
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic covalent polymer networks via combined nitroxide exchange reaction and nitroxide mediated polymerization†
Yixuan
Jia
 a,
Yannick
Matt
bcd,
Qi
An
a,
Isabelle
Wessely
bcd,
Hatice
Mutlu
ef,
Patrick
Theato
ef,
Stefan
Bräse
bcd,
Audrey
Llevot
*g and
Manuel
Tsotsalas
*a
a,
Yannick
Matt
bcd,
Qi
An
a,
Isabelle
Wessely
bcd,
Hatice
Mutlu
ef,
Patrick
Theato
ef,
Stefan
Bräse
bcd,
Audrey
Llevot
*g and
Manuel
Tsotsalas
*a
aInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Hermann-von Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: manuel.tsotsalas@kit.edu
bInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, D-76131 Karlsruhe, Germany
c3DMM2O – Cluster of Excellence (EXC-2082/1-390761711), Karlsruhe Institute of Technology (KIT), D-76131 Karlsruhe, Germany
dInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
eSoft Matter Synthesis Laboratory, Institute of Biological Interfaces III (IBG-3), Karlsruhe Institute of Technology (KIT), Hermann-von Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
fInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesser Str. 18, D-76131 Karlsruhe, Germany
gUniversity of Bordeaux, CNRS, Bordeaux INP, Laboratoire de Chimie des Polymères Organiques (LCPO), UMR 5629, 16 avenue Pey-Berland, F-33607, Pessac cedex, France. E-mail: Audrey.Llevot@enscbp.fr
First published on 3rd March 2020
Abstract
Although the control of chemical composition and macromolecular architectures of polymer networks is crucial to tailor their properties, the control and characterization of the crosslinking density and defects remains challenging. Therefore, new synthetic approaches are needed, which can, on the one hand dynamically tune the network structure and functionalization, and on the other hand facilitate characterization. The present study explores the combination of nitroxide exchange reaction (NER) and nitroxide mediated polymerization (NMP), in different sequences, to prepare structurally tailored and engineered macromolecular (STEM) networks with controlled strand lengths. The radical nature of the NER enables the precise monitoring of the reaction progress and determination of the defect ratio of the networks in a straightforward manner via electron paramagnetic resonance (EPR) spectroscopy. Additionally, the dynamic nature of the NER permits the disassembly of the networks and the determination of the strand length of the prepared networks by size exclusion chromatography (SEC). The final networks are also characterized by inverse size exclusion chromatography (ISEC) to determine and compare their mesh-size distributions. Thus, this study demonstrates that the combination of NER and NMP offers a versatile approach for the preparation of dynamic polymer networks with controlled and tunable structures.
Introduction
Nowadays, polymer networks find widespread applications in diverse areas due to their good mechanical properties and resistance.1–5 These properties are directly related to their structure and composition. However, modulating and characterizing the crosslinking density and reducing defects, such as dangling chain ends and loops, remain a challenge to be mastered by polymer scientists.6–11Traditionally, the topology of a polymer network is static and permanent after synthesis. Consequently, the polymer community showed growing interest in designing “smart” materials that can respond to changes in the environmental conditions, or be modified by post-synthetic functionalization.12,13 Recently, different concepts, such as covalent adaptable networks, structurally tailored and engineered macromolecular (STEM) networks, living additive manufacturing, and macromolecular metamorphosis, have emerged.14–17 All these developments are based on reversible dynamic covalent chemistries. Indeed, dynamic covalent bonds are capable of reversibly breaking and reforming, in the presence of a stimulus or in an autonomous fashion.18–20 Dynamic covalent polymers exhibit a dynamic behavior that enable them to reversibly assemble and disassemble under specific conditions, while remaining mechanically robust under ambient conditions.21–26 The dynamic nature of these processes facilitates the exchange of molecular components at equilibrium to achieve the thermodynamic minimum of the system, leading to highly ordered or even crystalline structures (as in the case of covalent organic frameworks, COFs).27,28 Additionally, the incorporation of reversible covalent bonds into the networks opens the door to a wide variety of post-synthetic modifications.29 The latter is of particular interest to alter the properties of a “parent” network to fulfill the requirements of new applications. With this objective, polymer networks incorporating latent initiator sites have been synthesized. In 2013, Johnson and co-workers reported the expansion of polymer networks incorporating latent trithiocarbonate initiators, which upon exposure to sunlight can trigger an in situ RAFT polymerization.30 This strategy has been further extended as living additive manufacturing to enable the photo-controlled insertion of monomers and crosslinkers into polymer gels.16 The group of Matyjaszewski developed the concept of STEM gels by immobilization of radical photoinitiators, directly available for growing polymer side chains under photoirradiation via free radical polymerization or photo atom transfer radical polymerization (ATRP). Such a post-synthetic modification enabled tuning of the hydrophobicity and mechanical properties of gels with spatio-temporal control.17,31 In this context, utilizing a dynamic covalent bond for network formation, which can also intrinsically initiate the transformation of the networks via the insertion of monomer units after the network formation, would be highly desirable to generate dynamic polymer networks in a straightforward manner. Advantageously, the introduction of reversible covalent bonds as crosslinking points would also provide a thermodynamic control of the crosslinking degree while ensuring the recyclability of the polymer networks. Hence, we propose that these requirements can be fulfilled by the implementation of the nitroxide exchange reaction in a versatile manner.
