
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical polymerization reactions for amplified biodetection signals†
Seunghyeon
Kim
 a and
Hadley D.
Sikes
*abc
a and
Hadley D.
Sikes
*abc
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: sikes@mit.edu
bProgram in Polymers and Soft Matter, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cAntimicrobial Resistance Integrated Research Group, Singapore-MIT Alliance for Research and Technology, 1 CREATE Way, Singapore 138602
First published on 4th February 2020
Abstract
Chemical reactions that provide amplified biodetection signals are essential in point-of-care diagnostics, a category of portable biosensors that should detect nanomolar to attomolar concentrations of clinically actionable biomarkers in bodily fluids without using advanced lab equipment. As an alternative to common signal amplification methods that use enzymes or nanoparticles, radical polymerization has been explored as an approach to sensitive biodetection because of the inherent amplification in the chain-growth process. Polymerization-based biodetection benefits from different types of initiation reactions and a wide variety of monomer choices, making it adaptable to diverse sensing conditions and detection methods. This review presents the many radical polymerization chemistries that have been implemented in biodetection platforms and evaluates their utility. First, we describe the principle of each polymerization-based biodetection and discuss its advantages and current limitations for practical use in the field. Then, we compare all of the methods in terms of performance, equipment-dependence, user-friendliness, and amplification time. Finally, we highlight exciting future directions and opportunities for developing practical biosensors that use radical polymerization reactions to generate signals.
 Seunghyeon Kim | Seunghyeon Kim obtained his BS degree in Chemical Engineering at Seoul National University in 2016. He received a Kwanjeong scholarship (2016–2020) and joined the group of Hadley Sikes at Massachusetts Institute of Technology as a PhD student. His research focuses on the development of photo-initiated polymerization reactions and their applications to biosensing for point-of-care diagnostics. |
 Hadley D. Sikes | Hadley D. Sikes is the Esther and Harold E. Edgerton associate professor of chemical engineering at the Massachusetts Institute of Technology and a PI in the Antimicrobial Resistance Interdisciplinary Research Group in Singapore's CREATE campus. She advises a team of researchers in the application of physical principles to design, synthesize, characterize and test molecules for utility in detecting and understanding disease. Hadley earned degrees in chemistry, a BS at Tulane University and a PhD at Stanford University, and trained as a postdoctoral scholar in chemical engineering at the University of Colorado, Boulder, and at the California Institute of Technology prior to joining the faculty at MIT. |
1. Introduction
Biosensors are analytical devices that specifically capture target biomolecules in complex fluids using bioreceptors, often nucleic acids and proteins, and transduce these molecular recognition events to observable signals. The signals can be directly produced from the biorecognition events by using label-free methods such as surface plasmon resonance (SPR),1 quartz-crystal microbalance (QCM),2 and surface-enhanced Raman scattering (SERS),3 or can be transduced indirectly by using detection probes conjugated with labels, including fluorophores, nanoparticles, and catalysts.4–8 One major application of biosensors is point-of-care diagnostics, where the portable biosensors detect disease-related biomarkers in bodily fluids near patients.9–11 In this setting, the biosensors should perform accurate and rapid biodetection without any help of sophisticated equipment in centralized laboratory. However, the concentrations of disease biomarkers in bodily fluids range from nanomolar (nM) to attomolar (aM),12 so it is very challenging to detect the signals from the small number of biorecognition events without the highly sensitive detectors.Alternatively, signals generated from the specific binding events can be amplified to enhance the sensitivity of biodetection.13 One approach is to couple molecular recognition events with enzymes or nanocatalysts that can catalytically convert multiple substrates to signaling molecules. For example, horseradish peroxidase (HRP)-conjugated detection probes are immobilized to where the target biomolecules are specifically captured. Then, an excess amount of substrate is added to the enzyme-functionalized test zone, and converted to colored, fluorescent, or chemiluminescent products, amplifying biodetection signals. Another signal amplification strategy is to increase the number of labels. Isothermal nucleic acid amplification techniques such as rolling circle amplification (RCA) can replicate specific nucleic acid sequence. By attaching this specific nucleic acid sequence to the detection probe, the presence of target biomolecules can be detected by replicating the sequence via RCA and hybridizing multiple fluorescently labeled complementary DNA sequences.14
Recently, radical polymerization has been explored as an alternative signal amplification tool in biodetection. The amplification results from chain-growth polymerization, in which one radical species reacts with hundreds to millions of monomers to generate a polymer. To couple this chain-growth polymerization with molecular recognition events, the essential components in each polymerization chemistry, such as initiators, chain transfer agents, or catalysts, are covalently bonded to a nucleic acid or protein detection probe, which recognizes a captured biomolecule. These labels are involved in either initiation or propagation, so polymers can be generated only in the presence of specific binding events. In addition to the inherent amplification of radical polymerization, different types of initiation reactions and a wide variety of monomer choices enable the polymerization-based biodetection to be versatile technique in terms of sensing conditions and detection methods.
Polymerization-based biodetection has been demonstrated with various polymerization chemistries, including atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT) polymerization, redox-initiated free radical polymerization, and photo-initiated free radical polymerization. Each of these polymerization chemistries has different characteristics that affect cost, operation conditions, user-friendliness, signal amplification time, and performance of biodetection, all of which are very important criteria for developing affordable, robust, easy-to-use, rapid, sensitive, and specific diagnostic tests. However, there has been no comprehensive review on polymerization-based biodetection that evaluates the nature of each polymerization chemistry from the practical point-of-view. Biodetection with specific polymerization methods such as ATRP, enzyme-mediated polymerization, and photo-initiated polymerization has been reviewed,15–18 providing an overview of sensitive biodetection using each polymerization chemistry. The previous review paper19 from our group compared photo-initiated free radical polymerization with other methods including ATRP, RAFT polymerization, and enzyme-mediated free radical polymerization as a signal amplification tool for biodetection and highlighted their relative merits, although not comprehensively. Thus, we hope this present review can evaluate and compare all of the radical polymerization chemistries that have been implemented in biosensing applications, helping to move polymerization-based biodetection towards real-world applications.
In this review, we consider various types of ATRP and RAFT polymerization, redox-initiated free radical polymerization, and photo-initiated free radical polymerization as signal amplification methods for biodetection. For each type of radical polymerization, we present the principle and nature of the method, describe its key applications in biodetection and numerous strategies to improve the biosensing performance, and discuss its advantages and limitations to overcome for practical use in the field. Finally, we compare all polymerization-based biodetection methods in terms of equipment-dependence, user-friendliness, amplification time, and performance.
2. Atom transfer radical polymerization (ATRP)
2.1 Normal ATRP
Normal atom transfer radical polymerization (ATRP) is one type of reversible-deactivation radical polymerization used in one of the earliest examples of polymerization-based biodetection. It employs initiation and reversible termination of propagating radicals (R−Pn˙) through a reversible homolytic halogen transfer between dormant species (R–X or R–Pn–X) and a transition metal (usually copper) complex in the lower oxidation state (Fig. 1A).20 The equilibrium of the reversible process favors deactivation of radical species,21 so ATRP can minimize bimolecular termination and chain transfer in radical polymerization and prolong the lifetime of growing chains into hours or longer.20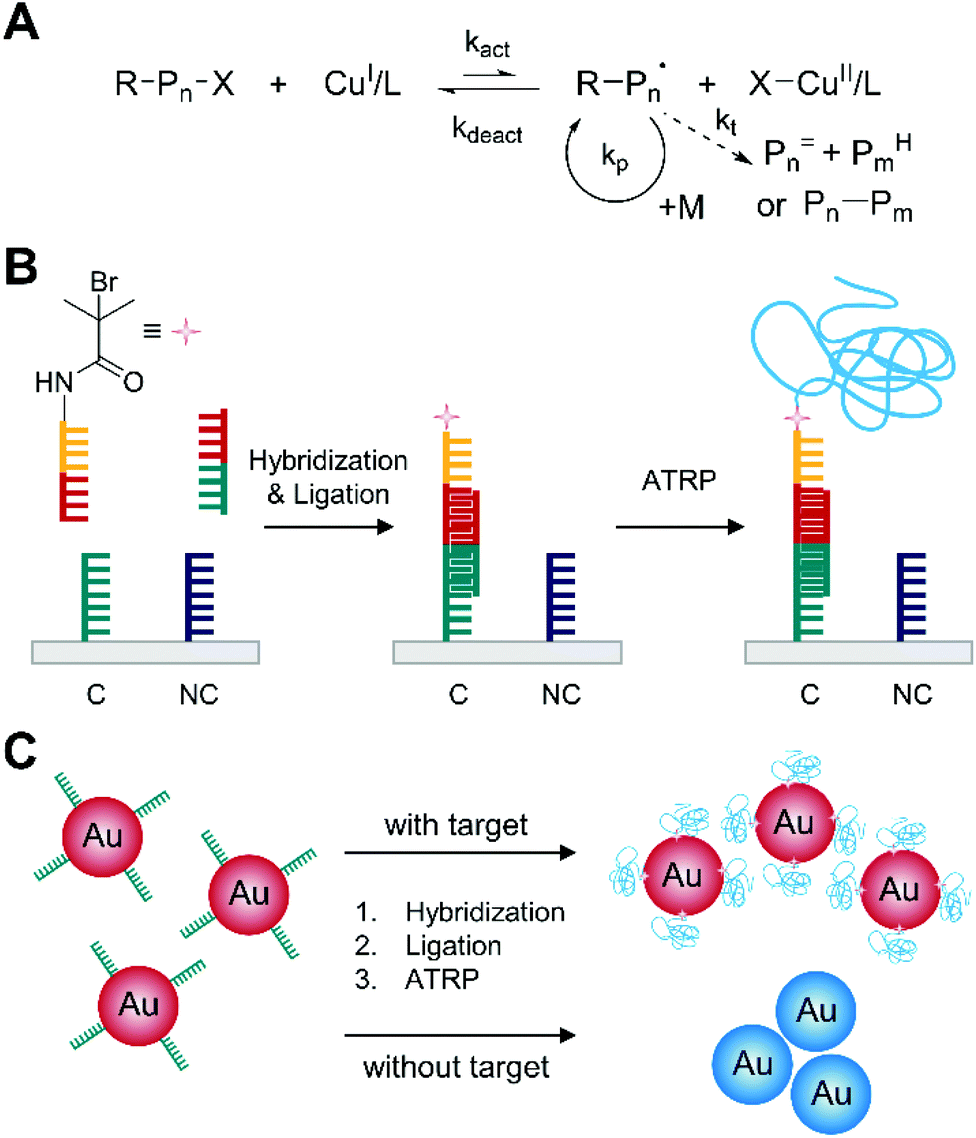 | ||
| Fig. 1 Biodetection with normal atom transfer radical polymerization (ATRP). (A) Schematic description of normal ATRP mechanism. Pn= and PmH are elimination and abstraction products, respectively, from termination by disproportionation. Pn–Pm is a recombination product. (B) Normal ATRP-based detection of DNA.22 Both complementary DNA, C, and non-complementary DNA, NC, are used as capture molecules, but only complementary DNA can capture the target DNA (half red and half green). The blue chain represents the polymer generated by ATRP. (C) Reverse colorimetric DNA detection assay integrated with normal ATRP.23 | ||
The reversible deactivation in ATRP renders it feasible to amplify biodetection signals by repetitive addition of thousands of monomer units to one initiator immobilized on the spot where target biomolecules are captured. He and coworkers first demonstrated that normal ATRP could amplify the biodetection signals of DNA (Fig. 1B).22 In this method, DNA hybridization and ligation reactions resulted in the immobilization of ATRP initiator-coupled oligonucleotides on a solid surface where 100 nM of target DNA recognized complementary capture DNA (C). Subsequently, these initiators were activated upon the addition of the ATRP monomer solution including 2-hydroxyethyl methacrylate (HEMA), CuCl, CuBr2, and 2,2′-bipyridyl (bpy), triggering the polymerization. Here, the monomer solution and initiator-functionalized surface were purged with argon because the activator, CuI/L, could be oxidized by oxygen, and CuBr2 was added as a deactivator to ensure the formation of thick film by controlling the radical production rate and minimizing radical–radical termination. After the 5 h ATRP reaction, the growth of poly(2-hydroxyethyl methacrylate) (PHEMA) changed the surface opacity, making spots with target DNA distinguishable to the naked eye. To further improve the sensitivity of the DNA detection, He and coworkers devised a second ATRP reaction to form branched polymers on the spots with target DNA. Specifically, a polymeric anchor layer (PHEMA) was formed in the first 30-minute ATRP, followed by coupling of additional 2-bromoisobutyryl bromide initiators to the hydroxyl groups on the side chains of PHEMA for 20 minutes. Then, the second ATRP grafted polymer brushes from the new initiators for another 30 minutes, drastically improving the visibility of DNA hybridization in much shorter time (1 h). Using this method, 1 nM of target DNA could be readily detected with the naked eye, and there was no interference from one-base-mismatch and three-base-mismatch DNA sequences.
In addition to opacity change resulted from polymer film, normal ATRP can induce color change upon biodetection. One method is to incorporate ATRP-based biodetection into gold nanoparticle-based aggregation assays as demonstrated by He and coworkers (Fig. 1C).23 In this approach, core–shell gold nanoparticles were formed upon DNA hybridization so that only gold nanoparticles without polymer shell could aggregate, inducing color change from red to blue. Specifically, ATRP initiator-coupled oligonucleotides were immobilized on gold nanoparticles only when target DNA was captured on the nanoparticles. In a purged monomer solution, the ATRP initiators caused polymerization to form polymer shell that could prevent aggregation of nanoparticles with target DNA. The nanoparticle without ATRP initiators, on the other hand, aggregated during ATRP step because of strong binding interactions between transition metal divalent ions, such as CuII, and phosphate groups of DNA. The demonstration of colorimetric detection of 1 μM target DNA showed drastic color difference between positive (red) and negative (blue) samples, but detection limit of the colorimetric DNA assay was not reported. Similar ATRP-based colorimetric biodetection method was extended to immunosensing by Liu and coworkers.24 In their approach, ATRP initiator-coupled anti-rabbit antibodies were immobilized on gold nanoparticles when rabbit antibodies presented in the sample. Subsequently, polymerization of HEMA was carried out with CuBr and bpy as the ATRP catalyst to form polymer shell on ATRP-immobilized gold nanoparticles and prevent aggregation of the nanoparticles. At optimal conditions, the signal intensity from UV-Vis spectrophotometer was proportional to log scale of rabbit IgG concentration within the range of 3.3–166.7 pM, and the calculated detection limit was 0.2 pM.
