
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers from sugars and unsaturated fatty acids: ADMET polymerisation of monomers derived from D-xylose, D-mannose and castor oil†
Marco
Piccini
 a,
David J.
Leak
b,
Christopher J.
Chuck
c and
Antoine
Buchard
*a
a,
David J.
Leak
b,
Christopher J.
Chuck
c and
Antoine
Buchard
*a
aCentre for Sustainable and Circular Technologies, Department of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bDepartment of Biology and Biochemistry, University of Bath, Bath BA2 7AY, UK
cDepartment of Chemical Engineering, University of Bath, Bath BA2 7AY, UK
First published on 13th March 2020
Abstract
α,ω-Unsaturated glycolipids derived from natural monosaccharides D-xylose and D-mannose, and from a castor oil derivative, 10-undecenoic acid, were synthesised and polymerised via acyclic diene metathesis (ADMET), using Grubbs second generation catalyst. The synthesis of these polymers, which combine a rigid isopropylidene-functionalised carbohydrate core with flexible unsaturated aliphatic chains, was confirmed by NMR spectroscopy and size exclusion chromatography (SEC). The effect of different parameters on the polymerisations were investigated, including temperature, catalyst loading, absence/presence of solvents, effect of a molecular weight moderator and mixing technique. Polyesters with high molecular weights (up to 71 kg mol−1) could be obtained in elevated yields (85%). These amorphous polymers were highly thermally (up to 355 °C) and hydrolytically (pH 7, 0, 14) stable, and showed relatively low glass transition temperatures (−28 to −8 °C), imparted by the flexible fatty acid chain. Deprotection of ketal groups on the polymer backbone was possible up to 72% and changed the material properties, leading to partial crystallinity and insolubility. These partially deprotected polymers allowed the production of transparent thin polymer films and were amenable to further functionalisation.
Introduction
The widespread and misguided use of oil-derived plastics is known to be causing a number of problems, namely environmental pollution, greenhouse gas emission and fossil resources depletion. To tackle these challenges, one solution may be the development of renewable (biobased) polymers with targeted chemical and/or biological degradability.To date, polymerisation of monomers derived from plant oils,1,2 terpenes3 and lignin4 has been reported extensively. However, for a large-scale replacement of fossil-based plastics, carbohydrates are regarded as the most promising feedstock,5 primarily due to their availability compared to other biomolecules, including from non-edible and waste sources. Sugars also display extensive structural and stereochemical diversity, making them perfect candidates for the synthesis of a variety of polymers. Moreover, their hydrophilicity and hydrolytic degradability, together with their absence of toxicity, make sugars a highly promising feedstock for the synthesis of environmentally benign polymers.
However, starch and sucrose, the main commercial carbohydrate sources, are classified as first generation feedstocks (i.e. sourced from crop plants), and cannot be truly considered sustainable if they compete with food use. Alternatively, the utilisation of lignocellulosic biomass, for example from agricultural waste, could prevent many of the issues associated with first generation feedstocks. Cellulose and lignin, two of the three main constituents of lignocellulosic materials, are promising source of chemicals and monomers, even if their depolymerisation (especially for lignin) may present significant challenges for a large-scale industrial process. Hemicellulose, the second most abundant biopolymer (20–30 wt% of lignocellulosic biomass), is conversely easily hydrolysed by acids, bases and enzymes, thanks to its amorphous random structure and its moderate molecular weight (75–450 kg mol−1), and is a promising sustainable source of monosaccharides such as D-xylose or D-mannose.
Significant research effort has focused on transforming monosaccharides into a variety of monomers and polymers through a multitude of chemical and biochemical techniques, the main ones being fermentation (e.g. polylactide via lactic acid,6 polyhydroxyalkanoates7), oxidation/reduction (mainly for acetalised alditols and isosorbide-based polymers),8–10 and dehydration (furan-based materials).11,12 Conversely, the incorporation of unmodified sugar cores (with or without the use of functional or protecting groups) into the main polymer chains (as opposed to glycopolymers, in which sugars are pendant groups to the main chain), has been reported in a limited number of studies. Among these, ring opening polymerisation (ROP) has been most frequently exploited.13 ROP of cyclic carbonates from monosaccharides has been pioneered by Gross and co-workers,14–17 and studied more in depth by the Wooley group.18–21 We also recently reported a series of studies on the ROP of cyclic carbonates synthesised from sugars and CO2.22–24 Other significant examples of sugar-based monomers polymerised via ROP are phosphodiesters25,26 and β-lactams.27–31
Beyond ROP and transesterification techniques, acyclic diene metathesis (ADMET) polymerisation is also a method of choice for the polymerisation of renewable monomers, including those derived from plant oils.2,32 In 2009, Meier and co-workers reported the first study of a sugar-based monomer polymerised via ADMET.33 In this work, a fully bio-based monomer was synthesised by esterification of isosorbide with 10-undecenoic acid. The latter is also a renewable building block, being derived from castor oil, and has been exploited in metathesis reactions thanks to its terminal olefin moiety.32 The resulting α,ω-diene, isosorbide diundecenoate, was polymerised in the melt using Grubbs first- and second-generation catalysts, albeit with moderate molecular weights (9–26 kg mol−1). A more detailed study of poly(isosorbide diundecenoate) was reported in 2015 by Reineke and co-workers.34 Together with isosorbide, the structural analogue glucarodilactone (GDL) was used as building block for the synthesis of the corresponding monomer and polymers. Moreover, the two different monomers were copolymerised in different ratios, yielding random copolymers. ADMET polymerisation, performed in toluene under vacuum using Grubbs second generation catalyst, yielded high molecular weight polymers (51–61 kg mol−1). GDL units were shown to impart enhanced thermal properties to the resulting polymers, while isosorbide units increased the polymer crystallinity making them more thermally and hydrolytically stable. While both homopolymers were brittle, copolymers were rubbery materials displaying shape memory effects. In a successive study, a number of copolymers with different GDL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isosorbide ratios were synthesised, and the effect on the polymer properties (thermal and hydrolytic stability, elastic behaviour) were assessed. Additionally, a new monomer was synthesised from 2,5-bis(hydroxymethyl)furan and incorporated in copolymers.35 A different study, published the same year by Cramail and coworkers, employed trehalose (a glucose dimer) for the synthesis of undecenoyl diesters.36 The esterification with vinyl undecenoate using immobilised Candida Antarctica Lipase B was performed selectively on primary hydroxyl groups, avoiding the need to protect secondary hydroxyls. Using Hoveyda–Grubbs second generation catalyst in THF under N2 atmosphere, ADMET homo- and copolymerisation with undecenoyl undecenoate was performed. Medium to low molecular weight polymers were obtained (2–13 kg mol−1). Homo- and copolymers exhibited semicrystalline behaviour; although no glass transition temperature was detected, two distinct melting points were observed in copolymers for the saccharide and lipid components. Polymers were also shown to self-assemble into micellar nanoparticles in water thanks to their amphiphilic nature. Besides ADMET, ring opening metathesis polymerisation (ROMP) was also reported for the polymerisation of lactonic sophorolipids37–40 and levoglucosenol.41
:isosorbide ratios were synthesised, and the effect on the polymer properties (thermal and hydrolytic stability, elastic behaviour) were assessed. Additionally, a new monomer was synthesised from 2,5-bis(hydroxymethyl)furan and incorporated in copolymers.35 A different study, published the same year by Cramail and coworkers, employed trehalose (a glucose dimer) for the synthesis of undecenoyl diesters.36 The esterification with vinyl undecenoate using immobilised Candida Antarctica Lipase B was performed selectively on primary hydroxyl groups, avoiding the need to protect secondary hydroxyls. Using Hoveyda–Grubbs second generation catalyst in THF under N2 atmosphere, ADMET homo- and copolymerisation with undecenoyl undecenoate was performed. Medium to low molecular weight polymers were obtained (2–13 kg mol−1). Homo- and copolymers exhibited semicrystalline behaviour; although no glass transition temperature was detected, two distinct melting points were observed in copolymers for the saccharide and lipid components. Polymers were also shown to self-assemble into micellar nanoparticles in water thanks to their amphiphilic nature. Besides ADMET, ring opening metathesis polymerisation (ROMP) was also reported for the polymerisation of lactonic sophorolipids37–40 and levoglucosenol.41
Inspired by these previous works, we herein exploit ADMET polymerisation for the synthesis of renewable polymers incorporating both monosaccharides and fatty acid moieties in the main polymer chain. Previously mentioned 10-undecenoic acid was employed as it is known to confer flexibility to polymer backbones – in contrast with previously reported purely sugar-based polymers, characterised by elevated glass transition temperatures (Tg) but brittle behaviour.22–24D-Xylose and D-mannose were selected as substrates as they are relatively underutilised and can be easily obtained from abundant hemicellulose, which could be in turn derived from waste sources. The structural and stereochemical complexity of the original sugars was preserved in the resulting polymers and its effects on properties studied. Moreover, the polyfunctionality of sugar derivatives was exploited for post-polymerisation modification and functionalisation.
