
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium and magnesium polymeric electrolytes prepared using poly(glycidyl ether)-based polymers with short grafted chains†
Marco
Viviani
a,
Niels Laurens
Meereboer
a,
Ni Luh Putu Ananda
Saraswati
ab,
Katja
Loos
 a and
Giuseppe
Portale
*a
a and
Giuseppe
Portale
*a
aMacromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: g.portale@rug.nl
bInorganic and Physical Chemistry Division, Faculty of Mathematics and Natural Sciences, Bandung Institute of Technology, Jalan Ganesha 10, 40132 Bandung, Indonesia
First published on 7th February 2020
Abstract
Recently, poly(allyl glycidyl ether) (PAGE) has attracted great interest as a polymer electrolyte for Li-ion transport with conductivity values well above that of the benchmark polyethylene oxide polymer at temperatures below 60 °C. Here, we prepared lithium and magnesium polyelectrolytes by using two novel PAGE-based matrixes containing thioether and sulfone functionalities located in a short side chain inserted by the chemical post-functionalization of PAGE. The synthesized PAGEs, poly(2-(ethyl thio) propyl glycidyl ether) (PEthioPGE) and poly(2-(ethyl sulfone) propyl glycidyl ether) (PEsulfoPGE), were all amorphous at any temperature with Tg between −80 °C and −30 °C. These polymers were used to formulate electrolytes with different Li and Mg salts. The impact of the side chain, used salt and temperature on the ionic conductivity was studied in detail. Ionic conductivities as high as 5.1 × 10−4 S cm−1 at 90 °C can be achieved using PAGE–LiTFSI and PEthioPGE–LiTFSI, values comparable to that achieved using PEO–LiTFSI with identical salt loading. When LiCl is used, PEthioPGE outperforms all other polymers including PEO with the highest conductivity value at 90 °C (1.1 × 10−5 S cm−1). Moreover, the studied complexes with magnesium salts showed promising ionic conductivities, comparable to those achieved using lithium and up to 4.1 × 10−4 S cm−1 at 90 °C for PAGE–Mg(TFSI)2. The results presented here highlight the possibility of tuning the structures and the complexation properties of poly(glycidyl ether)-based electrolytes towards both lithium and magnesium ions.
Introduction
Due to climate change and the related need to reduce the dependence on fossil fuels, development of alternative and renewable energy sources has drawn increasing attention. Harvesting renewable energy has several implications related to natural sources, making power generation less controllable and harder to manage in terms of energy distribution. Energy storage devices play a key role in overcoming these issues and secondary batteries are optimal systems due to their versatility.While the market for rechargeable batteries is still dominated by lead-acid batteries, the increasing demand for rechargeable portable and automotive devices has raised the production of lithium batteries (LiBs). LiBs are the best candidates for compact and high-performance applications due to their light weight, flexibility and high power density.1,2 Lithium is the lightest metal with the highest electrochemical reduction potential (−3.04 V vs. SHE), allowing the design of high energy density batteries.3,4 Apart from all these benefits, some drawbacks still need to be addressed: shortcuts caused by dendrite formation at the anode/electrolyte interface, reduced efficiency over time due to passivation layer growth at the electrode/electrolyte interface and the safety issues related to the intrinsic reactivity of metallic lithium.3 Another critical aspect is the balance between the natural abundance of lithium and the global demand for LiBs which is expected to increase significantly in the coming decades, especially due to automotive electrification.2,5 All these concerns related to lithium technology stimulated research on alternative metals to overcome its disadvantages and limitations.
Among the other mono- and multivalent cations, magnesium seems to be the most promising element due to its potentially higher volumetric specific capacity (3.8 A h cm−3vs. 2.0 A h cm−3). Although it has a lower reduction potential (−2.37 V vs. SHE) and a lower specific capacity (2.2 A h g−1vs. 3.9 A h g−1) compared to lithium, this alkaline earth metal is the eighth-most abundant element in the Earth's crust and its lower reactivity allows for making metal electrodes without forming dendrites.3,6–8 In the case of magnesium batteries (MgBs) the main disadvantage is the passivation of the anode which has a much more detrimental effect than in the LiBs due to the impermeability of the layer to the Mg2+ ions.9
While liquid electrolytes represent the benchmarks for both LiBs and MgBs in terms of conductivity, they are unfortunately volatile, flammable and pollutants, leading to safety problems in the manufacture, handling and disposal/recycling of the batteries. Compared to the LiBs, the MgBs have additional challenges regarding the stability of the electrolytes and their performances which make it difficult to adopt the same solutions as those used for the lithium batteries.6,7,10 In order to overcome all these complications, several approaches have been attempted spanning from organic gels to inorganic solid electrolytes.6,7,11
Among these approaches, polymer electrolytes (PEs) have shown potential for substituting small-molecule liquid electrolytes, allowing the development of safer solvent-free and better performing (metal anode) batteries.4,12 However, these benefits are offset by their reduced ionic conductivity, leading to a gap between the theoretical and accessible performances of these devices.13 Ever since Fenton et al.14 discovered that poly(ethylene oxide) (PEO) can solvate sodium iodide, polyether-based electrolytes have attracted significant interest. The peculiar solvation properties towards lithium and magnesium cations of this class of polymers are the result of the strong interaction between the ions and the oxygen atoms along the polymer chains resulting in a chelate structure which affects the segmental mobility and the transport mechanism of the final complexes.15–20 Despite their low Tg values, the residual crystallinity of PEO-based electrolytes is responsible for conductivity drop below their melting point (T < 65 °C) essentially limiting the practical use of these PEs. Research on alternative polymers with high conductivity at room temperature has a non-trivial solution. The presence of polar groups, necessary for endowing the polymers good solvation properties towards Li and Mg salts, is also responsible for chain stiffening and eventually crystallinity.
Polyethers, polycarbonates, polynitriles, polyesters, polyamines and polyalcohols have been explored as potential polymer electrolytes and a recent comprehensive review has summarized the past research results for lithium electrolytes.11 Polymer magnesium electrolytes were firstly designed based on the lithium approach. In this perspective, poly(ether)s17,21,22 and poly(vinyl alcohol)s23 and their derivatives have been attempted, but the coordination of magnesium di-cations with the oxygen atoms is stronger than that of lithium, reducing the overall conductivity of the resulting complexes and increasing the related Tg.24–26 Despite the fact that segmental mobility is considered to be one of the most important factors for high ionic conductivity, an increasing number of studies have highlighted that Tg is not always a vital parameter27–33 and also other possible transport mechanisms can contribute to ion mobility in the matrix.31–35 In the Salt in Polymer Electrolyte (SIPE) and dilute regimes, the interplay between chain polarity and stiffness is relevant, controlling the solvation mechanisms of the salts and their mobility through the polymer matrices.29,30,36
A class of poly(allyl glycidyl)ethers (PAGEs) proposed by Barteau et al.28 offered the opportunity to investigate the effect of several structural parameters on the resulting conductivity of the lithium-complexes. Interestingly, the conductivity did not scale monotonically with the Tg of the polymers suggesting additional contributions playing a major role in the ion mobility. Follow-up studies reported by Wheatle et al.29,30 confirmed the relevance of the polymer backbone polarity by comparing computational and experimental results obtained for different structures29 and by claiming the existence of an optimal compromise between ion solvation and segmental mobility that must be taken into account in designing novel PEs.30 Using different heteroatoms than oxygen in the polymer structure is a straightforward method to modify both chain mobility and polarity. Specifically, the presence of sulfur heteroatoms in combination with oxygen has been proved to give encouraging results for lithium transport, even though crystallinity is not prevented.27,37 Poly alkyl sulfides alone15 proved to be unsatisfactory and the reported higher reactivity of the thioether compared to that of the ether linkage limited the studies on these polymers. On the other hand, the co-presence of both elements led to the formation of complexes with improved electrochemical stability with both lithium and magnesium salts.27,38–40
Here we present the synthesis, characterization and transport properties of lithium and magnesium polymer electrolytes based on the post-modification of PAGE. The PAGE polymer has already been synthesized by other groups and shows interesting transport properties, surpassing those of PEO polymers at temperatures below 50 °C.28 Considering its low Tg (ca. −80 °C) and ease of modification due to the dangling allylic functionality,28 we chose allyl glycidyl ether (AGE) as the building block for a new class of fully amorphous polymer electrolytes with low Tg and improved solvation capability towards lithium and magnesium salts. Recently, high Mg2+-ion conductivity, Mg cycling efficiency and anodic stability have been achieved using MgCl2 salts in dialkyl sulfone electrolytes, motivating our endeavours to investigate both Li+- and Mg2+-ion transport in sulfone containing solid polymer electrolytes.41 Three different polymers have been synthesized and studied: pristine PAGE, poly(2-(ethylthio) propyl glycidyl ether) (PEthioPGE) and poly(2-(ethyl sulfone) propyl glycidyl ether) (PEsulfoPGE). Different oxidation states of the sulfur atom allowed for the investigation of the effect of polymer polarity on both chain mobility and ionic conductivity. Four different salts, namely lithium trifluoromethylsulfonimide (LiTFSI), lithium chloride (LiCl), magnesium trifluoromethylsulfonimide (Mg(TFSI)2) and magnesium chloride (MgCl2), were tested to assess the potential application of these new complexes as solvent-free electrolytes for both LiBs and MgBs.