As shown in above examples, normal ATRP could amplify biodetection signals by adding thousands of monomer units to immobilized initiator. However, many limitations have made this basic type of ATRP not feasible for practical use in the field. First, normal ATRP requires a few hours to form thick polymer film for sensitive biodetection because predominant deactivation reduces the rate of propagation. The fast reversible deactivation of active radical species in ATRP ensures high turnover number (the total number of monomers polymerized per total number of initiators), but it takes a long time to achieve it. Second, normal ATRP is not tolerant to oxygen because oxygen can scavenge the activator, CuI, and slow down the polymerization.20 Thus, the polymerization should be conducted under inert gas, which hinders the practical use of normal ATRP-based biodetection under atmospheric condition. Lastly, copper-based catalyst can cause high background noise in ellipsometric and colorimetric measurements because copper ions can be adsorbed to negatively charged molecules, such as DNA, reducing the sensitivity of biodetection.22,23,25,26
2.2 Activator generated by electron transfer ATRP (AGET ATRP)
Activator generated by electron transfer (AGET) ATRP is an oxygen-tolerant ATRP, in which reducing agents, such as ascorbic acid, remove oxygen in situ by reducing oxidatively stable CuII complex to generate active catalyst, CuI (Fig. 2A).27,28 Compared to other radical initiator-based approaches, such as reverse ATRP, simultaneous reverse and normal initiation (SR&NI) ATRP, and initiators for continuous activator regeneration (ICAR) ATRP, where free radicals reduce CuII complexes to active CuI complexes, AGET ATRP is not subject to non-specific free radical polymerization because it employs a reducing agent that does not form free radicals.29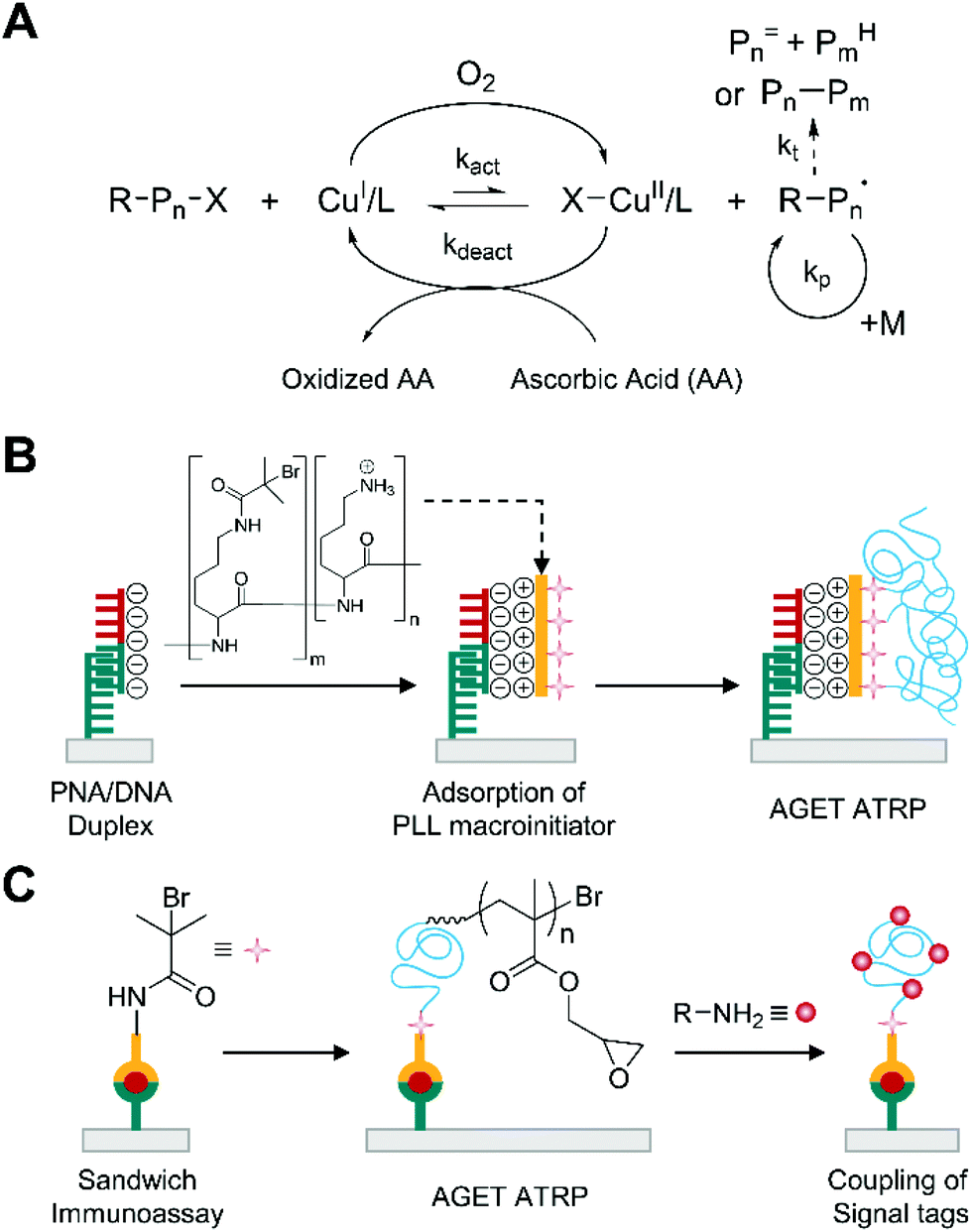 | ||
| Fig. 2 Biodetection with activator generated by electron transfer (AGET) ATRP. (A) Schematic description of AGET ATRP mechanism. Pn= and PmH are elimination and abstraction products, respectively, from termination by disproportionation. Pn–Pm is a recombination product. (B) AGET ATRP-based detection of DNA.31 Peptide nucleic acid (PNA) specifically forms duplex with target DNA (half red and half green), which then captures positively charged polylysine (PLL)-based macroinitiators by electrostatic interactions. The blue chains represent polymers generated by AGET ATRP. (C) AGET ATRP-based detection of protein.36,37 | ||
The high specificity to alkyl halide initiator and oxygen-tolerance of AGET ATRP allows sensitive biodetection without purging monomer solutions.29,30 He and coworkers first reported that the purge-free ATRP could amplify the biodetection signals upon DNA hybridization29 although it was a model half-sandwich assay where one-step binding events immobilized initiator-coupled target DNA. In this method, the hybridization of initiator-coupled single stranded DNA (ssDNA) with gold surface-immobilized peptide nucleic acid (PNA) resulted in the attachment of ATRP initiators to the surface. Subsequently, this initiator-immobilized surface was immersed into aqueous HEMA solution containing CuCl2 and tris(2-pyridylmethyl)amine (TPMA), which was the ligand that formed reasonably stable, but still reactive transition metal complexes. Polymerization was conducted at room temperature for 2 h in the sealed reaction vial immediately after addition of ascorbic acid. After thoroughly rinsing the surface with DMSO and methanol, the thickness of polymer film was measured by ellipsometry. There was a linear correlation between the film thickness and the amount of initiator-coupled ssDNA with a dynamic range over 10 nM–1 μM, achieving comparable limit of detection (≈0.2 pmol) to normal ATRP-based DNA detection without secondary amplification.22
To improve the sensitivity of AGET ATRP-based biodetection, the methods to increase the number of localized ATRP-initiators per captured target biomolecules were proposed. He and coworkers demonstrated the use of polylysine (PLL) as a carrier to localize multiple alkyl halide initiators to surface-immobilized PNA/DNA duplexes for additional signal amplification in AGET ATRP-based DNA detection (Fig. 2B).31 In this method, optimization of the number of ATRP initiators and residual amino groups per PLL chain was critical because over-modification could reduce electrostatic interaction of PLL chain with negatively charged captured DNA and accessibility of initiators because of steric hindrance. Furthermore, the ATRP initiator is hydrophobic, so the over-modification could increase non-specific adsorption of the macroinitiator to the gold surface, leading to high background noise. Using macroinitiators with the optimized ratio of ATRP initiators to amino groups, a linear correlation at 1 nM–100 nM DNA and a detection limit of 1 nM, or 3 fmol, were obtained by ellipsometry, which implies approximately 60-fold enhancement in detection limit compared to one-initiator-per-DNA case.29 For protein detection, however, PLL-based approach is not effective because not all proteins are as strongly negatively charged as DNA. To address this problem, Liu and coworkers demonstrated that the use of dual functional macroinitiators could improve the sensitivity of AGET ATRP-based protein detection.32 Inspired by dual functional macrophotoinitiators from Bowman group,33 Liu and coworkers used poly(acrylic acid-co-acrylamide) backbone and modify its side chain with ATRP initiators and streptavidin. The resultant macroinitiator was used to detect biotinylated antibodies immobilized on a gold surface after sandwich immunoassays. Using this macroinitiator, the optimal polymerization time was only ten minutes, which was enough to detect 67 pM human IgG with the naked eye. A linear correlation at 6.7 pM–670 nM human IgG and a detection limit of 0.9 pM were obtained by contact angle measurement.
AGET ATRP is a standalone method to detect biomolecules, but it can also be coupled with other quantitative biodectection tools such as electrochemical (EC) and electrochemiluminescence (ECL) sensors to enable ultrasensitive biodetection.34–38 Expanding on their earlier works,34,35 Liu and coworkers demonstrated that various signals tags for EC and ECL-based sensing could be coupled to a great amount of epoxy groups of poly(glycidyl methacrylate) (PGMA) generated by AGET ATRP upon molecular recognition events (Fig. 2C).36,37 Glycidyl methacrylate (GMA) was chosen for AGET ATRP-based protein detection because of its simple one-step coupling to amino-modified signal tags such as aminoferrocene (FcNH2), horseradish peroxidase (HRP), and 2-(diisopropylamino)ethylamine (DPEA). To couple these signal tags with molecular recognition events, AGET ATRP is carried out on initiator-functionalized solid substrates for 2 h with monomer solution including CuCl2, bpy, GMA, and ascorbic acid. After rigorous rinsing with acetone to remove unreacted monomers, the solid substrates are incubated with signal tag solutions for 5–10 h before EC or ECL-based sensing. Using these methods, ultrasensitive detection of clinically relevant antigens such as PSA and CEA was achieved (Table S1†) although all electrochemical measurements required inert atmosphere, diminishing the advantage of using AGET ATRP.
As can be seen in above examples, AGET ATRP successfully resolved the need for purging the monomer solution with inert gas in ATRP-based biodetection without sacrificing limit of detection in various assays. As a result, it has been used in various biodetection applications to amplify signals from molecular recognition events. However, long polymerization time, limited oxygen tolerance, and non-specifically adsorbed transition metal catalysts have limited the use of AGET ATRP in biodetection. To achieve high sensitivity, the polymerization time is often set as a few hours and coupling of signal tags for EC- and ECL-sensors takes additional few hours. Although the macroinitiator approaches can reduce the polymerization time significantly, large efforts to rapidly remove non-specifically bound macroinitiators should be made to minimize high background noise for ultrasensitive biodetection in a few minutes. Because of its limited oxygen tolerance, AGET ATRP requires sealed reactors to prevent continuous oxygen diffusion from atmosphere into the reaction system. Moreover, rigorous rinsing with organic solvents is required to remove unreacted monomers and residual catalysts for optimal signal-to-noise ratio, which might be inconvenient to end-users.
2.3 Electrochemically mediated ATRP (eATRP)
Electrochemically mediated ATRP (eATRP) is an advanced version of oxygen-tolerant ATRP compared to AGET ATRP in that polymerization rate can be readily controlled by electrochemical means.39 In the eATRP, a targeted fraction of the air stable CuII complexes are reduced to active CuI complexes by applying certain applied potential or current instead of using reducing agents such as ascorbic acid in AGET ATRP (Fig. 3A).39 The precise control on the ratio of [X–CuII/L]/[CuI/L] helps to keep the radical concentration low and suppress the disproportionation of CuI/L in water, reducing the rate of bimolecular termination and allowing smaller amount of catalyst loading compared to AGET ATRP in aqueous solution.40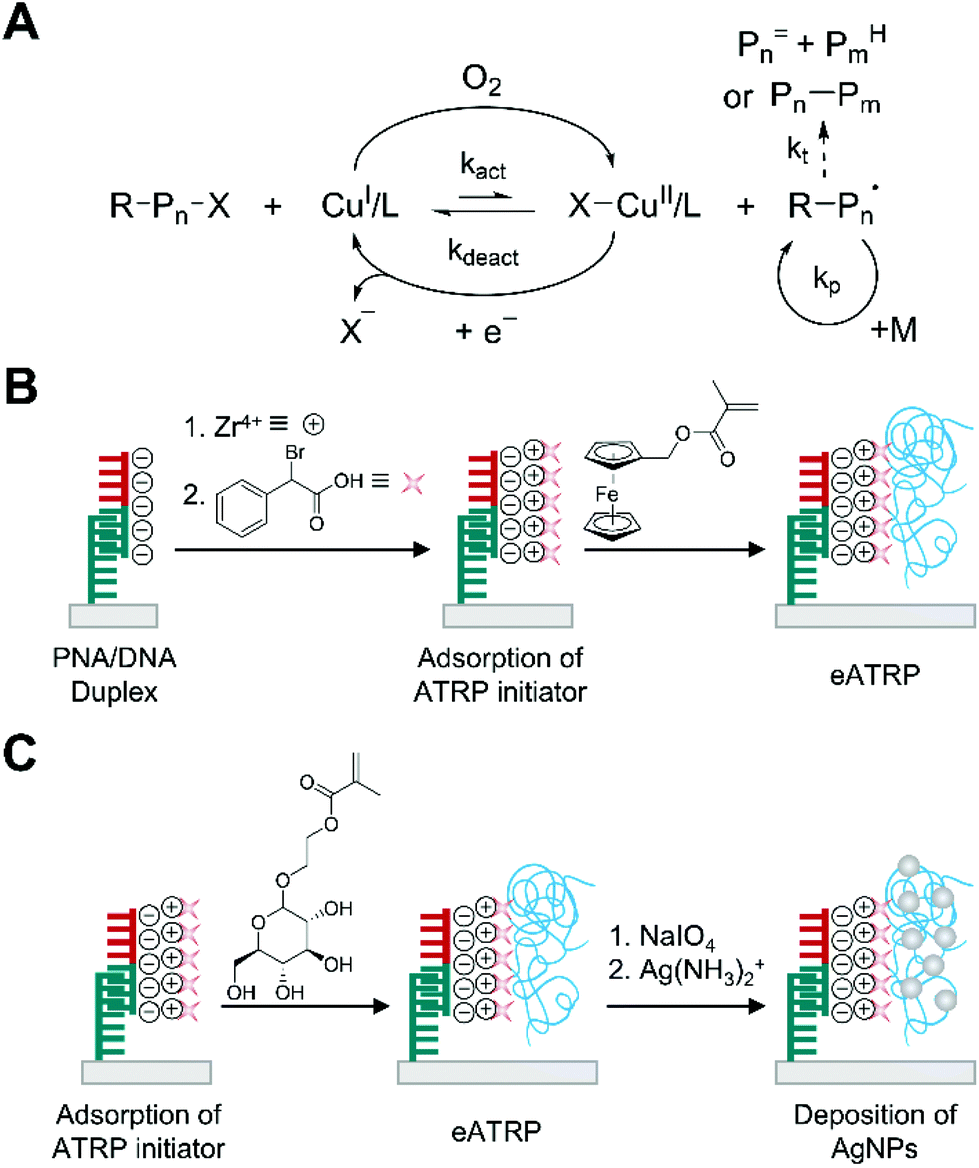 | ||
| Fig. 3 Biodetection with electrochemically-mediated ATRP (eATRP). (A) Schematic description of eATRP mechanism. Pn= and PmH are elimination and abstraction products, respectively, from termination by disproportionation. Pn–Pm is a recombination product. (B) eATRP-based electrochemical detection of DNA.41 Peptide nucleic acid (PNA) specifically forms duplex with target DNA (half red and half green). ATRP initiators are attached to phosphate groups of DNA through phosphate-Zr4+-carboxylate chemistry. The blue chains represent polymers generated by eATRP. (C) Silver deposition on polymer generated by eATRP to enhance sensitivity of electrochemical biodetection.44 | ||
With its well-controlled polymerization process, eATRP recently has been employed to generate electroactively labelled polymers for electrochemical detection of DNA and protein activity in much faster and efficient ways compared to previous methods.41–43 Hu and Wang et al. first demonstrated the use of eATRP to polymerize an electroactively labelled monomer, ferrocenylmethyl methacrylate (FMMA) from negatively charged spots of single stranded DNA (ssDNA) captured by PNA on a gold surface (Fig. 3B).41 In their demonstration, the captured DNA was treated with ZrOCl2 and α-bromophenylacetic acid (BPAA) sequentially to attach the ATRP initiators by using phosphate-Zr4+-carboxylate chemistry. From these immobilized initiators, FMMA started to grow through eATRP, where the activator, CuI/tris(2-dimethylaminoethyl)amine (Me6TREN) was generated from CuII/Me6TREN by electroreduction under potentiostatic conditions. It is interesting to note that the activators, CuI/Me6TREN, could be localized at the electrode surface because of electrostatic interaction, significantly enhancing the kinetics of polymerization. In their optimization study, Hu and Wang et al. found that the oxidation current from ferrocene tags increased drastically only during the first 20 minutes of eATRP presumably because of reduced accessibility of initiators as polymerization proceeded. Under optimal conditions including 30 minutes of eATRP time, a linear correlation between the logarithm of target ssDNA concentration and the oxidation current was obtained over 7 orders of magnitude from 0.1 fM to 0.1 nM. The limit of detection was calculated to be 0.072 fM.
The improvement in the limit of detection for ssDNA has been achieved by growing silver nanoparticles over the polymers generated by eATRP in the presence of ssDNA (Fig. 3C).44 Sun et al. used glycosyloxyethyl methacrylate (GEMA) as a monomer to localize a large number of polysaccharides to the specifically formed PNA/DNA duplexes via the same phosphate-Zr4+-carboxylate chemistry and eATRP as in the previous example. Subsequently, sodium periodate (NaIO4) oxidized the polysaccharides to polymerized aldehydes. Each aldehyde on the polymer chains could reduce silver ion to metallic silver, which further grew by Ostwald ripening. Under optimal conditions including 30 minutes of eATRP, 2 h of NaIO4 oxidation, and 1 h of silver mirror reaction, a linear correlation between the logarithm of target ssDNA concentration and the oxidation current was obtained over 6 orders of magnitude from 10 aM to 10 pM. The limit of detection was estimated to be 5 aM.