The exchange reaction between the nitroxides of different alkoxyamine derivatives (i.e. nitroxide exchange reaction (NER)) has been explored for the preparation of reversible and self-healing polymer networks.32–35 In these processes, the thermal C–O bond homolysis of alkoxyamines leads to two types of radicals: a transient carbon-centered radical and a persistent nitroxide radical. When the homolysis of an alkoxyamine is performed in the presence of additional nitroxide radicals, the thermodynamically favored mixed derivatives are obtained. Thus, diols, diepoxides, dialkynes, diacrylates or divinyl monomers incorporating an alkoxyamine unit were synthesized for the preparation of a broad range of polymer networks.36–39 The alkoxyamine divinyl monomer enabled the in situ polymerization of styrene to expand the network structure.40 We recently showed that the nitroxide radical exchange reaction can generate crosslinked dynamic polymer networks directly by stitching together multifold nitroxide and alkoxyamine molecular components of a defined structure. The resulting polymer networks are covalently crosslinked, dynamic in nature, self-healable, have a tunable crosslinking degree and can be easily recycled by modulating the reaction equilibrium.41 Furthermore, alkoxyamine moieties, as well-known initiators for nitroxide mediated polymerization (NMP), can control polymerization based on a thermally reversible capping and uncapping reaction during the polymerization of a monomer.42–46
In this report, we utilize the alkoxyamine bond as the key component for the synthesis of a dynamic polymer network. The alkoxyamine functional group within the network not only intrinsically plays the role of a dynamic bond providing recyclability, but also acts as an NMP initiator enabling network expansion.46 The NER and NMP will be combined in different sequences (Fig. 1): (i) regular nitroxide mediated polymerization of styrene using dialkoxyamine with the subsequent addition of a multitopic nitroxide, (ii) styrene polymerization and network formation using a one-pot approach and (iii) network formation followed by network expansion via the in situ polymerization of styrene. This versatile combination not only enables the preparation of polymer networks with a controlled mesh size (approach (i)) or the post-modification of an existing polymer network (approach (iii)), but also opens doors to a one-pot polymerization, which, to the best of our knowledge, has not been reported for NMP before.
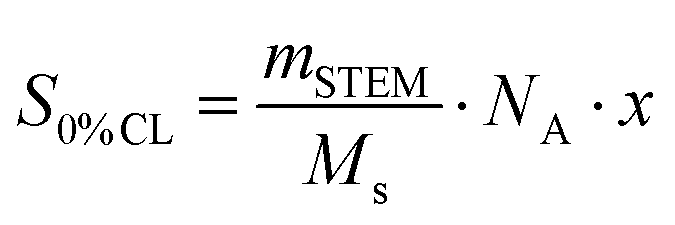 | (1) |
| CLE = 100 − 100·(SSTEM/S0% CL). | (2) |
