
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic poly(esteracetal)s as dual pH- and enzyme-responsive micellar immunodrug delivery systems†
Leon
Bixenmann
,
Judith
Stickdorn
and
Lutz
Nuhn
 *
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: lutz.nuhn@mpip-mainz.mpg.de; Tel: +49 (0) 6131-379-311
First published on 27th February 2020
Abstract
Poly(esteracetal)s are equipped with both acid-labile acetal and base-labile ester functionalities in their backbone, thus, providing dual pH-responsive degradation profiles. In this study, the cyclic esteracetal monomer 2-methyl-1,3-dioxan-4-one (MDO) was polymerised via a cationic ring-opening mechanism onto methoxy poly(ethylene glycol) (mPEG) affording amphiphilic mPEG-b-P(MDO) poly(esteracetal) block copolymers that self-assemble into well-defined monomodal micelles by direct hydration with aqueous buffers. Their pH-responsive behaviour towards rapid particle degradation was confirmed by DLS and 1H-NMR. Upon acidification or basification and lipase-mediated enzymatic hydrolysis, immediate polymer degradation and cargo release could be observed. As an example for further suitable applications, an amphiphilic immune stimulatory TLR-7 agonist (Adifectin) was co-formulated into mPEG-b-P(MDO) block copolymer micelles and its activity was investigated by TLR stimulation of RAW Blue macrophages. The degradable amphiphilic block copolymer was able to solubilize the drug more efficiently and retained its receptor activity. Consequently, mPEG-b-P(MDO) based amphiphilic poly(esteracetal)s can be considered as promising delivery systems for further immunodrugs and, thus, may contribute towards improving their translatability.
1. Introduction
Delivering pharmaceutically active molecules to their specific therapeutic target by using nanosized carrier systems has attracted major interest to overcome current limitations of drug therapies.1,2 Many hydrophobic drugs suffer from low solubility and unspecific binding which cause non-controllable toxicities.3,4 This includes novel promising anticancer or immune therapeutics which accumulate insufficiently at their target sites. As a result, their related off-target effects limit any clinical translatability. In that context, amphiphilic block copolymers may support the drugs when formulated into nanometre-sized micellar delivery systems. They are structurally composed of a hydrophilic corona and a hydrophobic core where the therapeutic compounds can be entrapped physically.2,5–7 The size, surface properties and responsive behaviour of the micelles can determine the accessibility and tissue accumulation of drugs.8 Many attempts have been made to translate this general principle into clinical practice including paclitaxel formulations such as Genexol-PM, which was approved in 2007 in South Korea for breast cancer.4,7,9In parallel, novel therapeutic antitumour concepts have evolved over the last decade, especially with respect to the role of the immune system in cancer progression. Scientists envision reinforcing immune reactions against cancer cells by delivering immune-stimulating therapeutics to the tumour microenvironment and its draining lymph nodes.6,10 Immunostimulants such as the Toll-like receptor (TLR) 7 agonist imiquimod are highly potent in inducing innate immune responses and result in an activation of a broad spectrum of antigen-presenting cells and sufficient activation of cytotoxic T-cells. Unfortunately, if administered systemically, uncontrolled inflammations are elicited also in healthy tissues leading to severe side effects.11 To overcome these dose-limiting toxicities, nanocarriers are considered as ideal delivery vehicles especially for such small molecular immunotherapeutics, e.g. to enhance their transport, release and activity in the lymph nodes.12–14 Consequently, a fundamental challenge is to design carriers with sufficient tissue mobility to lymph nodes that show at the same time appropriate uptake by relevant immune cells.
Recently reported findings imply that lymph nodes can exhibit acidic compartments that inhibit T-cell effector functions.15 Therefore, an acid triggered cleavage of the shielding corona followed by the release of immunotherapeutics might be a beneficial strategy towards lymph node targeted delivery of immune stimulatory drugs. As advanced multi-responsive class of materials, poly(esteracetal)s can provide such acid-cleavable features beyond classical ester hydrolysis (the latter commonly guarantees avoiding long-term particle accumulation by e.g. enzyme-mediated ester cleavage).
Poly(esteracetal)s are accessible by ring-opening polymerisation and are gaining growing interest especially towards the development of degradable polymers for sustainable plastics.16–18 In this respect, Hillmyer and coworkers have recently provided major contributions to a mechanistic understanding of their fabrication.17,18 In one of their seminal works, Hillmyer et al. applied cationic ring-opening polymerisation conditions to cyclic ester acetal monomers which prevented the expulsion of acetaldehyde.18 Using the acidic organocatalyst diphenyl phosphoric acid (DPP), they showed successful poly(esteracetal) formation with different end groups based on the applied alcohol initiator. However, to the best of our knowledge, a block copolymerisation with macroinitiators towards amphiphilic diblock copolymers has so far not been reported.
With the expansion of classical micellar drug carrier systems, we believe that micelles derived from poly(esteracetal) diblock copolymers are a promising platform for lymph node-responsive delivery of immunotherapeutics. The acid-degradable acetal group ensures rapid carrier degradation and cargo release in acidic environments such as in immunosuppressive cancers and their draining lymph nodes, while the ester functionalities further enable long-term enzymatic carrier clearance from the body. In this context, our work aims at providing first proof-of-concept studies on applying amphiphilic poly(esteracetal)s in immunodrug delivery. In this respect, the reported synthesis of poly(esteracetal) homopolymers initiated by small molecular alcohols was adopted and successfully modified towards amphiphilic block copolymerisation by using mPEG as a macroinitiator. The self-assembly behaviour of the resulting block copolymers was carefully investigated in aqueous media. Thereby, the dual pH-responsive and enzymatic degradation profiles of the polymers and their derived micelles could be confirmed by NMR spectroscopy, size exclusion chromatography and dynamic light scattering. To demonstrate the ability of solubilizing hydrophobic drug compounds, poly(esteracetal) micelles were loaded with a hydrophobic dye and an amphiphilic immune stimulatory TLR7 agonist. Primary in vitro cell studies revealed low carrier toxicities and successful TLR7 stimulation for the drug-loaded micelles (Fig. 1). Altogether, our findings confirm the high potential of amphiphilic poly(esteracetal)s in immunodrug delivery and may contribute towards improving the translatability of further promising immunodrugs.
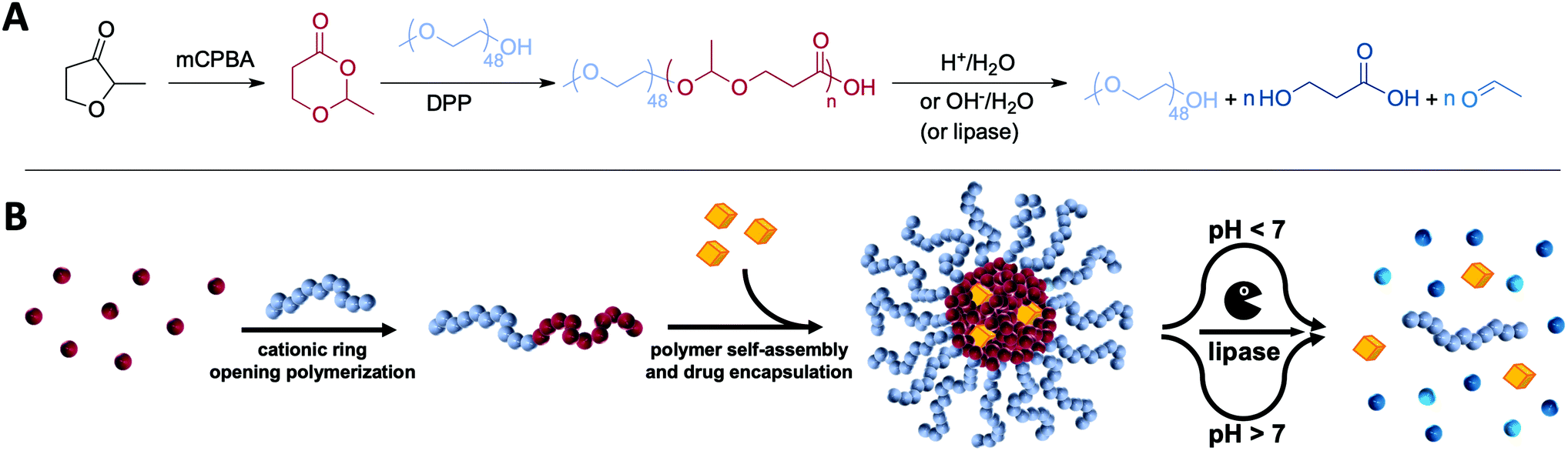 | ||
| Fig. 1 (A) Synthesis and dual pH-responsive (or enzymatic) degradation of amphiphilic poly(esteracetal) block copolymers. (B) Schematic illustration of the concept using amphiphilic poly(esteracetal) block copolymers and their derived micelles for pH- and lipase-responsive delivery of immunodrugs. | ||
2. Experimental section
2.1 Materials
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich or Fisher Scientific. 1,2-Dichloroethane (DCE) and dichloromethane (DCM) as solvents were dried by heating under reflux over calcium hydride for 3 h, and then stirred overnight at room temperature and distilled prior to use. The cyclic esteracetal monomer 2-methyl-1,3-dioxan-4-one (MDO) was synthesized from commercial 2-methyltetrahydrofuran-3-one, as has recently been reported.17 Prior to use, MDO was dried over calcium hydride overnight at room temperature (rt) and freshly distilled. Magnesium sulphate was obtained from Acros Organics and diphenylphosphoric acid (DPP) was purchased from Alfa Aesar. DPP was dried under high vacuum (0.05 mbar) overnight. DPP-catalyst stock solutions were prepared from dried DPP in either anhydrous 1,2-dichloroethane or dichloromethane. Polyethylene glycol was purchased from Fluka® Analytical. Prior to use, it was also dried by azeotropic distillation with toluene three times and then left under high vacuum (0.05 mbar) overnight. 120 mM phosphate buffer (10× PB) was prepared from sodium hydrogen phosphate (100 mM) and potassium dihydrogen phosphate (20 mM) diluted with Millipore water. Millipore water was obtained from a Milli-Q Reference A + System (water was used at a resistivity of 18.2 MΩ cm−1 and total organic carbon of < 5 ppm). Deuterated solvents were obtained from Deutero. Deuterated toluene was purchased from Sigma Aldrich. For enzymatic degradation studies, a recombinant lipase immobilized on acrylic resin beads was used (Novozyme 435 ≥ 5000 U g−1, recombinant, expressed in Aspergillus niger, from Sigma Aldrich). The solid support enables enzymatic polymer degradation studies both in organic (toluene) and aqueous environments. Dialysis was performed using Spectra/Por7 dialysis membranes with a molecular weight cut-off of 1000 g mol−1. Sterile Dulbecco's PBS, cell culture medium and supplements were purchased from Thermo Fisher. The RAW-Blue reporter cell line, QUANTI-Blue reagent and Adifectin (CL347) were purchased from Invivogen and handled as recommended by the supplier.2.2 Instrumentation
