Bispentafluorothiophenolate | 200/1 | 24 | 60000 |
145000 |
194.0 | 78.5 | 74 |
| 6 | Bispentafluorophenolate | 1000/1 | 24 | 24000 |
57000 |
192.5a | 81a | 76 |
ROPs initiated by alcohols
Since the HTm phase was discovered for REPs with cyclic tin catalysts, this finding raised the question, if the cyclic architecture is a prerequisite for its formation or, if linear poly(L-lactide)s may also yield this type of crystallites. It is conspicuous that this HTm phase was not observed in the numerous studies on syntheses and properties of linear poly(L-lactide)s (based on SnOct2 as catalyst) reported in the literature. To answer this question, several polymerizations were performed with SnOct2 as catalyst and alcohols as initiators (no. 1–3, Table 4). No crystallization was observed within 48 h for the experiments no. 2 and 3, but a few crystallites appeared within 48 h in the experiment with n-butanol (no. 1). After 72 h an almost complete crystallization of the reaction mixture was achieved, but the DSC measurements revealed that the LTm crystallites had been formed. However, when salicylic acid was added instead of an alcohol crystallization began within 24 h and was complete after 48 h (no. 4, Table 4). From previous studies it is known, that salicylic acid forms a cyclic Sn(II) catalysts which catalyzes REPs.28Hence these experiments demonstrate that REPs with cyclic tin catalysts distinctly favor formation of HTm polylactides.
This conclusion was confirmed by four more experiments (no. 5–10). DSTL, BuSnBuCa and BuSnBiph were used as catalysts, because it was demonstrated in previous publications that addition of excess alcohol changes the polymerization mechanism, so that linear polylactides are formed instead of cyclic ones.5,15,17n-Butanol or ethyl lactate were added as initiators in analogy to the experiments with SnOct2 (no.1–3). No crystallization was observed within 48 h although the neat catalysts yield the crystalline HTm crystallites within 24 h (s. Tables 1–3). These results suggest, that it is not the linear topology of polylactide chains which hinders formation of HTm crystallites but mainly the alcohol-initiated polymerization mechanism.
Only a hypothetical explanation of this finding can be forwarded at this time. This hypothesis purports that the OH end groups reduce the reactivity of the catalysts and reduce their mobility in the amorphous phase and along the surface of the crystallites for two reasons.
They undergo a temporary covalent binding to the Sn atoms, and they occupy additional d- or sp3d2 orbitals because their concentration is 5 to 10 times higher than that of the Sn atoms. Why a high transesterification activity and mobility of the catalyst is needed for the formation of the HTm morphology is discussed in section 3.5. A more detailed study of this aspect is in progress. It cannot be excluded that a broader variation of initiator and reaction conditions has the consequence, that also certain alcohol-initiated pol(L-lactide) will crystallize in the HTm form from the reaction mixture.
Polymerizations with ROPPOC catalysts
In a previous publication the ROPPOC synthesis of cyclic poly(L-lactide)s by means of the dibutyltin bismercaptides depicted in Scheme 5 was reported.19 | ||
| Scheme 5 Example of a transesterification (ring–ring equilibration) reaction involving loops on the surface of lamellar crystal. | ||
When the reaction time was extended to 22 h with the BuSnSBn catalyst (not described in the previous paper) the polymerization product crystallized at 160 °C and Tm's of 189.5 °C were found when Lac/Cat ratios of 400 and 1000/1 were applied. For the Lac/Cat = 200/1 the Tm was considerably lower and with a Lac/Cat ratio of 2000/1 no crystallization occurred. These results agree well with those obtained by means of DSTL as catalyst (Table 1). Yet, in the case of the BuSnSBn experiments the reaction products were mixtures of cyclic and a smaller fraction of linear poly(L-lactide)s, as shown for shorter reaction times in a previous publication.