Experimental
Materials and methods
Calcium hydride (Acros Organics 93%), potassium metal cubes in mineral oil (Sigma-Aldrich 99.5%), ethanethiol (Acros Organics 99+%), 3-chloroperbenzoic acid (MCPBA) (Sigma-Aldrich ≤77%), acetic acid glacial (Merck), and methanol (Macron Fine Chemicals ≥99.8%) were used as received. Naphthalene (Sigma-Aldrich 99%) was recrystallized from methanol, and dry tetrahydrofuran (THF) was collected from an MBraun SPS5 solvent purification system and used immediately afterwards. Potassium naphthalenide was prepared from potassium metal and recrystallized naphthalene in anhydrous THF by stirring for 24 h with a glass-coated stirring bar at room temperature under a nitrogen atmosphere. Benzyl alcohol (Merck ≥99.0%) was dried over calcium hydride and distilled prior to titration with potassium naphthalenide in THF. Allyl glycidyl ether (AGE) (Sigma-Aldrich ≥99%) was degassed through five freeze–pump–thaw cycles, dried over n-butyl magnesium chloride and distilled in a dried Schlenk tube. The purified AGE was used immediately afterwards. 2,2′-Azobis(2-methylpropionitrile) (AIBN) (Sigma-Aldrich ≥98%) was recrystallized twice from methanol prior to use.Synthesis of poly(allyl glycidyl ether) (PAGE)
PAGE was synthesized according to a previously reported procedure42 with minor modifications. Freshly distilled benzyl alcohol (1.34 mmol, 0.139 mL) was titrated with potassium naphthalenide in a pre-dried Schlenk tube under a nitrogen atmosphere. Potassium naphthalenide was added to the alcohol under stirring through a septum with a degassed syringe and needle until a green color was formed in the solution indicating the complete deprotonation of the alcohol. A pre-determined volumetric amount of AGE (202 mmol, 24 mL) was charged into the Schlenk tube with a degassed needle. The reaction mixture was allowed to stir at 35 °C for 24 hours. The polymerization was quenched with a deoxygenated mixture of acetic acid in methanol. The polymerization mixture was diluted with dichloromethane and passed through a short silica plug to remove salts. The volatiles were removed in vacuo.1H NMR (400 MHz, CDCl3): δ = 3.42–3.67 (broad m, 7H, –CH2–CH–O–, –CH2–O–CH2–), 3.95–4.03 (d, 2H, –O–CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.12–5.30 (dd, 2H, –CHCH2), 5.83–5.97 (broad m, 1H, –CHCH2). (Mn: 14
), 5.12–5.30 (dd, 2H, –CHCH2), 5.83–5.97 (broad m, 1H, –CHCH2). (Mn: 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1, PDI: 1.09).
500 g mol−1, PDI: 1.09).
Synthesis of poly(2-(ethylthio) propyl glycidyl ether) (PEthioPGE)
A solution of PAGE (0.053 mmol*, 6.00 gram), ethanethiol (0.263 mol, 18.9 mL), and AIBN (39.4 mmol, 6.47 gram) in anhydrous THF (80 mL) was prepared in a pre-dried Schlenk tube under a nitrogen atmosphere. The solution was refluxed for 24 hours and then quenched with methanol. The resulting polymer was extensively washed with methanol using a centrifuge (5×, 3 min, 4500 rpm).1H NMR (400 MHz, CDCl3): δ = 1.20–1.30 (broad m, 3H, –CH3), 1.78–1.90 (broad m, 2H, –CH2–CH2–CH2–), 2.48–2.64 (broad m, 4H, –CH2–S–CH2–), 3.35–3.65 (broad m, 7H, –CH2–CH–O–, –CH2–O–CH2–).*Repeating units AGE.