All 1H-Nuclear Magnetic Resonance (NMR) spectra were recorded at room temperature on a Bruker 250 MHz, 300 MHz, 400 MHz or 600 MHz FT NMR spectrometer (Bruker Avance III 250, Bruker Avance III HD 300, Bruker Avance II 400, or Bruker Avance III 600). Chemical shifts (δ) are provided in parts per million relative to TMS. NMR spectra were processed with MestReNova 11.0.4 from Mestrelab Research. Samples were prepared in respective deuterated solvents and their signals referenced to the residual non-deuterated solvent signal. Integrals of proton signals of the block copolymer were normalized to the integral of the ethylene glycol units of mPEG, which were set to 194 protons (Fig. S13†). This relative number of protons has been determined from pure 2 kg mol−1 mPEG by 1H-NMR referenced to its methyl end group. Diffusion Ordered Spectroscopy (DOSY) spectra were recorded at room temperature on a Bruker 400 MHz FT NMR spectrometer (Bruker Avance III HD 400) and processed using Bayesian DOSY Transformation (minimum: 1.00 × 10−8; maximum: 1.00 × 10−4; resolution factor: 1.00; repetition factor: 1; points in dimension: 64). The molecular weights of the polymers were determined using a size exclusion chromatography (SEC) set up composed of an Agilent Series 1100 with THF as the mobile phase (flow rate: 1 mL min−1) equipped with an SDV column. Samples were run with toluene as an internal standard. All measurements were performed using an RI (Agilent G1362A) and UV (275 nm) detector. SEC data were analysed via WinGPC Unity using a PEG calibration (Polymer Standards Service Mainz). Dynamic light scattering (DLS) and zeta potential measurements were performed on a Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, U.K.) equipped with a HeNe laser (λ = 633 nm) and detected at a scattering angle of 173° at 25 °C. The obtained data were processed by cumulant fitting for z-average and PDI as well as by CONTIN fitting for particle size distribution. Unless otherwise stated, dust was removed from the sample prior to each measurement by filtration through a GHP syringe filter (0.45 μm pore size, Acrodisc). The zeta potential was measured by laser Doppler electrophoresis using folded capillary cells (Malvern, DTS 1070). The effective zeta potential was calculated using the Smoluchowski approximation. MALDI-ToF mass spectra were acquired on a rapiflexTM MALDI-ToF/ToF mass spectrometer from Bruker equipped with a 10 kHz scanning smartbeam 3D laser (Nd: YAG at 355 nm) and a 10 bit 5 GHz digitizer. Measurements were performed in positive reflector mode using DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile) acid as a matrix. The data were processed with mMass and Origin 7.5/8.0. pH measurements were performed using a pH-measuring instrument (Seven Compact) manufactured by Mettler Toledo (standardized to pH 4, pH 7, pH 10). UV/Vis spectra were recorded on a Jasco V-630 spectrophotometer equipped with a Peltier thermostatted single cell holder (JASCO ETC-717) cooled by a water thermostat (A. Knüss Optronic V50) to guarantee measurement conditions at 25.0 °C.2.3 Synthesis of a 2-methyl-1,3-dioxan-4-one (MDO) monomer
According to the literature,17 the cyclic esteracetal-monomer 2-methyl-1,3-dioxan-4-one (MDO) was synthesized from racemic 2-methyldihydrofuran-3-one via Baeyer–Villiger oxidation. 77% m-CPBA (69.17 g; 309 mmol; 1.2 eq.) was dissolved in 600 mL methylene chloride (DCM), dried over MgSO4, and filtered. With 100 mL of DCM, residual MgSO4 was rinsed and filtered. The combined filtrates were transferred into a 2000 mL round bottom flask under a nitrogen atmosphere and cooled to 0 °C. 2-Methyltetrahydrofuran-3-one (25 mL; 257.2 mmol; 1 eq.) was added stepwise via a syringe over a time range of 30 minutes. The solution was then brought to room temperature and stirred for a further 21.5 h. The precipitated meta-chlorobenzoic acid was removed by repeated filtration at −78 °C. The volume of the solution was then concentrated to 60 mL under vacuum and filtered once more. The filtrate was washed with saturated sodium bicarbonate (8 × 20 mL) until no further carbon dioxide emission was evident. The aqueous fractions were extracted three times with DCM. The combined organic layers were washed with 10 mL of deionized water and subsequently dried over MgSO4. Dichloromethane was removed by distillation. The crude oil was transferred into a 50 mL round bottom flask, dried over calcium hydride for 12 h at room temperature and subsequently distilled via short path distillation apparatus (56 °C, 5 × 10−2 mbar). The product was obtained as a colourless liquid (17.58 g; 151.40 mmol; 59%) and stored at −17 °C. The purified product was characterised by 1H-, 13C-, COSY- and HSQC-NMR spectroscopy. All spectra are provided in Fig. S1–S4.†1H-NMR (400 MHz, CDCl3) δ [ppm] = 5.39 (q, J = 5.1, 1H; –O–CH(CH3)–O–), 4.17 (ddd, J = 11.5, 8.2, 2.3, 1H; –C(O)–CH2–CH2–O–), 3.91 (ddd, J = 11.4, 10.7, 5.4, 1H; –C(O)–CH2–CH2–O–), 2.75 (ddd, J = 17.8, 10.7, 8.2, 1H; –C(O)–CH2–CH2–O–), 2.56 (ddd, J = 17.9, 5.4, 2.3, 1H; –C(O)–CH2–CH2–O–), 1.45 (d, J = 5.2, 3H; –O–CH(CH3)–O–).
13C-NMR (100 MHz, CDCl3) δ [ppm] = 167.54 (–C(O)–); 101.17 (–O–CH(CH3)–O–); 63.33 (–C(O)–CH2–CH2–O–); 29.93 (–C(O)–CH2–CH2–O–); 21.12 (–O–CH(CH3)–O–).
2.4 Homopolymerisation of MDO
One day before polymerisation, pyrene butanol (35.15 mg; 0.13 mmol; 1/80 eq.) was weighed into a dried 5 mL Schlenk flask, dried by azeotropic distillation with toluene three times and further kept under high vacuum overnight. The polymerisation of MDO was carried out under a nitrogen atmosphere using dried glassware and a Teflon stir bar. MDO (1.0 mL; 10.25 mmol; 1 eq.) was added to pyrene butanol (volumetrically by using a 1 mL syringe). 0.27 mL of dichloroethane (DCE) was added as solvent. After complete solvation of all compounds, DPP was added from a prepared stock solution using a 1 mL syringe (200 μL of a 24.635 mM stock solution in DCE; 4.93 μmol; the amount corresponds to a ratio of 1/2080 DPP/MDO). Conversion of MDO was determined by 1H-NMR. After 24 h, polymerisation was quenched using triethylamine (3.4 μL dissolved in 1 mL of DCM; 5 eq.) and stirred for 10 minutes. The crude product was subsequently purified by repeating precipitation three times in 40 mL of a THF/n-hexane solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). The precipitate was isolated by centrifugation (4500 rpm, 20 min, 4 °C) and removal of the supernatant. The remaining pellet was dissolved in 1 mL of DCM and precipitated again. After drying under vacuum at room temperature, 237 mg of poly(pyrene butanol-PMDO) was obtained as a slightly yellowish, highly viscous liquid (19% yield relative to 100% of the monomer). The homopolymer was analysed by SEC, MALDI and 1H-NMR as well as 13C-NMR, COSY, HSQC and HMBC spectroscopy. All data are provided in Fig. S5–S12.†
:9). The precipitate was isolated by centrifugation (4500 rpm, 20 min, 4 °C) and removal of the supernatant. The remaining pellet was dissolved in 1 mL of DCM and precipitated again. After drying under vacuum at room temperature, 237 mg of poly(pyrene butanol-PMDO) was obtained as a slightly yellowish, highly viscous liquid (19% yield relative to 100% of the monomer). The homopolymer was analysed by SEC, MALDI and 1H-NMR as well as 13C-NMR, COSY, HSQC and HMBC spectroscopy. All data are provided in Fig. S5–S12.†
SECUV (THF, PEG calibration): Mn = 2400 g mol−1, PDI = 1.31.
1H-NMR (400 MHz, CDCl3) δ [ppm] = 8.33–7.79 (m, pyrene-H); 5.96 (q, J = 5.2, 1H; –O–CH(CH3)–O–); 4.78–4.64 (m, pyrene–(CH2)4–O–CH(CH3)–O–); 3.97–3.85 (m, 2H; –C(O)–CH2–CHH–O–); 3.84–3.71 (m, 2H; –C(O)–CH2–CHH–O–); 3.37 (t, J = 7.6 Hz; pyrene–(CH2)–(CH2)); 2.59 (m, 2H; –C(O)–CH2–CH2–O–); 2.09–2.00 (m; pyrene–(CH2)–(CH2)–(CH2)); 1.99–1.89 (m; pyrene–(CH2)2–(CH2)), 1.37 (d, J = 5.3, 3H; –O–CH(CH3)–O–).
13C NMR (100 MHz, CDCl3) δ [ppm] = 170.95 (–C(O)–); 96.51 (–O–CH(CH3)–O–); 64.42 (–C(O)–CH2–CH2–O–); 35.16(–C(O)–CH2–CH2–O–); 20.76 (–O–CH(CH3)–O–).
Polymerisation at 0 °C: yield 43%, SECUV (THF, PEG calibration): Mn = 3700 g mol−1, PDI = 1.79.
2.5 Block copolymerisation of MDO onto mPEG
As an example, the synthesis of mPEG48-b-P(MDO)50 is described. One day before polymerisation, 129.36 mg (64.9 μmol; 1/158 eq.) mPEG was weighed into a 10 mL Schlenk tube equipped with a stir bar. For drying, 1 mL of toluene was added and the suspension was stirred for 10 min. Afterwards, the solution was frozen in liquid nitrogen and dried under reduced pressure (0.05 mbar). This procedure was repeated three times and the dried polymer was then kept at 0.05 mbar overnight. Into the prepared Schlenk tube equipped with a stir bar and dried mPEG, MDO was added volumetrically via a purged syringe (1 mL; 10.25 mmol 1 eq.). All compounds were dissolved without adding an additional solvent. Subsequently, 150 μL catalyst stock solution was added volumetrically (concentration: 32.846 mM stock solution in dichloromethane; 4.93 μmol; the amount corresponds to a ratio of 1/2080 DPP:MDO). Then, polymerisation was conducted at room temperature. Conversion of MDO was tracked by 1H-NMR. After 23 h, further conversion was marginal (∼1 monomer unit per hour) and, therefore, the polymerisation was quenched with triethylamine (1.5 eq.). (Note that in order to find an appropriate purification method, previous solubility experiments with pyrene butanol initiated homopolymers were conducted. They indicated that homopolymers are slightly soluble in diethyl ether, whereas mPEG precipitated well.) Therefore, the crude product was repetitively precipitated in Et2O three times at −17 °C. Whereas the crude product of polymers with exceptionally shorter chain lengths precipitated directly, the longer chains precipitated only after cooling. For polymerisation conditions used at mPEG to monomer ratios of 95 and 126, the purified product was obtained as a colourless solid (yield: 31%), whereas for higher ratios the product was obtained as a viscous liquid. Summarized data of all reaction conditions applied to obtain mPEG48-b-PMDO block copolymers are summarized in Table 1. Each polymer was further characterised by 1H-NMR, 13C-NMR, COSY, HMBC and HSQC and diffusion ordered spectroscopy (DOSY), as well as SEC and MALDI-ToF MS (compare Fig. S14–S30†).
| χ targn | Con. [%] | χ prodn | M prodn [g mol−1] | M SECn [g mol−1] | Đ SEC | |
|---|---|---|---|---|---|---|
| χ targn: Initial monomer to initiator (= mPEG) ratio; con.: monomer conversion during polymerisation determined by 1H-NMR prior to polymer purification. χprodn: monomer to initiator (= mPEG) composition of the purified polymer determined by 1H-NMR using the resonance signals of the poly(esteracetal) proton and referencing them to the proton signals of the ethylene glycol units of PEG. Mprodn: calculated molecular weight based on the χprodn of the monomer and adding the molecular weight of the initiator (= 2 kDa mPEG). MSECn and ĐSEC: number average molecular weight and distribution determined by THF SEC using mPEG calibration. | ||||||
| mPEG48-b-P(MDO)15 | 95 | 44 | 15 | 3700 | 5000 | 1.46 |
| mPEG48-b-P(MDO)23 | 126 | 44 | 23 | 4700 | 6000 | 1.57 |
| mPEG48-b-P(MDO)50 | 158 | 55 | 50 | 7800 | 8800 | 1.65 |
| mPEG48-b-P(MDO)71 | 190 | 54 | 71 | 10200 |
10300 |
1.48 |
SEC (THF, PEG calibration): Mn = 8800 g mol−1, PDI = 1.65.