Results and discussion
Synthesis of monomers
α,ω-Dienes 1 and 2 were obtained through double esterification of 10-undecenoic anhydride with xylose- and mannose-derived diols respectively (Scheme 1). | ||
| Scheme 1 Synthesis of the α,ω-unsaturated glycolipid monomers 1 and 2. | ||
Protection of some of the hydroxyl groups of the natural monosaccharides was necessary to selectively obtain diesters, and these diols could be obtained either commercially (in the case of 1,2-O-isopropylidene-α-D-xylofuranose), or through acetalisation of commercial methyl α-D-mannopyranoside.22
10-Undecenoic anhydride was synthesised in a typical procedure by reaction of 10-undecenoic acid with acetic anhydride under reflux, and obtained in quantitative yield with no purification required.35 Esterification of both sugar diols catalysed by scandium(III) triflate (reported for the esterification of secondary alcohols with anhydrides),42 showed no reactants conversion. The combination of triethylamine (NEt3; stoichiometric amount) and 4-dimethylaminopyridine (DMAP; catalytic), known to promote the reactivity of deactivated alcohols in esterification reactions with anhydrides,43 was therefore applied and diesters 1 and 2 were obtained quantitatively after 20 minutes at room temperature in dichloromethane (DCM; which would have to be replaced for any large scale implementation due to toxicity concerns) (Scheme 1). 10-Undecenoic acid side-product was removed from the reaction mixture by adsorption on anionic exchange resin Amberlyst A26 OH form, allowing its potential recovery and reuse. The two glycolipid monomers 1 (from xylose) and 2 (from mannose) were obtained in elevated yields (82% and 86%, respectively). Interestingly, 13C NMR spectra of both monomers showed two distinct peaks for the carbonyl carbons (172.4 and 173.3 ppm for 1, 172.5 and 173.4 ppm for 2; see Fig. S2 and S5, ESI†), further confirming that 1 and 2 are diesters in which the ester moieties are located on chemically different (i.e. primary and secondary) hydroxyl groups of the sugars. It is worth emphasising that both monomers are predominantly obtained from renewable feedstocks (91.9 and 89.9 wt% of biobased content for 1 and 2, respectively); furthermore, the potential sourcing of acetone (diol protecting group) and methanol (anomeric hydroxyl protecting group in 2) from biomass (i.e. via fermentation) would lead to 100% biobased monomers.
Polymerisations
Among the various catalysts commercially available for ADMET polymerisation, Grubbs second generation catalyst (G-II; a ruthenium alkylidene complex), was selected because of its high stability to air and moisture, elevated tolerance to polar functional groups and high activity. Polymerisations were carried out under dynamic vacuum (0.1 mbar) in order to remove the ethylene formed as by-product in the metathesis reaction (which would be captured and used in any large scale implementation) and drive the equilibrium towards the formation of the polymers. Bulk polymerisation was preferred over solution polymerisation as it provides maximum monomer concentration, key for achieving high conversion and elevated molecular weights, and avoid solvent losses caused by reduced pressure.Based on shielding arguments, the pattern of this new signal, composed of a major signal at 5.30 ppm and a minor one at 5.25 ppm, suggested that the configuration of the new alkene is predominantly trans (ca. 75% for all polymer samples from relative integration), as expected from thermodynamic stability. This evidence was confirmed by similarly doubled NMR signals for allylic protons (1.90 ppm for trans, 1.95 ppm for cis) and carbons (32.5 and 27.2 ppm, respectively; Fig. S8†).
While monomer 1 is unsymmetric, no head-tail enchainment or variations thereof could be detected experimentally. However, the polymerisable moieties (alkene groups) being more than ten atoms apart from the xylose unit, no regioregularity of the polymer was expected. In addition, while this has not been thoroughly investigated in this study, as previously reported by Fokou and Meier,33 and as suggested here by the presence of several alkene signals in the 13C NMR spectra for all polymers, internal olefin isomerisation is likely to occur during ADMET polymerisation, in particular under the conditions used here (high temperature, G-II catalyst and low catalyst loading).
Polymers were purified by precipitation in cold methanol and isolated as brown materials. Molecular weight distributions were determined by size exclusion chromatography (SEC) in tetrahydrofuran (THF) using a refracting index (RI) detector calibrated with polystyrene standards. As observed by other authors for analogue polymers, Mn values thus obtained are likely to be underestimated.33–36 Molecular weight dispersities (ĐM) of 1.7–2.4, reflecting the theoretical value expected for polycondensation polymerisations, were obtained in all experiments (except for very low molecular weight polymers).
The effect of different parameters was assessed for the ADMET polymerisation of the xylose-derived monomer 1 (Scheme 2). The first set of experiments was carried out in bulk with 4.0 mol% of catalyst for 20 hours at three different temperatures (Table 1, entries 1–3). The polymerisation conducted at room temperature led to incomplete conversion and minimal yield, with formation of a significant amount of oligomers, soluble in methanol (entry 1). When the reaction was repeated at 60 °C (entry 2), monomer conversion was quantitative. A polymer with 64% yield and moderate molecular weight (Mn 14.4 kg mol−1, ĐM 2.0) was obtained, close to the theoretical value of 12.8 kg mol−1 obtained from Carothers equation when considering the catalyst as an additional monofunctional monomer. A further increase in temperature (90 °C; entry 3) did not have any influence, except for a limited increase in yield and a slightly broader dispersity. The reaction conducted at 60 °C was repeated in order to assess reproducibility, and consistent results were obtained (entry 4).
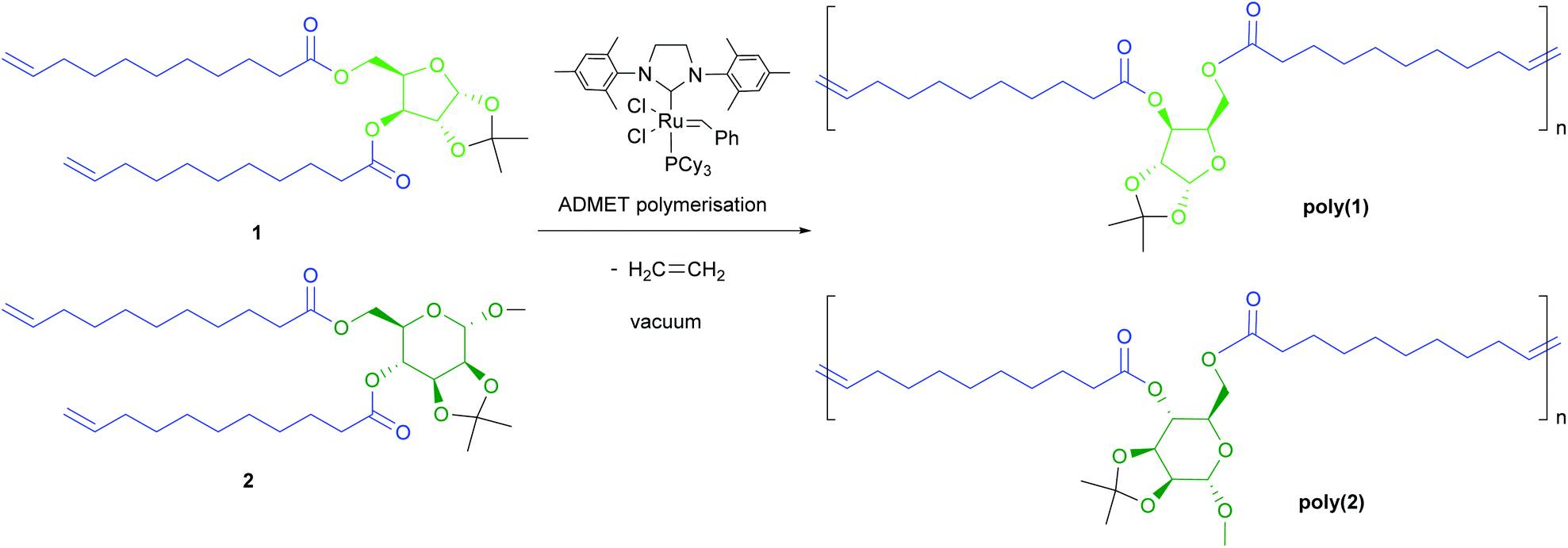 | ||
| Scheme 2 ADMET polymerisation of the α,ω-unsaturated glycolipid monomers 1 and 2. | ||