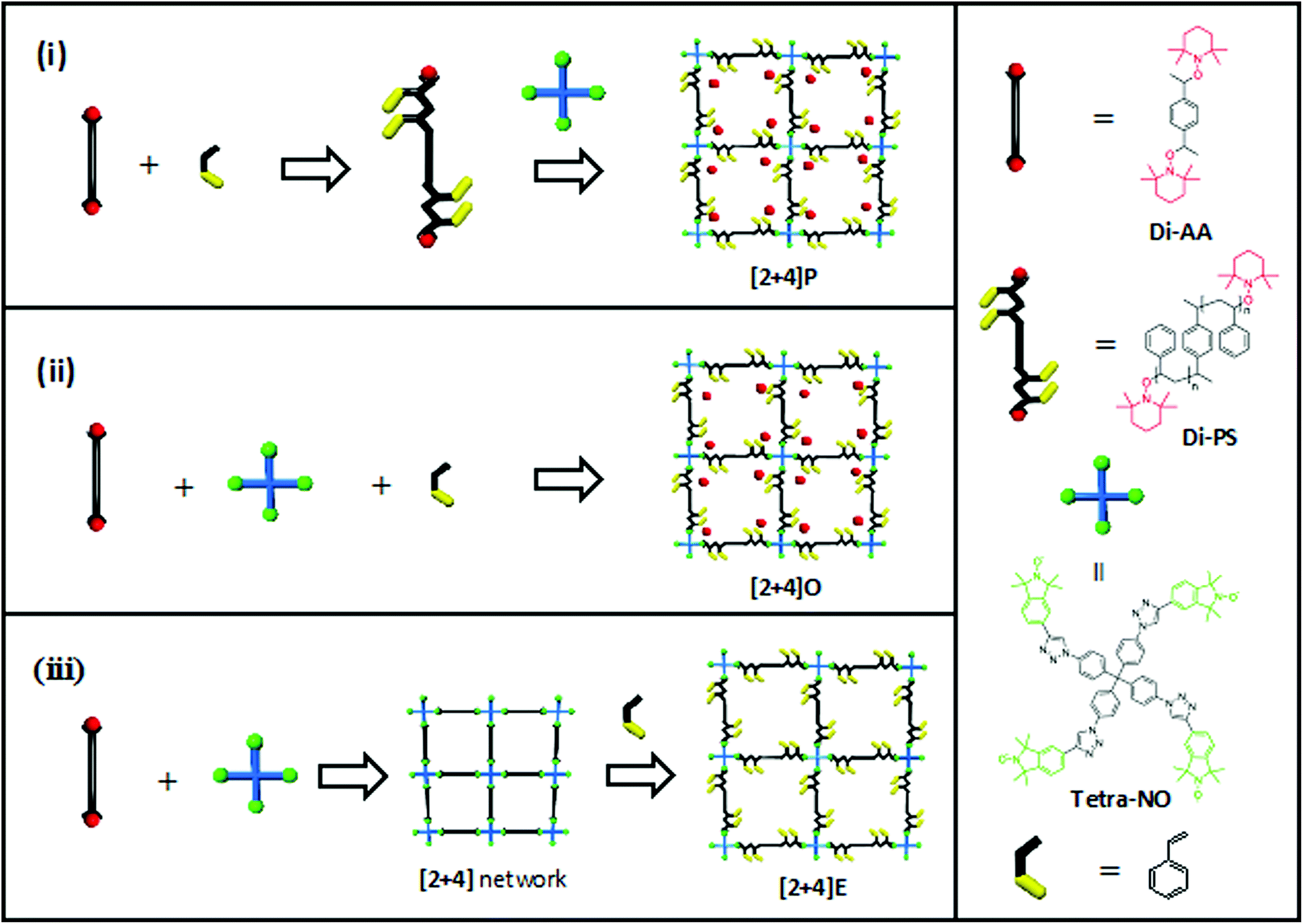 | ||
| Fig. 1 Different approaches for the synthesis of STEM networks. | ||
The efficiency of the different pathways is discussed in detail. Furthermore, the network characteristics were determined using differential scanning calorimetry (DSC), inverse size exclusion chromatography (ISEC), and electron paramagnetic resonance (EPR) spectroscopy. Last but not least, the decrosslinking products were characterized by size exclusion chromatography (SEC).
Results and discussion
Synthesis of the polymer networks
The spin count of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of the starting materials with 0% crosslinking is given as S0%CL, which can be calculated out of the mass mSTEM (10.4 mg) of the measured STEM network, with Ms being the molecular weight of the employed starting materials (Ms = 2·MnDiPS + MTetra-NO, 15742 g mol−1) and NA being the Avogadro constant. x refers to the number of spins per crosslinker (x = 4 in this case). SSTEM represents the EPR spin count of the investigated STEM network (8.8 × 1016). Thus, the calculated high CLE value of 94.5% confirmed the successful synthesis of a highly crosslinked network via NER (see the ESI† for the EPR spectrum). Moreover, it is generally recognized that introducing crosslinking points into a polymer restricts segmental mobility and hence increases the glass transition temperature (Tg).47,48 As a result, the increase of Tg measured by differential scanning calorimetry (DSC) from 83 °C for DiPS1 to 109 °C for [2 + 4]P, respectively, also implies the success of the network formation (Fig. 2a).
:2 mixture of the starting materials with 0% crosslinking is given as S0%CL, which can be calculated out of the mass mSTEM (10.4 mg) of the measured STEM network, with Ms being the molecular weight of the employed starting materials (Ms = 2·MnDiPS + MTetra-NO, 15742 g mol−1) and NA being the Avogadro constant. x refers to the number of spins per crosslinker (x = 4 in this case). SSTEM represents the EPR spin count of the investigated STEM network (8.8 × 1016). Thus, the calculated high CLE value of 94.5% confirmed the successful synthesis of a highly crosslinked network via NER (see the ESI† for the EPR spectrum). Moreover, it is generally recognized that introducing crosslinking points into a polymer restricts segmental mobility and hence increases the glass transition temperature (Tg).47,48 As a result, the increase of Tg measured by differential scanning calorimetry (DSC) from 83 °C for DiPS1 to 109 °C for [2 + 4]P, respectively, also implies the success of the network formation (Fig. 2a).