1H-NMR: (400 MHz, CDCl3) δ [ppm] = 5.96 (q, J = 5.3, 1H; –O–CH(CH3)–O–); 4.77 (q, J = 5.3; mPEG–O–CH(CH3)–O–); 3.75–3.85 (m, 1H; –C(O)–CH2–CHH–O–); 3.98–3.85 (m, 1H; –C(O)–CH2–CHH–O–); 3.64 (s, mPEG–(CH2–CH2–O–)); 3.38 (s; (–CH3) methoxy-PEG), 2.67–2.50 (m, 4H; –C(O)–CH2–CH2–O–), 1.37 (d, J = 5.3, 3H; –O–CH(CH3)–O–).
13C-NMR: (100 MHz, CDCl3) δ [ppm] = 170.91(–C(O)–); 96.44 (–O–CH(CH3)–O–); 70.61 (mPEG–(CH2–CH2–O–), 64.36 (–C(O)–CH2–CH2–O–); 35.08 (–C(O)–CH2–CH2–O–); 20.71 (–O–CH(CH3)–O–).
2.6 Self-assembly process of mPEG-b-poly(MDO)
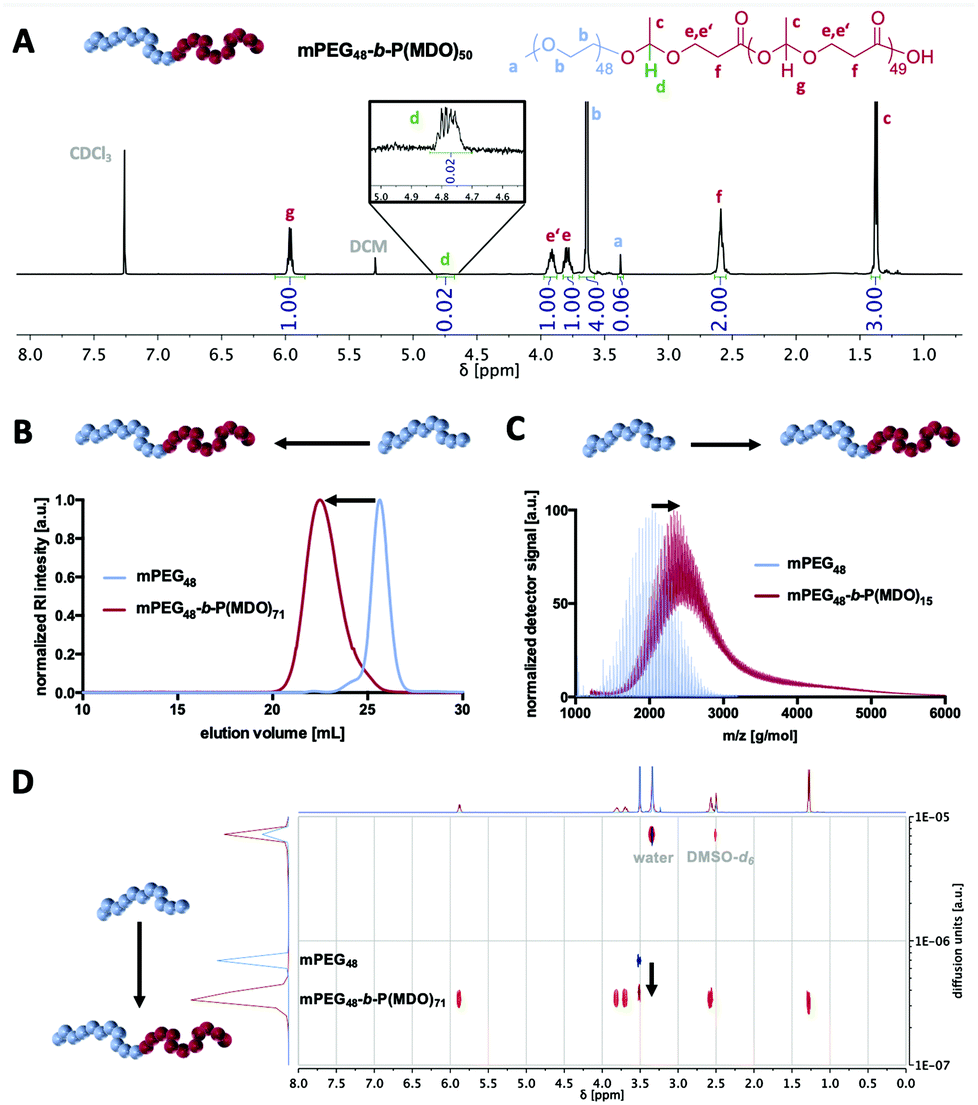 | ||
| Fig. 2 Characterisation of amphiphilic mPEG-b-PMDO block copolymers. A: 1H NMR (400 MHz) of mPEG48-b-PMDO50 in CDCl3. Resonance signals of the macroinitiator and the PMDO block as well as one single acetal proton between the PEG block and the first MDO monomer unit are annotated. B: SEC elugram of the mPEG48-b-PMDO71 block copolymer (red) and the mPEG48 macro-initiator (blue). C: MALDI-ToF mass spectra of the mPEG48-b-PMDO15 block copolymer (red) and the mPEG48 macroinitiator (blue). D: 1H-derived DOSY spectra (400 MHz) of the mPEG48-b-PMDO71 block copolymer (red) and the mPEG48 macroinitiator (blue). | ||
 | ||
| Fig. 3 Intensity size distribution plot of polymeric micelles prepared from mPEG-b-P(MDO) block copolymers of different hydrophobic block lengths via a direct hydration technique. | ||
2.7 Dual pH-responsive degradation studies
| pH(buffer of D2O) = pHmeasured − 0.41 |
Accordingly, the measured pD value of 7.5 correlates with a buffer with a pH of 7.1. After degradation, the pD dropped to a value of 7.0 which corresponds to a pH value of approx. 6.6.
After ten minutes of shaking, the dissolution process was assisted by sonication at 0 °C for 15 min. Subsequently, NMR analysis was conducted. The degradation was calculated by subtracting the relative amount of 3-hydroxypropanoic acid (determined by its proton signal referenced to the proton signal of the HO–CH2– group) from the degree of polymerisation determined in CDCl3.
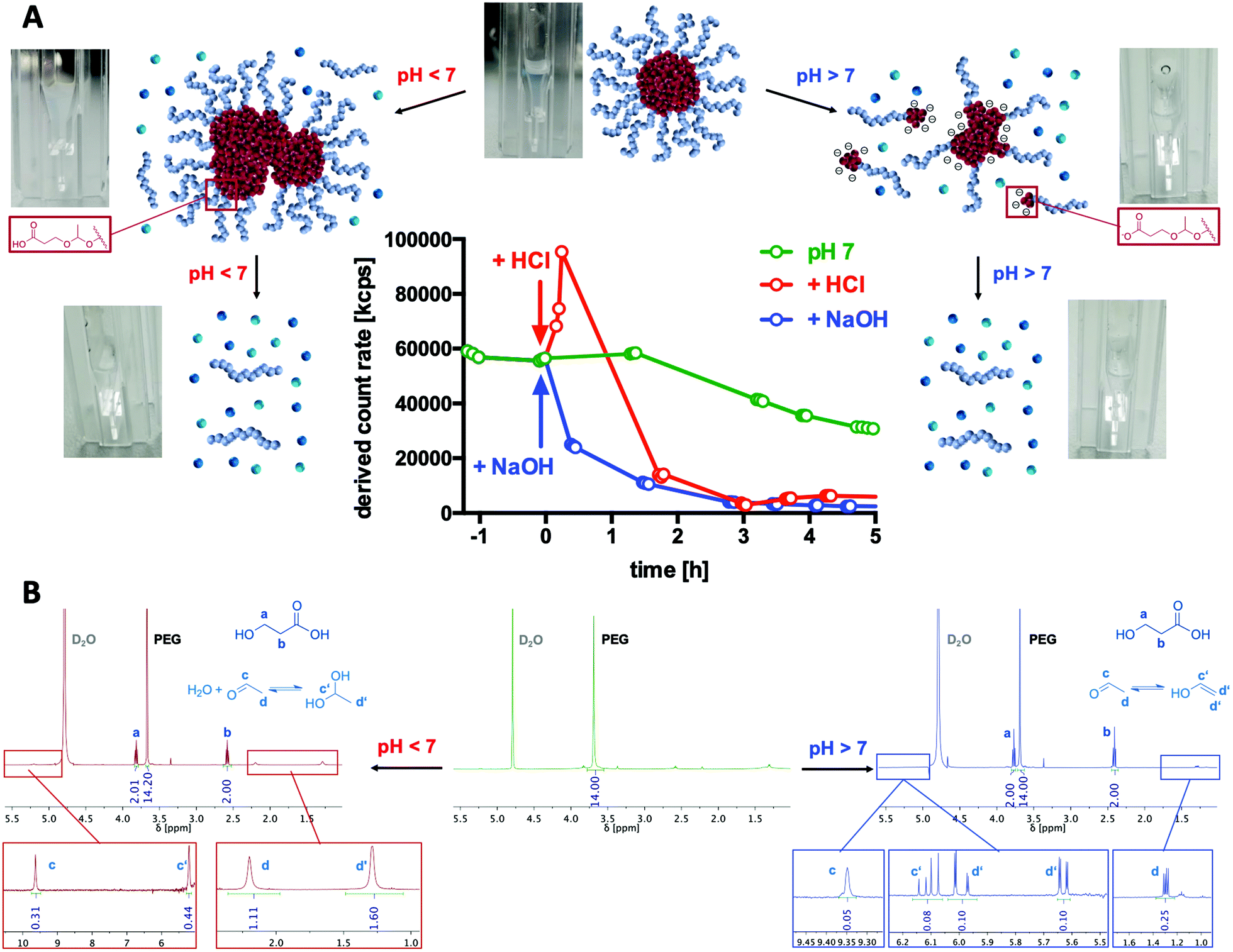 | ||
| Fig. 4 Dual pH-responsive degradation profile of mPEG-b-P(MDO) derived block copolymer micelles. (A) Scattering intensity observed using DLS (derived count rate) over time under acidic (pH = 5.7), neutral (pH = 7.2), and basic conditions (pH = 10.8) with photographs of the corresponding samples inside the cuvette and a proposed mechanism explaining the degradation and self-assembly behaviour. (B) 1H-NMR analysis (400 MHz) of micelles under neutral conditions and after acidic and basic degradation. Due to the suppressed molecular motion of the P(MDO) block inside the micellar core, the corresponding proton signals are diminished. Under acidic and basic conditions, micelles degrade into 3-hydroxypropionic acid and acetaldehyde (note that acetaldehyde can form hydrates under acidic conditions or further isomerize to enols and form aldol adducts at basic pH). | ||
2.8 Enzymatic degradation studies
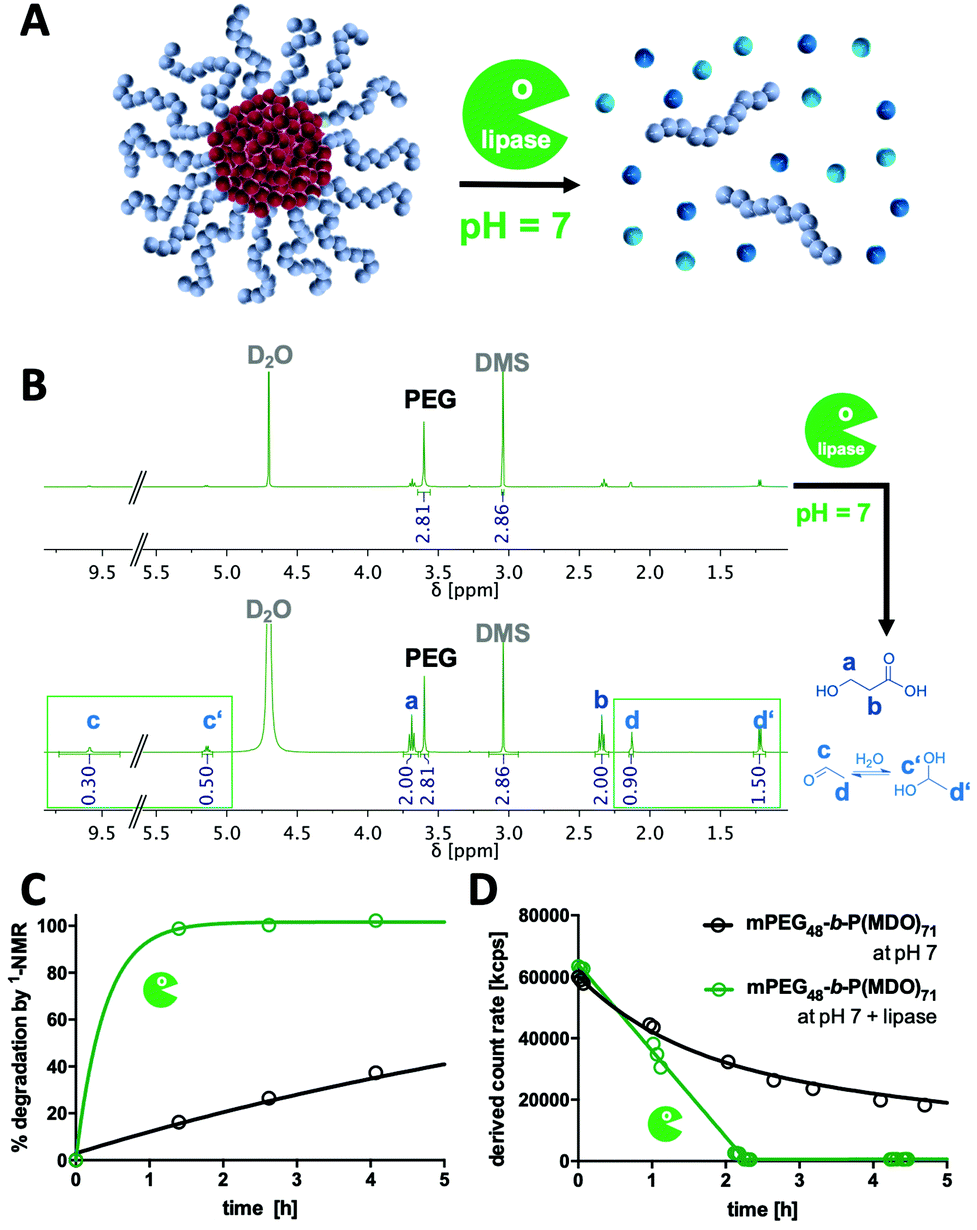 | ||
| Fig. 5 Lipase-catalysed enzymatic degradation profile of mPEG-b-P(MDO)71 derived block copolymer micelles. (A) Schematic representation of lipase mediated micelle degradation of the hydrophobic P(MDO) block at pH 7. (B) 1H-NMR analysis (400 MHz) of micelles under neutral conditions before and after the addition of lipase. Due to the suppressed molecular motion of the P(MDO) block inside the micellar core, the corresponding polymeric proton signals are first diminished; however, upon addition of lipase, micelles release 3-hydroxypropionic acid and acetaldehyde (or its hydrate form, respectively). (C) 1H-NMR kinetic analysis of accelerated enzymatic P(MDO) degradation for mPEG48-b-P(MDO)71 block copolymer micelles over time in 120 mM phosphate buffer. (D) Scattering intensity kinetics observed using DLS (derived count rate) of accelerated lipase-mediated P(MDO) degradation of mPEG48-b-P(MDO)71 block copolymer micelles over time in 120 mM phosphate buffer. | ||
2.9 Encapsulation and release of Nile Red into mPEG-b-poly(MDO) block copolymer micelles