As shown in above examples, the optimized reaction time of eATRP is much shorter than AGET ATRP, which could be because the ratio of [X–CuII/L]/[CuI/L] is precisely controlled by electrochemical means and localization of activators to electrode surface can enhance the kinetics of polymerization. Coupled with EC-sensors, eATRP-based biodetection methods showed very promising results with very long dynamic ranges and strikingly low detection limits for DNA detection. However, given that the target DNA has more than 20 phosphate groups on its chain, it is possible that the use of phosphate-Zr4+-carboxylate chemistry to immobilize ATRP initiators could lead to this ultrasensitivity. Therefore, it is expected that this novel platform would not provide the same sensitivity for detection of proteins. In addition to its limited applicability, eATRP's cumbersome reaction setup and limited oxygen tolerance should be addressed for its practical use in the field. The setup requires 10 mL aqueous solution including 20 vol% dimethylformamide (DMF) in an electrochemical cell, which can be difficult to handle by non-specialists. Moreover, the cell should not be exposed to air because eATRP is not tolerant to large amount of oxygen diffused from atmosphere. To address these issues, small volume (<100 μL) eATRP has been developed on a screen-printed electrode with enzyme-mediated deoxygenation, but the need for more than 1000 ppm of copper catalyst may diminish the benefits.45
2.4 Fluorogenic ATRP
Fluorogenic ATRP is a novel approach designed for polymerization-based detection of analytes to eliminate the need for time- and labor-intensive post-modification of polymers with signals tags and any instruments to detect the growth of polymer.46 Cooley and coworkers synthesized methacrylamide derivatives of polycyclic aromatic hydrocarbon (PAH) such as pyrene, anthracene, and acridine. These fluorogenic monomers were designed to be non-fluorescent until polymerized because of fluorescence quenching by the covalently linked α,β-unsaturated amide, but to recover its fluorescence when the unsaturated bonds participate in polymerization (Fig. 4A). Cooley and coworkers used another hydrophilic co-monomer, oligo(ethylene glycol) methyl ether methacrylate (OEGMA), to make the generated polymers water-soluble and prevent self-quenching of the fluorogenic monomers on the chains by inserting gaps between the fluorophores. In the fluorogenic ATRP, activators regenerated by electron transfer (ARGET) ATRP condition47 with CuIIBr2, TPMA, NaCl, and ascorbic acid was used to ensure that the polymerization was compatible with biological environment while using a reduced amount of copper catalyst compared to AGET ATRP. Under this condition, rigorous nitrogen purging of reaction mixture was required, otherwise trace amount of oxygen could lead to high background fluorescence by initiating polymerization without alkyl halide initiators. Using optimal conditions of the fluorogenic ATRP, Cooley and coworkers demonstrated the detection of streptavidin coated magnetic beads in aqueous solution with biotinylated ATRP initiators (Fig. 4B). After 24 h of the polymerization at 30 °C with the released ATRP initiators that had been specifically captured on the beads, fluorescence analysis indicated that sub-nanomolar detection limit was achieved.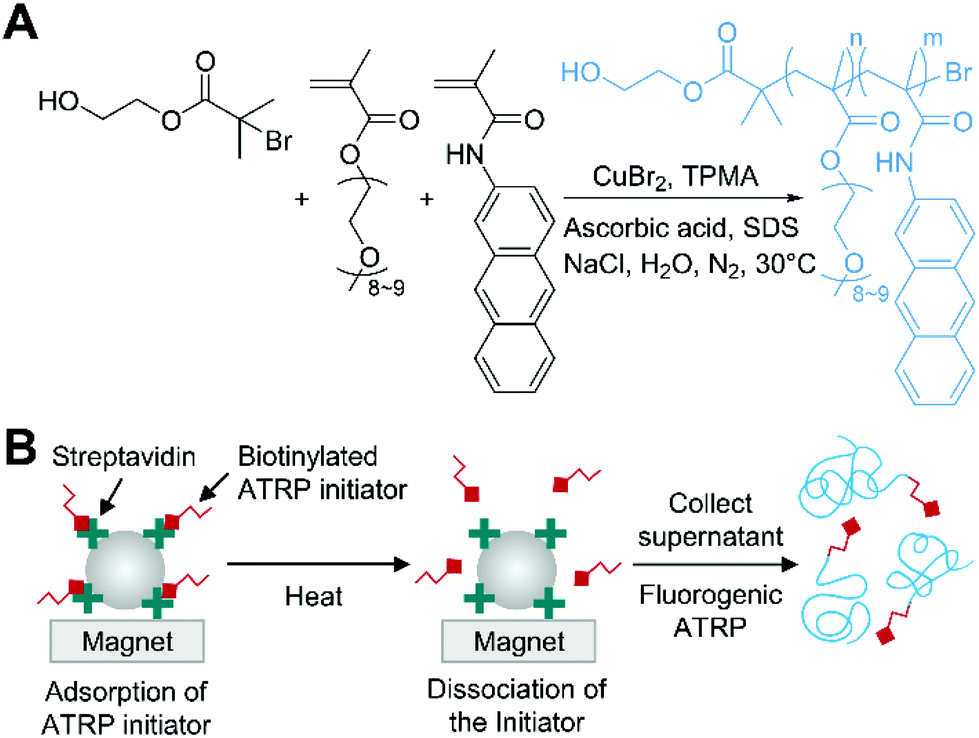 | ||
| Fig. 4 Biodetection with fluorogenic ATRP.46 (A) Schematic description of fluorogenic ATRP in aqueous media with anthracene monomer. (B) Fluorogenic ATRP-based detection of streptavidin-coated magnetic beads. The blue chains represent polymers generated by fluorogenic ATRP. | ||
Fluorogenic ATRP is a promising approach to sensitive and easy-to-use biodetection in that the fluorescence signals can be directly proportional to the degree of polymerization and it does not require any modification of resultant polymers with signal tags. However, the current platform needs several improvements to be ready for practical applications. First, the method is not tolerant to oxygen. The current fluorogenic ATRP requires rigorous nitrogen purging to perform the polymerization not only because oxygen can scavenge propagating radicals, but also because a trace amount of oxygen can initiate polymerization even in the absence of initiators, which leads to high background signals. Background signals are undesirable as they limit the sensitivity and responsive range of biodetection assays and can be interpreted as false positive results. Thus, efforts should be made to realize purge-free fluorogenic polymerization. Second, the reaction rates are slow. To enhance the reaction kinetics, increasing temperature to 60 °C was proposed,46 but the high temperature yielded high background signals even with rigorous degassing. Another way to solve this problem would be to synthesize water-soluble fluorogenic monomers. In the current formulation, the molar ratio of hydrophilic co-monomer, OEGMA, to fluorogenic monomers is approximately one thousand to one mainly because of low solubility of fluorogenic monomers in aqueous solution. Thus, strategies to design more hydrophilic fluorogenic monomers would be a promising future direction to improve the reaction kinetics by increasing the concentration of fluorogenic monomers in the reaction mixture.
2.5 Biocatalytic ATRP
Biocatalytic ATRP has been developed as a greener alternative to conventional ATRP. Conventional ATRP requires transition metal catalysts, often copper-based catalysts, which are toxic and difficult to remove from polymer products.48 In biocatalytic ATRP, the metal catalysts are replaced by metalloproteins, which have metal-containing active sites buried in their protein domains, allowing synthesis of metal-free polymers with controlled molecular weights.49–54 Moreover, biocatalytic ATRP with iron-containing proteins and bioinspired iron-based catalysts is especially advantageous to synthesize polymers for biological applications because iron has low toxicity and high biocompatibility compared to copper.55In addition to synthetic applications, biocatalytic ATRP can be used to detect clinically relevant biocatalysts via polymerization-based signal amplification. Bruns and coworkers recently proposed a new method to diagnose malaria using hemozoin-catalyzed ATRP (Fig. 5).56 Hemozoin is a biocrystal that consists of centrosymmetric μ-propionate dimers of heme. It is a powerful biomarker for malaria because hemozoin is generated during the digestion of hemoglobin by malaria parasites regardless of erythrocyte stages and type of Plasmodium species.57,58 Bruns and coworkers discovered that hemozoin could catalyze the polymerization of N-isopropylacrylamide (NIPAAm) with alkyl halide initiators presumably through an ATRP mechanism.56 Using this principle, they designed an assay to detect malaria parasites using precipitation of poly(N-isopropylacrylamide) (PNIPAAm) at 37 °C above its lower critical solution temperature (32 °C). In this assay, intact malaria parasites including hemozoin, hemoglobin, and free heme were isolated from blood samples spiked with varying amounts of P. falciparum-infected red blood cells (iRBCs) and were solubilized in basic solution (pH 13). Then, the solubilized biocatalysts were added to buffered monomer solution (final pH 6.9). Under the ARGET ATRP conditions, the rate of turbidity increase was proportional to the amount of iRBCs in blood and the detection limit was 9.7 iRBCs per μL, which corresponds to 4.7 nM hemozoin.56 Furthermore, the stability of each reagent was investigated for two months at 50 °C, showing no decrease in reactivity.
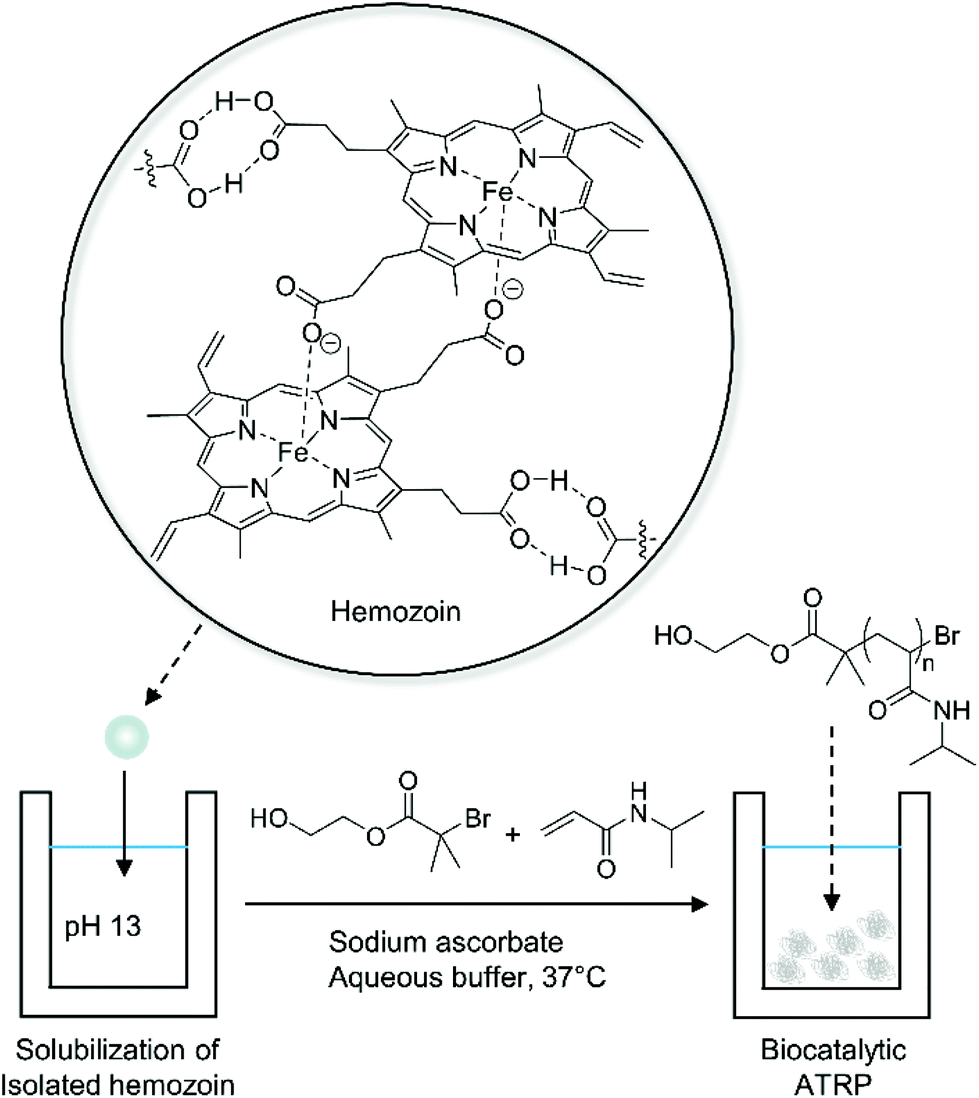 | ||
| Fig. 5 Biocatalytic ATRP of N-isopropylacrylamide (NIPAAm) for detection of hemozoin.56 | ||
Given that hemoglobin itself can catalyze ATRP, Bruns and coworkers also demonstrated the use of hemoglobin-catalyzed precipitation polymerization to quantify the amount of hemoglobin in biological fluids.59 In this method, the polymerization conditions were the same as the hemozoin-catalyzed ATRP56 except that sample preparation method and buffer pH were slightly different. The dose–response curve between logarithmic hemoglobin concentration and the rate of turbidity increase showed that the rate of turbidity increase was positively correlated with the concentration of hemoglobin although the rate reached a plateau at high concentration.59 According to authors, the plateau could appear likely because hemoglobin precipitated with PNIPAAm as polymerization proceeded. At low hemoglobin concentrations, the detection limit was 100 nM in buffer solution. In blood plasma and urine, on the other hand, the detection limit increased to 7.6 μM and 2.6 μM respectively, implying that the radical precipitation polymerization was affected by proteins and other biomolecules in the physiological fluids.
Biocatalytic ATRP is a new approach to detection of specific biomarkers with catalytic activity. As demonstrated by Bruns and coworkers, the biocatalyzed precipitation polymerization is a promising technology for point-of-care malaria56 and hemoglobin59 tests given that all reagents are stable during long-term storage at 50 °C and the method induces extinction changes depending on the concentration of biocatalysts. Although there are still many problems to be addressed for practical applications such as time- and labor-intensive isolation of the biocatalysts from blood samples and background polymerization, it is envisaged that more user-friendly methods for the isolation steps and more controlled polymerization with decreased background signals can be developed and additional clinically relevant biocatalysts may be identified.
3. Reversible addition–fragmentation chain transfer (RAFT) polymerization
3.1 Thermally initiated RAFT polymerization
Reversible addition–fragmentation chain transfer (RAFT) polymerization is another type of reversible-deactivation radical polymerization explored for polymerization-based biodetection to address several limitations of ATRP and broaden the biodetection platforms based on polymerization. The main problem in ATRP is the use of transition metal (usually copper) complexes. Non-specific adsorption of metal ions to surface-bound DNA molecules can lead to undesirable background polymerization that is not related to the target, and the toxicity of transition metal complexes could potentially limit the use of ATRP in future biomedical applications.25,26 RAFT polymerization, on the other hand, employs a fast reversible transfer of chain transfer agents (CTA), typically a thiocarbonylthio group (Z–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)S–R), between propagating radicals and dormant species (Z–C(S)S–R or Z–C(S)S–Pn) to prolong the lifetime of growing chains (Fig. 6A).60,61 This metal-free polymerization chemistry could make RAFT an alternative to ATRP in broader biosensing applications.
S)S–R), between propagating radicals and dormant species (Z–C(S)S–R or Z–C(S)S–Pn) to prolong the lifetime of growing chains (Fig. 6A).60,61 This metal-free polymerization chemistry could make RAFT an alternative to ATRP in broader biosensing applications.
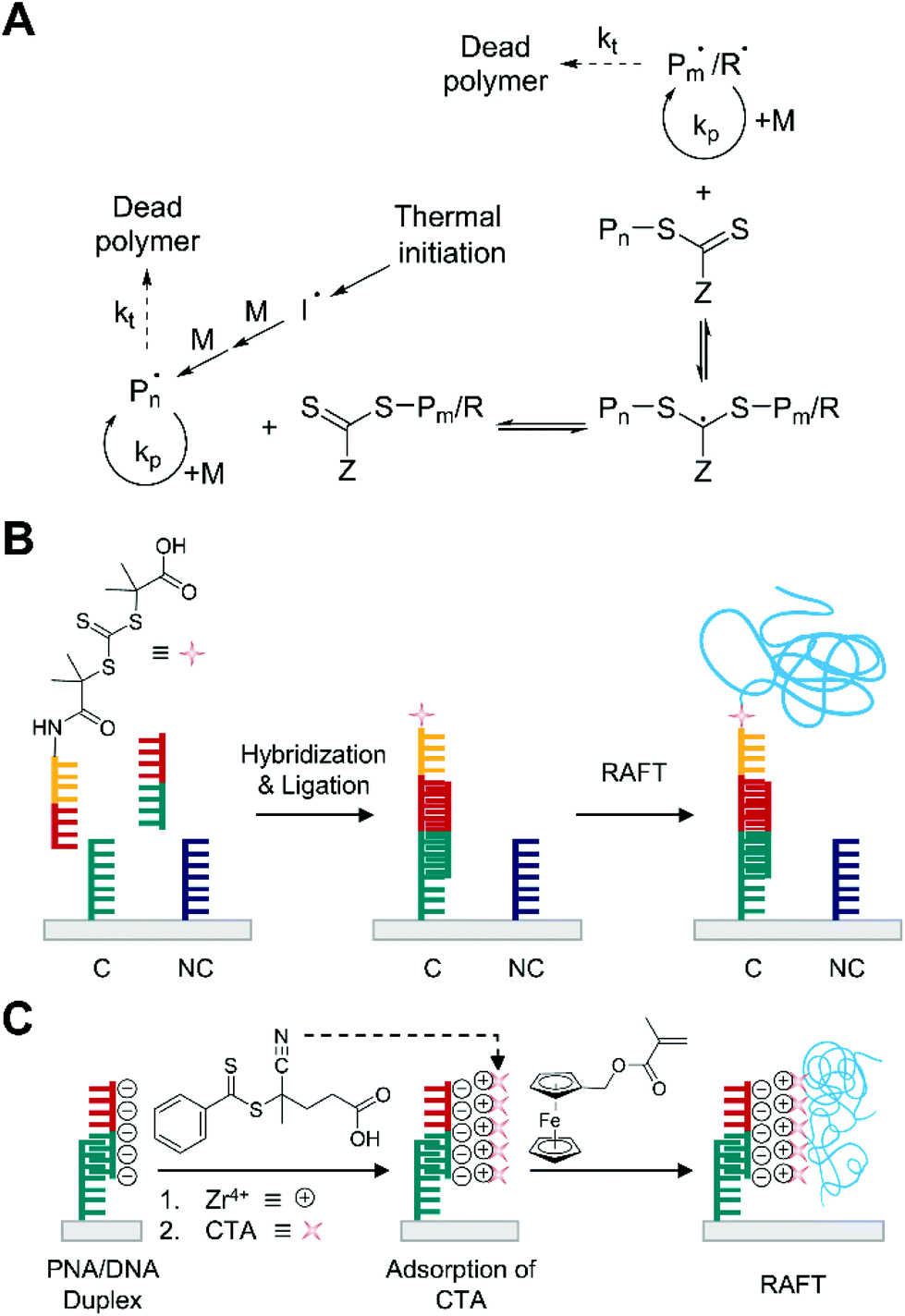 | ||
| Fig. 6 Biodetection with thermally initiated reversible addition–fragmentation chain transfer (RAFT) polymerization. (A) Schematic description of RAFT mechanism. Pn and Pm represent propagating chains growing from initiating radical (I˙) and R-group radical (R˙) from RAFT agent, respectively. The dead polymer includes disproportionation and recombination products. (B) RAFT-based detection of DNA.25 Both complementary DNA, C, and non-complementary DNA, NC, are used as capture molecules, but only complementary DNA can capture the target DNA (half red and half green). The blue chains represent polymers produced by thermally initiated RAFT polymerization. (C) Thermally initiated RAFT-based electrochemical detection of DNA.63 Peptide nucleic acid (PNA) specifically forms duplex with target DNA. Chain transfer agents (CTA) are attached to phosphate groups of DNA through phosphate-Zr4+-carboxylate chemistry. Thermally initiated RAFT polymerization grafts polymers (blue chains) from the CTAs. | ||
Thermally initiated RAFT polymerization has been employed in visual DNA sensing platforms, achieving lower background signals and more sensitive biodetection than ATRP-based methods.25,62 He and coworkers pioneered the use of surface-initiated RAFT to amplify biodetection signals upon DNA hybridization (Fig. 6B).25 In this method, CTA-conjugated detection probe was designed to be immobilized on the gold surface where the target DNAs were specifically hybridized to a capture probe, C. After 1 h of ligation, a nitrogen-purged aqueous monomer solution including OEGMA and azobisisobutyronitrile (AIBN) was added to the CTA-functionalized gold surface in a glass container. Thermal initiation at mild temperature (30 °C) allowed polymerization to occur although the initiation would be slow because AIBN has high decomposition temperature (64 °C) for 10 h half-life. With 1 μM of the target DNA, RAFT could generate a much thicker POEGMA film (≈80 nm) in two hours than the 15 nm film generated after 5 h using ATRP. As expected from the prolonged lifetime of growing chains in RAFT, a longer polymerization time (5 h) yielded much thicker film (≈200 nm) in the presence of 1 μM of the target DNA. A linear correlation between logarithmic DNA concentration and film thickness was demonstrated in the range 1 fM–1 μM while allowing even 1 fM target DNA to be easily detected by the naked eye.