 | ||
| Fig. 2 (a) DSC thermograms of the second heating cycle of the prepolymer DiPS1 and the [2 + 4]P network. (b) EPR spectra of the washing solution from the crude product in comparison with the fit. The reference spectra of Tetra-NO and TEMPO, which had a hyperfine coupling constant of 14.1 G and 15.5 G, respectively, were utilized for the fit. (c) Dissolution of the crosslinked network from the 2 + 4-combination via the addition of TEMPO in excess. (d) Comparison of SEC results of the decrosslinked [2 + 4]P, and the prepolymer DiPS1. | ||
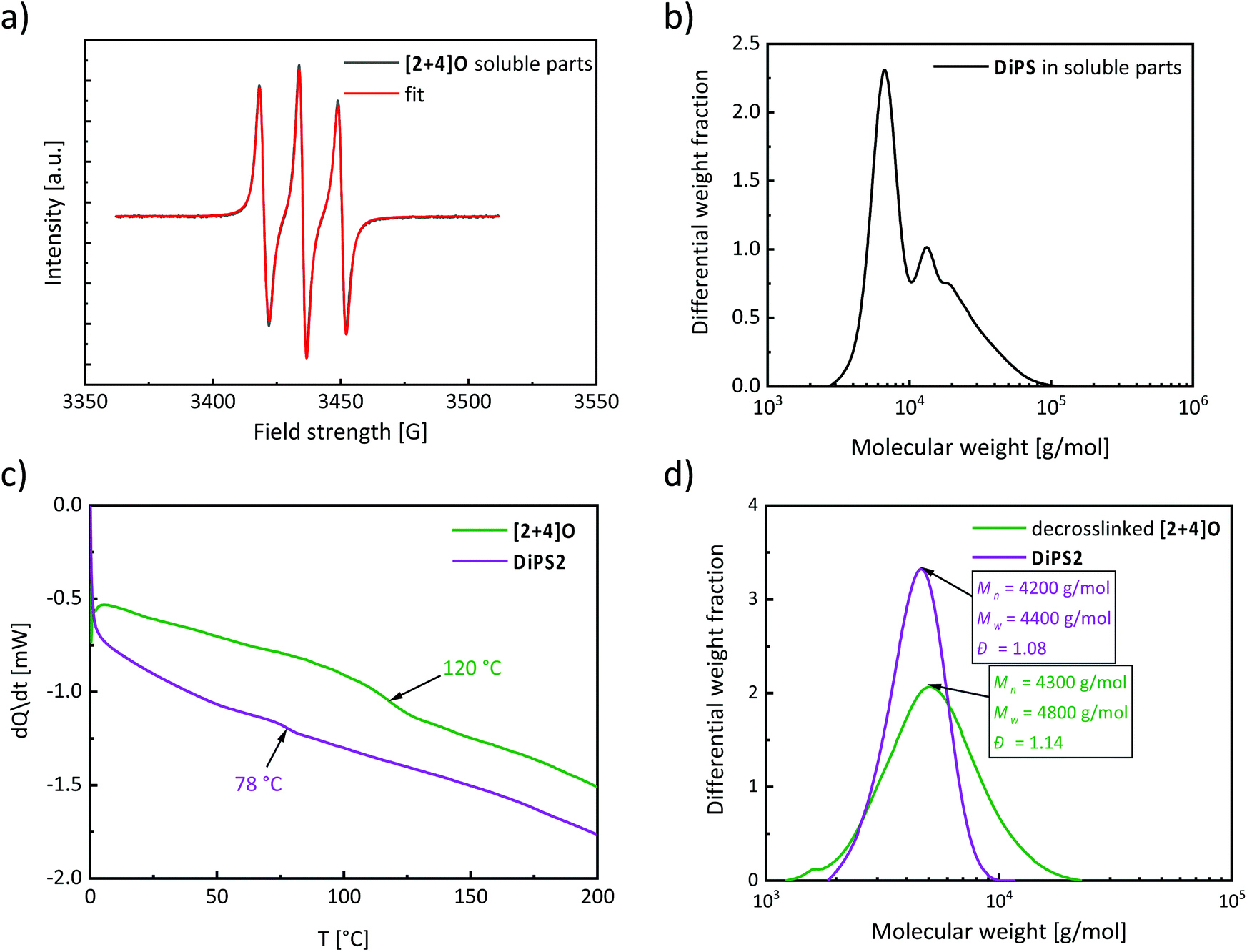
Besides the insoluble network, the ratio of free TEMPO (by-product of the network formation) to free isoindoline nitroxide in the washing solution was investigated by EPR measurements to provide a straightforward measure of the exchange rate. The fit of the EPR spectrum of the soluble part (Fig. 2b) with the reference spectra of Tetra-NO and TEMPO indicated a nitroxide exchange rate of 80%. Subsequently, the washing solution was precipitated in MeOH to recover unreacted DiPS1. A mass corresponding to 43% of the initial weight was gravimetrically calculated, indicating that 57% of DiPS1 reacted during crosslinking to form the network, which is concordant with the yield of 56% for the network formation.
With regard to the possibility to decompose the network e.g. by heating under ambient conditions for several days,41 the reversible character of the nitroxide exchange reaction also allows the recovery of the original components of the [2 + 4]P network by adding an excess amount of monomeric TEMPO nitroxide radicals (Fig. 2c) and heating in THF. The resulting solution contained the crosslinker Tetra-NO and polystyrene (called decrosslinked [2 + 4]P). After the precipitation of the decrosslinked [2 + 4]P in MeOH, the SEC analysis revealed a dispersity of Đ = 1.04, and a Mn of 7300 g mol−1, which is in excellent agreement with the values of the parent polymer DiPS1 (Mn = 7200 g mol−1 and Đ = 1.09) (Fig. 2d). This result proves the suitability of the “decrosslinking” method to recover the starting materials and to determine the molecular weight of the polymer strands in the networks that have been crosslinked via alkoxyamine moieties.