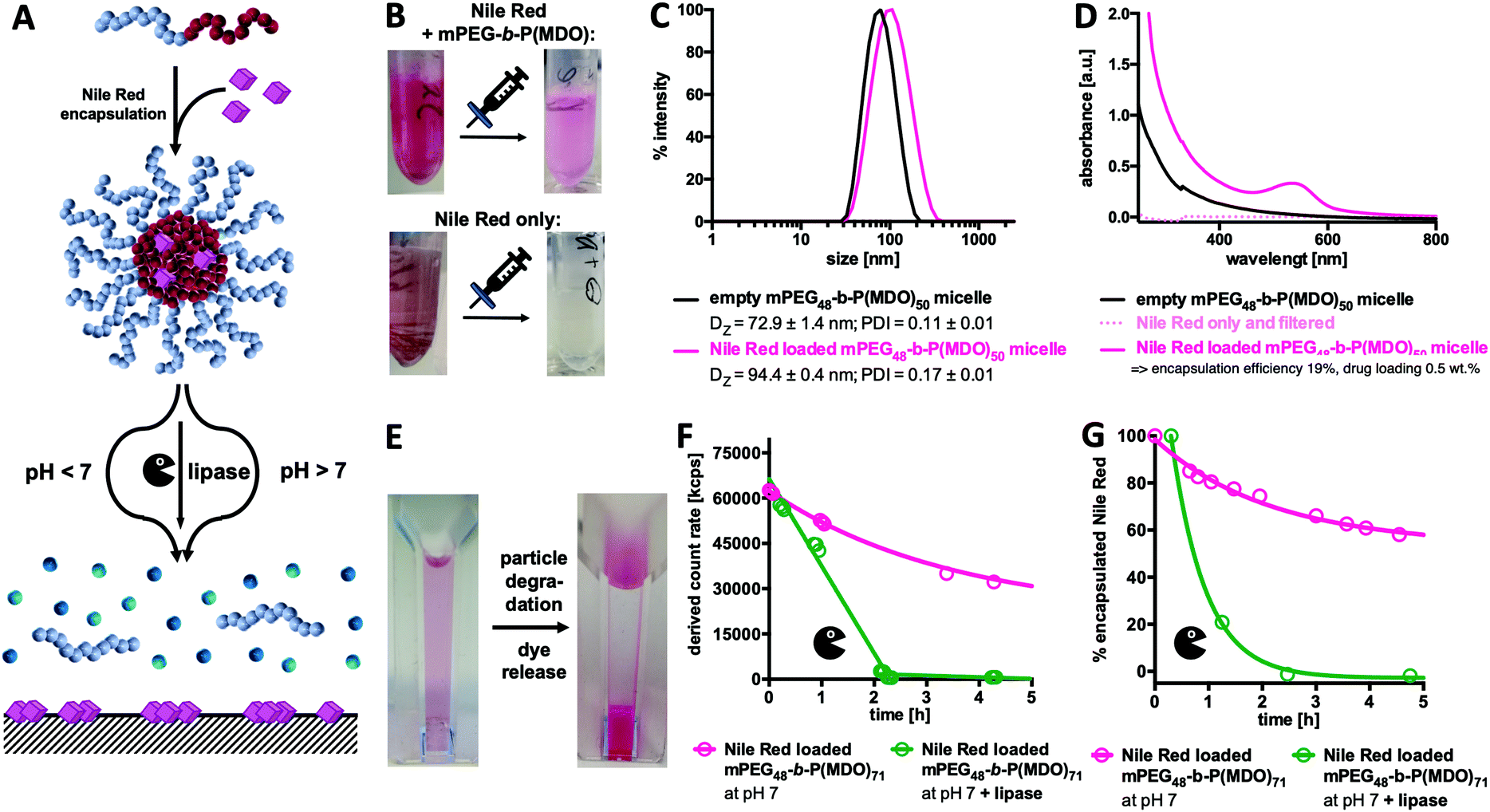 | ||
| Fig. 6 Nile Red formulated into mPEG48-b-P(MDO)50 micelles. (A) Schematic representation of Nile Red loading and release. Under acidic and basic conditions, micellar degradation is highly accelerated. Upon degradation, Nile Red is expected to be released and precipitate. (B) Nile Red formulated with and without the polymer. Pictures are shown before and after filtration (450 μm). Whereas Nile Red formulated with the polymer is stabilized in solution, Nile Red was solely removed by filtration leading to a clear colourless solution. (C) DLS measurement and (D) UV/Vis absorbance spectra of micelles formulated with and without Nile Red. (E) Photograph of Nile Red loaded particles before and after degradation. (F) Lipase-mediated enzymatic degradation of Nile Red loaded mPEG48-b-P(MDO)71 micelles followed by the light scattering count rate. (G) Lipase-mediated Nile Red release from mPEG48-b-P(MDO)71 micelles followed by UV-vis absorbance at 549 nm. | ||
2.10 Formulation of TLR 7 agonist Adifectin loaded mPEG-b-poly(MDO) block copolymer micelles
For formulations with the amphiphilic polymers, 500 μg Adifectin was diluted with 500 μL ethanol targeting a concentration of 1 mg mL−1. 128 μL of this stock solution was transferred each into two different 15 mL tubes, where one of them contained 4 mg of mPEG48-b-PMDO50. Both probes were subsequently freed from the solvent under reduced pressure, resulting in thin films covering the tube bottom. As described by the supplier, Adifectin itself was dissolved in ethanol, targeting a concentration of 10 μg μL−1 and vortexed. Once solubilized, the solution was diluted with 120 mM phosphate buffer to provide a 0.1 mM theoretical Adifectin solution and sonicated for 30 minutes. The formulation with the polymer was treated similarly. All samples were filtered through GHP-filters with a pore size of 0.45 μm. The prepared formulations were measured via DLS and the Adifectin concentration was quantified by UV/Vis spectroscopy.2.11 Quantification of Adifectin loaded into mPEG-b-poly(MDO) block copolymer micelles
In analogy to the quantification of Nile Red loading, the Adifectin concentration was determined by comparison to an external standard curve in ethanol (Fig. S61A + B†). Adifectin shows a peak maximum at 297 nm. The scattering absorbance of unloaded particles was subtracted from the UV/Vis absorption value obtained for the loaded particles (Fig. S61C†).2.12 Raw Blue macrophage cell culturing
RAW-Blue macrophages were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 0.02% Normocin and 0.01% Zeocin at 37 °C with 5% CO2 saturation.2.13 TLR 7 stimulation assay
Adifectin TLR receptor stimulation followed by NF-kB/AP-1 activation (detected by the secretion of embryonic alkaline phosphatase) was performed on RAW-Blue cells as recommended by the manufacturer (InvivoGen). RAW-Blue cells were seeded into 96-well plates at a density of 90000 cells per well in 180 μL culture medium. The cells adhered overnight and then they were treated in each well with 20 μL of the sample at given Adifectin concentrations (listed in Table S2†). After stimulation for 16 h, 50 μL of the supernatant from each well was collected and probed for secreted embryonic alkaline phosphatase (SEAP) using the QUANTI-Blue assay (InvivoGen). 150 μL QUANTI-Blue was added to each sample and incubated at 37 °C. Secretion of embryonic alkaline phosphatase levels were determined by measuring the optical density at 650 nm using the microplate reader. Activity was determined by an increase in the optical density relative to the negative control treated with PBS. The TLR 7/8 agonist 1-(4-(aminomethyl)benzyl)-2-butyl-1H-imidazo[4,5-c]quinolin-4-amine (IMDQ) served as a positive control, as used in previous experiments.12–14 A minimal interference between the 120 mM phosphate buffer and the SEAP assay was observed, due to an increased phosphate concentration. Therefore, the obtained results were corrected based on the contribution of the 120 mM phosphate buffer blank and the PBS blank. All experiments were conducted in quadruplicate (n = 4).