| Entry | Temp. (°C) | Catalyst (mol%) | Solvent | Vacuum (mbar) | Conversion (% (NMR)) | Yield (%) | SEC Mnb (kg mol−1) | Đ M | Theor. Mnc (kg mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerisation conditions: 1 (1 equiv., 0.25 g), G-II (0.04–0.005 equiv.), dynamic vacuum, 20 hours, magnetic stirring. b Calculated by SEC relative to polystyrene standards in THF eluent, ĐM = Mw/Mn. c Theorical Mn, calculated according to Carothers equation as MWrepeat unit × (1 + r)/(1 + r − 2 × r × conv.), where r = (N1)/(N1 + 2NG-II). d Replicate of entry 2. e Conducted in presence of methyl 10-undecenoate (0.08 equiv.). f Theorical Mn calculated according to Carothers equation (see note c) where r = (N1)/(N1 + 2(NG-II + Nmethyl 10-undecenoate)). g Conducted in presence of methyl 10-undecenoate (0.16 equiv.). h Conducted with mechanical overhead stirrer on 1.5 g of 1. | |||||||||
| 1 | rt | 4.0 | None | 0.1 | 80 | 2 | 5.0 | 1.4 | 2.1 |
| 2 | 60 | 4.0 | None | 0.1 | 100 | 64 | 14.4 | 2.0 | 12.8 |
| 3 | 90 | 4.0 | None | 0.1 | 100 | 74 | 14.1 | 2.3 | 12.8 |
| 4d | 60 | 4.0 | None | 0.1 | 100 | 66 | 14.8 | 1.7 | 12.8 |
| 5 | 60 | 2.0 | None | 0.1 | 100 | 66 | 17.7 | 2.0 | 25.2 |
| 6 | 60 | 1.0 | None | 0.1 | 97 | 77 | 14.8 | 1.9 | 12.5 |
| 7 | 90 | 1.0 | None | 0.1 | 100 | 85 | 25.5 | 2.0 | 50.0 |
| 8 | 90 | 0.5 | None | 0.1 | 100 | 84 | 20.6 | 1.9 | 99.4 |
| 9 | 60 | 4.0 | Toluene | 650 | 96 | 55 | 5.2 | 1.3 | 6.4 |
| 10 | 60 | 4.0 | Propylene carbonate | 50 | 100 | 81 | 15.3 | 2.1 | 12.8 |
| 11e | 60 | 1.0 | None | 0.1 | 100 | 83 | 15.8 | 1.9 | 6.0f |
| 12g | 60 | 1.0 | None | 0.1 | 100 | 86 | 11.8 | 1.7 | 3.4f |
| 13h | 90 | 0.5 | None | 0.2 | 100 | 70 | 71.4 | 2.2 | 99.4 |
| 14h | 90 | 0.1 | None | 0.2 | 100 | 82 | 68.2 | 2.4 | 495.2 |
| 15h | 90 | 0.5 | None | 0.2 | 100 | 83 | 71.6 | 2.3 | 99.4 |
The effect of decreasing catalyst loading was investigated as according to Carothers equation, decreasing the amount of excess monofunctional monomer (i.e. the catalyst) would lead to an increase in molecular weight of the polymers. At 60 °C, reduction to 2.0 mol% led to an increase in molecular weight (Mn 17.7 kg mol−1) as expected, and a slightly higher yield (entry 5). Further reduction to 1.0 mol% gave a Mn of 14.8 kg mol−1 (entry 6), due to incomplete (97%) conversion possibly caused by the lower reaction rate given by the lower catalyst loading. To surmount this limit, the polymerisation with 1.0 mol% of catalyst was repeated at 90 °C (entry 7). This experiment gave quantitative monomer conversion, yielding a polymer with a significantly enhanced molecular weight (Mn 25.5 kg mol−1). Using 0.5 mol% of catalyst (entry 8), a Mn of 20.6 kg mol−1 was obtained, without variation in yield (85%). This new Mn value, moderately lower compared to that obtained at 1.0 mol% of catalyst, was ascribed once more to the lower reaction rate due to the lower catalyst loading, as well as to mass-transfer limitations caused by the increased viscosity of the reaction medium. Regardless, these results demonstrated the possibility to achieve high molecular weight polymers from 1, in elevated yields, even with low catalyst loading.
To overcome limitations due to high viscosity in reaching high molecular weight, a series of polymerisations was carried out in solution instead than in bulk. As outlined earlier, the use of a solvent reduces the viscosity therefore improving mass transfer; however, the necessity of running the reaction under vacuum can lead to solvent losses. The choice of solvent, based on its boiling point, is therefore critical. In the following experiments, the working pressure was determined in such a way that the boiling point of the solvent used, at that pressure, was higher than the temperature at which the polymerisation was carried out, in order to minimise losses. In addition, a water condenser was used. A first attempt was conducted with 4.0 mol% of catalyst at 60 °C in toluene, its boiling point (110 °C) limiting the pressure to 650 mbar. Despite almost quantitative monomer conversion, modest isolated polymer yield and molecular weight were obtained (entry 9). A second reaction was carried out in propylene carbonate as solvent, chosen because of its high boiling point (240 °C) and its potential synthesis from CO2 and propylene oxide. This allowed the use of a much lower pressure (50 mbar), and the polymer thus obtained (Mn 15.3 kg mol−1; entry 10) was very similar to its counterpart obtained in bulk under similar conditions (entry 2).
Polymer chain length could be also manipulated via the use of monofunctional methyl 10-undecenoate as molecular weight moderator. At 60 °C, using 1.0 mol% of catalyst and 8.0 mol% of moderator (entry 11), the resulting Mn (15.8 kg mol−1) was very similar to that achieved when 4.0 mol% of catalyst was used without end-capping agent (entry 2; Mn 14.4 kg mol−1), with the advantage of using only 1.0 mol% of catalyst. Methyl 10-undecenoate is thought to end-cap both ends of growing chains,44 so relative integration of the signals generated in the 1H NMR spectrum of the polymer by the methoxy group (methyl 10-undecenoate, 3.61 ppm) and one of the sugar protons (5.88 ppm) was used to calculate a degree of polymerisation (≈33), resulting in a molecular weight of 16.2 kg mol−1, very similar to the value obtained by SEC/RI. A second experiment was then carried out with double the amount of end-capping agent (16.0 mol%; entry 12). The resulting polymer showed a Mn of 11.8 kg mol−1 by SEC.
A last series of experiments was aimed at overcoming mass transfer limitations when using low catalyst loadings. The experiments were performed with an overhead mechanical stirrer equipped with a vacuum seal (see Experimental section). With 0.5 mol% of catalyst, remarkably high Mn was obtained (71.4 kg mol−1, ĐM 2.2, entry 13), confirming the positive effect of the mechanical stirring. Further decreasing the catalyst loading to 0.1 mol% gave a very similar result (68.2 kg mol−1, entry 14) demonstrating that, even with very low catalyst-to-monomer ratios such as 1:1000 (and consequently reduced costs), it is possible to obtain polymers with elevated molecular weight. Moreover, decreasing the catalyst loading led to polymers with lighter colour, passing from dark brown (for 4.0 mol% of catalyst) to light yellow (with 0.1 mol%), therefore more suitable for commercial applications. Finally, with this set-up high molecular weight polymers could be obtained consistently (see replicate experiment at 0.5 mol% catalyst loading, Mn 71.6 kg mol−1, entry 15 vs. 71.4 kg mol−1, entry 13).