In addition to enabling visual biodetection, thermally initiated RAFT polymerization has been proposed as an alternative signal amplification strategy to ATRP for electrochemical biosensors.63,64 RAFT can reduce background signals from non-specific deposition of transition metals on electrodes, which may interfere with electrochemical measurements. Niu and coworkers demonstrated that thermally initiated RAFT polymerization could be exploited as a novel signal-amplifying tool for sensitive electrochemical detection of DNA (Fig. 6C).63 Specifically, target DNA was captured by PNA on a gold electrode, and the PNA/DNA duplexes were treated with ZrOCl2 and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPAD) sequentially to attach the CTA by using phosphate-Zr4+-carboxylate chemistry. The CTA-functionalized substrate was soaked in a purged monomer solution containing ferrocenylmethyl methacrylate (FcMMA) and 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044), which is selected as a thermal initiator because of its good water solubility and low decomposition temperature (44 °C) for 10 h half-life. After the 1.5 h RAFT polymerization at 47 °C, it was found that a greater extent of polymer-grafting to the electrode surface was observed than that obtained after eATRP. Under optimal conditions, the peak current was linearly correlated with the logarithmic DNA concentration over the range from 10 aM to 10 pM, and the detection limit was calculated to be 3.2 aM. This detection limit is approximately 20-fold more sensitive than a similar electrochemical biodetection platform amplified by eATRP.41
In light of these examples, RAFT polymerization is advantageous for sensitive biodetection compared to ATRP because it prevents interference from non-specifically bound metal complexes in signal analysis. However, the fact that thermally initiated RAFT requires high temperature and oxygen-free environment for rapid and effective initiation may limit its use in biodetection applications. To address these problems, enzyme-mediated deoxygenation65 or mild oxygen-tolerant initiation methods such as photoinduced electron/energy transfer (PET)-RAFT66 could be useful.
3.2 Electrochemically mediated RAFT (eRAFT) polymerization
Electrochemically mediated RAFT (eRAFT) polymerization has been recently explored in biosensing applications as an alternative to thermally initiated RAFT, which requires heating and temperature control during the polymerization. In eRAFT polymerization, electrical means such as applied potential or current enable the controlled production of radicals by reducing diazonium salts such as 4-bromobenzenediazonium (BrPhN2+) without affecting CTAs,67 so the polymerization can occur at ambient temperature.This mild reaction condition of eRAFT allows more convenient DNA biodetection platforms. Hu et al. demonstrated the use of eRAFT polymerization to amplify electrochemical signals resulting from the sequence-specific recognition of target DNAs (Fig. 7A).68 Specifically, CPAD, a carboxylate-containing chain transfer agent, was localized on a gold surface, where target DNA was captured by immobilized PNA, and a subsequent 1.5 h eRAFT polymerization grafted electroactively labelled polymer (PFcMMA) in the presence of CPAD on the surface. In the eRAFT step, initiating radicals were generated from electroreduction of BrPhN2+ at −0.3 V, which is far more positive than reduction peak potential (−1.0 V) of CPAD, to prevent irreversible cleavage of the weak C–S bond of the CTA via two-electron reduction that could generate anions instead of radicals (Fig. 7B).67 The resulting polymers localized numerous electroactive ferrocene tags on the gold electrode surface, improving the sensitivity of electrochemical DNA detection. Under optimal conditions, a linear correlation between logarithmic DNA concentration and sensing signals ranged from 10 aM to 10 pM, and a detection limit of 4.1 aM was achieved. The detection limit is similar to that of the electrochemical DNA sensor based on thermally initiated RAFT polymerization, but eRAFT polymerization enables the sensors to be operated under mild temperatures, which could make them more practical in the field.
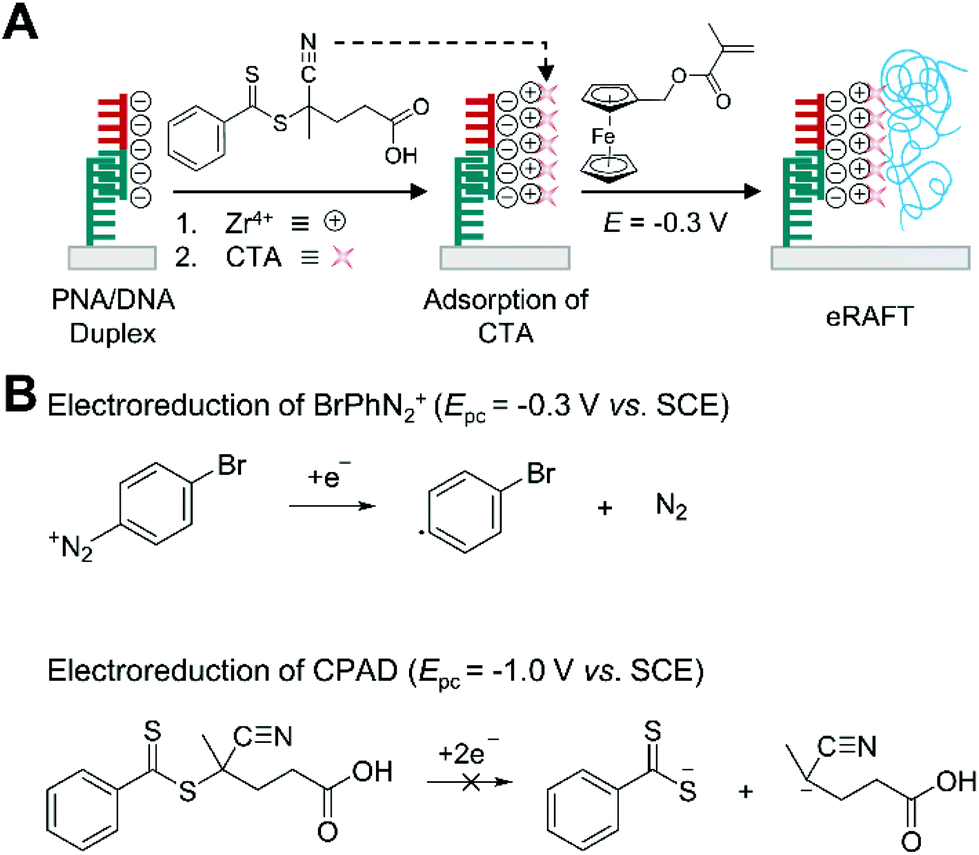 | ||
| Fig. 7 Biodetection with electrochemically mediated RAFT (eRAFT) polymerization. (A) eRAFT-based electrochemical detection of DNA.68 Peptide nucleic acid (PNA) specifically forms duplex with target DNA (half red and half green). Chain transfer agents (CTA) are attached to phosphate groups of DNA through phosphate-Zr4+-carboxylate chemistry. Electroreduction of 4-bromobenzenediazonium (BrPhN2+) generates radicals without affecting 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPAD). The blue chains represent polymers produced by eRAFT polymerization. (B) Electroreduction scheme and cathodic peak potentials (Epc) of BrPhN2+ and CPAD. | ||
The selective electroreduction of BrPhN2+ allowed RAFT polymerization-based DNA detection under mild temperature, and eRAFT yielded improved sensitivity compared with eATRP, likely because of its metal-free nature. Nonetheless, the need for a long polymerization time (1.5 h) and deoxygenation could limit the use of eRAFT in future biosensing applications. The rate of RAFT polymerization strongly depends on the rate of radical generation.69 Faster production of radicals may be achieved by applying more negative potentials, but the potential window could be too narrow to prevent electroreduction of CTAs.67,68 Moreover, the high reactivity of bromophenyl radicals (BrPh˙) causes electrografting of branched bromobenzenes on the electrode surface, which can decrease its conductivity and, thus, the rate of radical production under potentiostatic conditions.67 Therefore, it would be helpful in reducing the polymerization time to use galvanostatic conditions for eRAFT and optimize the applied currents for more efficient initiation. Successful deoxygenation in eRAFT polymerization-based biosensors may be the most crucial requirement for adoption. Currently, efforts have been made to find a good mediator that can be continuously reduced to reactive species at the electrode and then react with CTA to form propagating radicals.70 We envision that more effective mediators will be identified and promote faster and more oxygen-tolerant eRAFT polymerization while completely avoiding the CTA decomposition.
4. Redox-initiated free radical polymerization
4.1 Enzyme-mediated free radical polymerization (EFRP)
Enzyme-mediated free radical polymerization (EFRP) is a chain growth polymerization that employs enzymes to catalyze redox initiation reactions (Fig. 8A).71 The enzymatic redox initiation is direct when oxidized (Aox) or reduced enzymatic substrate (Bred) itself is initiating radicals (I˙). The indirect enzymatic redox initiation, on the other hand, requires additional redox reactions to generate radicals from oxidized (Aox) or reduced enzymatic substrate (Bred). Commonly used enzymes for EFRP are oxidoreductases, which catalyze the electron transfer from one molecule to another, such as horseradish peroxidase (HRP)72–74 and glucose oxidase.75,76 Among them, HRP has been the most widely used in EFRP because of its rapid radical production by reducing hydrogen peroxide while oxidizing a hydrogen-donating organic substrate in mild aqueous conditions.71,77 In many cases, glucose oxidase requires another redox initiation system such as Fenton chemistry because the enzyme uses oxygen as the oxidant to produce hydrogen peroxide, not radicals, but recently it has been confirmed that glucose oxidase can directly produce carbon-centered radicals by reducing N-hydroxy-5-norbornen-2,3-dicarboximide (HNDC).78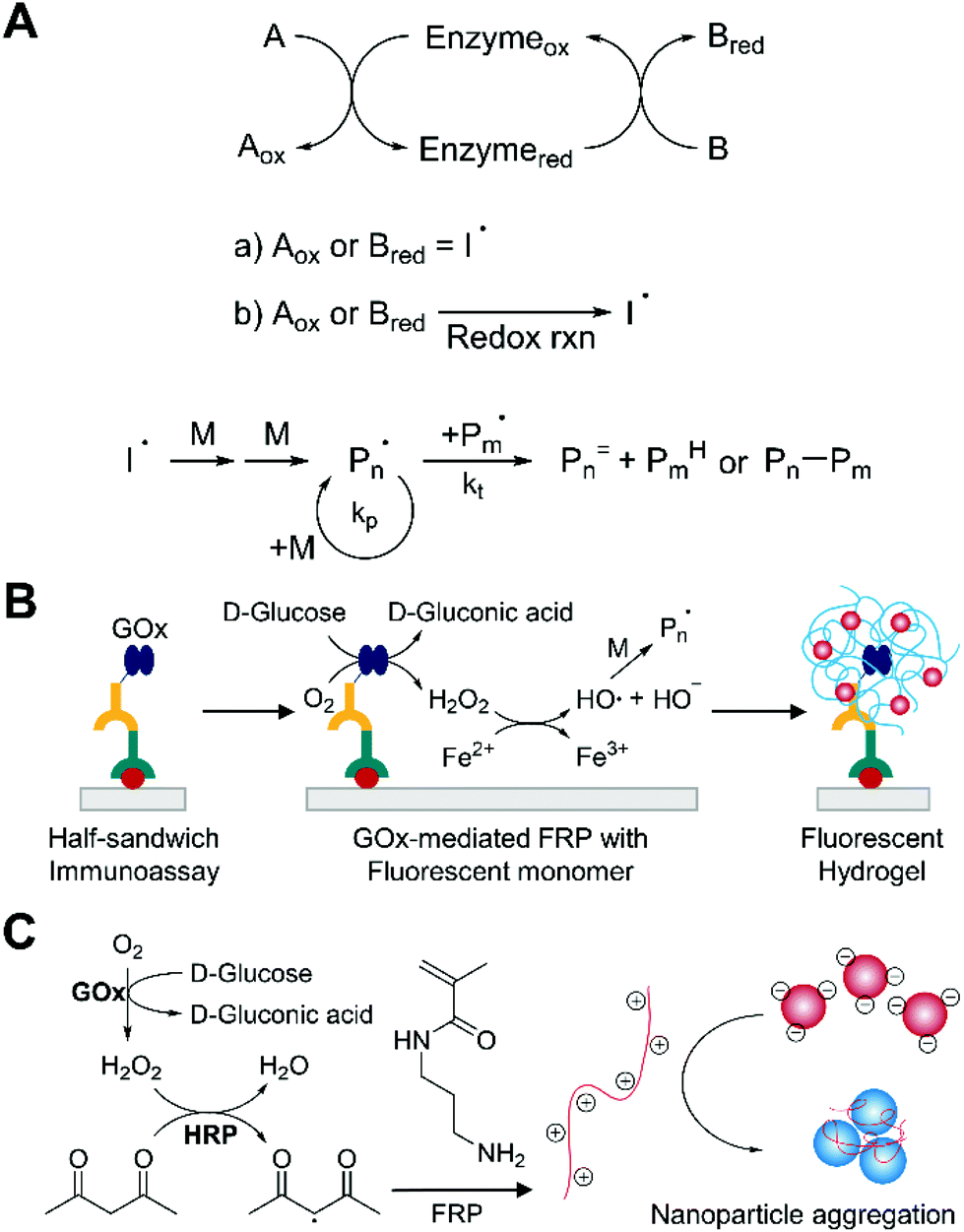 | ||
| Fig. 8 Biodetection with enzyme-mediated free radical polymerization (EFRP). (A) Schematic description of EFRP. (a) Direct enzymatic redox initiation. (b) Indirect enzymatic redox initiation. Free-radical polymerization is more subject to bimolecular termination than reversible-deactivation radical polymerization. (B) EFRP-based detection of protein.79 Specifically bound glucose oxidase (GOx) catalyzes the production of hydrogen peroxide (H2O2), which is converted to hydroxyl radicals by Fenton chemistry. The blue chains with red dots represent fluorophore-embedded polymer network generated by EFRP. (C) Gold nanoparticle-based aggregation assay integrated with EFRP.80 Horseradish peroxidase (HRP) produces free radicals using H2O2 and acetylacetone. The polymer (red chain with positive charges) induces aggregation of the negatively charged nanoparticles. | ||
Applications of EFRP to biodetection combine two signal amplification strategies to improve the sensitivity of biosensors. The methods employ the catalytic efficiency of enzymes to amplify the number of radicals and propagating chains per molecular recognition event. For example, Bowman and coworkers demonstrated the use of glucose oxidase as an indirect redox initiator for protein detection (Fig. 8B).79 Here, the enzymes selectively bound to the surface generated hydrogen peroxide from glucose and oxygen when they were contacted with an acrylate monomer solution including glucose, oxygen, Fe2+ salts, and fluorescently labeled monomer. The hydrogen peroxide was subsequently converted to hydroxyl radical via Fenton chemistry. In the propagation step, the fluorescent monomer units copolymerized with hydroxyethyl acrylate (HEA) and PEGDA, resulting in fluorescently-labelled hydrogels that could be detected visually or using a fluorescence measurement. By using the oxygen-consuming enzyme as a label, polymerization could occur even in the presence of atmospheric oxygen after the concentration of dissolved oxygen was reduced sufficiently by glucose oxidase. It is interesting to note that longer polymerization time provided lower detection limit without increasing non-specific signals. The authors pointed out that in contrast with other enzymatic amplification systems, EFRP-based biodetection is not challenged by false positive results even with longer amplification times. Because the amount of non-specifically bound enzymes is not enough to consume excess oxygen in the monomer solution on practical time scales, free radical quenching by oxygen totally inhibits the polymerization. Under optimal conditions including 4 h of polymerization time, authors demonstrated protein detection with transforming growth factor-beta (TGF-β) at concentrations >156 ng mL−1 (6.24 nM).
Given its ability for oxygen-tolerant radical polymerization, glucose oxidase has also been coupled with HRP for EFRP-based biodetection. Stevens and coworkers developed a new oxygen-tolerant colorimetric assay platform based on gold nanoparticle aggregation triggered by EFRP in the presence of HRP (Fig. 8C).80 In this formulation, glucose oxidase consumed oxygen and produced hydrogen peroxide (H2O2). Using this H2O2, HRP could oxidize acetylacetone and produce free radicals. When oxygen concentration decreased to sufficiently low level, polymerization of 3-aminopropyl methacrylamide could occur. The resultant polymers were positively charged, so the negatively charged nanoparticles were aggregated by electrostatic interactions between them, leading to change in color. The nanoparticle surface chemistry was designed to be tolerant to monomers at high concentration, but only be subject to aggregation by polymers as short as 27 monomer units. Under optimal conditions, the sensitivity for HRP detection was limited to single digit nanomolar concentration. The authors suggested that the low sensitivity was not because of the ability of HRP to generate radicals, but because of high background signals resulted from radical production in the absence of HRP. To test this hypothesis, they designed a reverse assay to detect catalase, which catalyzed decomposition of hydrogen peroxide. With catalase, hydrogen peroxide was consumed immediately after production by glucose oxidase, preventing polymerization and subsequent color change from the nanoparticle aggregation. This format permitted sensitive detection of catalase at low concentrations >3 pM.
EFRP is a promising approach for enhancing sensitivity of biodetection via increasing the number of radicals based on enzymatic catalysis and propagation from each radical. Using oxygen-consuming enzymes as labels achieves oxygen-tolerant polymerization and prevents false-positive results because small amounts of non-specifically bound enzymes cannot consume all of oxygen in monomer solutions. Compared to other reversible-deactivation radical polymerization methods, free radicals are not covalently attached to immobilized biomolecules, so surface-initiated EFRP may have to include formation of cross-linked polymer network to prevent loss of polymers during rinsing process. A main challenge of EFRP-based biodetection is maintaining the stability of enzymes during storage and polymerization. In addition to a loss of enzyme activity during storage, monomers, substrates, or radicals may deactivate enzyme labels and reduce the efficiency of the method.