2.14 Cell viability assay
50 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (0.5 mg mL−1 in PBS) was added to the RAW-Blue macrophages that were treated with the corresponding samples at given Adifectin/polymer concentrations. After MTT incubation for 3 h, the formazan crystals formed were dissolved by the addition of 100 μL of 10% m/v SDS/0.01 M HCl and incubated overnight at 37 °C. Quantification was done by measuring the absorbance at 570 nm using the microplate reader. Cell viability was calculated in relation to positive (blank PBS, referred to as 100% viability) and negative (10% DMSO, referred to as 0% viability) control samples. All experiments were conducted in quadruplicate (n = 4).3. Results and discussion
Among different classes of amphiphilic polymers used for drug delivery, only a few guarantee full fragmentation into small molecules upon exposure to external stimuli. Poly(esteracetal)s can provide such degradation features towards variation in pH both below and above physiological values. While the ester group in the backbone is prone to basic hydrolysis or enzymatic degradation, the additional acetal group enables acid triggered depolymerisation occurring in tumour microenvironments and their draining lymph nodes, as well as intracellularly inside endolysosomes. To the best of our knowledge, polymerising hydrophobic poly(esteracetal)s onto hydrophilic polymers providing amphiphilic poly(esteracetal)s has not been reported before and will be examined in this work with respect to applications in immunodrug delivery.3.1 MDO monomer synthesis
According to the literature,17 the cyclic esteracetal-monomer 2-methyl-1,3-dioxan-4-one (MDO) was synthesized from racemic 2-methyldihydrofuran-3-one via Baeyer–Villiger oxidation. The purified product was dried over calcium hydride and distilled under vacuum. The compound was obtained as a colourless liquid with a yield of 59%. The structure of the product could be validated by 1H-NMR, 13C-NMR, COSY- and HSQC-NMR spectroscopy and the results are shown in Fig. S1–S4.† As we observed that MDO slowly degraded over time into 3-hydroxypropanoic acid and acetaldehyde, it was stored at −18 °C and always distilled freshly prior to use.3.2 Homopolymerisation of poly(MDO)
Hillmyer et al. have previously evaluated the ring-opening homopolymerisation of MDO using diphenyl phosphoric acid (DPP) as an organic catalyst.18 Attributed to low thermodynamic driving forces for ring-opening of MDO and achieve equilibrium conditions during polymerisation, reaction conditions at high dilutions prohibit high monomer conversion. Therefore, Hillmyer et al. performed homopolymerisations in the bulk.18,20 These conditions seem to be primarily difficult for later block copolymerisations. Accordingly, MDO polymerisation conditions are required which take into account both solubility and dilution by a macroinitiator.To address this question, we initially tested polymerisation at 7 M monomer dilution using pyrene butanol as the initiator (compared to ∼10 M in the bulk).17 Polymer purification was performed as described by Hillmyer et al. via precipitation in hexane THF (9:1) and subsequent drying under reduced pressure at room temperature. Poly(pyrene butanol-P(MDO)) was characterised by NMR, SEC and MALDI-ToF MS (Fig. S5–S12†). 1H-NMR analysis confirmed successful ring-opening of the cyclic monomer. The characteristic quartet resonance signal at 5.96 ppm verified the incorporation of poly(esteracetal)s into the polymer backbone.17,18 Signals corresponding to the pyrene butanol end group were found in the 1H-NMR spectrum, too (with the assistance of the 1H-NMR derived COSY-NMR spectrum, because some relevant pyrene butanol signals were partially covered by other resonance signals – compare Fig. S7†).
By size exclusion chromatography (SEC) we observed reasonably narrow dispersities and in the elugrams UV absorption emerged nearly simultaneously with the refractive index (Fig. S11†), which confirmed that pyrene butanol was present as an end group on the poly(esteracetal)s (note that poly(esteracetal)s themselves are not UV active).
In accordance with that, we could find intact MDO monomer repeating units (116 g mol−1) by MALDI ToF mass spectrometry. A comparison with simulated mass peaks supported successful integration of one pyrene butanol as an end group onto mostly multimeric esteracetal moieties and only minor (one or two) integration of ester units into the backbone (Fig. S12†). Interestingly, comparatively small mass peaks matched the formation of cyclic homopolymers without any pyrene butanol end group. As rationalized by others, these concurring side reactions can hardly be suppressed completely under the applied reaction conditions.18,21 MDO can polymerise after self-initiation via an activated chain end mechanism into either homopolymers with an enol ester end group or a cyclized product. Based on their similar molecular weights MALDI-ToF MS cannot differentiate between polymers terminated by an enol ester end group and cyclized polymers. In fact, only a minor formation of such a product was found in the mass spectra (Fig. S12†); however, by 1H-NMR spectroscopy no enol ester signals were observed. Instead, only esteracetal protons of the cyclized side products were detectable (the signals overlap with the pyrene butanol end group derived acetal signal – Fig. S5 and S6†).
Taken together, the combined results from 1H-NMR, SEC and MALDI-ToF MS analyses confirmed that the synthesis of pyrene butanol initiated poly(MDO) can be achieved under slightly diluted conditions in the presence of solvents. For equilibrium polymerisation conditions, the overall observed conversion should decrease at higher dilution compared to bulk polymerisation. Alternatively, reduced temperatures would thermodynamically favour higher conversions. We, therefore, performed also polymerisations at 0 °C, and indeed higher conversions could be found at longer reaction times; yet, we also observed broader molecular weight distribution (PDI increased from 1.31 to 1.79). We attribute this to lower viscosities at these temperatures that might further be affected by the presence of polymers. The latter would be more relevant for block copolymerisations using mPEG macroinitiators.
To conclude, polymerisations at room temperature and monomer dilutions at 7 M seem to result in acceptable product formation and were, therefore, applied in the following block copolymerisation reactions.
3.3 Block copolymerisation of mPEG-b-poly(MDO)
A variety of amphiphilic block copolymers were synthesized by polymerising MDO onto mPEG48 (2000 g mol−1). mPEG is commonly used as a hydrophilic, non-toxic coating for nanomedicines, because its stealth-like properties prevent opsonisation and plasma protein binding, increase colloidal stability and prolong circulation half-life.22 For polymerising MDO onto it, we applied the previous conditions of homopolymerisation. With respect to high monomer concentrations for MDO ring-opening, the diluting effect of the macroinitiator mPEG needs to be taken into account. We, therefore, adapted the solvent volume targeting a concentration between 7 M and 8 M (the mPEG-density was assumed to contribute only by its solid density). It was necessary to dissolve the macroinitiator-mPEG first in the monomer to ensure sufficient mixing before the DPP catalyst was added (pre-dissolved in the remaining solvent).By varying the monomer to initiator ratio, we could target different poly(MDO) block lengths. Conversion of MDO was tracked by 1H-NMR. After 23 h, an equilibrium was reached (further conversion was marginal) and, consequently, the polymerisation was quenched with triethylamine. In order to find an appropriate purification method, previously conducted solubility experiments with pyrene butanol initiated homopolymers indicated that they are slightly soluble in diethyl ether, whereas mPEG does precipitate well. Therefore, the crude product was subsequently precipitated in Et2O three times at −17 °C. We obtained different PMDO block lengths at polymerisation degrees between 15 and 71 relative to the mPEG units. The obtained results are summarized in Table 1.
1H-, 13C- and 2D-NMR analyses confirmed the successful formation of mPEG-poly(esteracetal) block copolymers in all cases and the absence of major impurities. As an example, the 1H-NMR spectrum of mPEG48-b-P(MDO)50 is shown in Fig. 2A while all remaining spectra can be found in the ESI (Fig. S14–S29,† the degree of polymerisation of the MDO block was determined based on the integral of the poly(esteracetal) groups referenced to the ethylene glycol units of mPEG). Notably, the presence of exactly one single acetal proton signal can further be observed derived from the ring-opening of the first MDO monomer unit by mPEG.
We carefully used additional characterisation methods beyond 1H-NMR spectroscopy to prove successful block copolymer formation, as displayed in Fig. 2B–D. By size exclusion chromatography (SEC) and by DOSY NMR and MALDI-ToF MS analyses a distinct shift to higher molecular weights/slower diffusion species was achieved for all block copolymers, while signals of the remaining unmodified mPEG macroinitiator were generally absent (Fig. S16, S19, S22 and S25†).
In the MALDI-ToF mass spectra, repetitive MDO (116 g mol−1) and ethylene glycol (44 g mol−1) units can be found (Fig. S15, S18, S21 and S24†). By detailed analysis and comparison with simulated mass signals (Fig. S30†), several mass peaks can be annotated to block copolymer species and not be explained by non-modified mPEG or self-initiated P(MDO) homopolymers.