Synthesis of poly(2-(ethyl sulfone) propyl glycidyl ether) (PEsulfoPGE)
To a solution of PEthioPGE (2.27 mmol, 400 mg) in THF (6 mL), meta-chloroperbenzoic acid (11.34 mmol, 1.96 grams) was slowly added. The mixture was stirred for 24 hours at room temperature. The reaction mixture was concentrated and precipitated in cold methanol (liquid nitrogen cooled). Methanol was decanted and the polymer was washed with cold methanol extensively. Finally, the polymer was dried in vacuo.1H NMR (400 MHz, CDCl3): δ = 1.33–1.42 (broad m, 3H, –CH3), 2.00–2.15 (broad m, 2H, –CH2–CH2–CH2–), 2.96–3.12 (broad m, 4H, –CH2–SO2–CH2–), 3.42–3.65 (broad m, 7H, –CH2–CH–O–, –CH2–O–CH2–).Polyelectrolyte preparation
PAGE-like polymer electrolytes (PEs) were prepared in an MBraun nitrogen glove box, with both H2O and O2 concentrations less than 0.1 ppm. The preparation of the materials was done by mixing the pristine polymer with a calculated amount of salt. The salt concentration is referred here in terms of the molar ratio (r) of the lithium or magnesium atom to the ether oxygen atoms (EO) in the polymer backbone, [Li]/[EO] and [Mg]/[EO]. We used r = 0.06 which corresponds to 16 EO every lithium or magnesium atom, based on the added materials. This ratio was chosen because it is reported to correspond to the peak in the ionic conductivity for the PAGE–LiTFSI system.28 The samples used in this study are denoted using the notation [polymer name]–[salt name] (i.e. PAGE–LiTFSI). The lithium and magnesium salts were first vacuum-dried at 120 °C overnight to remove traces of water and then dissolved in a certain amount of extra pure anhydrous DMF solution before use. Approximately 0.2 g of pristine polymers was placed in glass vials and then mixed with the calculated amount of salt/DMF solution. The solutions were first heated at 60 °C under a N2 flow. The mixtures were then dried in the glove box-antechamber oven for at least 3 days at 70 °C under vacuum to completely remove the residual solvent and water. The dried samples were then transferred into the glove box and kept there before being characterized.Characterization
Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on a 400 MHz Varian (VXR) spectrometer at room temperature using CDCl3 as the solvent (13C NMR spectra are reported in Fig. S1–S3†). Gel Permeation Chromatography (GPC) was performed in DMF (containing 0.01 M LiBr) using a Viscotek GPCmax equipped with model 302 TDA detectors and two columns (Agilent Technologies-PolarGel-L and M, 8 μm 30 cm) at a flow rate of 1.0 mL min−1 and 50 °C. Narrow dispersity PMMA standards (Polymer Laboratories) were used for constructing a universal calibration curve applied for determining the molecular weights of the polymers (GPC traces of the pure polymers are reported in Fig. S4†). The Fourier Transform Infrared (FTIR) spectra of the polymers were recorded on a Bruker Vertex 70 spectrophotometer using 32 scans at a nominal resolution of 4 cm−1 using a diamond single reflection attenuated total reflectance (ATR) accessory. Thermogravimetric analysis (TGA) was conducted using a TA Instruments Discovery TGA5500 over the temperature range of 0 °C to 700 °C at a 10 °C min−1 heating rate in air. Differential scanning calorimetry (DSC) was performed using a TA Instruments DSC Q1000 calorimeter. The samples (approximately 5 mg) were placed in aluminium hermetic pans. DSC scans consisted of two heating and one cooling cycles over the temperature range of −80 °C to 100 °C. The first heating rate was 10 °C min−1, while the cooling and second heating rates were 5 °C min−1. The Tg values of all samples were determined as the inflection point on the second heating cycle. Ion transport properties were measured via Electrochemical Impedance Spectroscopy (EIS). Polymer electrolyte samples were prepared inside an MBraun nitrogen filled glove box using custom made conductivity cells designed similarly to those reported by Lascaud et al.43 Each polymer electrolyte was heated in a closed cell at 80 °C for about 15 minutes to ensure good contact between the electrodes. After cooling down at room temperature the loaded cell was transferred outside the glove box for impedance measurements. Temperature-dependent AC impedance measurements were performed using the Intermediate Temperature System (ITS/Bio-Logic) connected to a Bio-Logic SP-300 potentiostat. Impedance data were collected every 10 °C from 90 °C to 0 °C with an equilibration time of at least 1 hour at each temperature. The temperature was measured with an accuracy of ±1 °C by means of a thermocouple inserted in a well dug in the body of the top part of the cell. Impedance measurements were conducted in a through-plane two-electrode mode at an AC voltage of 20 mV and with the frequency ranging from 7 MHz to 1 Hz with 0 V DC bias. Data were acquired and analysed using the EC-Lab software (Bio-Logic). Conductivity values were calculated from complex impedance analysis (|Z| = Z′ + iZ′′ where Z′ and Z′′ are the real and the imaginary part of the impedance, respectively). Experimental data were analysed using the equivalent circuit represented in the inset in Fig. S14† and the bulk resistance (Rb) obtained from the fitting was used to calculate the sample conductivity (σ) according to eqn (1):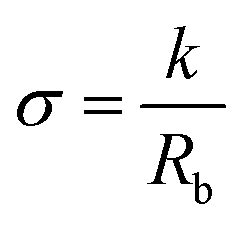 | (1) |
Results and discussion
Polymer matrix synthesis