With the more reactive catalysts BuSnSPF and BuSnOPF analogous experiments were performed in this work, whereas experiments with shorter polymerization times were described in the previous publications.18,19 At the longer polymerization times listed in Table 5 again crystalline polylactides were obtained and Tm's up to 194 °C with crystallinities above 80%. The mass spectra of the polylactides no. 5 and 6 represent two extreme cases (Fig. 6). In agreement with the previous results, the sample prepared with a low Lac/Cat ratio (no. 5) exclusively displayed peaks of cycles (Fig. 6A) whereas a predominance of linear chains was found for the polymerization with the high Lac/Cat ratio of 1000/1.18–20
 | ||
| Fig. 6 MALDI-TOF mass spectra of polylactiddes prepared (A) with BuSnSPF (no. 5, Table 5), (B) with BuSnOPF (no. 6, Table 5, L means linear chains). | ||
This correlation between catalyst concentration and content of cycles was found for all ROPPOC catalysts studied so far, but a satisfactory explanation of this strange correlation is still lacking. In summary, these results allow for the following conclusions:
(1) The ROPPC mechanism favors formation of HTm polylactides more than alcohol initiated ROPs.
(2) The presence of linear chains does not hinder the formation of HTm crystallites, at least, when the most reactive catalyst BuSnOPF is involved.
(3) The most reactive catalysts, BuSnSPF and BuSnOPF, yielded the products with the highest Tm's and the highest crystallinities.
Characterization of the HTm poly(L-lactide)s
At first it should be emphasized that the characterization of the HTm poly(L-lactide)s in this work is confined to the virgin reaction products. Any post-treatment, such as annealing at different temperatures has been avoided. Such studies including the transformation of the LTm-crystallites into the HTm version are close to completion and will be reported in the near future. Around 50 experiments with variation of catalyst, catalyst concentration, temperature and time were performed. Under optimized conditions LTm crystallites have been transformed into HTm crystallites and this transition is irreversible. Those experiments confirm what has been learned from the DSC measurements described above. Therefore, all these measurements demonstrate that the HTm crystallites are the thermodynamically most stable phase of poly(L-lactide)s. The LTm crystallites are the kinetically favored alternative, which is formed upon slow cooling of the melt and upon annealing around 120 °C or below. This fact is well known in the literature1–4,22,28–30 and it is documented in this work by Fig. 1 and by the annealing experiments at 120 °C/2 h listed in Table 4 and by the DSC curve (A) in Fig. 2.DSC data are usually presented in the literature without margin of error, therefore the scattering of the Tm values mentioned above (Table 1) deserve a short discussion. At least two reasons may be given. First, the usual calibration with indium and zinc may result in a margin of error of ±1 °C, because zinc may contain traces of ZnO on its surface. A second factor is the handling and properties of the specimens used for the measurements. In this work the virgin crystalline plaques directly resulting from the polymerization in bulk were used for all DSC (and X-ray) measurements, and these plaques had a thickness of approx. 3–4 mm. When crystalline powder scraped from the surface was measured, the Tm's were lower than those obtained from pieces picked out from the central bottom of the plaques by means of a special pincer. The difference amounted to approx. 1 °C for sample 3B, Table 1, 1.5 °C for 4B and 4C, Table 1 (194.5 versus 196 °C). This systematic deviation suggests that the perfection of morphology and crystallites structure have a gradient across the samples. This suggestion was confirmed by SAXS measurements. Nonetheless, the DSC data are clearly outside the thermal properties reported for annealed or isothermally crystallized linear poly(L-lactide)s.