Taken together, these results clearly demonstrate that poly(MDO)s can be attached with varying molecular weights onto mPEG and the resulting amphiphilic poly(esteracetal) block copolymers are further investigated for their self-assembly and drug encapsulation performance.
3.4 Block copolymer self-assembly
Generally, nanoparticles within a size range between 10 nm and 100 nm are considered as ideal drug carrier systems for several purposes including lymph node targeting after subcutaneous injection.13,23,24 For the latter, such particles would be large enough to prevent fast clearance by the bloodstream, but still small enough to provide sufficient diffusion through the water channels of the interstitium into the lymphatic system.Therefore, we investigated the influence of the hydrophobic chain length of the mPEG-b-poly(MDO) block copolymers on their self-assembly behaviour in aqueous media. Note that poly(MDO) homopolymers alone could not be solubilised in water after extensive mixing (Fig. S31†). Thus, it was at first intended to prepare block copolymer micelles via a simple dialysis-derived solvent switch method, which is most commonly used for micelle preparation.25,26 For this purpose, the block copolymers were dissolved in DMSO and dialyzed against water. Subsequent DLS measurement revealed aggregates with a size of approximately 100 nm (Fig. S32†). However, an unusually low scattering intensity implied preliminary degradation during the self-assembly process. In fact, when dialysis was performed for several days, no particles could be detected anymore and 1H-NMR proved polymer degradation to solely mPEG (Fig. S32†).
In this respect, an alternative preparation method had to be applied to access block copolymer micelles rapidly. It has been reported that for amphiphilic block copolymers with relatively low glass temperatures (Tg) for their hydrophobic blocks, micelles can be prepared via direct hydration.25 Interestingly, poly(MDO) homopolymers seem to provide such low Tg as the isolated polymer behaving like a highly viscous liquid (according to the literature,17 the Tg is around −30 °C). A direct hydration without the assistance of further organic solvents is a fast preparation method and would reduce preliminary hydrolysis as the hydrophobic domains are immediately forced into self-assembled and shielded structures in the aqueous environment. Thus, they would instantaneously be protected from hydrolysis compared to solvent exchange methods, where mixtures of organic and aqueous solvents are present during dialysis and cannot shield the hydrophobic block from degradation immediately.
For the direct hydration formulation, 120 mM phosphate buffer was directly added to the block copolymers and the subsequent dissolution and self-assembly process was assisted by ultrasonication at 0 °C. Indeed, DLS measurements proved the formation of micelles at different sizes depending on the length of the hydrophobic block. As shown in Fig. 3, micellar sizes could be varied from 26 nm to 141 nm depending on the molecular weight of the P(MDO) block. Moreover, the reproducibility of the direct hydration formulation method could further be validated by preparing micelles from mPEG48-b-P(MDO)50 independently three times. Subsequent DLS measurement always indicated almost identical micelles with similar sizes and distributions (Fig. S33†).
In addition, the critical micelle concentration (CMC) was determined for mPEG48-b-P(MDO)15 and mPEG48-b-P(MDO)50via pyrene fluorescence spectroscopy in order to compare the influence of the hydrophobic P(MDO) block on the self-assembly behavior. The intensities of pyrene's first and third vibronic band maxima I1 and I3 at 375 nm and 387 nm varied depending on the molar block copolymer concentration (Fig. S34 and S35†). The critical micelle concentration (CMC) was then estimated by plotting the ratio I3/I1versus logarithmic molar polymer concentration (Fig. S36 and S37†). Thereby, for the mPEG48-b-P(MDO)50 block copolymer with a 3.3-fold larger P(MDO) block, a 3.3-fold lower CMC could be determined, which confirms that block copolymer self-assembly is triggered by the P(MDO) block in aqueous media.
Altogether, the synthesized amphiphilic mPEG-b-P(MDO) block copolymers show an attractive self-assembling behaviour and their resulting micellar size ranges are ideal for drug delivery purposes including passive lymph node delivery.13,23
3.5 Dual pH-responsive degradation
To generally verify the dual pH-responsive degradation profile of mPEG-b-poly(MDO) block copolymers, micelles were exposed to acidic and basic conditions in aqueous media and analysed via1H-NMR. For this purpose, micelles were prepared from mPEG48-b-poly(MDO)15 in D2O. Under these conditions, the poly(MDO) block is not observable (Fig. 4B). However, 30 min after the addition of 5 vol% 1 M HCl or NaOH, more than 80% of the degradation products appeared in the 1H-NMR spectra. A measurement after 4 hours showed complete degradation, as shown in Fig. 4B. The micelles degrade into acetaldehyde, 3-hydroxypropanoic acid and mPEG48. Note that acetaldehyde can afterwards either form a hydrate under acidic conditions or isomerize to its enol and react further by aldol condensation under basic conditions (Fig. 4B). All of these degradation products could be found in the 1H-NMR spectra. In analogy to these observations, also the elugram of subsequent SEC measurements proved complete poly(MDO) block degradation to the remaining hydrophilic mPEG-block, as shown in Fig. S42.†To investigate the degradation of the block copolymers on the micellar self-assembly behaviour, intact micelles were prepared in 120 mM phosphate buffer and monitored by DLS under acidic, neutral and basic conditions. After the integrity of the micelles was monitored under neutral conditions for up to two hours, they were treated with either 10 vol% of 1 M HCL, phosphate buffer or 1 M NaOH, respectively. Thereby, the pH dropped to 5.7, remained constant at 7.2 or increased to 10.8. For the various pH values, a significant drop in the scattering intensity was found after 2 hours indicating that micelle degradation under both acidic and basic conditions was tremendously accelerated (Fig. 4A).
Interestingly, further analyses revealed that under acidic conditions micelles first formed micrometre-sized aggregates, as shown by an increase in scattering intensity (Fig. 4A). Corresponding DLS-derived size distribution curves can be found in Fig. S38.† This responsive aggregation was also visible to the naked eye due to the massive increase in turbidity, yet, it disappeared after 2 h completely. We hypothesize that this aggregation might be a consequence of preliminary hydrolysis of the stabilising PEG units at the interface between the hydrophobic cores and their hydrophilic coronas. Consequently, the hydrophobic domains are not sterically shielded anymore and, therefore, aggregate into large particles. To support this hypothesis, we performed zeta potential measurements immediately after lowering the pH and found an increase from slightly negative (−13 ± 6 mV) to neutral (−3 ± 7 mV) zeta potential values corresponding to a charge neutralization by protonation of the carboxylate groups formed (Fig. S40†). We schematically depict this behaviour in Fig. 4A.
In contrast, under basic conditions, micelles did not aggregate but gradually fragmentized into smaller particles, as found from a gradual decrease in the count rate (Fig. 4A) and from the monitored size distribution plots (Fig. S35†). Presumably, under basic conditions, the particles are slightly charged by the deprotonated carboxylic end groups, which prevent aggregation due to charge repulsion. This could again be confirmed by zeta potential measurements directly after increasing the pH, where slightly negative zeta potential values (−13 ± 6 mV) further dropped to more negative values (−25 ± 6 mV) which contributed to the colloidal stability of the degradation fragments formed in aqueous media (Fig. S40†).
According to preliminary published data suggesting acidic compartments to be present in compartments of lymph nodes,15 we want to pinpoint that such pH-responsive aggregation behaviour might be a promising strategy for tissue-selective particle trapping.
Micelle stability was also monitored over time under neutral conditions. In Fig. 4A, a gradual decrease in the light scattering count rate is observed for mPEG-b-P(MDO) block copolymers, too, although in a slower fashion. Retained micellar breakdown is desirable because degradation into hydrophilic small molecules with reasonably low molecular weight is expected to enable easy renal carrier clearance from the body, preventing long-term accumulation.27
Comparison of micelles derived from different P(MDO) block lengths showed that those with the shortest P(MDO) content were already degraded in a few hours (Fig. S41†). We were able to track their degradation kinetics via1H-NMR in phosphate-buffered D2O, too, as illustrated in Fig. S43 and S44.† Interestingly, the kinetics perfectly matched the timeframes of disassembly observed by DLS as shown in Fig. S41.† An increased particle stability, however, was confirmed for block copolymers with increasing hydrophobic block lengths (Fig. S41†). Micelles from mPEG48-b-P(MDO)50 exhibited a reasonable stability and combined with their size below 100 nm they were considered most promising for further delivery purposes.
3.6 Enzymatic degradation
Acid hydrolysis conditions can physiologically occur either intracellularly (e.g. in endolysosomes at pH levels of up to 5) or extracellularly (e.g. in immunosuppressive tumour microenvironments or lymph nodes27). Basic pH levels, however, are generally not reached under physiological conditions; yet, many hydrolytic esterase enzymes apply base-catalysed mechanisms to cleave ester bonds. Consequently, we studied poly(MDO) degradation behaviour in the presence of esterases and chose lipase as an enzyme with high ester-cleaving activity at hydrophilic–hydrophobic interfaces.In first trials, the enzymatic degradation was tested on non-self-assembling single mPEG-b-P(MDO) poly(esteracetal) block copolymers. Yet, this could only be conducted in the presence of organic solvents (Fig. S45†). For this purpose, a recombinant lipase immobilized on acrylic resin beads was used (Novozyme 435), which facilitates ester cleavage in both organic (toluene) and aqueous environments according to the manufacturer. Block copolymer degradation experiments were performed in toluene-d8 in order to monitor the formation of small molecular degradation products by 1H-NMR over time (Fig. S46†). As control samples, mPEG-b-P(MDO) block copolymers were analysed in toluene-d8 without an enzyme, too, and they proved high polymer stability in organic media over 1 week (Fig. S47†). In the presence of lipase, however, gradual P(MDO) block degradation was observed from an increase in the formation of acetaldehyde coupled with a decrease of the polymeric esteracetal resonance signal (Fig. S46†). According to the low amount of water present in the toluene reaction mixture, polymer degradation was not complete and reached around 50% after 90 h (Fig. S47†). These results motivated us to perform further lipase degradation experiments also in buffered aqueous media (Fig. 5).
For this purpose, mPEG-b-P(MDO)71 micelles were prepared in H2O or D2O (supplemented with 120 mM phosphate at pH 7) by the direct hydration method (Fig. 5A). After the addition of lipase Novozyme 435, accelerated release of 3-hydroxypropionic acid and acetaldehyde (and its hydrate form, respectively) could be detected by 1H-NMR (Fig. 5B). Kinetic monitoring of the 1H-NMR samples over time revealed that already within the first 1.5 h almost all P(MDO) was hydrolysed into its small molecular degradation products (Fig. 5C), while in the control sample without an enzyme these hydrolysis products could be found gradually (after 90 h the expected amount of degradation products could be observed, Fig. S48†).