| Entry | Catalyst (mol%) | Yield (%) | SEC Mnb (kg mol−1) | Đ M | Theor. Mnc (kg mol−1) |
|---|---|---|---|---|---|
| a Polymerisation conditions: Mannose monomer 2 (1 equiv., 0.25 g), G-II (0.001–0.04 equiv.), bulk, 90 °C, 20 hours, 0.1 mbar, magnetic stirring. Conversion (NMR): 100%. b Calculated by SEC relative to polystyrene standards in THF eluent, ĐM = Mw/Mn. c Theorical Mn, calculated according to Carothers equation as MWrepeat unit × (1 + r)/(1 + r − 2 × r × conv.), where r = (N2)/(N2 + 2NG-II) and conv. = 1. d Conducted with mechanical overhead stirrer on 1.0 g of 2. | |||||
| 1 | 4.0 | 77 | 18.0 | 2.2 | 14.0 |
| 2 | 1.0 | 92 | 49.5 | 2.3 | 54.4 |
| 3 | 0.5 | 96 | 32.4 | 2.4 | 108.3 |
| 4d | 0.1 | 83 | 60.8 | 3.5 | 539.3 |
Characterisation of polymers
Thermal characterisation was conducted on both xylose- and mannose-derived polymers poly(1) and poly(2); for each polymer, two samples with different Mn were analysed (Table 3). In particular, the analysis of poly(1) was conducted on samples of Mn 71.4 kg mol−1 (poly(1)-71, Table 1, entry 13) and 17.7 kg mol−1 (poly(1)-18, Table 1, entry 5). For poly(2), the samples with Mn 49.5 kg mol−1 (poly(2)-50, Table 2, entry 2) and 18.0 kg mol−1 (poly(2)-18, Table 2, entry 1) were analysed.| Polymer | T d5% (°C) | T inf (mass loss)c (°C (%)) | Residual massd (%) | T g (°C) | T m (°C) | |
|---|---|---|---|---|---|---|
| a Temperature at which 5% mass loss is observed. b Temperature at which maximum mass loss is observed for the considered decomposition step. c Relative mass loss for the considered decomposition step. d Residual mass at 600 °C. e Glass transition temperature determined for the second heating cycle. f Melting temperature determined for the second heating cycle. g Not detected. h An additional decomposition step was observed with Tinf 196 °C (17.7% mass loss). | ||||||
| Poly(1)-71 | 332 | 340 (60.4) | 442 (23.5) | 15.6 | −14 | n.d.g |
| Poly(1)-18 | 351 | 365 (68.2) | 437 (16.5) | 13.0 | −28; −16 | n.d.g |
| Poly(1)-depr2h | 192 | 317 (48.5)h | 442 (21.5)h | 12.2 | n.d.g | 48 |
| Poly(2)-50 | 355 | 400 (70.2) | 441 (16.8) | 9.9 | −19 | n.d.g |
| Poly(2)-18 | 340 | 399 (68.1) | 440 (15.4) | 12.7 | −24; −8 | n.d.g |
| Poly(2)-depr2h | 256 | 391 (75.1) | 446 (18.1) | 6.7 | −10 | 49 |
Thermogravimetric analysis (TGA) showed that polymers were highly thermally stable, with temperature of decomposition (Td5%) in the range of 330–355 °C when the measure was conducted under argon (Fig. 1a, Table 3), and very similar when tested in air (Td5% 318 °C for poly(1)-71; Fig. S12†). Interestingly, the nature of the sugar core showed an impact on polymers thermal stability, with mannose-based poly(2) being more stable than xylose-containing poly(1). Moreover, thermogravimetric profiles obtained for both poly(1) and poly(2) showed two distinct thermal degradation steps, especially for poly(2). A first decomposition, with maximum at 340–365 °C for poly(1) and 400 °C for poly(2), corresponded to 60–70% mass loss. A second decomposition, with maximum close to 440 °C for all polymers, was associated with a smaller (16–23%) mass loss. This interesting behaviour was ascribed to the separate carbohydrate and lipid components of the polymers. In particular, the first decomposition step was attributed to the sugar cores which, being dissimilar, decompose at different temperatures. The second step, very similar for both polymers, was assigned to the 10-undecenoic acid building block. This interpretation is corroborated by the higher thermal stability of hydrocarbons over carbohydrates. Finally it was shown that, while for poly(2) different Mn had no effect in the thermal decomposition, poly(1)-18 was more stable compared to poly(1)-71 (Td5% 351 and 332 °C, respectively).
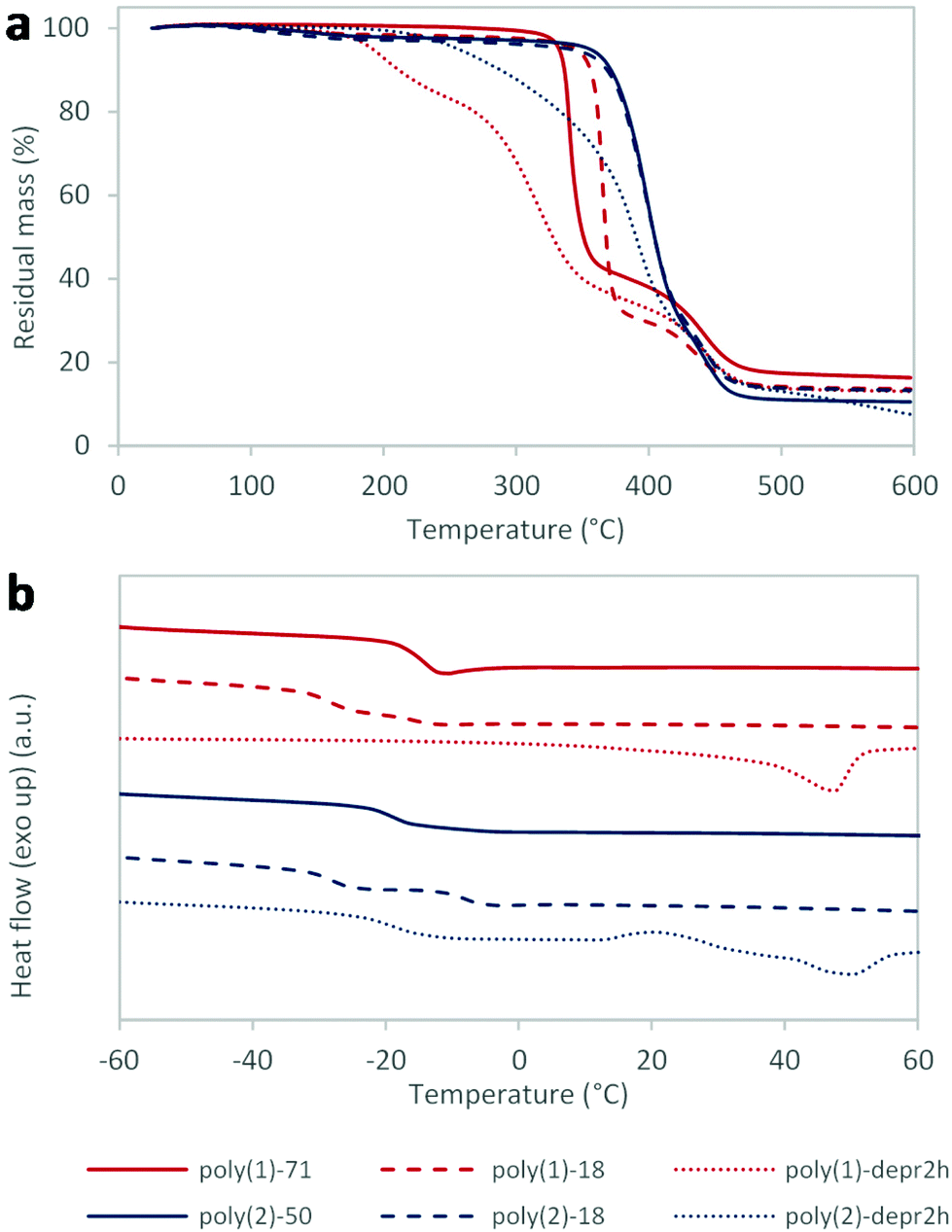 | ||
| Fig. 1 Thermal analyses of selected poly(1) and poly(2) samples: (a) thermogravimetric analysis (TGA); (b) differential scanning calorimetry (DSC) (2nd heating cycle). | ||
Differential scanning calorimetry (DSC), performed between −75 and 150 °C on two sequential cycles, while revealing no melting temperatures, showed glass transition temperatures (Tg) ranging from −28 to −8 °C (Fig. 1b, Table 3), as expected from polymers incorporating long fatty acid chains. The effect of different molecular weights was noticeable, with lower Mn giving lower Tg as expected. Interestingly, two distinct Tg were observed for poly(1)-18 and poly(2)-18, while just one transition was detected for the higher molecular weight polymers. Different sugar cores did not impart significantly different Tg between poly(1) and poly(2), conversely to what had been observed for alditol-based polyesters.10
Hydrolytic stability testing was assessed under neutral, acidic and alkaline aqueous conditions. Finely divided polymer samples were placed in deionised water, HCl 1.0 mol L−1 and NaOH 1.0 mol L−1 and stirred for 60 days at room temperature. After this time, polymers were re-analysed via SEC, showing in all cases no apparent change in Mn, thus demonstrating that polymers are stable under hydrolytic conditions (Table S1, ESI†). Further NMR spectroscopy analysis of the supernatant solutions showed complete absence of any degradation products.