Pennings and coworkers studied the relationship between crystallization temperature (Tc) and Tm under conditions of isothermal crystallization and found a significant dependence of Tm on Tc.29,30 For example, they found at the standard heating rate of 10 K min−1, that a Tc of 160 °C yields single crystals having a Tm around 183–185 °C or a Tm of 171–173 °C for a Tc of 130 °C. Unfortunately, those authors did not list precise Tm data but published diagrams of limited accuracy. Nonetheless, their Tm's are about 10 or even 20 °C below the values presented in Tables 2 or 3 for samples prepared at 160 or 130 °C. However, the Tm found by Pennings and coworkers at a Tc of 160 °C is higher than the values obtained in this work by annealing of catalyst-free poly(L-lactide)s at 160 °C or obtained by other groups1–4 for commercial samples containing a poisoned catalyst (see Purapol in Table 6). The main reason for this difference is that Pennings observed the melting of relatively large single crystals under the optical microscope. Yet, in this work and other publications DSC measurements of samples containing crystallites of varying size and perfection were measured. The Tm is here defined (as usual) as the minimum of the melting endotherm and thus, is necessarily several degrees C lower than the Tm of an individual, rather perfect single crystal. Hence, this comparison clearly demonstrates that the results obtained in this work are not a copy of the isothermal crystallization as reported by Pennings and coworkers.
| Standard morphology and crystallites | HTm morphology and crystallites | ||||
|---|---|---|---|---|---|
| Sample | T m (°C) | L (nm) | Sample | T m (°C) | L (nm) |
| No. 1, Table 4 | 178.5 | 21 | No 4B, Table 1 | 195.5 | 29 |
| No. 2B, Table 4 | 173.5 | 20 | No. 4C, Table 1 | 194.5 | 29 |
| No. 5B, Table 4 | 179.0 | 18 | No 5B, Table 1 | 193.5 | 31 |
| No. 6B, Table 4 | 179.0 | 20 | No. 4, Table 3 | 193.5 | 33 |
| No. 7B, Table 4 (2 h/120 °C) | 170.5 | 15 | No. 9, Table 3 | 192.5 | 33 |
| No. 7B, Table 4 (1 d/120 °C) | 171.0 | 15 | No, 13, Table 3 | 194.5 | 32 |
| No. 8B, Table 4 (2 h/120 °C) | 170.0 | 16 | No. 4, Table 4 | 192.5 | 25 |
| No. 8B, Table 4 (1 d/120 °C) | 170.5 | 16 | No: 5, Table 5 | 194.0 | 26 |
| Purapol: (1 d/120 °C) | 177.5 | 18 | No. 6, Table 5 | 192.5 | 30 |
| Purapol: (1 d/160 °C) | 180.5 | 21 | |||
In addition to Tm's, the DSC measurements also yield melting enthalpies as interesting source of information. The data listed in Tables 1–5 indicate that those samples showing the highest Tm's also gave high ΔHm's. Based on a maximum ΔHm of −106 J g−1, which seems to be the widely agreed value for 100% crystallinity, crystallinities up to 89% may be calculated.31 Pennings and numerous other researchers30,32 used a ΔHm max value of −93 J g−1, which was first published by Fischer at al.33 But from the data in Tables 1 it is clear that this value is too low, because otherwise samples such as 4c and 5A, B would have crystallinities around 100%.
Extremely high Tm's (>200 °C) and ΔHm's were reported by Hoogsteen et al. for poly(L-lactide) fibers.34 Yet in this case the morphology is a frozen-in phase formed by the shear forces and termed “constraint lamellae” by those authors. Other authors used the term “frustrated morphologies” for such kinetically controlled morphologies.35 In contrast, the HTm morphology and crystallites reported here are formed under thermodynamic control without any mechanical motion quite analogous to isothermal crystallization.
WAXS patterns of six polylactides including the commercial, linear Purapol L105 were recorded (Fig. S7†) and found to be almost identical (with slight variation of the crystallinity). Neither variation of catalyst, temperature or topology nor LTmversus HTm crystallites had any influence on the crystal modification. In all cases the a-modification first described by DeSantis and Kovacs was obtained.21 This result is in perfect agreement with the pertinent literature36 according to which the α-modification is the thermodynamically most staple modification and usually formed >120 °C in the absence of mechanical stress. In other words, the WAXS patterns do not allow for a differentiation between LTm and HTm crystals or between linear and cyclic topology.