The synthetic route towards the polymers used in this study is outlined in Scheme 1. Benzyl alcohol deprotonated by potassium naphthalenide was used as the initiator for the anionic ring-opening polymerization of AGE. To obtain the quantitative conversion of AGE and to minimize isomerization reactions, the reaction temperature was kept at 35 °C for 24 hours. The polymerization was terminated by adding acidified methanol.42 In this way, narrow disperse PAGE was obtained (14.5 kg mol−1, Đ = 1.05). The signals corresponding to the protons of PAGE in the 1H NMR spectrum, depicted in Fig. 1a, are in good agreement with literature reports.28,42 Subsequently, using a thiol–ene click reaction, the allyl functionality in PAGE was used for introducing side chains containing sulfur heteroatoms. Using 1H NMR spectroscopy, the successful full conversion of the allyl moieties is confirmed by the disappearance of the signals in the regions 5.12–5.30 and 5.83–5.97 ppm corresponding to the CHCH2 and CHCH2 protons, respectively. The signals appearing at 2.48–2.64 ppm are characteristic of the –CH2–S–CH2– protons surrounding the thioether. The sulfone derivative of this polymer, PEsulfoPGE, was then synthesized by a quantitative oxidation method using m-chloroperbenzoic acid. The efficiency of this reaction was demonstrated by the complete downfield shift of the –CH2–S–CH2– protons to 2.96–3.12 ppm caused by an increased shielding of the sulfone groups. In addition, infrared spectroscopy further confirmed the formation of PEsulfoPGE, showing the characteristic sulfone stretching vibration at 1100 and 1300 cm−1 and sulfone bending vibration of C–S at around 800–750 cm−1 (Fig. 1b).44
 | ||
| Scheme 1 Synthetic route towards poly(allyl glycidyl ether) (PAGE), poly(2-(ethyl thio) propyl glycidyl ether)(PEthioPGE) and poly(2-(ethyl sulfone) propyl glycidyl ether) (PEsulfoPGE). | ||
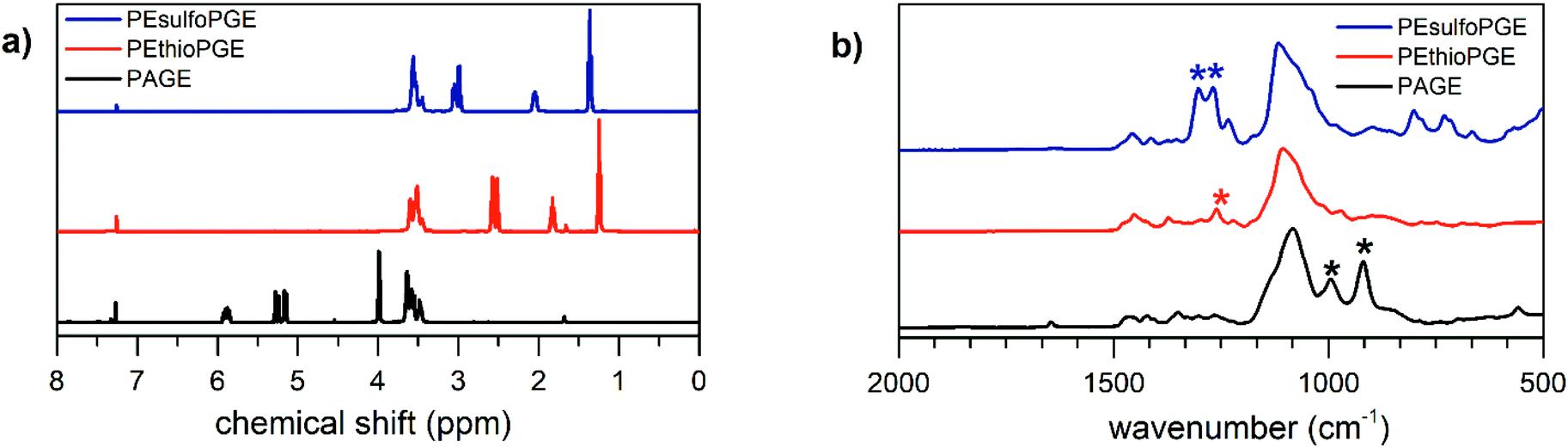 | ||
| Fig. 1 (a) 1H NMR spectra of PAGE, PEthioPGE and PEsulfoPGE. (b) FTIR spectra of the three different polymers. The stars indicate the positions of the characteristic peaks related to the functional groups: 917 and 995 cm−1 bending of monosubstituted alkenes for PAGE, 1270 cm−1 –CH2– wagging of aliphatic thioether for PEthioPGE, 1270–1300 cm−1 –SO2–asymmetric stretching vibration of PEsulfoPGE, with marks on the peculiar vibrations of the respective functional groups. | ||
Polymer electrolyte preparation and characterization
Polymer electrolytes (PEs) were prepared by adding lithium and magnesium salts to the polymers. In addition to the conventional TFSI salts (LiTFSI and Mg(TFSI)2), due to the recently reported high dielectric constants of these kinds of polymers29 and the presence of the sulfur heteroatoms located on the side chains, we have also decided to investigate complexes with LiCl and MgCl2. The PEs were prepared by dissolving LiTFSI, LiCl, Mg(TFSI)2 and MgCl2 in the three different polymers inside a nitrogen-filled glove box. This precaution is needed to avoid moisture absorption by the electrolyte which could affect ionic conductivity measurements. The molar ratio of the lithium or magnesium ions to the oxygen atoms in the repeating unit of the host polymer is crucial for the overall electrolyte conductivity. In this research, the molar ratio, [Li]/[EO] or [Mg]/[EO], was kept fixed at 0.06, which is considered to be optimal.26,28,29,45 This value means that there is one lithium or magnesium ion every sixteen ether oxygen atoms of the polymer. All salts showed good solubilization inside the polymer matrix except chlorides which were partially insoluble at room temperature, but were readily soluble as soon as the temperature was increased.The incorporation of the Li and Mg salts into the polymer structure was studied by FTIR. Due to the hygroscopic nature of the salts and due to the instrumental limitations to perform the measurements under an inert atmosphere, some complexes presented peaks of water (around 3400 and 1600 cm−1) that has been adsorbed only during the short time of the measurements (ESI Fig. S6–S9†). It is generally accepted that when forming a polymer electrolyte, the cations are solvated by coordination to oxygen atoms in the host polymer backbone.46 FTIR spectra presented in Fig. 2 clearly show the miscibility of lithium and magnesium ions in the polymers through the change in peaks belonging to the ethereal CH2–O–CH2 stretching around 1120 cm−1. In the case of LiTFSI and Mg(TFSI)2 salts, the anion absorption peaks also demonstrate the TFSI dissociation in all polymers. In particular, the vibrational frequencies of the anion in pure salts that appear at wavenumbers 800, 770 and 746 cm−1 shift to 787, 760 and 739 cm−1 in the polymer electrolytes, respectively (ESI Fig. S10†). The shifts of the peaks at 1197, 1140 and 1060 cm−1 to lower wavenumbers together with the change in the multiplicity of the peak at 1084 cm−1 are also correlated to the dissociation of the salt in the polymer matrix. These observations are in agreement with what was previously reported by other authors.27,28,47,48 In addition, the TFSI bands at around 600 cm−1, 614 cm−1 and 656 cm−1 are representative of the convoluted δ(SNS) vibrations of the transoid and cisoid forms, respectively, which are visible only in a fully dissociated scenario, where a conformational equilibrium is possible.49 In contrast, for the salt form, only the peak at 618 cm−1 is visible.
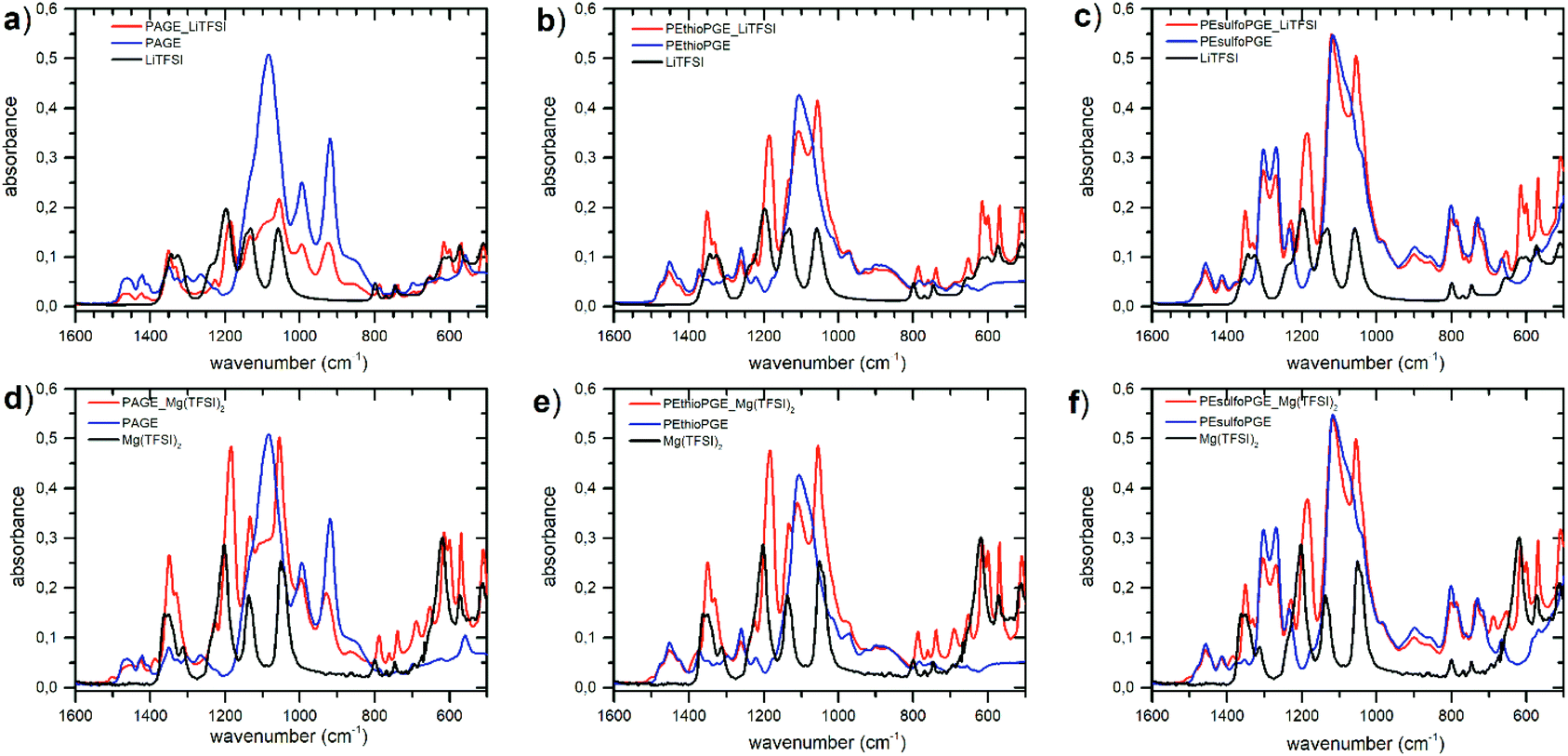 | ||
| Fig. 2 FTIR spectra of the neat salt, neat polymer and electrolyte complex for (a) PAGE–LiTFSI, (b) PEthioPGE–LiTFSI, (c) PEsulfoPGE–LiTFSI, (d) PAGE–Mg(TFSI)2, (e) PEthioPGE–Mg(TFSI)2 and (f) PEsulfoPGE–Mg(TFSI)2. | ||
As expected at this concentration,50 neither magnesium ions nor lithium ions form ion pairs in any complex. This is represented by a single peak at 740 cm−1 for magnesium50 and at 652 cm−1 for lithium.51,52 From the IR spectra, it appears quite clear that good solvation of the metal TFSI salts occurs in all matrixes.