Polymer deprotection, production and characterisation of films
Deprotection of ketal groups on poly(1) and poly(2) were run under typical conditions with a trifluoroacetic acid/water mixture added to a dichloromethane solution of the polymers (see Experimental section and Scheme 3).16,22 For poly(1), NMR analysis showed that the degree of deprotection increased over time, based on the relative integration of isopropylidene methyl protons (s, 3H, 1.48 ppm) and methylene protons on the 10-undecenoic acid chain (m, 4H, 2.31 ppm) (Fig. S13–15†). After 24 hours, 72% of ketal groups were deprotected and the material was still polymeric, albeit with a decrease in Mn (19.9 vs. 28.3 kg mol−1), which may be due to partial hydrolysis of ester linkages. While, after precipitation in methanol, the material was found still soluble in THF, after drying the polymer proved insoluble in common solvents (THF, CHCl3, acetone, DMSO, DMF, H2O, (CF3)2CHOH). Its solubilisation in the ionic liquid 1-ethyl-3-methylimidazolium acetate was very slow (3 days for complete dissolution). This unexpected behaviour may be due to the formation of an extensive network of hydrogen bonds across the polymer chains, similar to that found in natural polysaccharides such as cellulose. | ||
| Scheme 3 Strategy for the post-polymerisation deprotection and functionalisation of poly(1) by reaction with chlorodiphenylphosphine. | ||
Deprotection of 37% of ketal groups (achieved in 6 hours) produced, after solvent evaporation, a rubbery polymer which was found to form a gel in THF. Again, this behaviour was attributed to a H-bond network, preventing solubilisation of the polymer. Similar gel behaviour was observed after 2 hours, for which 20% deprotection was observed. Deprotection was also conducted on poly(2) for 2 hours. DSC analysis of poly(1) and poly(2) after 2 hours of ketal deprotection (named poly(1)-depr2h and poly(2)-depr2h; Mn 35.5 and 30.0 kg mol−1 respectively (Table S2†)) showed for both the appearance of a melting temperature (48 and 49 °C, respectively), suggesting the formation of a certain degree of crystallinity (Fig. 1b, Table 3). For poly(1)-depr2h, in contrast to what previously observed, no Tg was detected. On the other hand, for poly(2)-depr2h a Tg of −10 °C was measured. TGA of partially deprotected polymers showed reduced thermal stability compared with their precursors, being Td5% 192 vs. 332–351 °C for poly(1)-depr2hvs.poly(1), and 256 vs. 340–355 °C for poly(2)-depr2hvs.poly(2). This reduced thermal stability may stem from the presence of reactive free hydroxyl groups, while differences observed between poly(1)-depr2h and poly(2)-depr2h may be due to the nature of the sugar core as well as the degree of deprotection.
Finally, ketal deprotection was attempted directly on monomer 1 in order to assess if ADMET polymerisation could be performed on deprotected monomers, bearing free hydroxyl groups. However, the deprotection reaction, conducted under the same conditions used for deprotection of the polymers, showed hydrolysis of the ester and degradation of the sugar core, likely due to the strong acidic conditions; the deprotected monomer was not therefore isolated.
The reactivity of the newly revealed hydroxyl groups on the polymer chains was tested by means of functionalisation with chlorodiphenylphosphine (see Experimental part and Scheme 3). 31P{1H} NMR spectroscopy analysis of the isolated novel polymer showed the appearance of two sets of two signals in the region 111–119 ppm (Fig. S17†), consistent with the expected formation of Ph2P-OCHRR′ bonds. The presence of two sets of signals is tentatively attributed to the α and β anomers of the sugar ring. 1H–31P{1H} HMBC experiments revealed coupling of the 31P{1H} signals with the 1H sugar signals (Fig. S18†), confirming the localisation of the phosphine groups on the polymer structure, and consequently the reactivity of its hydroxyl groups. Moreover, the material remained polymeric (Mn 25.4 kg mol−1vs. 31.0 kg mol−1 for poly(1) sample used, see Fig. S19†), corroborating the post-polymerisation modification strategy and opening new possibilities for functional polymers.
Polymer films were produced by casting a polymer solution (100 g L−1 in THF) in a PTFE dish. Unmodified poly(1) was found unsuitable for the purpose as, although forming homogeneous films, those were not self-standing and behaved as highly viscous fluids. Film casting was therefore attempted on partially deprotected polymers immediately after precipitation in methanol, when the materials still retained some solubility in THF. Films were produced from poly(1)-depr2h and poly(2)-depr2h (Fig. 2 and S20†). These films were self-standing, flexible and transparent. Water and oil contact angles (CA) were measured for the two films. Values obtained for water were found in the range of 70–85° (Table S4†), while dodecane did not form droplets but rapidly wetted the polymer surface creating a thin oil film. Moreover, in all cases, water CA were observed to decrease linearly over time (Fig. S24†).
 | ||
| Fig. 2 Film casted from poly(1)-depr2h. | ||
These observations suggest that both xylose- and mannose-derived polymers show amphiphilic behaviour, likely due to the dual nature imparted by the carbohydrate and fatty acid building blocks. Mechanical properties of polymer films were investigated by means of uniaxial tensile testing. Films exhibited low Young moduli (31–64 MPa), typical of rubbery materials, low elongation at break and tensile strength values (7–12% and 2–3 MPa, respectively; Table S3†). Notwithstanding the poor tensile properties of these polymers, the ability of producing transparent films represents a significant improvement compared to purely sugar-based polymers, whose typical brittleness has so far prevented their shaping into any form in our hands.
Conclusions
In the present work, two novel monomers were synthesised from readily available sugars – xylose and mannose – and an unsaturated fatty acid, 10-undecenoic acid, with a renewable content of around 90 wt%. These monomers, bearing two terminal olefin groups, were polymerised via acyclic diene metathesis (ADMET) polymerisation with a ruthenium alkylidene complex, Grubbs second generation catalyst, under dynamic vacuum (0.1 mbar). For the polymerisation of the xylose-based monomer 1, the effect of several parameters was investigated, including temperature, catalyst loading, presence of solvents and molecular weight moderator, mixing technique. The best result was obtained when the polymerisation was performed in bulk with mechanical stirring at 90 °C with 0.5 mol% of catalyst, yielding a polymer with a molecular weight (Mn) of 71.4 kg mol−1 and a dispersity of 2.2. The best reaction conditions were then applied to the polymerisation of the mannose-based monomer 2, obtaining again high molecular weight (Mn 60.8 kg mol−1). Thermal analysis of the polymers showed them to be stable up to ∼350 °C with two characteristic decomposition steps. Glass transition temperatures ranged from −28 to −8 °C. While TGA revealed that different sugar cores imparted different stability to the polymers, with poly(2) more thermally stable than poly(1), DSC revealed no significant differences between xylose- and mannose-containing polymers. Polymers were found to be stable in water at pH 0, 7 and 14 for as long as 60 days. Partial deprotection of ketal moieties, revealing hydroxyl groups, modified the polymer properties without significantly affecting molecular weights. New melting temperatures were observed, indicative of semicrystallinity, and the solubilisation in common solvents was prevented. This allowed the production of transparent thin films with amphiphilic behaviour, but low elongation at break and poor tensile strength. The hydroxyl groups of these partially deprotected polymers were shown to be reactive and used for further post-polymerisation functionalisation.Experimental
Materials
Methyl α-D-mannopyranoside and 1,2-O-isopropylidene-α-D-xylofuranose were purchased from Carbosynth, Compton (Berkshire, UK) and used as received; all the other reagents were purchased from Sigma Aldrich and Fisher Scientific and used as supplied.All the solvents were supplied by VWR and used without further purification.
Characterization
1H NMR (13C {1H} NMR) spectra were recorded at 25 °C in CDCl3 on a Bruker AVANCE III spectrometer operating at 400 (100) MHz. Chemical shifts (δ) were reported in parts per million (ppm) relative to the residual protiated solvent.LC-MS was performed with an Agilent High Performance Liquid Chromatography (HPLC) unit coupled to a Bruker Daltonik MicroToF electrospray time-of-flight (ESI-TOF) mass spectrometer.
Size exclusion chromatography (SEC) was performed with an Agilent 1260 Infinity GPC/SEC system, equipped with two columns PLgel 5 μm MIXED-D 300 × 7.5 mm, and a guard column PLgel 5 μm MIXED Guard 50 × 7.5 mm kept at 35 °C, and a refractive index (RI) detector. SEC-grade THF was used as eluent (1.0 mL min−1). The system was calibrated with a set of polystyrene standards.
Differential scanning calorimetry (DSC) was performed on a TA Instruments DSC Q20, loading ∼5 mg of polymer in a 10 μL Tzero aluminium pan with lid with an identical empty cell as reference. The analysis was performed as two successive cooling–heating cycles between −75 and 150 °C at a rate of 10.0 °C min−1 under nitrogen; glass transition and melting temperatures were calculated from the second heating cycle.