The EPR analysis of the washing solution revealed an exchange ratio of 70%, indicating a high nitroxide exchange rate (Fig. 3a). The washing solution was also precipitated in MeOH to recover any linear or hyperbranched polystyrene, which was not incorporated into the network. The mass of the precipitate corresponded to 52% of the initial weight of styrene. Since the yield of the [2 + 4]O network was 37%, around 10% of styrene did not react, or formed oligomers of low molecular weight that did not precipitate in MeOH. The SEC chromatogram of the precipitate showed a bimodal distribution, with one peak centered at low molecular weight, e.g. 6400 g mol−1, and the other one at higher molecular weight, i.e. 17000 g mol−1 (Fig. 3b).
 | ||
| Fig. 3 (a) EPR spectra of the washing solution from the crude product in comparison with the fit. The reference spectra of Tetra-NO and TEMPO, which had a hyperfine coupling constant of 14.1 G and 15.5 G, respectively, were utilized for the fit. (b) SEC results of the soluble parts. (c) DSC thermogram of [2 + 4]O in comparison with that of the reference DiPS2. (d) SEC results of the decrosslinked [2 + 4]O and the reference DiPS2. | ||
After the disassembly of the [2 + 4]O network in the presence of an excess of TEMPO, a strand length of 4300 g mol−1 with a dispersity of Đ = 1.14 was measured by SEC (Fig. 3c: decrosslinked [2 + 4]O). The low value of the dispersity indicates a good control of the polymerization. The molecular weight of the decrosslinked [2 + 4]O is two times lower than that of the reference DiPS1, which was synthesized in solution under identical conditions (e.g. temperature and reaction time) in the absence of a crosslinker. This difference can be explained by the higher nitroxide concentration (i.e. the additional presence of Tetra-NO) during the synthesis of [2 + 4]O, which leads to a decrease in the reaction kinetics, and to a reduced degree of freedom, as the monomer can diffuse only after the alkoxyamine is immobilized in the polymer network.
The glass transition temperature Tg of the [2 + 4]O network was determined by DSC at 120 °C. For comparison, an additional linear polystyrene DiPS2, exhibiting a Mn of 4200 g mol−1 that is similar to the polymer network strand length (i.e. 4300 g mol−1), was synthesized and characterized by DSC. The higher Tg of the [2 + 4]O network compared to the linear reference (i.e. Tg of 78 °C) further confirms the success of the crosslinking reaction (Fig. 3d).
 | ||
| Fig. 4 (a) DSC thermogram of the [2 + 4] network before extension, the [2 + 4]E network after extension and the reference DiPS1. (b) SEC results of the decrosslinked [2 + 4]E and the reference polymer DiPS1. | ||
Since TEMPO has been already removed in the NER reaction step, the washing solution of the network was EPR silent (see the ESI†). The precipitation of the MeOH washing solution of the [2 + 4]E-network resulted in a solid with 41% of the initial weight, meaning that maximum 59% of styrene was incorporated into the polymer (for SEC data of the precipitate, see the ESI†). These values are in good agreement with the 52% yield of the [2 + 4]E-network. The presence of polystyrene in the soluble part could be attributed to a combination of three phenomena: (i) a chain transfer (to a monomer or a solvent) during NMP, leading to the free radical polymerization of styrene outside the network, (ii) the formation of loops once dynamic equilibrium of the NER is reached, which can result in the dissociation of fragments from the polymer network, (iii) a disproportionation (“H-transfer”) side reaction, which causes the decomposition of the alkoxyamine chain ends into hydroxylamine and a dead polystyrene chain, and thus, a potential disconnection from the network.49,50
Following the same dissolution procedure as for [2 + 4]P, by adding TEMPO in excess, the [2 + 4]E network revealed individual strands with a Mn of 7200 g mol−1 and a dispersity of Đ = 1.54 (Fig. 4b). The molecular weight is in the range of the reference prepolymer DiPS1, synthesized using a similar alkoxyamine/styrene ratio. The slightly higher dispersity compared to the other two STEM networks could be explained by two aspects. On the one hand, the heterogeneous dispersion of the monomer in the network (diffusion issues) leads to different amounts of monomer insertion close to the surface as compared to inside of the swollen framework. On the other hand, the efficiency of NMP (in regard of the polymerization rate and control over dispersity) critically depends on the homolysis rate of the alkoxyamine initiator C–O bond.46,51 In the network extension approach, the alkoxyamine that initiates the NMP exhibits an isoindoline and not a piperidine structure. Although the slightly higher stability of the isoindoline alkoxyamine compared to that of the TEMPO alkoxyamine is an advantage for the nitroxide exchange reaction,7 the lower dissociation constant of the C–O bond compared to that of TEMPO results in a lower performance as the NMP initiator, which might also explain the higher dispersity of the polymer obtained by the extension approach.52,53
Moreover, the dynamic behavior of the networks synthesized from the first two approaches ([2 + 4]P and [2 + 4]O) was confirmed by performing an additional network extension step. The SEC data of the dissolved networks [2 + 4]PE and [2 + 4]OE, after further extension, revealed a significant increase from 7200 g mol−1 to 79700 g mol−1 for [2 + 4]P and from 4300 g mol−1 to 444900 g mol−1 for [2 + 4]O, thus highlighting the possibility to post-functionalize the networks by in situ polymerization (for more details, see the ESI†).