For the chloride complexes, it is not possible to rely on the TFSI anion shifts and other characteristics peaks must be taken into account. In these cases, we can refer to the peaks at around 1390 cm−1, characteristic alteration in the wagging frequency of CH2–O that depends on the type of polymer–salt combination, and at around 690 cm−1, representing a particular interaction of magnesium with the polymer chain.
The PAGE–LiCl complex did not show any visible shifts or changes in the characteristic IR spectra of pristine PAGE, but both PEthioPGE and PEsulfoPGE show evidence of interactions with the salt (Fig. S5†). PEthioPGE shows some additional bands between 615 cm−1 and 500 cm−1. The increase of the intensity of the peak at around 1354 cm−1 and the absence of a water signal in the rest of the spectra suggest an interaction of the chain with the lithium salt. In the case of PEsulfoPGE, the splitting of the peak at 666 cm1 together with the peak at 1389 cm−1 is in good agreement with the partial dissolution of LiCl due to chain complexation. Finally, for the MgCl2 complexes, PEthioPGE shows no changes in the spectra with the salt, while PAGE only shows a slight increase in the intensity of the peak at around 1390 cm−1 and a shift of the peak at 690 cm−1 as previously observed in the case of Mg(TFSI)2. Conversely, PEsulfoPGE shows signs of a stronger interaction with MgCl2 due to the appearance of a new peak at 690 cm−1 and the inversion of the intensity of the two peaks at 1380 and 1350 cm−1 (Fig. S5†). Our observation is in agreement with previous studies on both lithium and magnesium demonstrating that sulfone oxygen atoms exhibit an increased stronger interaction with the metal cations when compared to ether oxygens.39,53
However, despite the observed interaction found via FTIR, in general, both LiCl and MgCl2 solubility in the polymers is lower than the TFSI salts, and this is also confirmed by the low conductivity values observed for the chloride complexes (vide infra). Ion transport in a polymeric matrix is possible due to two main mechanisms. The first is the ion migration by hopping from one chain to another, predominantly in the amorphous phase. The second is the whole chain diffusion together with the complexed ions.18,54,55 A good segmental mobility of the chain is thus crucial to obtain high conductivity values, and hence fully amorphous polymers with low Tg perform better than crystalline or glassy polymers. Therefore, we have conducted DSC analysis to determine Tg and any possible traces of crystallinity (ESI Fig. S11, S12 and Table S1†). The three neat polymers do not present any crystallization peak and their Tg values are well below room temperature ranging from −78 to −37 °C. While PEthioPGE presents a Tg value very similar to PAGE, the Tg value of PEsulfoPGE is about 40 °C higher, as a result of the increased polarity of the sulfone containing side chains.
The higher oxidation state of the sulfur atoms induces a stronger dipolar association of the chains and a reduction of the segmental mobility with a consequent increase in the Tg value. Such a trend was also observed by Sarapas et al.27 for polymers with different architectures but with the same functional groups along the chain.
The addition of LiTFSI and Mg(TFSI)2 salts to the polymers further increases the Tg of the polyelectrolyte complex as a consequence of ion solvation. It is generally accepted that in polyether systems the cations are solvated by coordination to the oxygen atoms in the host polymer backbone, leading to an increase in the chain stiffness.46 This explanation is also supported by the shifts of the FTIR peaks as mentioned above. Interestingly, while the addition of LiCl does not cause a shift in the glass transition for any polymer, the addition of MgCl2 shows a clear Tg increase only for the PEsulfoPGE–MgCl2 complex. This is an indication that the PEsulfoPGE polymer is able to efficiently solubilize MgCl2via the sulfone groups. The comparison of the Tg values of the chloride-complexes with their related FTIR traces corroborates that the weak interactions shown by PAGE and PEthioPGE have no effect on the mobility of the chains. The chloride salts are thus only partially soluble in the host matrix and only the PEsulfoPGE–MgCl2 complex shows signs of high solvation.