Thermogravimetric analysis (TGA) was performed on a Setaram Setsys Evolution TG-MS, loading ∼8 mg of polymer in a 170 μL alumina crucible and heating the sample under argon from 30 to 600 °C at a rate of 10.0 °C min−1.
Uniaxial tensile testing was performed on an Instron 3369 universal testing machine equipped with 50 N pneumatic grips. Tests were conducted on rectangular polymer strips (20 × 4 × 0.25 mm) at a rate of 5.0 mm min−1. For each polymer film, 5 replicates were conducted and results reported as average ± standard error.
Synthesis of monomers
1,2-O-Isopropylidene-α-D-xylofuranose (1.0 g, 5.26 mmol, 1.0 equiv.) was added to a 50 mL round bottom flask with 10-undecenoic anhydride (4.61 g, 13.15 mmol, 2.5 equiv.), triethylamine (1.60 g, 15.77 mmol, 3.0 equiv.) and dichloromethane (20 mL). The mixture was stirred at room temperature until homogeneous, then 4-dimethylaminopyridine (0.19 g, 1.58 mmol, 0.3 equiv.) was added. The reaction, monitored via TLC (petroleum ether–ethyl acetate 9
 :1; phosphomolybdic acid stain), was completed after 20 minutes. Amberlyst A26 OH form (20 g) was added to remove the excess 10-undecenoic acid and stirred for 30 minutes. The mixture was filtered, the solvent removed in vacuo and the crude product purified via column chromatography (silica gel, eluent petroleum ether–ethyl acetate 99:1 to 95:5) to give a colourless oil. Yield 82%. 1H NMR (400 MHz, CDCl3, δ in ppm): 1.23–1.33 (m, 23H, C(C
:1; phosphomolybdic acid stain), was completed after 20 minutes. Amberlyst A26 OH form (20 g) was added to remove the excess 10-undecenoic acid and stirred for 30 minutes. The mixture was filtered, the solvent removed in vacuo and the crude product purified via column chromatography (silica gel, eluent petroleum ether–ethyl acetate 99:1 to 95:5) to give a colourless oil. Yield 82%. 1H NMR (400 MHz, CDCl3, δ in ppm): 1.23–1.33 (m, 23H, C(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3)2, H-9-H-13), 1.47 (s, 3H, C(C3)2), 1.54–1.57 (m, 4H, H-8), 1.95–2.01 (m, 4H, H-14), 2.24–2.29 (m, 4H, H-7), 4.11–4.23 (m, 2H, H-5), 4.43–4.46 (m, 2H, H-2, H-4), 4.86–4.96 (m, 4H, H-16), 5.21 (d, J = 3.0 Hz, 1H, H-3), 5.70–5.80 (ddt, J = 16.9, 10.2, 6.7 Hz, 4H, H-15), 5.89 (d, J = 3.7 Hz, 1H, H-1). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.8 (C-8), 26.2 (C(
3)2, H-9-H-13), 1.47 (s, 3H, C(C3)2), 1.54–1.57 (m, 4H, H-8), 1.95–2.01 (m, 4H, H-14), 2.24–2.29 (m, 4H, H-7), 4.11–4.23 (m, 2H, H-5), 4.43–4.46 (m, 2H, H-2, H-4), 4.86–4.96 (m, 4H, H-16), 5.21 (d, J = 3.0 Hz, 1H, H-3), 5.70–5.80 (ddt, J = 16.9, 10.2, 6.7 Hz, 4H, H-15), 5.89 (d, J = 3.7 Hz, 1H, H-1). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.8 (C-8), 26.2 (C(![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3)2), 26.7 (C(H3)2), 28.8–29.2 (C-9-C-13), 33.7 (C-14), 34.0 (C-7), 61.1 (C-5), 75.8 (C-3), 76.8 (C-4), 83.3 (C-2), 104.9 (C-1), 112.2 ((CH3)2), 114.1 (C-16), 114.1 (C-16′), 139.1 (C-15), 139.1 (C-15′), 172.4 (C-6), 173.3 (C-6′). MSm/z [M + Na]+ calc. for C30H50O7Na 545.3449, found 545.3483. Elemental analysis calc. for C30H50O7: C 68.93%, H 9.64%; found C 65.11%, H 8.90%.
H3)2), 26.7 (C(H3)2), 28.8–29.2 (C-9-C-13), 33.7 (C-14), 34.0 (C-7), 61.1 (C-5), 75.8 (C-3), 76.8 (C-4), 83.3 (C-2), 104.9 (C-1), 112.2 ((CH3)2), 114.1 (C-16), 114.1 (C-16′), 139.1 (C-15), 139.1 (C-15′), 172.4 (C-6), 173.3 (C-6′). MSm/z [M + Na]+ calc. for C30H50O7Na 545.3449, found 545.3483. Elemental analysis calc. for C30H50O7: C 68.93%, H 9.64%; found C 65.11%, H 8.90%.
Methyl 2,3-O-isopropylidene-α-D-mannopyranoside, (1.23 g, 5.26 mmol, 1.0 equiv.), synthesised according to a previously reported procedure,22 was added to a 50 mL round bottom flask with 10-undecenoic anhydride (4.61 g, 13.15 mmol, 2.5 equiv.), triethylamine (1.60 g, 15.77 mmol, 3.0 equiv.) and dichloromethane (20 mL). The mixture was stirred at room temperature until homogeneous, then 4-dimethylaminopyridine (0.19 g, 1.58 mmol, 0.3 equiv.) was added and the mixture stirred for 20 minutes. Work-up was carried out as for 1, obtaining 2 as a colourless oil. Yield 86%. 1H NMR (400 MHz, CDCl3, δ in ppm): 1.20–1.33 (m, 23H, C(C
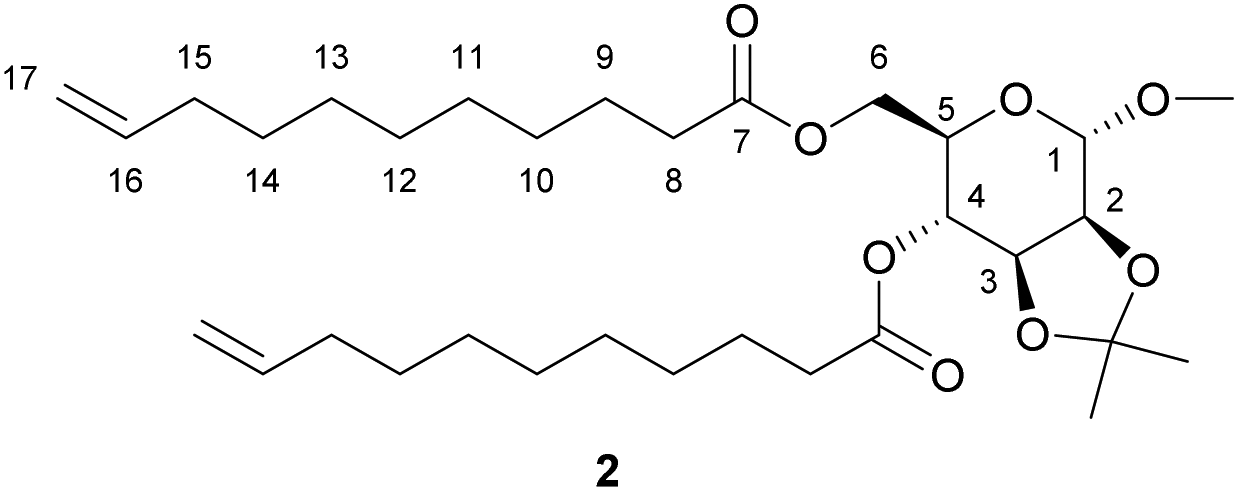 3)2, H-10–H-14), 1.51–1.58 (m, 7H, C(C3)2, H-9), 1.95–2.01 (m, 4H, H-15), 2.25–2.30 (m, 4H, H-8), 3.35 (s, 3H, OC3), 3.77 (ddd, J = 10.4, 5.8, 2.5 Hz, 1H, H-3), 4.04–4.16 (m, 4H, H-6, H-2, H-5), 4.86–5.02 (m, 6H, H-1, H-17, H-4), 5.76 (ddtd, J = 16.9, 10.1, 6.7, 0.8 Hz, 2H, H-16). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.7 (C-9), 26.3 (C(H3)2), 27.6 (C(H3)2), 28.8–29.2 (C-10–C14), 33.7 (C-15), 34.0 (C-8), 34.1 (C-8′), 55.0 (OH3), 62.3 (C-6), 66.2 (C-3), 69.1 (C-4), 75.6 (C-2), 75.9 (C-5), 98.1 (C-1), 109.9 ((CH3)2), 114.1 (C-17), 139.1 (C-16), 172.5 (C-7), 173.4 (C-7′). MSm/z [M + Na]+ calc. for C32H54O8Na 589.3711, found 589.3730. Elemental analysis calc. for C32H54O8: C 67.81%, H 9.60%; found C 67.76%, H 9.75%.