4.2 Redox-pair mediated free radical polymerization (RFRP)
Redox pair-mediated free radical polymerization (RFRP) is a chain growth polymerization where electron transfer reactions between redox pairs produce initiating radicals. Commonly used redox pairs for RFRP in aqueous media include inorganic oxidants–inorganic reductants such as persulfate–Ag+, peroxides-reducing agents such as hydrogen peroxide–Fe2+, and organic–inorganic redox pairs such as primary alcohol–Ce4+ (Fig. 9A).81,82 Among these types of pairs, the involvement of organic reducing agents is of growing interest in RFRP-based biodetection likely because of straightforward coupling of multiple organic redox-active groups to captured biomolecules.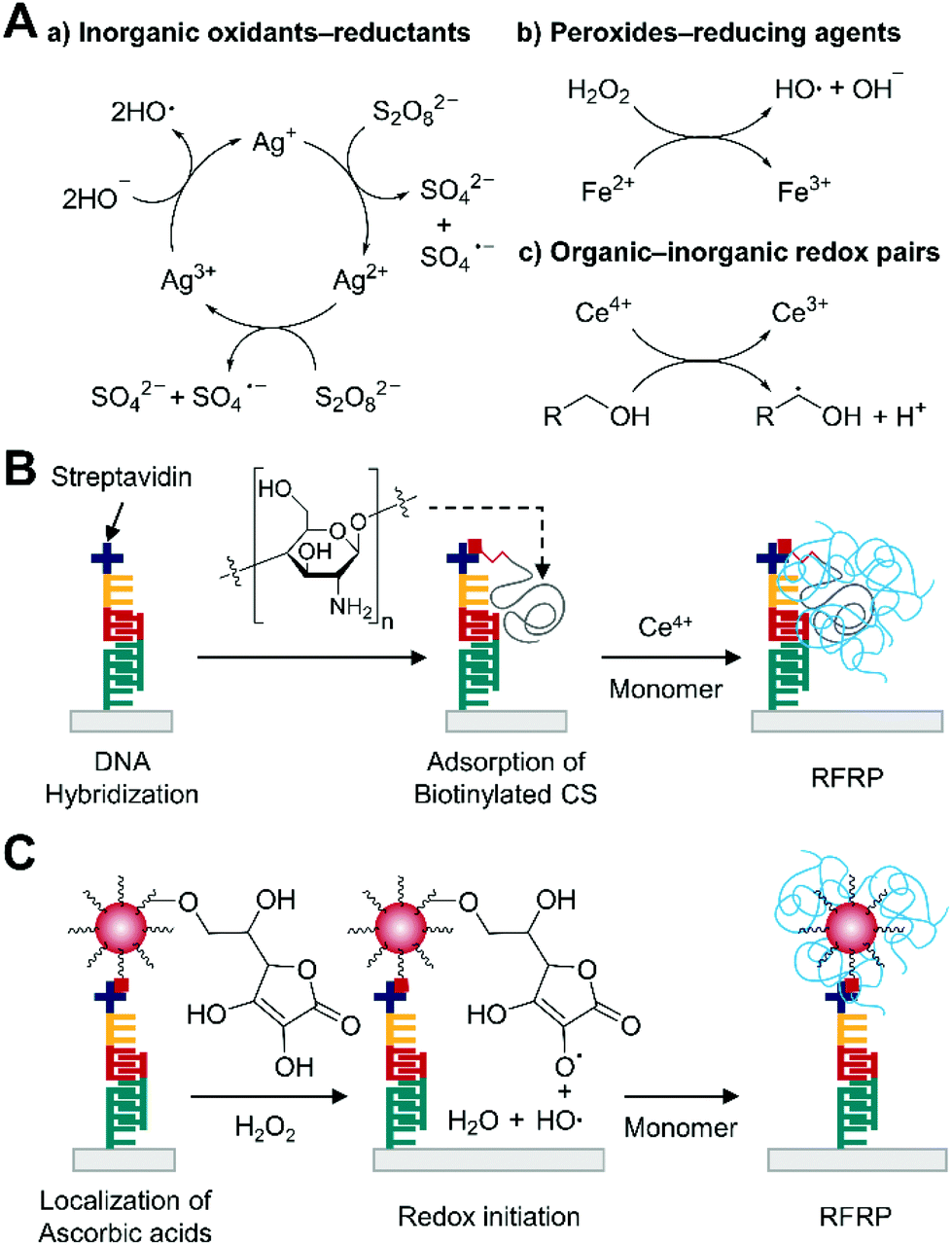 | ||
| Fig. 9 Biodetection with redox pair-mediated free radical polymerization (RFRP). (A) Examples of redox pairs and initiation mechanisms. (B) RFRP-based detection of DNA with the primary alcohol–Ce4+ redox pair.84 Biotinylated high molecular weight chitosan (CS) is localized to binding region upon DNA hybridization, providing many primary alcohol groups for redox initiation with Ce4+. The blue chains represent polymers generated by RFRP. (C) RFRP-based detection of DNA with the ascorbic acid–H2O2 redox pair.85 Polymeric nanoparticles (red dot) localize ascorbic acids to the binding site to produce polymers (blue chains) by RFRP. | ||
Unlike enzyme labels, these organic functional groups usually undergo non-catalytic redox reactions, limiting the number of initiating radicals per molecular recognition event less than one if only one functional group is linked to a captured biomolecule. Therefore, many strategies for localizing multiple organic redox-active groups per binding event have been proposed. For example, Yang and coworkers used ATRP to selectively localize PHEMA upon protein binding or DNA hybridization on gold substrates.83 The localized primary alcohol groups were used for redox initiation with Ce4+ to graft poly(acrylamide) brushes from the PHEMA. The successive 4 h ATRP and 2 h RFRP with primary alcohol–Ce4+ could generate visible polymer film in the presence of 0.2 nM of streptavidin or 5 nM target DNA with model half-sandwich assays where initiator-coupled target molecules were captured by immobilized binders. However, long ATRP reaction times and additional operation steps associated with the use of metal catalyst such as inert gas purging and rigorous rinsing could limit its use in the field. Thus, Yang and coworkers improved the system by replacing ATRP-based grafting of hydroxyl functional polymers with dual functional macroinitiator approach.84 In this approach, authors devised the use of chitosan, which is a linear polysaccharide densely decorated with primary alcohol groups, as a carrier for hydroxyl groups to target-bound region (Fig. 9B). By coupling binding molecules such as biotin to the high molecular-weight chitosan, this approach became more advantageous than the previous ATRP method because it could readily localize to captured target molecules thousands of hydroxyl groups that can be subsequently used with Ce4+ and monomers for 2 h RFRP. Using the same half-sandwich DNA hybridization assay format as described previously, this Ce4+/chitosan-based polymerization approach proved its lower visible detection limit (0.1 nM) than that (5 nM) of the previous successive ATRP–RFRP method. However, in full sandwich assay format, the visible detection limit slightly increased to 1 nM likely because dissociation of target DNA during incubation in following assay steps could reduce the overall binding efficiency of chitosan to captured target DNA.
To further enhance the sensitivity of RFRP-based DNA detection, Yang and coworkers employed biotinylated polymeric nanoparticles with the hydrophilic shell that was functionalized with organic reducing agents, ascorbic acid, which could pair up with hydrogen peroxide for redox reactions (Fig. 9C).85 The carrier of ascorbic acid could be localized to captured target by successive streptavidin–biotin interactions, significantly increasing the number of reducing agents per binding event. Then, the immobilized ascorbic acid was incubated with monomer solution containing HEA, PEGDA, glucose, and glucose oxidase for 40 minutes, during which hydrogen peroxide generated by glucose oxidase was reduced by the ascorbic acid, producing hydroxyl radicals. The initiating radicals were not covalently linked to the binding surface, so PEGDA was required to create cross-linked hydrogels that could remain attached to the surface after rinsing. Under optimal conditions, visible detection limit of target DNA in full sandwich assay format improved to 5 pM, indicating 200-fold enhancement in detection limit. This drastic improvement can be attributed to many factors. First, the amount of organic reducing agents per binding event can significantly increase by using the polymeric nanoparticle as a carrier for ascorbic acid. Second, the hydrogen peroxide–ascorbic acid pair does not participate in self-termination of propagating radicals as the Ce4+–primary alcohol pair does.81 Third, gluconic acid generated by glucose oxidase can reduce the solution pH, which enhances the kinetics of redox reactions and HEA polymerization.85
5. Photo-initiated free radical polymerization
5.1 UV-initiated free radical polymerization (UV-initiated FRP)
Ultraviolet light-initiated free radical polymerization (UV-initiated FRP) is a chain growth polymerization initiated by photochemical reactions under ultraviolet light. The generation of radicals from photoexcited initiators occurs in two pathways: cleavage (type I) and hydrogen abstraction (type II) (Fig. 10A).86 The type I photoinitiators are often substituted aromatic carbonyl compounds such as benzoin, which undergo homolytic α-cleavage under UV light. The cleavage reaction is unimolecular and fast, so bimolecular quenching by monomers or oxygen is negligible. In type II photoinitiation, on the other hand, an aromatic ketone such as benzophenone generates radicals through bimolecular hydrogen abstraction, so the process is not only slower, but also less efficient than the type I photoinitiation because bimolecular quenching of the excited photoinitiators can decrease the quantum yield. The initiating radicals from these processes react with vinyl monomers in solution, initiating polymerization. Since rapid response and high sensitivity are important design objectives in biodetection, a type I photoinitiator can be more suitable to be used in biodetection with UV-initiated FRP.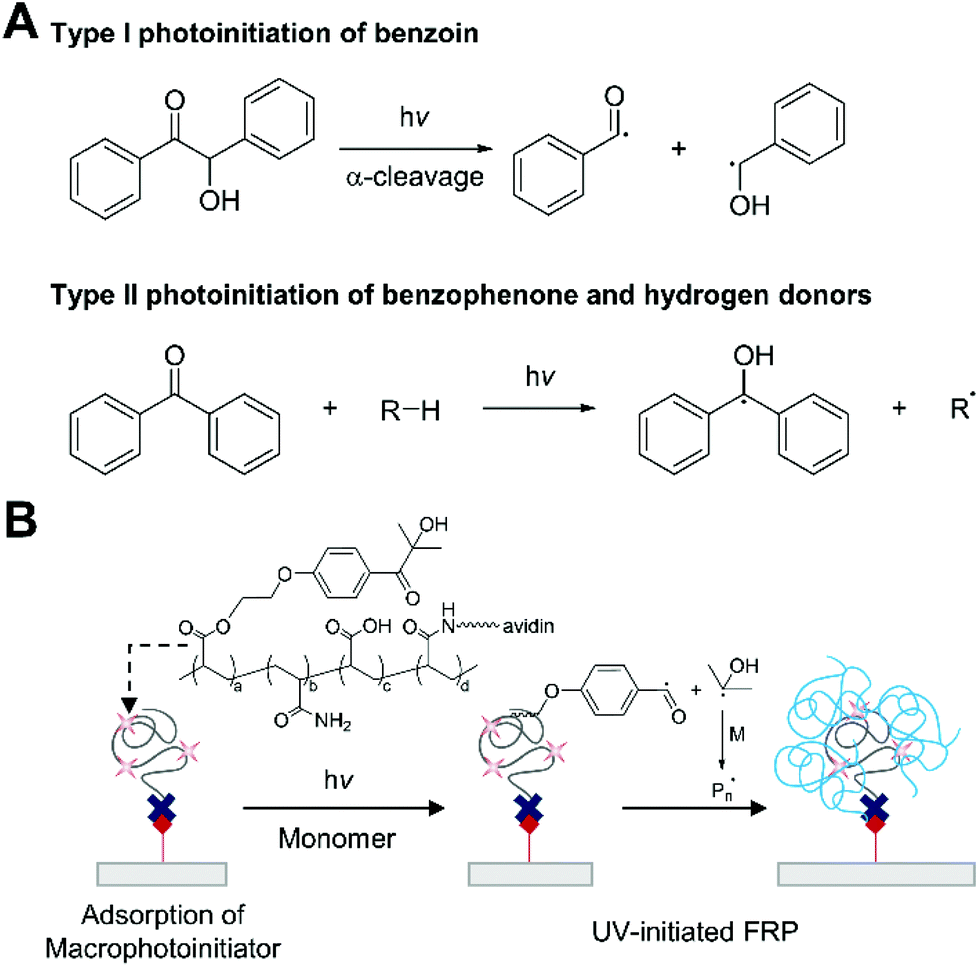 | ||
| Fig. 10 Biodetection with ultraviolet light-initiated free radical polymerization (UV-initiated FRP). (A) Examples of type I and type II photoinitiation under UV-light irradiation. (B) Detection of biotin with UV-initiated FRP.33 Irgacure 2959-based macrophotoinitiators are localized to biotin functionalized surface via biotin–avidin binding interactions. The immobilized Irgacure 2959 undergoes type I photoinitiation under UV-light, producing free radicals. The blue chains represent polymers generated by UV-initiated FRP. | ||
As a proof-of-concept study, Bowman and coworkers demonstrated a biodetection platform based on UV-initiated FRP (Fig. 10B).33 In this first demonstration, Irgacure 2959, a water-soluble type I photoinitiator, and neutravidin as a binding protein were coupled to poly(acrylic acid-co-acrylamide) to make dual functional macrophotoinitiators, which allowed localization of more than 100 initiators per molecular recognition event. To this photoinitiator-functionalized surface, was added argon-purged monomer solution including 97 wt% HEA and 3 wt% ethyleneglycol dimethacrylate (EGDMA), both of which were inhibitor-free, and the monomer solution was irradiated with 5 mW cm−2, 365 nm light. A ten-minute irradiation created visible polymer films from spots with one thousand (10 zmol) biotinylated oligonucleotides. One advantage of this technique is that false positive signals can be reduced by adjusting irradiation time. From a practical standpoint, non-specific binding can always occur with more complex samples or binding pairs with lower specificity and affinity than biotin–neutravidin pair, so it is crucial to suppress the signals generated by non-specific binding. In UV-initiated FRP, non-specific binding signals can be eliminated by controlling the irradiation time, reducing false positive results.
Expanding the proof-of-concept study, the authors also tested the applicability of the UV-initiated FRP-based biodetection in clinically relevant flu tests, in which influenza viruses from crude lysates were subtyped.87 In this work, either anti-influenza A or anti-influenza B monoclonal antibodies were conjugated to the macrophotoinitiators instead of neutravidin. Thus, the macrophotoinitiators could be localized to a positive control zone and to specific binding regions, one for Flu A and another for Flu B, depending on the types of influenza viruses in each sample. Using the same monomer solution and irradiation conditions as used in the previous study except for irradiation time (12 minutes), visible polymer films were created on the zones with macrophotoinitiators, achieving the same limit of detection as enzyme-based amplification used in the commercial flu tests.
While the UV-initiated FRP-based biodetection provided rapid and unambiguous response with high sensitivity in the proof-of-concept study, several problems have limited its further development and practical use in the field. First, it requires oxygen- and inhibitor-free environment to achieve sensitive biodetection. Although more than 100 photoinitiators can be coupled to one molecular recognition event by using the macrophotoinitiator, oxygen and other inhibitors can readily scavenge propagating radicals, significantly reducing degree of polymerization and consequently increasing detection limit. Second, resultant hydrogels are not stable enough to remain intact upon rinsing and post processing, thus limiting its analysis to qualitative visual inspection.88 The weak mechanical strength of the hydrogels was attributed to small amount (3 wt%) of cross-linker in monomer solution. The small diacrylate monomer fraction was inevitable to minimize non-specific polymerization because the diacrylate monomer could result in hydrogel under UV light even without macrophotoinitiators on test zone.89 Third, the use of toxic monomers could also limit the use of UV-initiated FRP for biodetection. Less toxic monomers such as PEG acrylates cannot be used because they are subject to faster non-specific polymerization under UV light.89
5.2 Vis-initiated free radical polymerization (Vis-initiated FRP)
Visible light-initiated free radical polymerization (Vis-initiated FRP) is a chain growth polymerization that initiated by visible light-induced photochemical reactions. The majority of visible light photoinitiators are type II photoinitiators, which undergo bimolecular reactions to produce radicals instead of unimolecular reactions, because the energy of visible light is not high enough to induce homolytic bond cleavage as with type I photoinitiation under UV light. Because two-component type II photoinitiation (photoinitiator and electron/hydrogen donor) are less efficient than type I photoinitiation owing to slower kinetics and side reactions,90 another component (electron acceptor) is added to produce additional radicals and improve the quantum yield of initiation. In the three-component systems, some photoinitiators such as xanthene dyes can be regenerated in the catalytic cycle91 and behave as photoredox catalysts, allowing rapid free radical polymerization with a low amount of the catalysts and low intensity of light. Thus, photoredox catalysis is suitable for biodetection with Vis-initiated FRP because the systems can generate multiple radicals per photoredox catalyst using inexpensive LEDs as light sources.In Vis-initiated FRP-based biodetection, eosin Y has been used as a preferred photoredox catalyst among other xanthene dyes because its high radical quantum yield92 and easy conjugation to binding molecules88 renders it feasible to be used as a very efficient signal tag. Moreover, eosin Y with tertiary amine such as triethanolamine (TEOA) can eliminate oxygen and other inhibitors in aqueous monomer solution via photoredox catalysis,91 which enables Vis-initiated FRP to occur with inhibitor-stabilized PEGDA and N-vinylpyrrolidone (NVP) under atmospheric reaction conditions. As demonstrated in Fig. 11A, the proposed mechanism for the photoredox catalysis begins with a single-electron transfer from TEOA to triplet state of eosin Y under green light.93 Energy transfer to oxygen could occur, but, in conventional monomer solutions, initial concentration of TEOA is approximately three orders-of-magnitude higher than that of dissolved oxygen, so the oxidation of TEOA is the most favorable route.93 The produced TEOA and eosin Y radicals can quantitatively reduce oxygen to superoxide94,95 and regenerate ground state of eosin Y, completing the photocatalytic cycle. When oxygen concentration decreases to a sufficiently low level, the newly generated TEOA radicals can finally initiate free radical polymerization.