The thermal stability of both the polymers and the complexes was studied by thermogravimetric analysis (TGA). TGA thermograms showed good thermal stability for all three homopolymers up to 350 °C (ESI Fig. S13 and Table S2†). According to this result, all three polymers will be thermally stable in the temperature range of polymer electrolyte battery applications.
Ion transport properties
The transport properties of the prepared Li and Mg polymer complexes were measured using electrochemical impedance spectroscopy (EIS). Two typical Nyquist plots of the PAGE–Mg(TFSI)2 complex at two different temperatures are reported in Fig. S14.† The conductivity can be calculated using the bulk resistance extracted from the high-frequency semicircle intercept determined according to the fitting curve using an equivalent circuit shown in the inset in Fig. S14.† The performance of each polymer complex was tested by measuring the resistance as a function of temperature in the range 90–0 °C. For the PEsulfoPGE–chloride complex, appreciable conductivity could be measured only down to 20–30 °C. The dependence of conductivity on temperature is summarized in Fig. 3 and 4. We found that the conductivity σ(T) vs. temperature plot can be well described using the Vogel–Tammann–Fulcher (VTF) equation (eqn (2)) as usually reported in the literature for polymeric solid electrolytes:43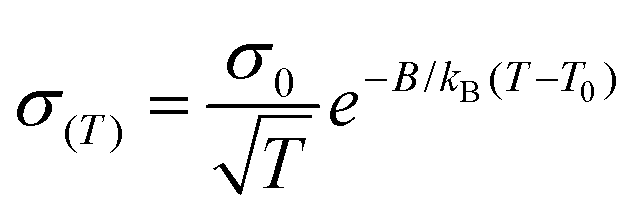 | (2) |
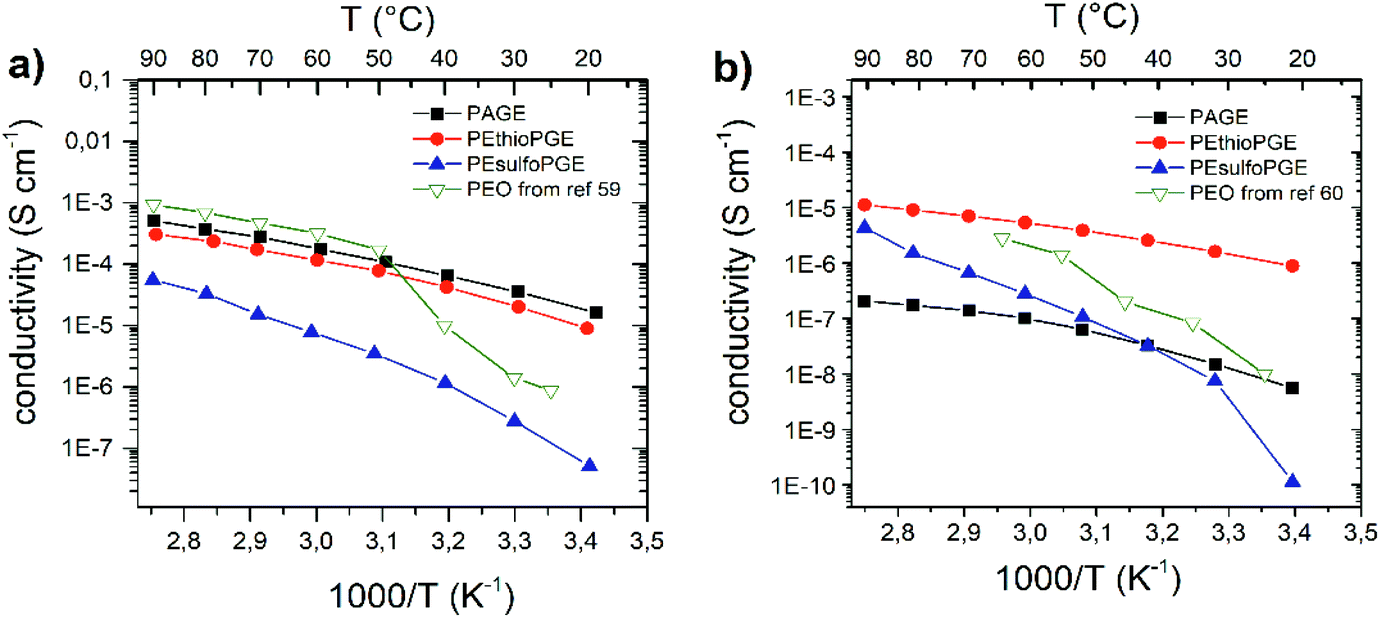 | ||
| Fig. 3 Temperature-dependent conductivity for (a) LiTFSI and (b) LiCl polymer complexes. The available data for PEO–LiTFSI from ref. 59 and PEO–LiCl from ref. 60 are also reported in (a) and (b), respectively, for comparison. | ||
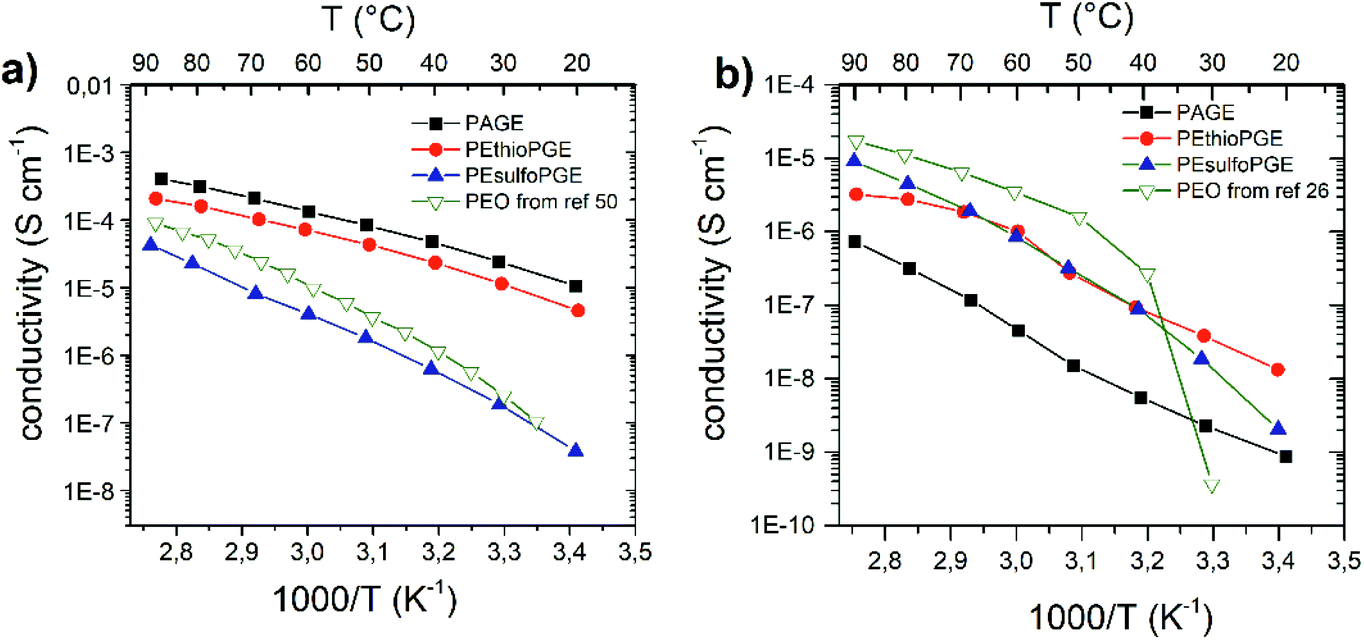 | ||
| Fig. 4 Temperature-dependent conductivity of (a) Mg(TFSI)2 and (b) MgCl2 polymer complexes. The available data for PEO9–Mg(TFSI)2 from ref. 50 and PEO16–MgCl2 from ref. 26 are also reported in (a) and (b), respectively, for comparison. | ||
VTF plots for the TFSI and chloride complexes are reported in the ESI (Fig. S15 and S16†). As expected, the conductivity of all studied complexes shows a linear increase in the ln(σ) vs. (T − T0)−1 plots with increasing temperature (moving from right to left). In the case of polymer–LiTFSI complexes, the highest conductivity values are found for PAGE (5.1 × 10−4 S cm−1 at 90 °C), closely followed by PEthioPGE (3.0 × 10−4 S cm−1). PEsulfoPGE complexes show a significantly lower conductivity (5.5 × 10−5 S cm−1). The conductivity value found for the PAGE–LiTFSI complex confirms the value reported previously by Barteau et al. for a similar polymer electrolyte but with a higher molecular weight.28 Interestingly, the thioether polymer exhibits comparable values of ionic conductivity. This may be explained by the good complexation of the sulfur atom and the Li-ions as commonly reported for ion-conducting glasses.27,57 Due to the amorphous nature of our polymers, the sudden conductivity drop observed below 50 °C for the PEO–LiTFSI complex due to the crystallization of PEO is not observed (Fig. 3). In the case of PEsulfoPGE, the lower conductivity could be ascribed to the lower Li/O ratio considering the sulfone oxygen, which leads also to a higher degree of coordination resulting in lower segmental mobility (higher Tg).
Interestingly, the trend in the ionic conductivities of the LiCl complexes is different from that of LiTFSI: PEthioPGE > PEsulfoPGE > PEO > PAGE. The best performance of PEthioPGE–LiCl is also combined with the lowest pseudo-activation energy (Table 1). However, the overall conductivities of the LiCl complexes remain one order of magnitude lower than those of the LiTFSI ones, in agreement with the lower solubility of the salt in the polymeric matrixes as observed experimentally by FTIR and DSC. The poor fit of the VTF behaviour for PAGE confirms that almost no interaction between the polymer and salt occurs. The differences in the trends between the conductivities of the LiTFSI and LiCl complexes may also reflect the differences in the interactions of the two anions with the host polymers.30,56,58 The strong dipole interaction of the sulfone group plays a major role in the conductivity of PEsulfoPGE, outperforming even PAGE which has a lower Tg of about 40 °C.