3)2, H-10–H-14), 1.51–1.58 (m, 7H, C(C3)2, H-9), 1.95–2.01 (m, 4H, H-15), 2.25–2.30 (m, 4H, H-8), 3.35 (s, 3H, OC3), 3.77 (ddd, J = 10.4, 5.8, 2.5 Hz, 1H, H-3), 4.04–4.16 (m, 4H, H-6, H-2, H-5), 4.86–5.02 (m, 6H, H-1, H-17, H-4), 5.76 (ddtd, J = 16.9, 10.1, 6.7, 0.8 Hz, 2H, H-16). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.7 (C-9), 26.3 (C(H3)2), 27.6 (C(H3)2), 28.8–29.2 (C-10–C14), 33.7 (C-15), 34.0 (C-8), 34.1 (C-8′), 55.0 (OH3), 62.3 (C-6), 66.2 (C-3), 69.1 (C-4), 75.6 (C-2), 75.9 (C-5), 98.1 (C-1), 109.9 ((CH3)2), 114.1 (C-17), 139.1 (C-16), 172.5 (C-7), 173.4 (C-7′). MSm/z [M + Na]+ calc. for C32H54O8Na 589.3711, found 589.3730. Elemental analysis calc. for C32H54O8: C 67.81%, H 9.60%; found C 67.76%, H 9.75%.
Polymerisations
1 H NMR (400 MHz, CDCl3, δ in ppm): 1.22–1.26 (m, 23H, C(C
 3)2, H-9–H-13), 1.47 (s, 3H, C(C3)2), 1.50–1.58 (m, 4H, H-8), 1.88–1.96 (m, 4H, H-14), 2.24–2.29 (m, 4H, H-7), 4.11–4.23 (m, 2H, H-5), 4.42–4.46 (m, 2H, H-2, H-4), 5.21 (d, J = 3.1 Hz, 1H, H-3), 5.26–5.37 (m, 2H, H-15), 5.89 (d, J = 3.8 Hz, 1H, H-1). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.8–24.9 (C-8), 26.2 (C(H3)2), 26.7 (C(H3)2), 27.2–27.2 (C-14′), 28.9–29.6 (C9–C-13), 32.5 (C-14), 34.0 (C-7), 61.1 (C-5), 75.8 (C-3), 76.8 (C-4), 83.4 (C-2), 104.9 (C-1), 112.2 ((CH3)2), 130.2–130.3 (C-15), 172.3 (C-6), 173.3 (C-6′).
3)2, H-9–H-13), 1.47 (s, 3H, C(C3)2), 1.50–1.58 (m, 4H, H-8), 1.88–1.96 (m, 4H, H-14), 2.24–2.29 (m, 4H, H-7), 4.11–4.23 (m, 2H, H-5), 4.42–4.46 (m, 2H, H-2, H-4), 5.21 (d, J = 3.1 Hz, 1H, H-3), 5.26–5.37 (m, 2H, H-15), 5.89 (d, J = 3.8 Hz, 1H, H-1). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.8–24.9 (C-8), 26.2 (C(H3)2), 26.7 (C(H3)2), 27.2–27.2 (C-14′), 28.9–29.6 (C9–C-13), 32.5 (C-14), 34.0 (C-7), 61.1 (C-5), 75.8 (C-3), 76.8 (C-4), 83.4 (C-2), 104.9 (C-1), 112.2 ((CH3)2), 130.2–130.3 (C-15), 172.3 (C-6), 173.3 (C-6′).
1 H NMR (400 MHz, CDCl3, δ in ppm): 1.22–1.29 (m, 23H, C(C
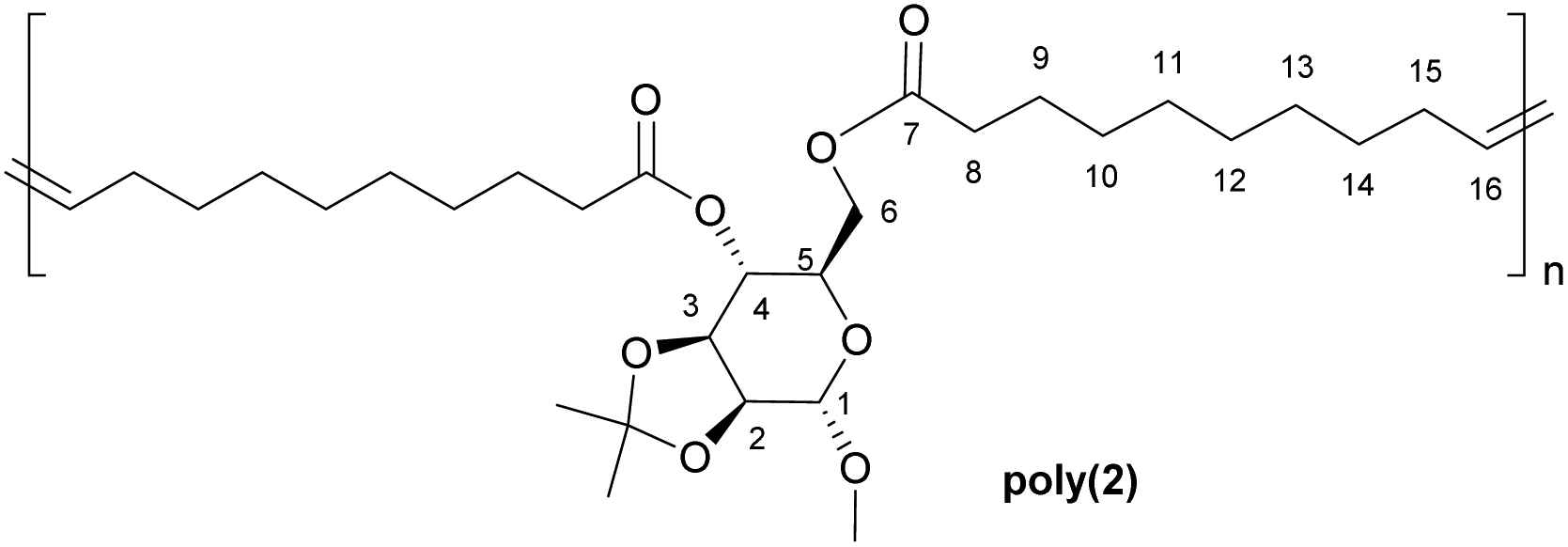 3)2, H-10–H-14), 1.51–1.59 (m, 7H, C(C3)2, H-9), 1.89–1.99 (m, 4H, H-15), 2.25–2.29 (m, 4H, H-8), 3.34 (s, 3H, OCH3), 3.76 (ddd, J = 10.4, 5.8, 2.5 Hz, z1H, H-3), 4.04–4.08 (m, 2H, H-6), 4.13 (dt, J = 7.2, 5.2 Hz, 2H, H-2, H-5), 4.91 (s, 1H, H-1), 4.99 (ddd, J = 10.4, 7.6, 1.8 Hz, 1H, H-4), 5.27–5.36 (m, 2H, H-16). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.7 (C-9), 26.3 (C(H3)2), 27.2 (C-15′), 27.6 (C(H3)2), 29.1–29.6 (C10–C14), 32.5 (C-15), 34.1–34.2 (C-8), 54.9 (OCH3), 62.4 (C-6), 66.3 (C-3), 69.2 (C-4), 75.7 (C-5), 75.9 (C-2), 98.1 (C-1), 109.9 ((CH3)2), 130.2–130.3 (C-16), 172.5 (C-7), 173.3 (C-7′).
3)2, H-10–H-14), 1.51–1.59 (m, 7H, C(C3)2, H-9), 1.89–1.99 (m, 4H, H-15), 2.25–2.29 (m, 4H, H-8), 3.34 (s, 3H, OCH3), 3.76 (ddd, J = 10.4, 5.8, 2.5 Hz, z1H, H-3), 4.04–4.08 (m, 2H, H-6), 4.13 (dt, J = 7.2, 5.2 Hz, 2H, H-2, H-5), 4.91 (s, 1H, H-1), 4.99 (ddd, J = 10.4, 7.6, 1.8 Hz, 1H, H-4), 5.27–5.36 (m, 2H, H-16). 13C {1H} NMR (100 MHz, CDCl3, δ in ppm): 24.7 (C-9), 26.3 (C(H3)2), 27.2 (C-15′), 27.6 (C(H3)2), 29.1–29.6 (C10–C14), 32.5 (C-15), 34.1–34.2 (C-8), 54.9 (OCH3), 62.4 (C-6), 66.3 (C-3), 69.2 (C-4), 75.7 (C-5), 75.9 (C-2), 98.1 (C-1), 109.9 ((CH3)2), 130.2–130.3 (C-16), 172.5 (C-7), 173.3 (C-7′).