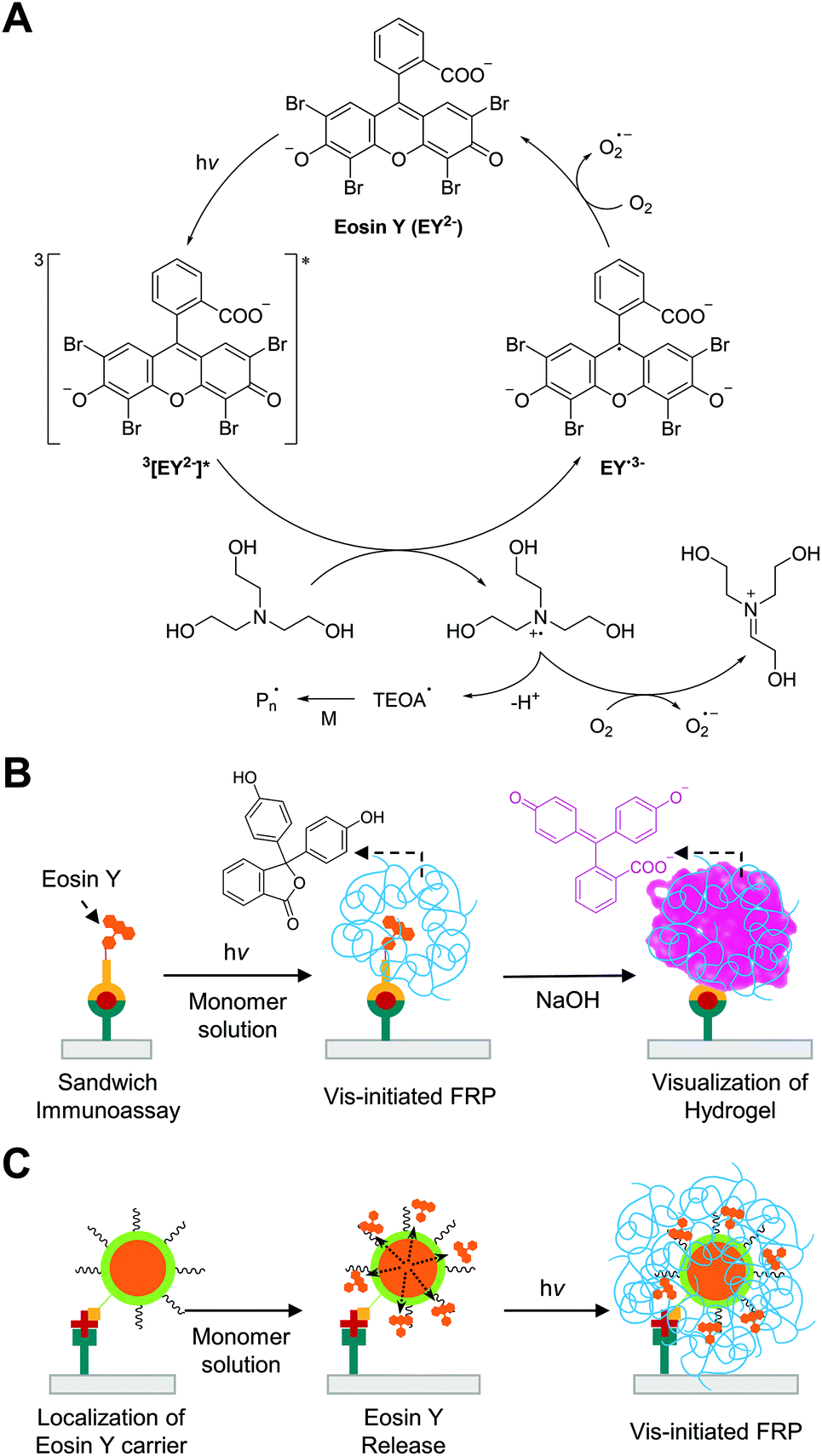 | ||
| Fig. 11 Biodetection with visible light-initiated free radical polymerization (Vis-initiated FRP). (A) Schematic description of photocatalytic initiation mechanism with eosin Y and triethanolamine (TEOA). (B) Detection of protein with Vis-initiated FRP.101 Eosin Y is localized to binding region upon the formation of sandwich immunocomplexes. Resultant hydrogel (blue chains) from Vis-initiated FRP entraps colorless phenolphthalein, which can immediately turn into pink upon addition of NaOH solution. (C) The use of eosin Y-loaded liposome (orange core and green shell) to enhance the sensitivity of biodetection with Vis-initiated FRP.108 The blue chains represent hydrogel produced by Vis-initiated FRP. | ||
While the eosin Y-based initiation system has high oxygen- and inhibitor-tolerance, the concentration of eosin Y in a monomer solution should be at least sub-micromolar to mitigate oxygen inhibition in the presence of atmospheric oxygen. In the early stage of Vis-initiated FRP-based biodetection developed by Bowman and coworkers,88,96–98 an inert atmosphere to prevent continuous oxygen diffusion into monomer solution was required for polymerization because the photoinitiation only occurred near the binding region where eosin Y-conjugated probes were immobilized on functionalized glass microscope slides by specific interactions. The amount of eosin Y coupled to molecular recognition events, in these methods, was too small compared to that of dissolved oxygen in monomer solution exposed to air, so oxygen inhibition could not be overcome. The first air-tolerant and rapid (<5 minutes) Vis-initiated FRP-based biodetection was demonstrated by Kuck and coworker although compositions of the monomer solutions were not specified.99 Later Sikes and coworker systematically examined the addition of sub-micromolar concentrations of free eosin Y to monomer solutions to solve the oxygen inhibition problem in atmospheric reaction conditions.100 Variation of the concentrations of free eosin Y in aqueous monomer solutions containing 200 mM PEGDA, 150 mM TEOA, and 100 mM NVP revealed that including 0.3–0.7 μM eosin Y in the monomer solutions could lead to rapid consumption (35–100 s) of dissolved oxygen through photoredox catalysis, followed by hydrogel formation if interfacial binding events had occurred. The hydrogels indicative of binding events at arrays printed on microscope slides could be visualized by eosin Y staining, so very high-contrast results could be obtained while not compromising the assay sensitivity99,100 compared to the same format with inert gas purging.88 The free eosin Y included in the monomer solutions could result in non-specific polymerization with excessive irradiation, but appropriate irradiation time could always be determined to limit the hydrogel formation to biodetection.
Vis-initiated FRP-based biodetection has been adapted in paper-based colorimetric tests to address limitations of other existing enzyme- and nanoparticle-based methods such as long color development time (20–30 minutes), false positive results, and low visual contrast. Sikes and coworkers demonstrated the integration of the rapid and oxygen-tolerant Vis-initiated FRP with a paper-based immunoassay to enhance visual contrast between negative and positive results (Fig. 11B).101 In this method, sandwich immunoassay for detection of Plasmodium falciparum histidine-rich protein 2 (PfHRP2) localized eosin Y on specific binding region using eosin Y-conjugated reporter antibody. Phenolphthalein-included monomer solution was used to create colored hydrogel in the presence of the antigen in samples instead of using eosin Y staining after hydrogel formation. Because eosin Y stains paper even in the absence of a hydrogel, post-polymerization staining could not be used to distinguish positive from negative results. The pH of the phenolphthalein-included monomer solution was maintained below 8 to make phenolphthalein present in its colorless form and prevent its competition with eosin Y for absorption of green light. After Vis-initiated FRP, the phenolphthalein entrapped in a polymer network turned pink immediately when 0.5 M NaOH solution was added to the surface. In addition to this immediate color development, accurate time-keeping before and after polymerization is not required to obtain optimal signals as opposed to other enzymatic and nanoparticle-based colorimetric methods, which can significantly reduce the possibility of false positive results.102 Under optimal conditions including 90 seconds of irradiation time, 7.2 nM of PfHRP2 could be detected by the naked eye using Vis-initiated FRP.101 The same method was also used to detect biomarkers for periodontal disease, matrix metalloproteinase-8 (MMP-8) and -9 (MMP-9), achieving detection limit of 15.4 nM and 5.0 nM, respectively.103
The formation of phenolphthalein-entrapped hydrogels could realize rapid and selective staining on paper-based immunoassays. However, the phenolphthalein-leaching from the hydrogel occurred while washing away unreacted monomer solution, causing the colorimetric results to depend on rinsing and storing time in alkaline solution before analysis. This limitation has been addressed by covalently linking phenolphthalein to the polymer network. Sikes and coworker replaced phenolphthalein in the monomer solution with phenolphthalein monomer (N-(2-hydroxy-5-(1-(4-hydroxyphenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl)-benzyl)acrylamide) to form phenolphthalein-conjugated hydrogel.104 In the same PfHRP2 immunoassay format, the hydrogel generated from 65 nM or more antigen successfully maintained its pink color in alkaline solution even after several drying and rehydration cycles, but hydrogels produced from smaller concentrations (13 and 26 nM) of antigen could not remain pink because not many phenolphthalein monomers could participate in the copolymerization. The main reason for this limitation is insolubility of the phenolphthalein monomer. Therefore, efforts should be made to synthesize more water-soluble phenolphthalein monomer or other dye-conjugated monomers that can be visualized by physical or chemical stimuli.
Another limitation of Vis-initiated FRP-based biodetection is insufficient detection limit (∼nM) for early diagnosis of many diseases. Eosin Y-based macrophotoinitiators have been employed to localize a greater amount of eosin Y to specific binding region as demonstrated in other biodetection systems.31–33,105 However, poor solubility106 and fluorescence self-quenching107 of the eosin Y-based macrophotoinitiators increased non-specific binding signals and decreased the radical quantum yield, reducing the benefits of this approach. To address these problems, Sikes and coworker demonstrated the use of liposomes as eosin Y-carrier (Fig. 11C).108 In this approach, concentrated eosin Y was loaded in biotinylated liposomes to be delivered to specific binding region where streptavidin was immobilized through molecular recognition. When the monomer solution was added to the liposome-bound cellulose surface, one liposome could release thousands of encapsulated eosin Y, contributing to rapid photoredox catalysis. The incorporation of liposomes improved the sensitivity by 30-fold and the detection limit by 3-fold compared to conventional method. Both the sensitivity and the detection limit were not improved as much as expected with the number of eosin Y released from one liposome, which could be because of amplified background noise. Non-specific interactions between the liposomes and biofunctionalized paper might also contribute to amplifying background noise, reducing the signal-to-noise ratio of the method. Thus, it would be a promising future direction to design eosin Y-carriers that are not subject to non-specific interactions with paper to further enhance the sensitivity of biodetection based on Vis-initiated FRP.
6. Comparison of polymerization-based biodetection methods and future goals
Various radical polymerization chemistries, including ATRP, RAFT polymerization, redox-initiated free radical polymerization, and photo-initiated free radical polymerization, have been employed to amplify biodetection signals. As summarized above, each polymerization-based biodetection has different characteristics that could affect cost, operation conditions, user-friendliness, signal amplification time, and performance, all of which are essential considerations for developing practical biodetection platforms. Thus, comparison of all polymerization-based biodetection methods in terms of the considerations should be a useful guide for both development of new polymerization chemistries for biodetection and incorporation of them into biodetection platforms. Readers might expect to see which one is the best among all polymerization methods, but that is very difficult because the methods have not been compared in the same biodetection system and different applications may weigh the importance of each criterion differently. Rather than selecting the best method to date, the purpose of this comparison is to assess the state of the field and suggest future directions to realize practical polymerization-based biosensors for point-of-care diagnostics. In this context, we hope to draw attention to the present capabilities and the future development needs of polymerization-based biodetection.Signal amplification methods for point-of-care diagnostics should be free of laboratory equipment and inert gas purging for portability and user-friendliness of diagnostic tests. However, most of the current polymerization-based biodetection methods in literature do not meet these criteria. As presented in Fig. 12, electricity-powered LED arrays, electrochemical cells, and hot plates or temperature controllers have been required for rapid and controlled initiation of photo-initiated FRP, electrochemically mediated ATRP and RAFT, thermally initiated RAFT and biocatalytic precipitation ATRP. Inert gas purging and sealed reactors should be employed for RAFT polymerization, normal ATRP, Fluorogenic ATRP, and RFRP without glucose oxidase. For polymerization methods with limited oxygen tolerance such as eATRP, AGET ATRP, and biocatalytic precipitation ATRP, oxygen diffusion from atmosphere needs to be prevented by using sealed reactors. EFRP and RFRP are only the two examples with no demand for equipment and sealed reactors, and both methods overcome oxygen inhibition with glucose oxidase and generate free radicals via redox initiations.
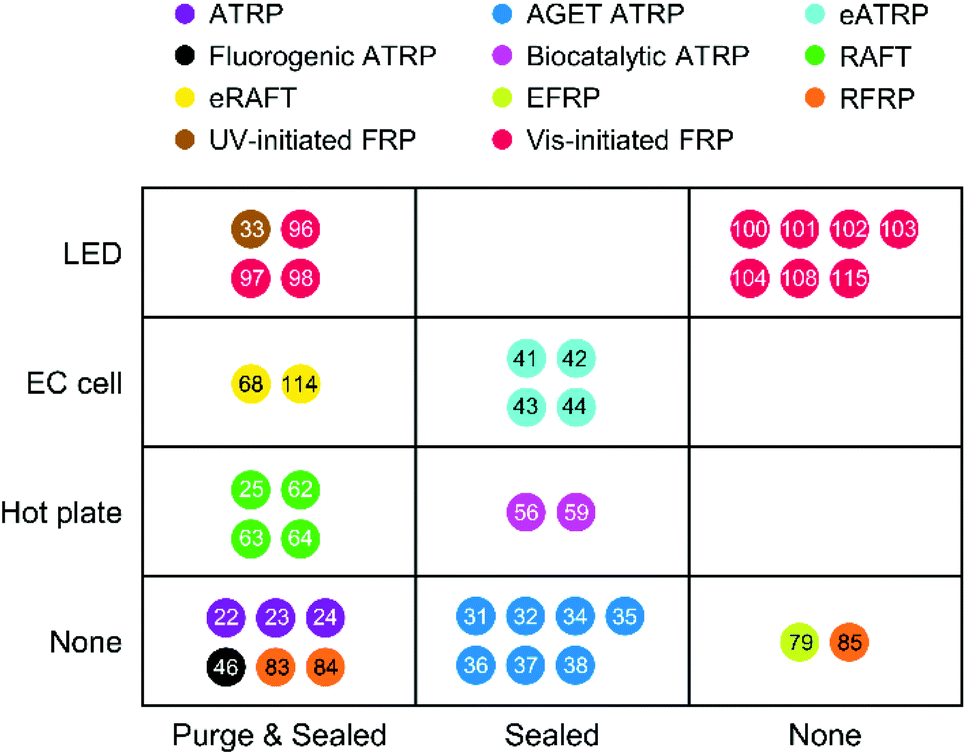 | ||
| Fig. 12 Required equipment and deoxygenation conditions for polymerization-based biodetection methods in literature. The colored dots with numbers show different types of polymerization methods and corresponding references. ATRP: Normal ATRP. RAFT: thermally initiated RAFT. Each row represents required equipment for the initiation of radical polymerizations. LED: Light-emitting diode. EC cell: Electrochemical cell. Hot plate includes any temperature controllers. Each column represents required deoxygenation conditions for radical polymerizations. Purge & sealed: Inert environment. Sealed: Inert gas purging is not required, but sealed reactors should be used to prevent oxygen diffusion from atmosphere. Extracted from Table S2 in ESI.† | ||
In the future, polymerization-based biodetection should be able to stop relying on sophisticated equipment and inert gas purging. Recently, miniaturized devices to replace lab equipment are being developed, which could potentially replace electricity-powered LED arrays, electrochemical cells, and hot plates or temperature controllers with portable, battery-powered or smartphone-based devices.109–112 Thus, initiation processes with external stimuli such as light, voltage or current, and heat can maintain their practicality. The polymerization methods requiring inert gas purging may be excluded from practical applications, but the demands for sealed reactors as in oxygen-tolerant ATRP methods could be resolved with simple methods such as plastic sealing bags or sealing tapes.
Even if a diagnostic test is portable and user-friendly, an hour-long signal amplification time or high limit of detection compared to clinically relevant levels of target molecules can be a major drawback that precludes use. A trade-off between amplification time and detection limit exists in each polymerization-based biodetection method, so plotting these two values of each approach could be useful to compare different approaches and draw future directions. As shown in Fig. 13, the amplification time ranges from 102 seconds to 105 seconds depending on polymer chemistry in each method. Among these methods, Vis-initiated FRP is the fastest because of its rapid initiation and propagation steps. The rapid initiation is attributed to inclusion of sub-micromolar eosin Y in monomer solution, leading to rapid consumption of dissolved oxygen through photoredox catalysis. Moreover, free radical propagation rapidly forms hydrogels near interfaces where specific binding events have concentrated reagents coupled to eosin Y. Other free radicals methods such as RFRP show faster propagation kinetics than reversible-deactivation polymerization, but their initiation kinetics are slower than that of Vis-initiated FRP, so their amplification time is not as short as Vis-initiated FRP. Amplification time of reversible-deactivation radical polymerization-based biodetection is at least 30 minutes unless macroinitiator increases the amount of initiators per specific binding event. Except for the method coupled with silver nanoparticles,44 electrochemically mediated ATRP achieves faster amplification than other reversible-deactivation radical polymerizations because the ratio of the amount of activators to the amount of deactivators can be precisely controlled and the activators are electrostatically localized to the electrode surface.41 In general, the amplification time could be reduced, but reducing the time would increase the limit of detection. In some cases, post-modification time is much longer than actual polymerization time as in AGET ATRP-based electrochemical biodetection,34,35,38 where electroactive molecules are coupled to the generated polymer. Recent advances41–43,63,64,68 demonstrate the use of electroactive monomers such as FMMA to reduce the amplification time.