Characteristics of the polymer networks
As demonstrated above, three different routes enabled the synthesis of dynamic polymer networks by combination of NMP and NER. In this section, a deeper characterization of the networks is presented to determine their key characteristics, such as the glass transition temperature, swelling degree and the mesh size distribution.The mesh size of the synthesized STEM networks was determined by size-exclusion chromatography in the inverse mode (i.e. ISEC). In this technique, the different STEM networks serve as the stationary phase and a range of well-defined polystyrene standards (13 standards) with known number-average molecular weights were eluted in THF (Fig. 5). The resulting standard elution volumes were plotted against the PS standard molecular weights (Fig. 6a). This plot enabled the calculation of the mesh sizes of the synthesized STEM networks (Fig. 6b and the ESI†) using the slit-like pore model of the software PSS Porocheck version 2.5. Accordingly, the [2 + 4]P network exhibited a mesh size radius, Pms, of 7.6 ± 1.7 nm, [2 + 4]O of 4.2 ± 1.1 nm and [2 + 4]E of 9.8 ± 2.3 nm, respectively (Fig. 7a). These results were compared with the molecular weights of the strands determined by depolymerization. With the lowest Mn of 4300 g mol−1 after depolymerization, [2 + 4]O showed the smallest mesh size.
 | ||
| Fig. 5 Schematic illustration of the stationary phase ([2 + 4]-STEM networks) in the SEC column. The small molecules can permeate the network and stay there for a corresponding time period, while the molecules that are larger than the mesh size of the network flow past and elute first. | ||
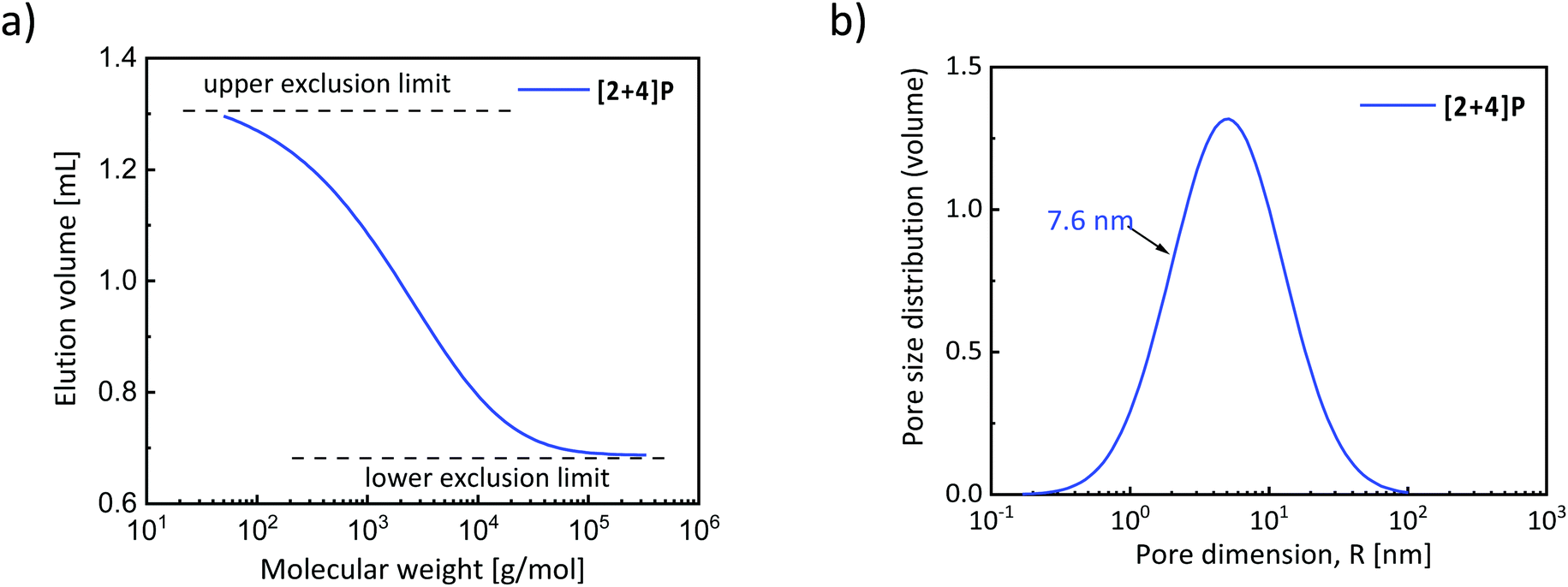 | ||
| Fig. 6 (a) ISEC plot obtained using [2 + 4]P as the stationary phase; the elution volume of the PS standards is plotted against their molecular weights; a curve with a single distinct exclusion limit is characteristic of a monomodal mesh size distribution of the materials of the stationary phase. (b) The monomodal mesh size distribution of [2 + 4]P obtained from the plot of (a), by using the slip pore model of the software PSS Porocheck version 2.5. | ||