Hydrolytic stability testing, deprotection and film casting
Hydrolytic stability testing was performed in closed vials on 25 mg of polymer divided in ∼1 mm pieces, to which 2.5 mL of the respective solution (deionised water, HCl 1.0 mol L−1, NaOH 1.0 mol L−1) were added, and the heterogeneous mixture was stirred at room temperature for 60 days. The polymer was then removed from the solution, rinsed with deionised water and left to dry prior to SEC analysis. Water was evaporated from the supernatant solution under reduced pressure, the residual solid was dissolved in D2O and analysed via1H NMR.Ketal deprotection was performed according to typical procedures, dissolving 1.0 g of polymer in 10 mL of dichloromethane and successively adding a mixture of trifluoroacetic acid (10 mL) and deionised water (2 mL).16,22 When the desired conversion was obtained, the polymer was precipitated dropwise into 100 mL of cold methanol.
After precipitation, the mixture was centrifuged, the supernatant removed and the resulting polymer dissolved in THF (100 g L−1). Film casting was performed transferring 6.0 mL of said solution, previously filtered through cotton wool, in a flat-bottomed PTFE evaporating dish (diam. 40 mm) and leaving it until THF was completely evaporated, leaving a homogeneous transparent polymer disc ∼0.25 mm thick.
Post-polymerisation hydroxyl functionalisation
Poly(1) (31.0 kg mol−1; 50 mg; 0.11 mmol repeat unit equiv.) was subjected to partial ketal deprotection for 3 hours according to the procedure described above. The resulting material was then dissolved in THF (1 mL); triethylamine (100 mg, 0.99 mmol, 9.0 equiv. vs. a fully-deprotected polymer) and 4-dimethylaminopyridine (24 mg, 0.20 mmol, 2.0 equiv.) were added, then chlorodiphenylphosphine (60 mg, 0.27 mmol, 2.5 equiv.) was added dropwise under an argon atmosphere. The vial was sealed and stirred at r.t. overnight. Successively, the modified polymer was precipitated in cold methanol (30 mL), centrifuged and the supernatant removed, yielding a white solid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Analytical facilities were provided through the Material and Chemical Characterisation Facility (MC2) at the University of Bath. This work was supported by the Engineering and Physical Sciences Research Council EP/L016354/1 (Studentship to MP, CDT in Sustainable Chemical Technologies). AB acknowledges Roger and Sue Whorrod (Whorrod Research Fellowship) and the Royal Society (UF/160021 fellowship) for research funding.References
- M. A. R. Meier, in Green Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction, ed. R. T. Mathers and M. A. R. Meier, Wiley-VCH, 2011, pp. 9–27 Search PubMed.
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS.
- A. J. D. Silvestre and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 17–38 Search PubMed.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- H. Tsuji, in Bio-Based Plastics: Materials and Applications, ed. S. Kabasci, John Wiley & Sons, Ltd, 2014, pp. 171–239 Search PubMed.
- P. J. Hocking and R. H. Marchessault, in Biopolymers from Renewable Resources, ed. D. L. Kaplan, Springer Berlin Heidelberg, Berlin, Heidelberg, 1998, pp. 220–248 Search PubMed.
- J. A. Galbis and M. G. García-Martín, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 89–114 Search PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- E. Zakharova, A. Martínez de Ilarduya, S. León and S. Muñoz-Guerra, Des. Monomers Polym., 2017, 20, 157–166 CrossRef CAS PubMed.
- A. Gandini, in Green Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction, ed. R. T. Mathers and M. A. R. Meier, Wiley-VCH, 2011, pp. 29–56 Search PubMed.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202–229 CrossRef CAS.
- G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- X. Chen and R. A. Gross, Macromolecules, 1999, 32, 308–314 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799–2802 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 3891–3897 CrossRef CAS.
- R. Kumar, W. Gao and R. A. Gross, Macromolecules, 2002, 35, 6835–6844 CrossRef CAS.
- K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P.-J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- A. T. Lonnecker, Y. H. Lim and K. L. Wooley, ACS Macro Lett., 2017, 6, 748–753 CrossRef CAS.
- S. E. Felder, M. J. Redding, A. Noel, S. M. Grayson and K. L. Wooley, Macromolecules, 2018, 51, 1787–1797 CrossRef CAS.
- Y. Song, X. Ji, M. Dong, R. Li, Y.-N. Lin, H. Wang and K. L. Wooley, J. Am. Chem. Soc., 2018, 140, 16053–16057 CrossRef CAS PubMed.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Köhn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef CAS.
- G. L. Gregory, E. M. Hierons, G. Kociok-Köhn, R. I. Sharma and A. Buchard, Polym. Chem., 2017, 8, 1714–1721 RSC.
- G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2017, 8, 2093–2104 RSC.
- Y.-Y. T. Tsao and K. L. Wooley, J. Am. Chem. Soc., 2017, 139, 5467–5473 CrossRef CAS PubMed.
- Y.-Y. T. Tsao, T. H. Smith and K. L. Wooley, ACS Macro Lett., 2018, 7, 153–158 CrossRef CAS.
- E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- S. L. Chin, Q. Lu, E. L. Dane, L. Dominguez, C. J. McKnight, J. E. Straub and M. W. Grinstaff, J. Am. Chem. Soc., 2016, 138, 6532–6540 CrossRef CAS PubMed.
- R. Xiao, E. L. Dane, J. Zeng, C. J. McKnight and M. W. Grinstaff, J. Am. Chem. Soc., 2017, 139, 14217–14223 CrossRef CAS PubMed.
- R. Xiao, J. Zeng and M. W. Grinstaff, ACS Macro Lett., 2018, 7, 772–777 CrossRef CAS.
- M. W. Grinstaff, A. S. Balijepalli, R. C. Sabatelle, M. Chen and B. Suki, Angew. Chem., Int. Ed., 2020, 59, 704–710 CrossRef PubMed.
- L. M. de Espinosa and M. A. R. Meier, in Organometallics and Renewables, ed. M. A. R. Meier, B. M. Weckhuysen and P. C. A. Bruijnincx, Springer, Berlin, Heidelberg, 2012, pp. 1–44 Search PubMed.
- P. A. Fokou and M. A. R. Meier, J. Am. Chem. Soc., 2009, 131, 1664–1665 CrossRef CAS PubMed.
- W. C. Shearouse, L. M. Lillie, T. M. Reineke and W. B. Tolman, ACS Macro Lett., 2015, 4, 284–288 CrossRef CAS.
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Polym. Chem., 2017, 8, 3746–3754 RSC.
- G. Hibert, E. Grau, D. Pintori, S. Lecommandoux and H. Cramail, Polym. Chem., 2017, 8, 3731–3739 RSC.
- W. Gao, R. Hagver, V. Shah, W. C. Xie, R. A. Gross, M. F. Ilker, C. Bell, K. A. Burke and E. B. Coughlin, Macromolecules, 2007, 40, 145–147 CrossRef CAS.
- E. Zini, M. Gazzano, M. Scandola, S. R. Wallner and R. A. Gross, Macromolecules, 2008, 41, 7463–7468 CrossRef CAS.
- Y. Peng, J. Decatur, M. A. R. Meier and R. A. Gross, Macromolecules, 2013, 46, 3293–3300 CrossRef CAS.
- Y. Peng, D. J. Munoz-Pinto, M. Chen, J. Decatur, M. Hahn and R. A. Gross, Biomacromolecules, 2014, 15, 4214–4227 CrossRef CAS PubMed.
- T. Debsharma, F. N. Behrendt, A. Laschewsky and H. Schlaad, Angew. Chem., 2019, 131, 6790–6793 CrossRef.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2015, 3, 662–667 CrossRef CAS.
- E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129–161 RSC.
- A. Rybak and M. A. R. Meier, ChemSusChem, 2008, 1, 542–547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and MS spectra of monomers and polymers, SEC traces, ketal deprotection analysis via NMR and plots vs. time, post-polymerisation hydroxyl functionalisation data, hydrolytic stability testing data, tensile properties of polymer films, contact angle data for polymer films. See DOI: 10.1039/c9py01809c |
| This journal is © The Royal Society of Chemistry 2020 |