
Effects of solvents, additives, and π-allyl ligand structures on the polymerization behavior of diazoacetates initiated by π-allylPd complexes†

Abstract
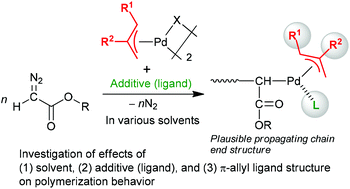
In the polymerization of diazoacetates initiated by π-allylPd-based initiating systems, the effects of solvents, additives, and π-allyl ligand structures on the polymerization behavior were investigated. As a result, the polymerization in the presence of pyridine or its derivatives as an additive was found to afford a polymer with relatively narrow molecular weight distribution compared to that with π-allylPdCl alone. Furthermore, we have demonstrated that Pd complexes with π-allyl ligands with a variety of substituents are capable of polymerizing diazoacetates in a similar manner to the parent unsubstituted π-allylPdCl, and the tacticity of the resulting polymers is affected by the structure of the π-allyl ligands.
- This article is part of the themed collection: Polymer Chemistry Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