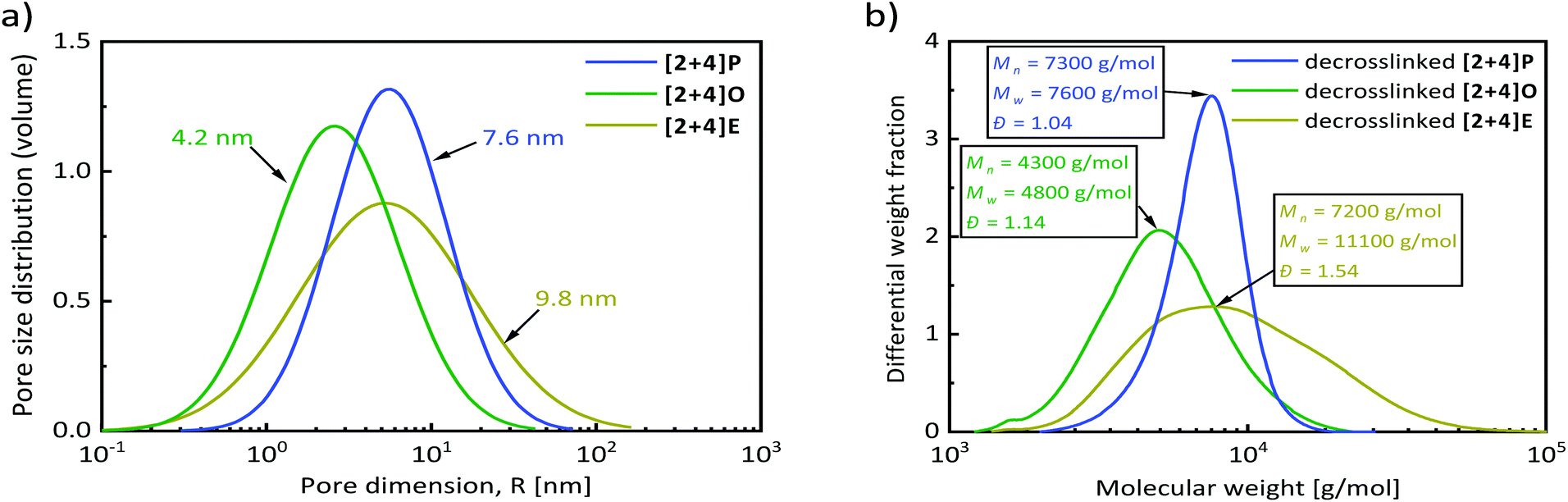 | ||
| Fig. 7 (a) Mesh size distribution of three different [2 + 4]-STEM networks determined from ISEC investigations. (b) SEC results of the decrosslinked polystyrene from different [2 + 4]-STEM networks. | ||
[2 + 4]P and [2 + 4]E showed a similar Mn of around 7200 g mol−1, but [2 + 4]E exhibited a higher weight average molecular weight, Mw of 11100 g mol−1versus 7800 g mol−1 for [2 + 4]P. The Mw is particularly sensitive to the presence of higher molecular weight molecules, whereas the number average is very sensitive to the presence of lower molecular weight molecules. With a similar Mn but a higher Mw, the depolymerized [2 + 4]E had more chains with a higher molecular weight than [2 + 4]P. As a result, the mesh size calculated from ISEC analyses was higher (Fig. 7b).
Moreover, the mesh sizes of the synthesized networks, determined by ISEC, were in good agreement with their swelling ability (see Table 1 for a summary of the essential parameters of the STEM networks). The lowest swelling degree was attributed to the polymer network with the shortest mesh size (1790%), [2 + 4]O, while the swelling degrees and mesh sizes of [2 + 4]P (3940%) and [2 + 4]E (3530%) fall in a similar range. Fox and Loshaek, and others, have developed several models showing that in crosslinked polymers, Tg is a function of the crosslinking density and thus the mesh size.54 For most of the studied systems, including polystyrene/divinylbenzene, the Tg increased with the crosslinking density.55 The Tg of the polymers developed in this study followed this trend. Indeed, the network with the shortest mesh size (highest crosslinking density), [2 + 4]O, exhibited the highest Tg of 120 °C, due to a restricted chain mobility, while [2 + 4]P and [2 + 4]E had a longer mesh size and accordingly slightly lower Tg of 109 °C and 108 °C, respectively.