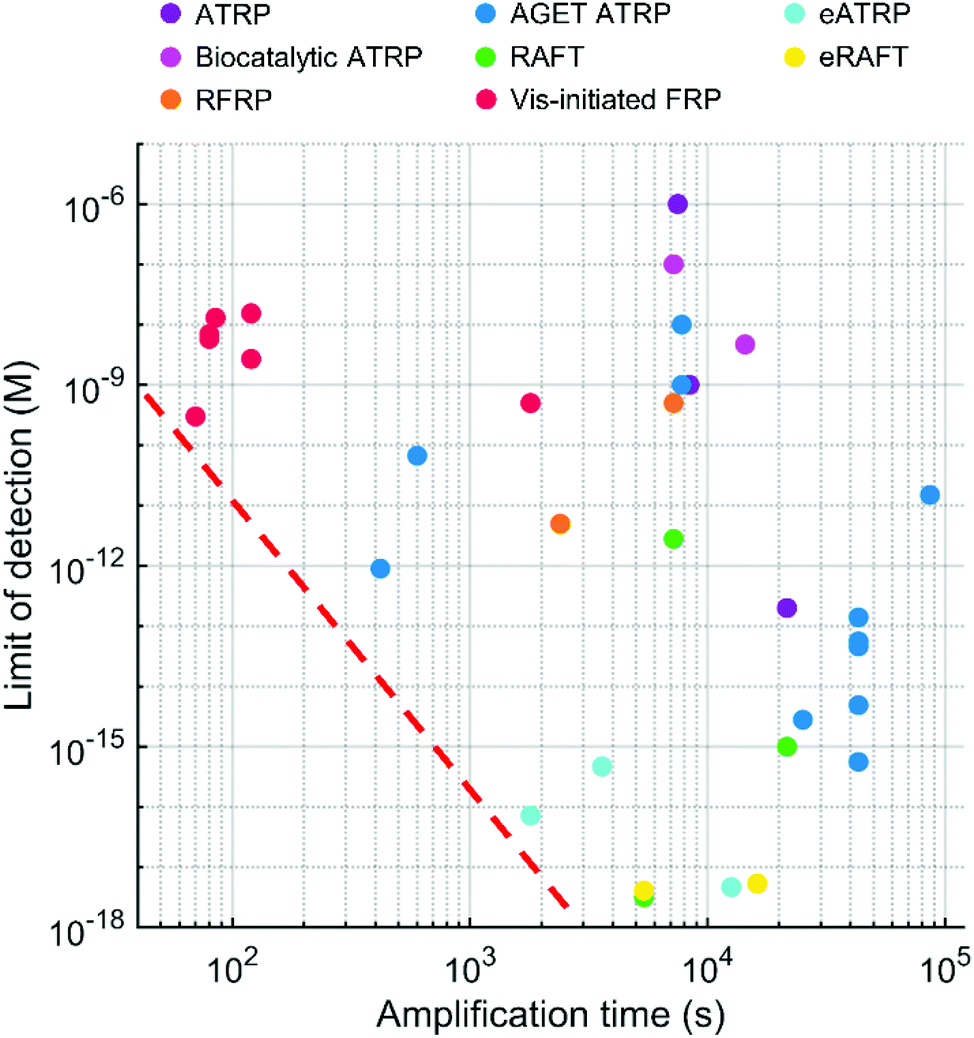 | ||
| Fig. 13 Reported limit of detection (M) and amplification time (s) of polymerization-based biodetection methods in literature. Colored dots represent different types of polymerization methods. ATRP: Normal ATRP. RAFT: Thermally initiated RAFT. The red dotted line indicates the lower bound, under which the amplification time and limit of detection of biosensors are not achieved yet, but potentially more practical. This plot considers only sandwich DNA or protein binding assays where target molecules are immobilized between capture binders and detection binders, which excludes half-sandwich assays. As an exception, solution-phase assays of hemozoin and hemoglobin (biocatalytic ATRP) are included. Amplification time includes time for inert gas purging if required, polymerization, and coupling signaling molecules. Rinsing time before and after polymerization is not considered. Extracted from Table S1 in ESI.† | ||
The limit of detection spans 12 orders of magnitude (Fig. 13). The values are not specific to polymer chemistry in each method, but affected by many other factors such as analyte, detection methods, and rinsing solvent/buffer (Table S1†). Each analyte is specific to certain binders whose equilibrium dissociation constants (Kd) are varied, so the capture efficiencies of target molecules can be significantly different. The sensitivity of detection methods would also contribute to a considerable difference in limit of detection. Two extreme cases are visual detection and electrochemical detection (Fig. S1†). The former one uses the naked eye to detect visible polymer film, which is suitable for diagnostic tests in resource-limited settings.101,113 This instrument-free detection includes all dots with the limit of detection higher than 1 nM. The latter one employs electrochemical methods to observe changes in the presence of electroactive polymer grafting, which includes all dots with the limit of detection lower than 1 fM. Using sensitive detectors such as electrochemical workstations can enhance the limit of detection, but the portability and user-friendliness of biosensors would be damaged. Recently, smartphone-based electrochemical detection is in development.111 The sensitivity of the portable devices is not comparable to the bench-top equipment yet, but this approach has a great potential to realize sensitive portable biosensors. In most papers, organic solvents were used for rinsing biodetection zones to remove unreacted monomers and minimize background signals, which could lower the limit of detection (Fig. S2†). Organic solvents such as alcohols, acetone, DMSO, and DMF should be better than aqueous buffer solutions for washing away hydrophobic monomers and signaling molecules adsorbed non-specifically. However, the use of organic solvents is limited in point-of-care diagnostic tests because of their difficult handling and toxicity.
As discussed above, the amplification time and the limit of detection of polymerization-based biodetection depend on various factors such as the nature of polymerization, post-modification reactions, binding affinity (Kd) of target-binder interactions, detection methods, and rinsing solvent/buffer. Thus, the unexplored yet potentially more practical amplification time and limit of detection for point-of-diagnostic tests (under the red line in Fig. 13) can be achieved by considering those factors. To reduce the amplification time, it would be helpful to enhance kinetics of polymerization and minimize post-modification of grafted polymers. Employing high affinity binders for target molecules and incorporating sensitive, affordable, and portable detectors in biosensors would be promising future directions to lower the limit of detection. Although organic rinsing solutions are more effective than aqueous solutions to improve limit of detection, it should be avoided to use organic solvents for user-friendliness of biosensors.
7. Conclusion
This review has summarized diverse radical polymerization chemistries for amplifying biodetection signals and compared them from the practical point of view. Combining various initiation reactions and functional monomers, polymerization-based biodetection has been coupled with different operation conditions and detection methods. Recent advances have demonstrated either short amplification time (<100 s) or ultralow limit of detection (∼aM), but none of the current polymerization methods have achieved both rapid and ultrasensitive biodetection while not compromising its portability and user-friendliness. Many opportunities exist to combine relative merits of the polymerization methods and address their limitations for developing more practical biodetection platforms. Given that biosensors consist of many components such as sample preparation, molecular recognition, signal amplification, and signal analysis, interdisciplinary research efforts are strongly encouraged to develop real-world applications of polymerization-based biodetection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation Singapore under its Antimicrobial Resistance IRG administered by the Singapore Alliance for Research and Technology. HDS also acknowledges support from an Esther and Harold E. Edgerton professorship. SK acknowledges support from the Kwanjeong Educational Foundation.Notes and references
- J. Homola, Surface Plasmon Resonance Sensors for Detection of Chemical and Biological Species, Chem. Rev., 2008, 108(2), 462–493 CrossRef CAS PubMed.
- S. K. Vashist and P. Vashist, Recent Advances in Quartz Crystal Microbalance-Based Sensors, J. Sens., 2011, 571405 Search PubMed.
- K. C. Bantz, A. F. Meyer, N. J. Wittenberg, H. Im, Ö. Kurtuluş, S. H. Lee, N. C. Lindquist, S. H. Oh and C. L. Haynes, Recent Rrogress in SERS Biosensing, Phys. Chem. Chem. Phys., 2011, 13(24), 11551–11567 RSC.
- X. Feng, L. Liu, S. Wang and D. Zhu, Water-Soluble Fluorescent Conjugated Polymers and Their Interactions with Biomacromolecules for Sensitive Biosensors, Chem. Soc. Rev., 2010, 39(7), 2411–2419 RSC.
- R. E. Wang, Y. Zhang, J. Cai, W. Cai and T. Gao, Aptamer-Based Fluorescent Biosensors, Curr. Med. Chem., 2011, 18(27), 4175–4184 CrossRef CAS PubMed.
- Y. Song, W. Wei and X. Qu, Colorimetric Biosensing Using Smart Materials, Adv. Mater., 2011, 23(37), 4215–4236 CrossRef CAS PubMed.
- H. Yang, Enzyme-Based Ultrasensitive Electrochemical Biosensors, Curr. Opin. Chem. Biol., 2012, 16, 422–428 CrossRef CAS PubMed.
- M. Holzinger, A. Le. Goff and S. Cosnier, Nanomaterials for Biosensing Applications: A Review, Front. Chem., 2014, 2, 1–10 CAS.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Point of Care Diagnostics: Status and Future, Anal. Chem., 2012, 84(2), 487–515 CrossRef CAS PubMed.
- M. Zarei, Portable Biosensing Devices for Point-Of-Care Diagnostics: Recent Developments and Applications, Trends Anal. Chem., 2017, 91, 26–41 CrossRef CAS.
- E. T. S. G. da Silva, D. E. P. Souto, J. T. C. Barragan, J. de F. Giarola, A. C. M. de Moraes and L. T. Kubota, Electrochemical Biosensors in Point-of-Care Devices: Recent Advances and Future Trends, ChemElectroChem, 2017, 4(4), 778–794 CrossRef CAS.
- S. O. Kelley, What Are Clinically Relevant Levels of Cellular and Biomolecular Analytes?, ACS Sens., 2017, 2(2), 193–197 CrossRef CAS PubMed.
- S. Goggins and C. G. Frost, Approaches Towards Molecular Amplification for Sensing, Analyst, 2016, 141(11), 3157–3218 RSC.
- B. Schweitzer, S. Wiltshire, J. Lambert, S. O'Malley, K. Kukanskis, Z. Zhu, S. F. Kingsmore, P. M. Lizardi and D. C. Ward, Immunoassays with Rolling Circle DNA Amplification: A Versatile Platform for Ultrasensitive Antigen Detection, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(18), 10113–10119 CrossRef CAS PubMed.
- Y. Wu, W. Wei and S. Liu, Target-Triggered Polymerization for Biosensing, Acc. Chem. Res., 2012, 45(9), 1441–1450 CrossRef CAS PubMed.
- S. Zhou, L. Yuan, X. Hua, L. Xu and S. Liu, Signal Amplification Strategies for DNA and Protein Detection Based on Polymeric Nanocomposites and Polymerization: A Review, Anal. Chim. Acta, 2015, 877, 19–32 CrossRef CAS PubMed.
- K. H. Malinowska and M. A. Nash, Enzyme- and Affinity Biomolecule-Mediated Polymerization Systems for Biological Signal Amplification and Cell Screening, Curr. Opin. Biotechnol., 2016, 39, 68–75 CrossRef CAS PubMed.
- E. Peris, M. J. Bañuls, Á. Maquieira and R. Puchades, Photopolymerization as a Promising Method to Sense Biorecognition Events, Trends Anal. Chem., 2012, 41, 86–104 CrossRef CAS.
- K. Kaastrup and H. D. Sikes, Using Photo-Initiated Polymerization Reactions to Detect Molecular Recognition, Chem. Soc. Rev., 2016, 45(3), 532–545 RSC.
- K. Matyjaszewski and J. Xia, Atom Transfer Radical Polymerization, Chem. Soc. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- W. Tang, N. V. Tsarevsky and K. Matyjaszewski, Determination of Equilibrium Constants for Atom Transfer Radical Polymerization, J. Am. Chem. Soc., 2006, 128(5), 1598–1604 CrossRef CAS PubMed.
- X. Lou, M. S. Lewis, C. B. Gorman and L. He, Detection of DNA Point Mutation by Atom Transfer Radical Polymerization, Anal. Chem., 2005, 77(15), 4698–4705 CrossRef CAS PubMed.
- X. Lou, C. Wang and L. He, Core–Shell Au Nanoparticle Formation with DNA–Polymer Hybrid Coatings Using Aqueous ATRP, Biomacromolecules, 2007, 8, 1385–1390 CrossRef CAS PubMed.
- H. Shi, L. Yuan, Y. Wu and S. Liu, Colorimetric Immunosensing via Protein Functionalized Gold Nanoparticle Probe Combined with Atom Transfer Radical Polymerization, Biosens. Bioelectron., 2011, 26(9), 3788–3793 CrossRef CAS PubMed.
- P. He, W. Zheng, E. Z. Tucker, C. B. Gorman and L. He, Reversible Addition-Fragmentation Chain Transfer Polymerization in DNA Biosensing, Anal. Chem., 2008, 80(10), 3633–3639 CrossRef CAS.
- W. Zheng and L. He, Particle Stability in Polymer-Assisted Reverse Colorimetric DNA Assays, Anal. Bioanal. Chem., 2009, 393(4), 1305–1313 CrossRef CAS PubMed.
- K. Min, H. Gao and K. Matyjaszewski, Preparation of Homopolymers and Block Copolymers in Miniemulsion by ATRP Using Activators Generated by Electron Transfer (AGET), J. Am. Chem. Soc., 2005, 127(11), 3825–3830 CrossRef CAS PubMed.
- K. Min, W. Jakubowski and K. Matyjaszewski, AGET ATRP in the Presence of Air in Miniemulsion and in Bulk, Macromol. Rapid Commun., 2006, 27(8), 594–598 CrossRef CAS.
- H. Qian and L. He, Surface-Initiated Activators Generated by Electron Transfer for Atom Transfer Radical Polymerization in Detection of DNA Point Mutation, Anal. Chem., 2009, 81(11), 4536–4542 CrossRef CAS PubMed.
- H. Qian and L. He, Detection of Protein Binding Using Activator Generated by Electron Transfer for Atom Transfer Radical Polymerization, Anal. Chem., 2009, 81(23), 9824–9827 CrossRef CAS PubMed.
- H. Qian and L. He, Polymeric Macroinitiators for Signal Amplification in AGET ATRP-Based DNA Detection, Sens. Actuators, B, 2010, 150(2), 594–600 CrossRef CAS.
- L. Xu, L. Yuan and S. Liu, Macroinitiator Triggered Polymerization for Versatile Immunoassay, RSC Adv., 2014, 4(1), 140–146 RSC.
- H. D. Sikes, R. R. Hansen, L. M. Johnson, R. Jenison, J. W. Birks, K. L. Rowlen and C. N. Bowman, Using Polymeric Materials to Generate an Amplified Response to Molecular Recognition Events, Nat. Mater., 2008, 7(1), 52–56 CrossRef CAS PubMed.
- Y. Wu, S. Liu and L. He, Electrochemical Biosensing Using Amplification-by-Polymerization, Anal. Chem., 2009, 81(16), 7015–7021 CrossRef CAS PubMed.
- Y. Wu, S. Liu and L. He, Activators Generated Electron Transfer for Atom Transfer Radical Polymerization for Immunosensing, Biosens. Bioelectron., 2010, 26(3), 970–975 CrossRef CAS PubMed.
- Y. Wu, H. Shi, L. Yuan and S. Liu, A Novel Electrochemiluminescence Immunosensor via Polymerization-Assisted Amplification, Chem. Commun., 2010, 46(41), 7763–7765 RSC.
- Y. Wu, S. Liu and L. He, Polymerization-Assisted Signal Amplification for Electrochemical Detection of Biomarkers, Analyst, 2011, 136(12), 2558–2563 RSC.
- Y. Wu, P. Xue, K. M. Hui and Y. Kang, A Paper-Based Microfluidic Electrochemical Immunodevice Integrated with Amplification-by-Polymerization for the Ultrasensitive Multiplexed Detection of Cancer Biomarkers, Biosens. Bioelectron., 2014, 52, 180–187 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Electrochemically Mediated Atom Transfer Radical Polymerization, Science, 2011, 332(1), 81–84 CrossRef CAS PubMed.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Electrochemically Mediated Atom Transfer Radical Polymerization (eATRP), Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- Q. Hu, Q. Wang, G. Sun, J. Kong and X. Zhang, Electrochemically Mediated Surface-Initiated de Novo Growth of Polymers for Amplified Electrochemical Detection of DNA, Anal. Chem., 2017, 89(17), 9253–9259 CrossRef CAS PubMed.
- Q. Hu, Q. Wang, J. Kong, L. Li and X. Zhang, Electrochemically Mediated in Situ Growth of Electroactive Polymers for Highly Sensitive Detection of Double-Stranded DNA without Sequence-Preference, Biosens. Bioelectron., 2018, 101, 1–6 CrossRef CAS PubMed.
- Q. Hu, Q. Wang, C. Jiang, J. Zhang, J. Kong and X. Zhang, Electrochemically Mediated Polymerization for Highly Sensitive Detection of Protein Kinase Activity, Biosens. Bioelectron., 2018, 110, 52–57 CrossRef CAS PubMed.
- H. Sun, J. Kong, Q. Wang, Q. Liu and X. Zhang, Dual Signal Amplification by eATRP and DNA-Templated Silver Nanoparticles for Ultrasensitive Electrochemical Detection of Nucleic Acids, ACS Appl. Mater. Interfaces, 2019, 11(31), 27568–27573 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, Preparation of Well-Defined Polymers and DNA-Polymer Bioconjugates via Small-Volume eATRP in the Presence of Air, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS.