These findings could be well correlated with the scattering intensities observed during DLS kinetic experiments in the presence of lipase. The count rates of the formulated mPEG-b-P(MDO)71 micelles dropped within 2 h dramatically (Fig. 5D), while a gradual decrease was found for the control samples only over a longer time frame (Fig. S49†). Interestingly, in analogy to the DLS kinetic experiments in the presence of NaOH, no increase in turbidity or scattering intensity was found during lipase-mediated degradation in contrast to the acid-catalysed degradation process at lower pH (Fig. 4A). Consequently, lipase-mediated P(MDO) degradation probably follows a similar base-catalysed hydrolysis process, as hypothesized in Fig. 4A (right), and confirms the unique dual pH-responsive versatility of mPEG-b-P(MDO)-based micelles for drug delivery purposes.
3.7 Loading and release studies
Ideal micellar-drug delivery systems should be able to encapsulate drugs inside their cores and release them upon desired stimuli that arise at their targeted site of action. To investigate the encapsulation and release performance of mPEG48-b-P(MDO)50 derived block copolymer micelles, Nile Red as a hydrophobic and water-insoluble dye was encapsulated to mimic hydrophobic drug model compounds (Fig. 6). Interestingly, the fraction of encapsulated Nile Red can be quantified by UV/Vis spectroscopy, while non-encapsulated Nile Red precipitates in water (solubility < 1 μg mL−1) and can be filtered off easily (Fig. S50 and S51†).28–31Micelles formulated with Nile Red were prepared via an adapted direct hydration method targeting a theoretical loading of 2.5 wt% of Nile Red. For this purpose, a polymer film containing Nile Red was first prepared from organic solvent and evaporated prior to direct hydration of the mixture. After sonication, the particles were filtered in order to remove the non-encapsulated, precipitated dye (a schematic representation of the preparation is shown in Fig. S50†). After filtration, pink solutions without any visible precipitation were obtained. In comparison, applying the same procedure to pure Nile Red but lacking the amphiphilic polymer led to a clear colourless solution which also showed no characteristic absorbance band for Nile Red (compare Fig. 6B and Fig. S50†). The loaded micelles were subsequently analysed by DLS and an increase in size confirmed successful dye encapsulation (Fig. 6C).
Moreover, Nile Red loading could be quantified by UV/Vis spectroscopy applying an external Nile Red standard curve determined in ethanol (Fig. S51 and S52†). Pure polymer micelles served as a reference for absorbance due to particle scattering (Fig. 6D). Based on the observed findings, a dye loading efficiency of 19% was achieved for the mPEG48-b-P(MDO)50 derived block copolymer micelles, and it corresponds to a dye loading of 0.5 wt%. Depending on the length of the P(MDO) block, drug loading and encapsulation efficiency could also be varied (Fig. S53†). For mPEG48-b-P(MDO)50, only 3.8% of the dye could be encapsulated affording a 0.1 wt% dye load, while for the larger mPEG48-b-P(MDO)71 polymers with higher hydrophobic to hydrophilic ratios, 36% of the dye was formulated into the corresponding micelles affording a 0.9 wt% dye load, as determined by UV/Vis spectroscopy (Fig. S53†).
In addition, zeta potential measurements were conducted showing that dye loading hardly affected the zeta potential of the Nile Red loaded mPEG48-b-P(MDO) micelles. Unloaded micelles provided a slightly negative zeta potential of −13 ± 6 mV that barely increased upon dye loading to −10 ± 10 mV, which is not significantly different. This behaviour can be explained by the rather low Nile Red loading below 1 wt%.
Subsequent drug release measurements were conducted and they correspond to previous particle degradation (compare Fig. 4A and 5A). After decreasing the pH by addition of HCl, Nile Red loaded micelles again started to aggregate instantaneously, whereas under basic conditions aggregation was not observed (compare Fig. S55†). Consequently, under acidic conditions, DLS studies cannot be performed properly to correlate them with Nile Red release. We, therefore, followed the process visually with the naked eye (Fig. S56†) and observed a massive increase in turbidity within the first 6 hours. Simultaneously, released Nile Red starts to adsorb to the bottom of the DLS cuvette. After one day, all drug-loaded micelles were fully degraded and solutions became colourless with the precipitated dye at the bottom (Fig. 6E and Fig. S56†).
As the UV absorbance of Nile Red is influenced by its solvation,28–31 UV/Vis spectroscopy can also be used as a tool to monitor its release from the drug carrier over time. However, as the decrease in pH significantly influenced particle aggregation, UV/Vis spectroscopy could not be applied to those formulations to monitor dye release at acidic pH. Alternatively, enzymatic degradation in the presence of lipase at pH 7 did not afford particle aggregation (compare Fig. S57†) and was, therefore, applied to study dye release kinetics by both UV/Vis spectroscopy and DLS. Similarly to the findings shown in Fig. 5D, a rapid decrease in the light scattering count rate was found for the samples in the presence of lipase, confirming again similar degradation behaviour for both empty and dye loaded micelles (Fig. 6F). In parallel, UV/Vis spectroscopy confirmed what was observed visually with the naked eye (Fig. S57†). A rapid decrease in the Nile Red absorbance maximum around 549 nm (compare Fig. 6D) is found for the dye loaded micelles in the presence of lipase (Fig. 6G) corresponding to the enzyme-mediated release of Nile Red from the micelles. Without lipase only a sustained degradation of mPEG48-b-P(MDO)50 derived block copolymer micelles (Fig. 6F) corresponding to a gradual release of encapsulated Nile Red over time (Fig. 6G) was detected at neutral pH.
3.8 Application as an immune drug delivery system
Prior to further investigations as suitable drug carriers, the in vitro cytotoxicity of the mPEG48-b-P(MDO)50 micelles was studied by MTT-assay on Raw Blue macrophages. Fig. 7D indicates that concentrations up of 400 μg mL−1 did not influence the viability (compared to PBS buffer as a negative control and 10% DMSO as a positive control). Encouraged by these results, an immunostimulating TLR agonist could be formulated into mPEG48-b-P(MDO)50 in analogy to Nile Red.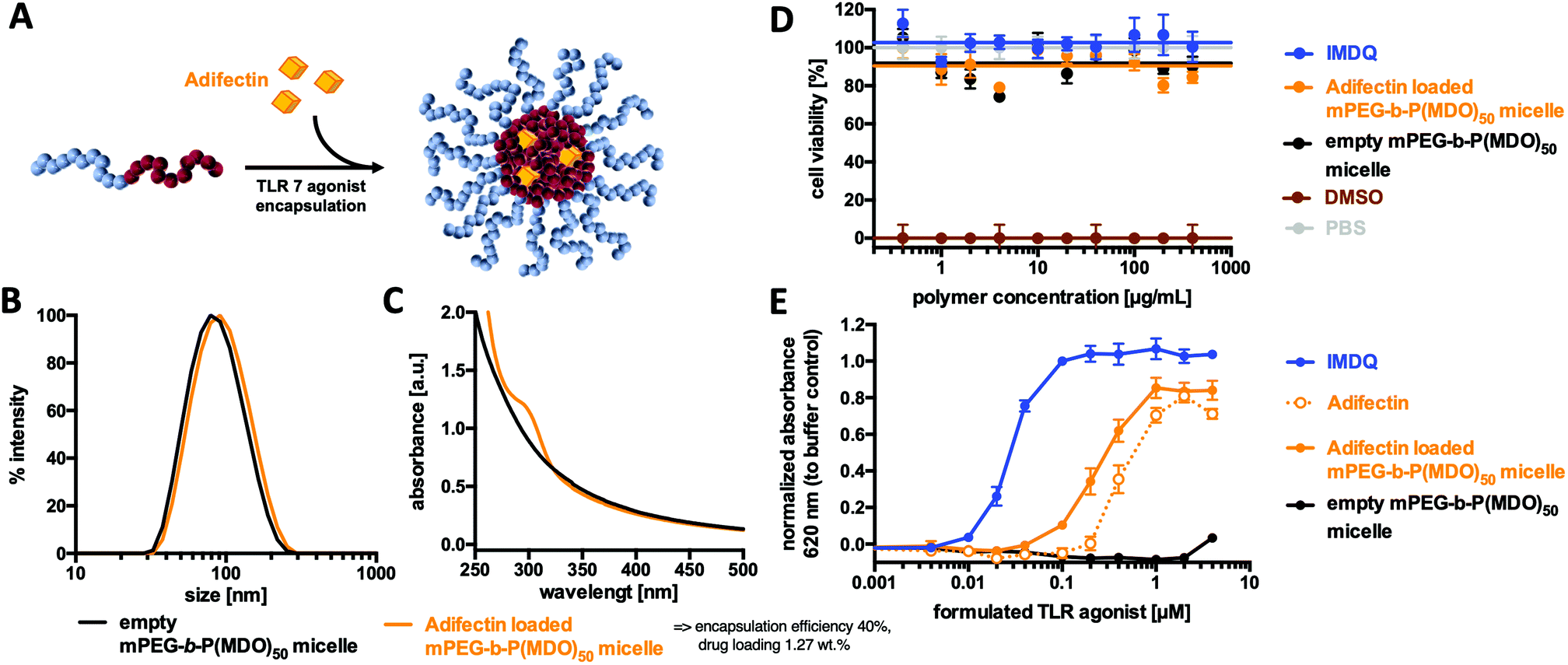 | ||
| Fig. 7 Application of P(MDO) derived micelles as an immunodrug delivery system: (A) Schematic representation of drug loading of a TLR 7 agonist into micelles for drug delivery purposes. (B) Normalized micellar size distribution plot and (C) UV/vis absorbance spectra of micelles with and without Adifectin. (D) Cellular toxicity analysis of mPEG48-b-PMDO50 on Raw Macrophages determined by MTT assay (n = 4). (E) TLR stimulation assay on RAW Blue macrophages of Adifectin formulated with and without mPEG48-b-PMDO50. The imiquimod-analogue IDMQ serves as a positive control and empty micelles provide blank probes with no immunostimulation (n = 4). | ||
For this purpose, the amphiphilic TLR 7 agonist Adifectin was selected. Its molecular structure is composed of a hydrophilic TLR 7 interacting head group, a basic spermine linker (which might favour additional interaction with the negatively charged carboxylate end groups of the mPEG48-b-P(MDO)50 block copolymers) and two hydrophobic phytanyl groups that guarantee interaction with the hydrophobic cores of the mPEG48-b-P(MDO)50 derived micelles (compare Fig. S58†). It was fabricated into a polymer film with mPEG48-b-P(MDO)50 and then rehydrated with 120 mM phosphate buffer. Adifectin itself can also be formulated into water according to the manufacturer's guidelines assisted by ethanol. In both cases with and without mPEG48-b-P(MDO)50, all samples were sonicated and filtered through GHP-filters of 0.45 μm pore size prior to use.
Subsequent DLS measurement showed only a slight increase in micellar size for the loaded micelles, which can be seen as a first indication of successful immunodrug loading (Fig. 7B – note that Adifectin is an amphiphile and might probably be more incorporated into the particle interface than into the core, thus only slightly contributing to an increase in size). In addition, the UV/Vis spectrum in Fig. 7C further proves the successful formulation of Adifectin into the micelles. The drug load could be quantified by applying an external calibration curve (Fig. S61†), where micelles prepared without Adifectin served as a blank sample.