More informative were the SAXS curves, which revealed for all samples studied so far, that two reflections representing 1st and 2nd order of the long period were detectable (Fig. 7 and Fig. S5 + S6 in the ESI†). The existence of a 2nd order reflection indicates a high degree of order inside the lamellar stacks. On the average, the occurrence of the SAXS reflections of the HTm samples was lower than that of the low-Tm samples as exemplarily demonstrated in Fig. 6. The lower occurrence is most likely a consequence of a denser packing of the HTm crystallites due to their smooth surface in combination with a loss of amorphous phase and consequently with a higher degree of crystallinity.
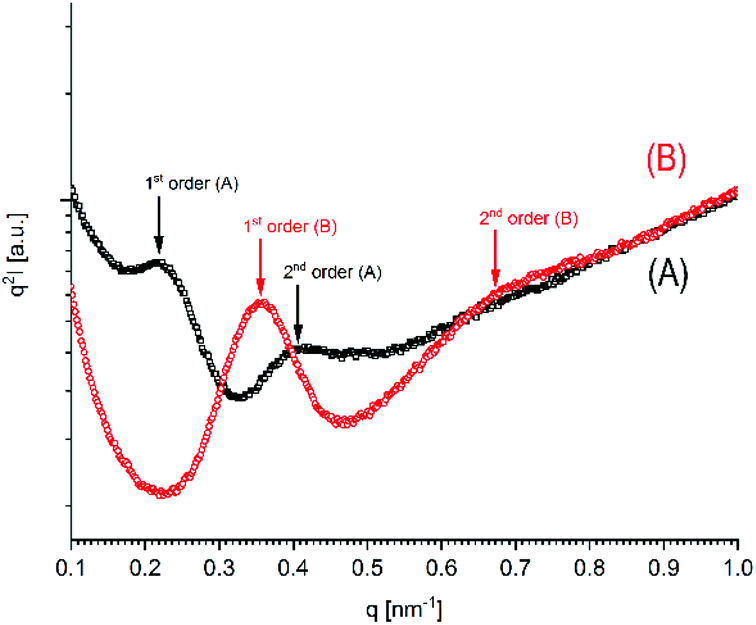 | ||
| Fig. 7 SAXS curves of poly(L-lactide)s prepared with DSTL: (A) (no. 4A, Table 1); (B) no. 5B. Table 4. | ||
These SAXS measurements allow for the following preliminary interpretation (a more detailed study will follow). The L-values derived from the SAXS curves of relatively low-melting crystallites (Tm < 181 °C) fall into the range of 15–21 nm. Such L-values were obtained for relatively low molecular weight samples crystallized at 120 or 160 °C (e.g. no. 1, 2, 5B, 6B, 8B, Table 4) and also for the high molecular weight (Mw ∼ 140000 g mol−1) commercial “Purapol L105”, regardless if annealed at 120 or 160 °C (Table 6, left column). The same range of L-values was reported by other authors for poly(L-lactide) crystallized below 120 °C (and yielding the conformationally disordered α′-crystallites).37,38
Hence, this range of L-values may serve as reliable basis for comparisons with the L-data of HTm-crystallites (Table 6, right column). For samples prepared with cyclic tin catalysts (Tables 1–3) L-values of 29–33 nm are characteristic, but L-values as low as 26 or 25 nm were also found for polylactides prepared with less reactive catalysts. These results allow for two conclusions. First, the HTm crystallites possess a greater lamellar thickness than the LTm crystallites, and this higher thickness is one important reason for the higher Tm's. Second, the L-values scatter over a broader range than the Tm's suggesting that still another factor contributes to their perfection. This assumption agrees with the Gibbs–Thomson eqn (1) which correlates the Tm with the properties of a large ideal crystal ( ,
, 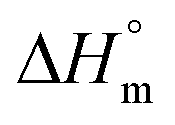 ), with the lamellar thickness (L), the density of the crystalline phase (ρ) and with the “fold surface free energy” (σe).