| After depolymerization (strand) | STEM networks | |||||
|---|---|---|---|---|---|---|
|
Mna [g mol−1] |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) wa [g mol−1]
wa [g mol−1] |
Đ | Swelling degreeb [%] | Mesh size radius Pmsc [nm] |
T
gd [°C] |
|
| a Determined by SEC relative to polystyrene standards in THF as the eluent. b Calculated after 5 days of swelling in THF. c Determined by ISEC. d Determined by DSC with a heating/cooling rate of 5 K min−1 from −50 °C to 200 °C. | ||||||
| [2 + 4]P | 7200 | 7800 | 1.04 | 3940 | 7.6 ± 1.7 | 109 |
| [2 + 4]O | 4300 | 4800 | 1.14 | 1790 | 4.2 ± 1.1 | 120 |
| [2 + 4]E | 7200 | 11100 |
1.54 | 3530 | 9.8 ± 2.3 | 108 |
Conclusions
In this article, we describe the synthesis of dynamic polymer networks with a controlled network structure and molecular composition via the combination of the nitroxide exchange reaction with the nitroxide mediated polymerization. The dynamic equilibrium in the nitroxide exchange reaction allows tuning the crosslinking degree and the complete dissolution of the STEM networks, which is highly attractive for recycling and characterization of the network structure. The possibility to disassemble the network at the crosslinking points greatly facilitates the characterization of the networks (e.g. determining the strand length by SEC), which is usually a great challenge in polymer network formation. Together with the intrinsic possibility to determine the defect density in the networks by EPR spectroscopy, the networks prepared by the combination of NER and NMP provide full and facile characterization. Furthermore, the described system allows for modulating the network properties, via NMP, initially by the choice of the chemical structure of the monomer or after synthesis by post-modification (polymerization in situ), or via variation of the crosslinking degree by modulating the equilibrium in the NER, thereby creating dynamic and post-functionalizable polymer networks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dominique Moock and Birgit Huber are gratefully acknowledged for their support in the analysis of the polymer networks. We acknowledge the SFB 1176 funded by the German Research Council (DFG) in the context of project C5.References
- J.-P. Pascault and R. J. J. Williams, in Thermosets, Elsevier, 2017 Search PubMed.
- P. J. Flory, Chem. Rev., 1944, 35, 51–75 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef.
- I. Wessely, V. Mugnaini, A. Bihlmeier, G. Jeschke, S. Bräse and M. Tsotsalas, RSC Adv., 2016, 6, 55715–55719 RSC.
- D. Estupiñán, C. Barner-Kowollik and L. Barner, Angew. Chem., Int. Ed., 2018, 57, 5925–5929 CrossRef PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- M. A. Gauthier, M. I. Gibson and H. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. J. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS PubMed.
- A. Beziau, A. Fortney, L. Fu, C. Nishiura, H. Wang, J. Cuthbert, E. Gottlieb, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Polymer, 2017, 126, 224–230 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 1585 CrossRef PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- J. Cuthbert, T. Zhang, S. Biswas, M. Olszewski, S. Shanmugam, T. Fu, E. Gottlieb, T. Kowalewski, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2018, 51, 9184–9191 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- J. Cuthbert, A. Beziau, E. Gottlieb, L. Fu, R. Yuan, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2018, 51, 3808–3817 CrossRef CAS.
- H. Otsuka, Polym. J., 2013, 45, 879 CrossRef CAS.
- C. e. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- F. Wang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2012, 22, 13076–13084 RSC.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 3936–3943 CrossRef CAS.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- S. Telitel, Y. Amamoto, J. Poly, F. Morlet-Savary, O. Soppera, J. Lalevée and K. Matyjaszewski, Polym. Chem., 2014, 5, 921–930 RSC.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2009, 42, 8733–8738 CrossRef CAS.
- Q. An, I. D. Wessely, Y. Matt, Z. Hassan, S. Bräse and M. Tsotsalas, Polym. Chem., 2019, 10, 672–678 RSC.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- C. J. Hawker, J. Am. Chem. Soc., 1994, 116, 11185–11186 CrossRef CAS.
- C. J. Hawker, G. G. Barclay, A. Orellana, J. Dao and W. Devonport, Macromolecules, 1996, 29, 5245–5254 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- K. Ueberreiter and G. Kanig, J. Chem. Phys., 1950, 18, 399–406 CrossRef CAS.
- S. Loshaek, J. Polym. Sci., 1955, 15, 391–404 CrossRef CAS.
- G. Gryn'ova, C. Y. Lin and M. L. Coote, Polym. Chem., 2013, 4, 3744–3754 RSC.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- E. Drockenmuller and J.-M. Catala, Macromolecules, 2002, 35, 2461–2466 CrossRef CAS.
- J. P. Blinco, K. E. Fairfull-Smith, A. S. Micallef and S. E. Bottle, Polym. Chem., 2010, 1, 1009–1012 RSC.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- H. Stutz, K. Illers and J. Mertes, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1483–1498 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic and characterization details (EPR, SEC, ISEC, DSC, NMR, ATR-FTIR, and EI-MS). See DOI: 10.1039/c9py01878f |
| This journal is © The Royal Society of Chemistry 2020 |