- Z. T. Allen, J. R. Sackey-Addo, M. P. Hopps, D. Tahseen, J. T. Anderson, T. A. Graf and C. B. Cooley, Fluorogenic Atom Transfer Radical Polymerization in Aqueous Media as a Strategy for Detection, Chem. Sci., 2019, 10(4), 1017–1022 RSC.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Aqueous ARGET ATRP, Macromolecules, 2012, 45(16), 6371–6379 CrossRef CAS.
- G. Kali, T. B. Silva, S. J. Sigg, F. Seidi, K. Renggli and N. Bruns, ATRPases: Using Nature's Catalysts in Atom Transfer Radical Polymerizations, ACS Symp. Ser., 2012, 1100, 171–181 CrossRef CAS.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Horseradish Peroxidase as a Catalyst for Atom Transfer Radical Polymerization, Macromol. Rapid Commun., 2011, 32(21), 1710–1715 CrossRef CAS PubMed.
- T. B. Silva, M. Spulber, M. K. Kocik, F. Seidi, H. Charan, M. Rother, S. J. Sigg, K. Renggli, G. Kali and N. Bruns, Hemoglobin and Red Blood Cells Catalyze Atom Transfer Radical Polymerization, Biomacromolecules, 2013, 14(8), 2703–2712 CrossRef CAS PubMed.
- C. Fodor, B. Gajewska, O. Rifaie-Graham, E. A. Apebende, J. Pollard and N. Bruns, Laccase-Catalyzed Controlled Radical Polymerization of N-Vinylimidazole, Polym. Chem., 2016, 7(43), 6617–6625 RSC.
- K. Renggli, N. Sauter, M. Rother, M. G. Nussbaumer, R. Urbani, T. Pfohl and N. Bruns, Biocatalytic Atom Transfer Radical Polymerization in a Protein Cage Nanoreactor, Polym. Chem., 2017, 8(14), 2133–2136 RSC.
- Y.-H. Ng, F. di. Lena and C. L. L. Chai, PolyPEGA with Predetermined Molecular Weights from Enzyme-Mediated Radical Polymerization in Water, Chem. Commun., 2011, 47(22), 6464–6466 RSC.
- Y.-H. Ng, F. d. Lena and C. L. L. Chai, Metalloenzymatic Radical Polymerization Using Alkyl Halides as Initiators, Polym. Chem., 2011, 2(3), 589–594 RSC.
- A. Simakova, M. Mackenzie, S. E. Averick, S. Park and K. Matyjaszewski, Bioinspired Iron-Based Catalyst for Atom Transfer Radical Polymerization, Angew. Chem., Int. Ed., 2013, 52(46), 12148–12151 CrossRef CAS PubMed.
- O. Rifaie-Graham, J. Pollard, S. Raccio, S. Balog, S. Rusch, M. A. Hernández-Castañeda, P. Y. Mantel, H. P. Beck and N. Bruns, Hemozoin-Catalyzed Precipitation Polymerization as an Assay for Malaria Diagnosis, Nat. Commun., 2019, 10(1), 1369 CrossRef PubMed.
- D. J. Sullivan Jr., Hemozoin : A Biocrystal Synthesized during the Degradation of Hemoglobin, Biopolymers Online, 2005, 9, 129–163 Search PubMed.
- L. M. Coronado, C. T. Nadovich and C. Spadafora, Malarial Hemozoin: From Target to Tool, Biochim. Biophys. Acta, 2014, 1840(6), 2032–2041 CrossRef CAS PubMed.
- J. Pollard, O. Rifaie-Graham, S. Raccio, A. Davey, S. Balog and N. Bruns, Biocatalytically Initiated Precipitation Atom Transfer Radical Polymerization (ATRP) as a Quantitative Method for Hemoglobin Detection in Biological Fluids, Anal. Chem., 2020, 92, 1162–1170 CrossRef CAS PubMed.
- C. L. Mccormick and A. B. Lowe, Aqueous RAFT Polymerization: Recent Developments in Synthesis of Functional Water-Soluble (Co)Polymers with Controlled Structures, Acc. Chem. Res., 2004, 37(5), 312–325 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Living Radical Polymerization by the RAFT Process, Aust. J. Chem., 2005, 58(6), 379–410 CrossRef CAS.
- P. He, E. Z. Tucker, C. B. Gorman and L. He, Chemical Amplification for In-Gel DNA Detection, Anal. Methods, 2011, 3(11), 2463–2468 RSC.
- Q. Hu, D. Han, S. Gan, Y. Bao and L. Niu, Surface-Initiated-Reversible-Addition-Fragmentation-Chain-Transfer Polymerization for Electrochemical DNA Biosensing, Anal. Chem., 2018, 90(20), 12207–12213 CrossRef CAS PubMed.
- Q. Hu, J. Kong, D. Han, Y. Bao, X. Zhang, Y. Zhang and L. Niu, Ultrasensitive Peptide-Based Electrochemical Detection of Protein Kinase Activity Amplified by RAFT Polymerization, Talanta, 2020, 206, 120173 CrossRef CAS PubMed.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Highly Controlled Open Vessel RAFT Polymerizations by Enzyme Degassing, Macromolecules, 2014, 47(24), 8541–8547 CrossRef CAS.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, PET-RAFT Polymerisation: Towards Green and Precision Polymer Manufacturing, Chem. Commun., 2018, 54(50), 6591–6606 RSC.
- Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu and K. Matyjaszewski, Electrochemically Mediated Reversible Addition-Fragmentation Chain-Transfer Polymerization, Macromolecules, 2017, 50(20), 7872–7879 CrossRef CAS PubMed.
- Q. Hu, J. Kong, D. Han, L. Niu and X. Zhang, Electrochemical DNA Biosensing via Electrochemically Controlled Reversible Addition-Fragmentation Chain Transfer Polymerization, ACS Sens., 2019, 4(1), 235–241 CrossRef CAS PubMed.
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization - A User Guide, Macromolecules, 2017, 50(19), 7433–7447 CrossRef CAS.
- F. Lorandi, M. Fantin, S. Shanmugam, Y. Wang, A. A. Isse, A. Gennaro and K. Matyjaszewski, Toward Electrochemically Mediated Reversible Addition-Fragmentation Chain-Transfer (eRAFT) Polymerization: Can Propagating Radicals Be Efficiently Electrogenerated from RAFT Agents?, Macromolecules, 2019, 52(4), 1479–1488 CrossRef CAS.
- S. R. Zavada, T. Battsengel and T. F. Scott, Radical-Mediated Enzymatic Polymerizations, Int. J. Mol. Sci., 2016, 17(2), 195 CrossRef PubMed.
- O. Emery, T. Lalot, M. Brigodiot and E. Maréchal, Free-Radical Polymerization of Acrylamide by Horseradish Peroxidase-Mediated Initiation, J. Polym. Sci., Part A: Polym. Chem., 1997, 35(15), 3331–3333 CrossRef CAS.
- A. Singh, D. Ma and D. L. Kaplan, Enzyme-Mediated Free Radical Polymerization of Styrene, Biomacromolecules, 2000, 1(4), 592–596 CrossRef CAS PubMed.
- B. Kalra and R. A. Gross, Horseradish Peroxidase Mediated Free Radical Polymerization of Methyl Methacrylate, Biomacromolecules, 2000, 1(3), 501–505 CrossRef CAS PubMed.
- H. Iwata, Y. Hata, T. Matsuda and Y. Ikada, Initiation of Radical Polymerization by Glucose Oxidase Utilizing Dissolved Oxygen, J. Polym. Sci., Part A: Polym. Chem., 1991, 29(8), 1217–1218 CrossRef CAS.
- R. Shenoy, M. W. Tibbitt, K. S. Anseth and C. N. Bowman, Formation of Core-Shell Particles by Interfacial Radical Polymerization Initiated by a Glucose Oxidase-Mediated Redox System, Chem. Mater., 2013, 25(5), 761–767 CrossRef CAS PubMed.
- F. Hollmann and I. W. C. E. Arends, Enzyme Initiated Radical Polymerizations, Polymers, 2012, 4, 759–793 CrossRef.
- T. Su, Z. Tang, H. He, W. Li, X. Wang, C. Liao, Y. Sun and Q. Wang, Glucose Oxidase Triggers Gelation of N-Hydroxyimide-Heparin Conjugates to Form Enzyme-Responsive Hydrogels for Cell-Specific Drug Delivery, Chem. Sci., 2014, 5(11), 4204–4209 RSC.
- B. J. Berron, L. M. Johnson, X. Ba, J. D. McCall, N. J. Alvey, K. S. Anseth and C. N. Bowman, Glucose Oxidase-Mediated Polymerization as a Platform for Dual-Mode Signal Amplification and Biodetection, Biotechnol. Bioeng., 2011, 108(7), 1521–1528 CrossRef CAS PubMed.
- A. J. Gormley, R. Chapman and M. M. Stevens, Polymerization Amplified Detection for Nanoparticle-Based Biosensing, Nano Lett., 2014, 14(11), 6368–6373 CrossRef CAS PubMed.
- A. S. Sarac, Redox Polymerization, Prog. Polym. Sci., 1999, 24, 1149–1204 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., 4th edn, 2004, pp. 216–217 Search PubMed.
- D. Zhuang, H. Shen, G. Liu, C. Yu and J. Yang, A Combining Signal Amplification of Atom Transfer Radical Polymerization and Redox Polymerization for Visual Biomolecules Detection, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(19), 2791–2799 CrossRef CAS.
- D. Zhuang, F. Wen, Y. Cui, T. Tan and J. Yang, Chitosan/Ce(IV) Redox Polymerization-Based Amplification for Detection of DNA Point Mutation, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(13), 1929–1937 CrossRef CAS.
- Y. Cui, D. Zhuang, T. Tan and J. Yang, Highly Sensitive Visual Detection of Mutant DNA Based on Polymeric Nanoparticles-Participating Amplification, RSC Adv., 2016, 6(116), 115238–115246 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Photoinitiated Polymerization: Advances, Challenges, and Opportunities, Macromolecules, 2010, 43(15), 6245–6260 CrossRef CAS.
- H. D. Sikes, R. Jenison and C. N. Bowman, Antigen Detection Using Polymerization-Based Amplification, Lab Chip, 2009, 9(5), 653–656 RSC.
- R. R. Hansen, H. D. Sikes and C. N. Bowman, Visual Detection of Labeled Oligonucleotides Using Visible-Light-Polymerization-Based Amplification, Biomacromolecules, 2008, 9(1), 355–362 CrossRef CAS PubMed.
- T. Scherzer, Photopolymerization of Acrylates without Photoinitiators with Short-Wavelength UV Radiation: A Study with Real-Time Fourier Transform Infrared Spectroscopy, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(4), 894–901 CrossRef CAS.
- N. Zivic, M. Bouzrati-Zerelli, A. Kermagoret, F. Dumur, J. P. Fouassier, D. Gigmes and J. Lalevée, Photocatalysts in Polymerization Reactions, ChemCatChem, 2016, 8(9), 1617–1631 CrossRef CAS.
- H. J. Avens and C. N. Bowman, Mechanism of Cyclic Dye Regeneration During Eosin-Sensitized Photoinitiation in the Presence of Polymerization Inhibitors, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6083–6094 CrossRef CAS PubMed.
- M. V. Encinas, A. M. Rufs, S. G. Bertolotti and C. M. Previtali, Xanthene Dyes/Amine as Photoinitiators of Radical Polymerization: A Comparative and Photochemical Study in Aqueous Medium, Polymer, 2009, 50(13), 2762–2767 CrossRef CAS.
- A. Aguirre-Soto, K. Kaastrup, S. Kim, K. Ugo-Beke and H. D. Sikes, Excitation of Metastable Intermediates in Organic Photoredox Catalysis: Z-Scheme Approach Decreases Catalyst Inactivation, ACS Catal., 2018, 8(7), 6394–6400 CrossRef CAS.
- K. Kaastrup, A. Aguirre-Soto, C. Wang, C. N. Bowman, J. W. Stansbury and H. D. Sikes, UV-Vis/FT-NIR in Situ Monitoring of Visible-Light Induced Polymerization of PEGDA Hydrogels Initiated by Eosin/Triethanolamine/O2, Polym. Chem., 2016, 7, 592–602 RSC.
- A. Aguirre-Soto, S. Kim, K. Kaastrup and H. D. Sikes, On the Role of N-Vinylpyrrolidone in the Aqueous Radical-Initiated Copolymerization with PEGDA Mediated by Eosin Y in the Presence of O2, Polym. Chem., 2019, 10(8), 926–937 RSC.
- L. M. Johnson, R. R. Hansen, M. Urban, R. D. Kuchta and C. N. Bowman, Photoinitiator Nucleotide for Quantifying Nucleic Acid Hybridization, Biomacromolecules, 2010, 11, 1133–1138 CrossRef CAS PubMed.
- H. J. Avens and C. N. Bowman, Development of Fluorescent Polymerization-Based Signal Amplification for Sensitive and Non-Enzymatic Biodetection in Antibody Microarrays, Acta Biomater., 2010, 6(1), 83–89 CrossRef CAS PubMed.
- H. J. Avens, E. L. Chang, A. M. May, B. J. Berron, G. J. Seedorf, V. Balasubramaniam and C. N. Bowman, Fluorescent Polymeric Nanocomposite Films Generated by Surface-Mediated Photoinitiation of Polymerization, J. Nanopart. Res., 2011, 13(1), 331–346 CrossRef CAS.
- L. Kuck and A. Taylor, Photopolymerization as an Innovative Detection Technique for Low-Density Microarrays, BioTechniques, 2008, 45(2), 179–186 CrossRef CAS PubMed.
- K. Kaastrup and H. D. Sikes, Polymerization-Based Signal Amplification under Ambient Conditions with Thirty-Five Second Reaction Times, Lab Chip, 2012, 12(20), 4055–4058 RSC.
- A. K. Badu-Tawiah, S. Lathwal, K. Kaastrup, M. Al-Sayah, D. C. Christodouleas, B. S. Smith, G. M. Whitesides and H. D. Sikes, Polymerization-Based Signal Amplification for Paper-Based Immunoassays, Lab Chip, 2015, 15(3), 655–659 RSC.
- S. Lathwal and H. D. Sikes, Assessment of Colorimetric Amplification Methods in a Paper-Based Immunoassay for Diagnosis of Malaria, Lab Chip, 2016, 16(8), 1374–1382 RSC.
- E. H. Yee, S. Lathwal, P. P. Shah and H. D. Sikes, Detection of Biomarkers of Periodontal Disease in Human Saliva Using Stabilized, Vertical Flow Immunoassays, ACS Sens., 2017, 2(11), 1589–1593 CrossRef CAS PubMed.
- S. Kim and H. D. Sikes, Phenolphthalein-Conjugated Hydrogel Formation under Visible-Light Irradiation for Reducing Variability of Colorimetric Biodetection, ACS Appl. Bio Mater., 2018, 1(2), 216–220 CrossRef CAS.
- J. K. Lee, B. W. Heimer and H. D. Sikes, Systematic Study of Fluorescein-Functionalized Macrophotoinitiators for Colorimetric Bioassays, Biomacromolecules, 2012, 13(4), 1136–1143 CrossRef CAS PubMed.
- J. K. Lee and H. D. Sikes, Balancing the Initiation and Molecular Recognition Capabilities of Eosin Macroinitiators of Polymerization-Based Signal Amplification Reactions, Macromol. Rapid Commun., 2014, 35(10), 981–986 CrossRef CAS PubMed.
- K. Kaastrup and H. D. Sikes, Investigation of Dendrimers Functionalized with Eosin as Macrophotoinitiators for Polymerization-Based Signal Amplification Reactions, RSC Adv., 2015, 5(20), 15652–15659 RSC.
- S. Kim and H. D. Sikes, Liposome-Enhanced Polymerization-Based Signal Amplification for Highly Sensitive Naked-Eye Biodetection in Paper-Based Sensors, ACS Appl. Mater. Interfaces, 2019, 11(31), 28469–28477 CrossRef PubMed.
- D. Dellal, E. Yee, S. Lathwal, H. Sikes and J. Gomez-Marquez, Low-Cost Plug and Play Photochemistry Reactor, HardwareX, 2018, 3, 1–9 CrossRef.
- A. F. D. Cruz, N. Norena, A. Kaushik and S. Bhansali, A Low-Cost Miniaturized Potentiostat for Point-of-Care Diagnosis, Biosens. Bioelectron., 2014, 62, 249–254 CrossRef CAS PubMed.
- A. C. Sun and D. A. Hall, Point-of-Care Smartphone-Based Electrochemical Biosensing, Electroanalysis, 2019, 31(1), 2–16 CrossRef CAS.
- E. A. Phillips, T. J. Moehling, K. F. K. Ejendal, O. S. Hoilett, K. M. Byers, L. A. Basing, L. A. Jankowski, J. B. Bennett, L.-K. Lin and L. A. Stanciu, et al., Microfluidic Rapid and Autonomous Analytical Device (MicroRAAD) to Detect HIV from Whole Blood Samples, Lab Chip, 2019, 19, 3375–3386 RSC.
- S. Nayak, N. R. Blumenfeld, T. Laksanasopin and S. K. Sia, Point-of-Care Diagnostics: Recent Developments in a Connected Age, Anal. Chem., 2017, 89(1), 102–123 CrossRef CAS PubMed.
- H. Sun, W. Xu, B. Liu, Q. Liu, Q. Wang, L. Li, J. Kong and X. Zhang, Ultrasensitive Detection of DNA via SI-eRAFT and in Situ Metalization Dual-Signal Amplification, Anal. Chem., 2019, 91, 9198–9205 CrossRef CAS PubMed.
- S. Lathwal and H. D. Sikes, A Method for Designing Instrument-Free Quantitative Immunoassays, Anal. Chem., 2016, 88(6), 3194–3202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01801h |
| This journal is © The Royal Society of Chemistry 2020 |