), with the lamellar thickness (L), the density of the crystalline phase (ρ) and with the “fold surface free energy” (σe).
 | (1) |
At this point the role of the catalyst comes into play. The catalyst can certainly not penetrate the crystallites and therefore, it cannot modify the conformational order or defects inside the crystallites, but it can modify the surface of the crystallites and the neighboring amorphous phase by transesterification. This transesterification processes may have three effects: (a) the homogenization of the loops, (b) a removing of the tie-molecules between different crystallites and between crystallites and amorphous phase and (c) an enhancement of the thickness of the crystallites.
Scheme 5 illustrates a transesterification mechanism which in the case of cyclic polylactides may also be understood as ring-ring equilibration. Transesterification of large loops, tie-molecules and rings in the amorphous phase produces lactide and higher cyclic oligomers that can, in turn, play the role of monomers which support the growth of the lamellar thickness. Fig. 8 illustrates part of this process.
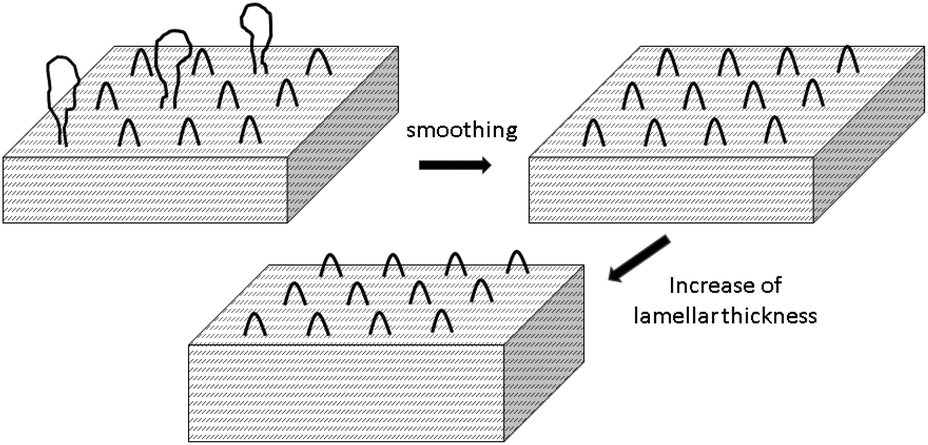 | ||
| Fig. 8 Model of the reactions underlying the formation of HTm crystallites. | ||
A high mobility and transesterification activity of the catalyst along the surfaces of the crystallites is essential for formation of the HTm poly(L-lactide)s. This hypothesis also allows for a hypothetical interpretation of the finding that alcohol initiated ROPs are unfavorable for the formation of HTm polylactides. The OH end groups sticking out from the flat surfaces of the crystalline lamellae can form covalent bonds with the catalysts (known from the polymerization mechanism, Scheme 2), and even when those bonds only have a short live-time, they reduce the mobility of the catalyst along the surface of the lamellae and reduce their effectiveness. Furthermore, this hypothesis and the experimental data presented above allow for an explanation, why the HTm morphology and crystallites have not been discovered before.
The presence of a reactive transesterification catalyst is required, and its concentration should whether be too low nor too high (typically Lac/Cat = 500–1500). Commercial polylactides containing poisoned SnOct2 catalysts cannot form HTm crystallites regardless of time and temperature.
Alcohol-initiated ROPs of L-lactide underlaying almost all studies of physical properties of poly(L-lactide) are unfavorable as synthetic method, and although the presence of a small fraction of linear chains does not hinder formation of HTm poly(L-lactide), only polymerization methods favoring formation of cyclic polylactides yielded HTm crystallites so far. Formation of HTm crystallites occurs preferentially in the temperature range of 130–160 °C. Long reaction times (20–50 h) are needed for an almost complete formation of HTm polylactides. In contrast, the typical reaction times needed for SnOct2-catalyzed polymerizations of L-lactide at temperatures above 130 °C amounts to 1–3 h.