Subsequently, the ability of in vitro immune cell TLR 7 activation was determined by incubating RAW-Blue macrophages as the TLR 7 reporter cell line with different poly(esteracetal)/Adifectin concentrations (Table S2†). Interestingly, the formulation containing the amphiphilic polymer was more potent compared to the free drug, although the polymer solely did not show any immune stimulation (Fig. 7E). Additional quantification of the sample without a block copolymer by UV/Vis spectroscopy, as shown in Fig. S61,† revealed that the increased TLR stimulation activity results from an enhanced ability to solubilise Adifectin during preparation. In fact, the presence of the amphiphilic mPEG48-b-P(MDO)50 block copolymer promotes the drug's solubility in water and, thus, its accessibility to the TLR.
Retrospective correction of the determined Adifectin concentration according to Fig. S61† showed that formulations with mPEG48-b-P(MDO)50 micelles did not lose Adifectin's receptor activity and remained almost as potent as the free drug alone (Fig. S60 and Table S3†).
Furthermore, there was no indication of decreased cell viability on macrophages after treatment with the polymer formulation, as determined by MTT assay (Fig. 7D). Taken together, these findings suggest that amphiphilic TLR agonists were safely and efficiently formulated by the mPEG-b-P(MDO)50 block copolymers and retained their drug activity on the TLR receptor in a micellar form. Consequently, the dual pH-degradable mPEG48-b-P(MDO)50 block copolymer system can be considered as a highly promising approach for such immunodrug molecules towards possible lymph node focused delivery strategies.
4. Conclusion
To the best of our knowledge, for the first time a variety of amphiphilic poly(esteracetal) diblock copolymers could successfully be synthesized and studied for their potential as immunodrug carrier systems.For this purpose, reaction conditions for the cationic ring-opening polymerisation of cyclic esteracetal monomer 2-methyl-1,3-dioxan-4-one were optimised and then applied for block copolymer synthesis with methoxy poly(ethylene glycol) (mPEG) as the macroinitiator. Successful amphiphilic mPEG-b-P(MDO) block copolymer formation could be verified by several NMR and mass spectroscopy techniques as well as SEC analysis. By varying the monomer to initiator ratio, it was possible to obtain block copolymers with different hydrophobic block lengths between 15 and 71 monomer units.
All synthesized block copolymers could be self-assembled into well-defined monomodal micelles by direct hydration. Their sizes were highly reproducible under these conditions and varied between 26 nm and 141 nm, covering the relevant size range for immunodrug delivery purposes (e.g. passive lymph node accumulation). A dual pH-responsive behaviour towards rapid particle degradation was confirmed by DLS and 1H-NMR. While micelles remained stable at pH 7.2, HCl or NaOH addition as well as exposure to lipase led to immediate degradation.
In subsequent drug loading studies, Nile Read as a hydrophobic guest molecule could be loaded and released from the degradable amphiphilic block copolymers efficiently. Finally, first in vitro studies were conducted where no cellular toxicities were found on macrophages. An amphiphilic TLR-7 agonist (Adifectin) was co-formulated into the mPEG-b-P(MDO) block copolymer micelles and its activity was investigated by TLR stimulation of RAW Blue macrophages. The degradable amphiphilic block copolymer was able to solubilize Adifectin more efficiently and retained the drug's activity on the TLR receptor.
These findings suggest that mPEG-b-P(MDO) can be utilized for providing optimized delivery opportunities for promising immunodrug molecules, also towards possible lymph node focused delivery. In our ongoing research endeavours, we will focus on enhancing drug loading and micellar stability at neutral pH to prevent premature drug release before reaching its target site.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Monika Schmelzer for SEC measurements, Hans Joachim Räder and Stefan Türk for MALDI-ToF MS measurements, and Petra Kindervater for NMR measurements. Moreover, the authors want to gratefully acknowledge financial support by the DFG through the Emmy-Noether program and the SFB 1066 Project B04. Open Access funding was provided by the Max Planck Society.References
- (a) A. P. Singh, A. Biswas, A. Shukla and P. Maiti, Signal Transduction Targeted Ther., 2019, 4, 33 CrossRef PubMed; (b) S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed; (c) Y. Zhang, Y. Huang and S. Li, AAPS PharmSciTech, 2014, 15, 862–871 CrossRef CAS PubMed; (d) A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS PubMed; (e) R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed; (f) E. H. Chang, J. B. Harford, M. A. W. Eaton, P. M. Boisseau, A. Dube, R. Hayeshi, H. Swai and D. S. Lee, Biochem. Biophys. Res. Commun., 2015, 468, 511–517 CrossRef CAS PubMed; (g) S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discov., 2014, 13, 655–672 CrossRef CAS PubMed.
- M. Talelli, M. Barz, C. J. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed.
- (a) H. Chen, C. Khemtong, X. Yang, X. Chang and J. Gao, Drug Discovery Today, 2011, 16, 354–360 CrossRef CAS PubMed; (b) Á. Tarcsay and G. M. Keserű, J. Med. Chem., 2013, 56, 1789–1795 CrossRef PubMed.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23, 7794–7803 CrossRef CAS PubMed.
- (a) K. S. Lee, H. C. Chung, S. A. Im, Y. H. Park, C. S. Kim, S.-B. Kim, S. Y. Rha, M. Y. Lee and J. Ro, Breast Cancer Res. Treat., 2008, 108, 241–250 CrossRef CAS PubMed; (b) T. Chida, Y. Miura, H. Cabral, T. Nomoto, K. Kataoka and N. Nishiyama, J. Controlled Release, 2018, 292, 130–140 CrossRef CAS PubMed; (c) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef; (d) N. Nishiyama, Y. Matsumura and K. Kataoka, Cancer Sci., 2016, 107, 867–874 CrossRef CAS PubMed.
- G. M. Lynn, R. Laga and C. M. Jewell, Cancer Lett., 2019, 459, 192–203 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- (a) H. Jiang, Q. Wang and X. Sun, J. Controlled Release, 2017, 267, 47–56 CrossRef CAS PubMed; (b) H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed; (c) N. J. Butcher, G. M. Mortimer and R. F. Minchin, Nat. Nanotechnol., 2016, 11, 310–311 CrossRef CAS PubMed; (d) Z. Zhao, A. Ukidve, V. Krishnan and S. Mitragotri, Adv. Drug Delivery Rev., 2019, 143, 3–21 CrossRef CAS PubMed; (e) K. Miyata, R. J. Christie and K. Kataoka, React. Funct. Polym., 2011, 71, 227–234 CrossRef CAS.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 CrossRef CAS PubMed.
- (a) J. Azzi, Q. Yin, M. Uehara, S. Ohori, L. Tang, K. Cai, T. Ichimura, M. McGrath, O. Maarouf, E. Kefaloyianni, S. Loughhead, J. Petr, Q. Sun, M. Kwon, S. Tullius, U. H. von Andrian, J. Cheng and R. Abdi, Cell Rep., 2016, 15, 1202–1213 CrossRef CAS PubMed; (b) E. Hong and M. A. Dobrovolskaia, Adv. Drug Delivery Rev., 2019, 141, 3–22 CrossRef CAS PubMed.
- (a) B. J. Ignacio, T. J. Albin, A. P. Esser-Kahn and M. Verdoes, Bioconjugate Chem., 2018, 29, 587–603 CrossRef CAS PubMed; (b) G. M. Lynn, R. Laga, P. A. Darrah, A. S. Ishizuka, A. J. Balaci, A. E. Dulcey, M. Pechar, R. Pola, M. Y. Gerner, A. Yamamoto, C. R. Buechler, K. M. Quinn, M. G. Smelkinson, O. Vanek, R. Cawood, T. Hills, O. Vasalatiy, K. Kastenmüller, J. R. Francica, L. Stutts, J. K. Tom, K. A. Ryu, A. P. Esser-Kahn, T. Etrych, K. D. Fisher, L. W. Seymour and R. A. Seder, Nat. Biotechnol., 2015, 33, 1201–1210 CrossRef CAS PubMed; (c) P. J. Pockros, D. Guyader, H. Patton, M. J. Tong, T. Wright, J. G. McHutchison and T.-C. Meng, J. Hepatol., 2007, 47, 174–182 CrossRef CAS PubMed; (d) P. Savage, V. Horton, J. Moore, M. Owens, P. Witt and M. E. Gore, Br. J. Cancer, 1996, 74, 1482–1486 CrossRef CAS PubMed.
- L. Nuhn, N. Vanparijs, A. de Beuckelaer, L. Lybaert, G. Verstraete, K. Deswarte, S. Lienenklaus, N. M. Shukla, A. C. D. Salyer, B. N. Lambrecht, J. Grooten, S. A. David, S. de Koker and B. G. de Geest, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8098–8103 CrossRef CAS PubMed.
- S. Van Herck, K. Deswarte, L. Nuhn, Z. Zhong, J. P. Portela Catani, Y. Li, N. N. Sanders, S. Lienenklaus, S. De Koker, B. N. Lambrecht, S. A. David and B. G. De Geest, J. Am. Chem. Soc., 2018, 140, 14300–14307 CrossRef CAS PubMed.
- S. Keller, J. T. Wilson, G. I. Patilea, H. B. Kern, A. J. Convertine and P. S. Stayton, J. Controlled Release, 2014, 191, 24–33 CrossRef CAS PubMed.
- H. Wu, V. Estrella, P. Enriquez-Navas, A. El-Kenawi, S. Russell, D. Abrahams, A. Ibrahim-Hashim, D. Longo, Y. Reshetnyak, K. Luddy, M. Damaghi, S. R. Pillai, M. Beatty, S. Pilon-Thomas, P. Swietach and R. J. Gillies, Lymph Nodes Inhibit T-cell Effector Functions Locally by Establishing Acidic Niches, bioRxivorg, 2019 DOI:10.1101/689604.
- (a) R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC; (b) S. A. Miller and R. T. Martin, U.S. Pat., WO2010/065641, 2010 Search PubMed; (c) A. E. Neitzel, L. Barreda, J. T. Trotta, G. W. Fahnhorst, T. J. Haversang, T. R. Hoye, B. P. Fors and M. A. Hillmyer, Polym. Chem., 2019, 10, 4573–4583 RSC; (d) A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- A. E. Neitzel, M. A. Petersen, E. Kokkoli and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 1156–1160 CrossRef CAS.
- A. E. Neitzel, T. J. Haversang and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11747–11755 CrossRef CAS.
- A. K. Covington, M. Paabo, R. A. Robinson and R. G. Bates, Anal. Chem., 1968, 40, 700–706 CrossRef CAS.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409–1437 CrossRef CAS.
- (a) S.-D. Li and L. Huang, J. Controlled Release, 2010, 145, 178–181 CrossRef CAS PubMed; (b) K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- A. Schudel, D. M. Francis and S. N. Thomas, Nat. Rev. Mater., 2019, 4, 415–428 CrossRef.
- C. Oussoren, J. Zuidema, D. J. A. Crommelin and G. Storm, Biochim. Biophys. Acta, Biomembr., 1997, 1328, 261–272 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290–8291 CrossRef CAS PubMed.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- P. Greenspan and S. D. Fowler, J. Lipid Res., 1985, 26, 781–789 CAS.
- J. Bigot, B. Charleux, G. Cooke, F. Delattre, D. Fournier, J. Lyskawa, L. Sambe, F. Stoffelbach and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01716j |
| This journal is © The Royal Society of Chemistry 2020 |