| Polymer | Salt | [EO]/[M] | r = [M]/[O] | T g (°C) | B (eV) |
|---|---|---|---|---|---|
| PAGE | LiTFSI | 16 | 0.06 | −54.2 | 0.100 |
| PEthioPGE | LiTFSI | 16 | 0.06 | −64.5 | 0.120 |
| PEsulfoPGE | LiTFSI | 16 | 0.03 | −17.7 | 0.118 |
| PAGE | LiCl | 16 | 0.06 | −76.1 | 0.144 |
| PEthioPGE | LiCl | 16 | 0.06 | −73.8 | 0.098 |
| PEsulfoPGE | LiCl | 16 | 0.03 | −36.5 | 0.189 |
| PAGE | Mg(TFSI)2 | 16 | 0.06 | −50.7 | 0.107 |
| PEthioPGE | Mg(TFSI)2 | 16 | 0.06 | −59.0 | 0.174 |
| PEsulfoPGE | Mg(TFSI)2 | 16 | 0.03 | −17.8 | 0.117 |
| PAGE | MgCl2 | 16 | 0.06 | −77.0 | 0.272 |
| PEthioPGE | MgCl2 | 16 | 0.06 | −75.8 | 0.238 |
| PEsulfoPGE | MgCl2 | 16 | 0.03 | −9.7 | 0.126 |
Recently, high Mg2+-ion conductivity and high Mg cycling efficiency have been achieved using MgCl2 salts in dialkyl sulfone electrolytes, motivating our endeavours to investigate Mg2+-ion conductivity in the synthesized sulfone containing solid polymer electrolytes.39,41 In Fig. 4 we report the transport properties of the Mg(TFSI)2 and MgCl2 complexes. For the TFSI complexes, the same trend observed for Li is found. Interestingly, the conductivity values are comparable to those of the LiTFSI complexes and, except for the more rigid PEsulfoPGE, they are all higher than the corresponding PEO complex with a higher salt concentration (see Fig. 4). The highest conductivity is found for the PAGE complex (4.1 × 10−4 S cm−1 at 90 °C), followed by PEthioPGE (2.1 × 10−4 S cm−1). For the MgCl2 complexes, a behaviour different from that of the LiCl case is observed. The conductivity scales as PEsulfoPGE > PEthioPGE > PAGE. The inversion between PEsulfoPGE and PEthioPGE can be attributed to the stronger interaction between the sulfone oxygen atoms and the magnesium cations, needed here to dissociate the less soluble MgCl2 salt. In fact, the Tg of the PEsulfoPGE–MgCl2 complex is the highest measured for all the electrolytes, even higher than the same polymer with the Mg(TFSI)2 counter ion (Table 1).
The VTF plot also shows that PEthioPGE–MgCl2 deviates from linearity, suggesting the low solubility of the salt inside this polymer, in agreement with the FTIR results (ESI Fig. S16†). In general, all MgCl2 complexes have conductivities comparable to the corresponding LiCl complexes, although a steeper decrease at lower temperatures is observed due to the reduced solubility of the Mg salt. Although PEO performs better above 65 °C, all synthesized polymers show higher conductivity below 50 °C, as they do not exhibit crystallization behaviour typical of PEO. Moreover, although in the case of both PEthioPGE and PEsulfoPGE the ratio between the ions and the total functional groups is less than that in the case of PAGE, their conductivities with both lithium and magnesium chlorides are higher than that of the unsaturated polyether. This could also be attributed to the contribution from the anion as reported for other PEO complexes.20,26,61,62 In order to evaluate the impact of the anion mobility on the overall conductivity, transference number measurements and battery tests will be performed and published in a separate study.
The impact of the host–polymer polarity is crucial and previous work based on simulations demonstrated that a balance between ion solvation and chain interaction exists.29,30 In our case, we proved experimentally that an increase in the dipole strength of the polymer backbone indeed promotes the solubilisation and dissociation of salts such as LiCl and MgCl2 but it also negatively affects the conductivity of TFSI-salts. The latter could be interpreted as an effect of the reduced mobility of the polymer due to strong interchain dipolar interaction which in turn affects the ion mobility through the electrolyte.
Conclusions
In this work, we demonstrated a practical and efficient way to synthesize poly(allyl glycidyl ether)-based polymer electrolytes with different types of functional short side chains, creating a new library of potential ion-conducting polymers. Poly(2-(ethylthio) propyl glycidyl ether (PEthioPGE) and poly(2-(ethyl sulfone) propyl glycidyl ether (PEsulfoPGE) were successfully synthesized starting from poly(allyl glycidyl ether) via a sequential “thiol–ene” click reaction and oxidation. Fully amorphous polymers with Tg well below room temperature and thermal stability up to 300 °C were obtained. The increase in the oxidation state of the sulfur atom and the overall polarity of the side chain resulted in reduced segmental mobility with Tg varying from −78 °C for PAGE to −37 °C for PEsulfoPGE. All three synthesized polymers showed promising ion-conducting properties when mixed with different lithium and magnesium salts. Generally, PAGE shows better ion transport when LiTFSI is used, while the polymers containing functional polar side chains exhibit better ionic conductivities than PAGE when LiCl is used. Interestingly, owing to the high polarity of the polymers and the presence of the functional side chains, very good ionic conductivities are also reported here for complexes made with Mg(TFSI)2 and MgCl2 salts. Conductivity values competitive with the analogous PEO systems were measured. In some cases such as when mixed with Mg(TFSI)2, the reported ionic conductivities outperform that of the benchmark PEO polymer. Conductivities as high as 4.1 × 10−4 S cm−1 at 90 °C were registered. These aspects, together with the intrinsic absence of crystallization for both the neat polymers and the related Li and Mg complexes, constitute a great advantage for the future development of new polymeric materials for low and room temperature battery applications. In particular, applications where higher safety standards are required would benefit from the absence of liquid electrolytes, without dramatically affecting the performances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financed via the start-up budget made available by the Zernike Institute for Advanced Materials. The authors acknowledge the Group of Photophysics and OptoElectronics of the Zernike Institute of Advanced Materials of the University of Groningen for giving access to their glove box for the preparation of the electrolytes. Albert J. J. Woortman is acknowledged for GPC analysis and Jur van Dijken is acknowledged for TGA measurements.Notes and references
- P. Meshram, B. D. D. Pandey and Abhilash, Resour. Policy, 2019, 60, 9–22 CrossRef.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- Z. Ma, D. R. MacFarlane and M. Kar, Batteries Supercaps, 2019, 2, 115–127 CrossRef.
- J.-M. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- R. Deivanayagam, B. J. Ingram and R. Shahbazian-Yassar, Energy Storage Mater., 2019, 21, 136–153 CrossRef.
- R. Mohtadi and F. Mizuno, Beilstein J. Nanotechnol., 2014, 5, 1291–1311 CrossRef PubMed.
- P. Vanysek and D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, 70th edn, 1989 Search PubMed.
- D. Aurbach, I. Weissman, Y. Gofer and E. Levi, Chem. Rec., 2003, 3, 61–73 CrossRef CAS PubMed.
- J. Song, E. Sahadeo, M. Noked and S. B. Lee, J. Phys. Chem. Lett., 2016, 7, 1736–1749 CrossRef CAS PubMed.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- Z. I. Takehara, J. Power Sources, 1997, 68, 82–86 CrossRef CAS.
- D. T. Hallinan and N. P. Balsara, Annu. Rev., 2013, 43, 503 CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- D. F. Shriver, B. L. Papke, M. A. Ratner, R. Dupon, T. Wong and M. Brodwin, Solid State Ionics, 1981, 5, 83–88 CrossRef CAS.
- M. A. Ratner and D. F. Shriver, Chem. Rev., 1988, 88, 109–124 CrossRef CAS.
- A. Patrick, M. Glasse, R. Latham and R. Linford, Solid State Ionics, 1986, 18–19, 1063–1067 CrossRef CAS.
- J. M. Parker, P. V. Wright and C. C. Lee, Polymer, 1981, 22, 1305–1307 CrossRef CAS.
- M. Gauthier, D. Fauteux, G. Vassort, M. Duval, P. Ricoux, D. Muller, P. Rigaud, M. B. Armand, D. Deroo, A. Bélanger, M. Duval, P. Ricoux, J.-M. Chabagno, D. Muller, P. Rigaud, M. B. Armand and D. Deroo, J. Electrochem. Soc., 1985, 132, 1333–1340 CrossRef CAS.
- A. V. Chadwick, J. H. Strange and M. R. Worboys, Solid State Ionics, 1983, 9–10, 1155–1160 CrossRef CAS.