Finally, it should be mentioned that the examination of the virgin reaction products by SAXS revealed gradients of crystallinity and orientation of the stakes of crystallites. However, these phenomena were not only observed for HTm samples but also for the low melting sample no. 1 Table 4 prepared with SnOct2. Therefore, it is clear that this complex aspect of the overall morphology of larger specimens is not a characteristic of the HTm phase, but a consequence of the reaction conditions used in this work.
Optical purity
It is well known for decades that the optical purity has a significant influence on Tm and mechanical properties of pol(L-lactide)s and it is also well known that SnOct2, the catalyst used for the technical production of polylactides, does not cause racemization up to 180 °C for a reaction time of a few hours. Furthermore, it was previously demonstrated in a comparison of 14 different catalytically active metals (in the form of salts or organometal compounds)39 that only tin and bismuth salts enabled racemization-free polymerizations. In our first paper dealing with REP of L-lactide by means of DSTL5 optical rotation measurements were performed which demonstrated that racemization was absent at 160 °C and reaction times of several hours. In this work the polylactides prepared with four other cyclic tin catalysts were characterized by optical rotation measurements: no. 3 and 11, Table 3, no. 4, Table 4 and no. 5 and 6, Table 5. For all samples, molar rotations [α] around −155.0/−156.5° measured under standard conditions (25 °C, Na D-line) were found indicating an optical purity of 99.5% or higher.40 Furthermore, high resolution 13C NMR spectra of five samples have been recorded with a signal-to-noise ratios > 200/1 (no.5B, Table 1, no.1B, Table 3, no.1 and 4, Table 4 and no.5, Table 5), because 13C NMR signals (mainly CO and CH) are sensitive to variations of the stereosequence.35 All NMR spectra were identical, and the signals were perfectly symmetrical as illustrated by a representative example in Fig. S8 in the ESI.† No signals of tetrads based on D–L/L–D sequences were detectable. In summary, the results and interpretations presented in this work are not affected by racemization.Conclusions
On the basis of both DSC and WAXS plus SAXS measurements this work demonstrates that cyclic and linear poly(L-lactide)s can form high-melting crystallites which are different from the standard (LTm) crystallites known from linear poly(L-lactide)s for many decades. These new HTm poly(L-lactide)s are characterized by relatively thick lamellar crystallites having a smooth surface, but it is not based on a new crystal modification. The well-known, thermodynamically stable α-modification was found for all samples subject to WAXS measurements Furthermore, it may be concluded that the standard morphology and crystallites known from linear poly(L-lactide)s are the kinetically favoured phase, whereas the new HTm phase is the thermodynamically more stable alternative. The formation of the new crystallites is favoured by a combination of several optimized experimental parameters, such as a temperature range between 130 and 160 °C, reaction times around or above 20 h, Lac/Cat rations between 500/1 and 1500/1 and presence of a reactive transesterification catalyst typically favouring formation of cyclic polylactides. These catalysts can modify the flat surface of the crystallites, so that homogenized, thermodynamically favourable surfaces are formed. Surprisingly it was found that alcohol initiated ROPs are highly unfavourable for the formation of the new crystallites. Finally, it may be concluded that the polymerization mechanism and, above all, the reactivity of the catalyst are decisive for the formation of the HTm crystallites and not the topology of the polylactides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Federal Institute for Materials Research and Testing for technical support, R. Laging (BAM) for performing numerous SEC and MALDI-TOF measurements and, S. Bleck (TMC, University Hamburg) for DSC measurements.Notes and references
- D. M. Bigg, Annu. Tech. Conf. Soc. Plast. Eng., 1996, 30, 2028–2039 Search PubMed.
- J. R. Dorgan, J. Janzen, M. P. Clayton, S. B. Hait and D. M. Knauss, J. Rheol., 2005, 49, 607–619 CrossRef CAS.
- K. Jamshidi, S. H. Hyon and Y. Ikada, Polymer, 1988, 29, 2229–2234 CrossRef CAS.
- J. J. Kolstad, J. Appl. Polym. Sci., 1996, 62, 1079–1091 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2017, 8, 1589–1596 RSC.
- M. H. Chisholm, J. C. Gallucci and H. F. Yin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15315–15320 CrossRef CAS PubMed.
- D. A. Culkin, W. H. Jeong, S. Csihony, E. D. Gomez, N. R. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS PubMed.
- W. Jeong, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2007, 129, 8414–8415 CrossRef CAS PubMed.
- H. R. Kricheldorf, N. Lomadze and G. Schwarz, Macromolecules, 2008, 41, 7812–7816 CrossRef CAS.
- A. V. Prasad, L. P. Stubbs, M. Zhun and Y. H. Zhu, J. Appl. Polym. Sci., 2012, 123, 1568–1575 CrossRef CAS.
- X. Y. Zhang and R. M. Waymouth, ACS Macro Lett., 2014, 3, 1024–1028 CrossRef CAS.
- E. Piedra-Arroni, C. Ladaviere, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13306–13309 CrossRef CAS PubMed.
- N. Sugai, S. Asai, Y. Tezuka and T. Yamamoto, Polym. Chem., 2015, 6, 3591–3600 RSC.
- N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 105, 158–166 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2017, 218, 1700274 CrossRef.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Eur. Polym. J., 2019, 116, 256–264 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3767–3775 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 952–960 CrossRef CAS.
- P. Desantis and A. J. Kovacs, Biopolymers, 1968, 6, 299–306 CrossRef CAS PubMed.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1915–1925 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 749–759 CrossRef CAS.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2017, 218, 1600331 CrossRef.
- G. Benecke, W. Wagermaier, C. H. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Muller-Buschbaum, M. Trebbin, S. Forster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2017, 218, 1700114 CrossRef.
- H. Kricheldorf, S. M. Weidner and F. Scheliga, Eur. Polym. J., 2019, 116, 256–264 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2019, 119, 37–44 CrossRef CAS.
- B. Kalb and A. J. Pennings, Polymer, 1980, 21, 607–612 CrossRef CAS.
- K. Kishore, R. Vasanthakumari and A. J. Pennings, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 537–542 CrossRef CAS.
- R. Vasanthakumari and A. J. Pennings, Polymer, 1983, 24, 175–178 CrossRef CAS.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Kolloid Z. Z. Polym., 1973, 251, 980–990 CrossRef CAS.
- W. Hoogsteen, A. R. Postema, A. J. Pennings, G. Tenbrinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642 CrossRef CAS.
- H. R. Kricheldorf, C. Boettcher and K. U. Tonnes, Polymer, 1992, 33, 2817–2824 CrossRef CAS.
- B. Lotz, in Synthesis, Structure and Properties of Poly (lactic acid), Springer, 2017, pp. 273–302 Search PubMed.
- T. Y. Cho and G. Strobl, Polymer, 2006, 47, 1036–1043 CrossRef CAS.
- T. Kawai, N. Rahman, G. Matsuba, K. Nishida, T. Kanaya, M. Nakano, H. Okamoto, J. Kawada, A. Usuki, N. Honma, K. Nakajima and M. Matsuda, Macromolecules, 2007, 40, 9463–9469 CrossRef CAS.
- H. R. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497–502 CrossRef CAS.
- F. Chabot, M. Vert, S. Chapelle and P. Granger, Polymer, 1983, 24, 53–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01908a |
| This journal is © The Royal Society of Chemistry 2020 |