- K. Chen and D. F. Shriver, Chem. Mater., 1991, 3, 771–772 CrossRef CAS.
- V. Di Noto, S. Lavina, D. Longo and M. Vidali, Electrochim. Acta, 1998, 43, 1225–1237 CrossRef CAS.
- S.-K. Jeong, Y.-K. Jo and N.-J. Jo, Electrochim. Acta, 2006, 52, 1549–1555 CrossRef CAS.
- M. Jaipal Reddy and P. P. Chu, J. Power Sources, 2002, 109, 340–346 CrossRef CAS.
- J. Sharma and S. A. Hashmi, J. Solid State Electrochem., 2013, 17, 2283–2291 CrossRef CAS.
- L. L. Yang, A. R. McGhie and G. C. Farrington, J. Electrochem. Soc., 1986, 133, 1380 CrossRef CAS.
- J. M. Sarapas and G. N. Tew, Macromolecules, 2016, 49, 1154–1162 CrossRef CAS.
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef CAS.
- B. K. Wheatle, S. Mogurampelly, J. R. Keith, N. A. Lynd and V. Ganesan, ACS Macro Lett., 2017, 6, 1362–1367 CrossRef CAS.
- B. K. Wheatle, N. A. Lynd and V. Ganesan, ACS Macro Lett., 2018, 7, 1149–1154 CrossRef CAS.
- H. Every, F. Zhou, M. Forsyth and D. MacFarlane, Electrochim. Acta, 1998, 43, 1465–1469 CrossRef CAS.
- M. Forsyth, J. Sun, D. R. Macfarlane and A. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 341–350 CrossRef CAS.
- M. Forsyth, J. J. Sun and D. R. MacFarlane, Solid State Ionics, 1998, 112, 161–163 CrossRef CAS.
- A. Sato, T. Okumura, S. Nishimura, H. Yamamoto and N. Ueyama, J. Power Sources, 2005, 146, 423–426 CrossRef CAS.
- H. Shimomoto, T. Uegaito, S. Yabuki, S. Teratani, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Solid State Ionics, 2016, 292, 1–7 CrossRef CAS.
- M. A. Webb, Y. Jung, D. M. Pesko, B. M. Savoie, U. Yamamoto, G. W. Coates, N. P. Balsara, Z.-G. G. Wang and T. F. Miller III, ACS Cent. Sci., 2015, 1, 198–205 CrossRef CAS PubMed.
- P. Johansson, Polymer, 2001, 42, 4367–4373 CrossRef CAS.
- J. Alvarado, M. A. Schroeder, M. Zhang, O. Borodin, E. Gobrogge, M. Olguin, M. S. Ding, M. Gobet, S. Greenbaum, Y. S. Meng and K. Xu, Mater. Today, 2018, 21, 341–353 CrossRef CAS.
- L. C. Merrill and J. L. Schaefer, Chem. Mater., 2018, 30, 3971–3974 CrossRef CAS.
- L. C. Merrill and J. L. Schaefer, Front. Chem., 2019, 7, 1–10 CrossRef PubMed.
- S.-J. Kang, S.-C. Lim, H. Kim, J. W. Heo, S. Hwang, M. Jang, D. Yang, S.-T. Hong and H. Lee, Chem. Mater., 2017, 29, 3174–3180 CrossRef CAS.
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS PubMed.
- S. Lascaud, M. Perrier, A. Vallée, S. Besner, J. Prud'homme and M. Armand, Macromolecules, 1994, 27, 7469–7477 CrossRef CAS.
- G. Socrates, Infrared and Raman characteristic group frequencies: Tables an charts, John Wiley & Sons, Ltd, 3rd edn, 2004 Search PubMed.
- M. A. G. Martins and C. A. C. Sequeira, J. Power Sources, 1990, 32, 107–124 CrossRef CAS.
- A. Vallée, S. Besner and J. Prud'Homme, Electrochim. Acta, 1992, 37, 1579–1583 CrossRef.
- I. Rey, J. C. C. Lassègues, J. Grondin and L. Servant, Electrochim. Acta, 1998, 43, 1505–1510 CrossRef CAS.
- S. J. Wen, T. J. Richardson, D. I. Ghantous, K. A. Striebel, P. N. Ross and E. J. Cairns, J. Electroanal. Chem., 1996, 408, 113–118 CrossRef.
- M. Herstedt, M. Smirnov, P. Johansson, M. Chami, J. Grondin, L. Servant and J. C. Lassègues, J. Raman Spectrosc., 2005, 36, 762–770 CrossRef CAS.
- A. Bakker, S. Gejji, J. Lindgren, K. Hermansson and M. M. Probst, Polymer, 1995, 36, 4371–4378 CrossRef CAS.
- M. Deepa, S. A. Agnihotry, D. Gupta and R. Chandra, Electrochim. Acta, 2004, 49, 373–383 CrossRef CAS.
- S. P. Gejji, C. H. Suresh, K. Babu and S. R. Gadre, J. Phys. Chem. A, 2002, 103, 7474–7480 CrossRef.
- X. Bogle, R. Vazquez, S. Greenbaum, A. V. W. Cresce and K. Xu, J. Phys. Chem. Lett., 2013, 4, 1664–1668 CrossRef CAS PubMed.
- A. Nitzan and M. A. Ratner, J. Phys. Chem., 1994, 98, 1765–1775 CrossRef CAS.
- S. D. Druger, A. Nitzan and M. A. Ratner, J. Chem. Phys., 1983, 79, 3133–3142 CrossRef CAS.
- N. A. Stolwijk, M. Wiencierz, C. Heddier and J. Kösters, J. Phys. Chem. B, 2012, 116, 3065–3074 CrossRef CAS PubMed.
- J. H. Kennedy, Z. Zhang and H. Eckert, J. Non-Cryst. Solids, 1990, 123, 328–338 CrossRef CAS.
- D. B. Shah, K. R. Olson, A. Karny, S. J. Mecham, J. M. DeSimone and N. P. Balsara, J. Electrochem. Soc., 2017, 164, A3511–A3517 CrossRef CAS.
- M. Chintapalli, T. N. P. Le, N. R. Venkatesan, N. G. Mackay, A. A. Rojas, J. L. Thelen, X. C. Chen, D. Devaux and N. P. Balsara, Macromolecules, 2016, 49, 1770–1780 CrossRef CAS.
- X. Q. Yang, H. S. Lee, C. Xiang, J. McBreen, L. S. Choi and Y. Okamoto, in Proceedings of Lithium Polymer Batteries Symposium, ed. J. Broadhead and B. Scrosati, San Antonio, 1996, vol. 96, pp. 146–153 Search PubMed.
- J. E. Weston and B. C. H. Steele, Solid State Ionics, 1981, 2, 347–354 CrossRef CAS.
- P. Ravn Sørensen and T. Jacobsen, Electrochim. Acta, 1982, 27, 1671–1675 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR of pure polymers, GPC traces of polymers, FTIR spectra of polymer electrolytes, FTIR comparison of the vibrational frequencies of the TFSI anion in polymer electrolytes and in the LiTFSI salt, DSC curves of polymer electrolytes, TGA of pure polymers, summary of Tg and thermal properties of polymer electrolytes and pristine polymers, typical Nyquist plots of Mg(TFSI)2-PAGE and the equivalent circuit used to fit the data for extracting the bulk resistance and Vogel–Tammann–Fulcher (VTF) plots of polymer electrolytes. See DOI: 10.1039/c9py01735f |
| This journal is © The Royal Society of Chemistry 2020 |