
Functionalization of NaGdF4 nanoparticles with a dibromomaleimide-terminated polymer for MR/optical imaging of thrombosis†

Abstract
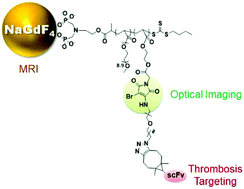
Herein, we report the development of a thrombosis-targeted molecular imaging probe with magnetic resonance (MR) and optical dual-modality capacity using dibromomaleimide (DBM)-bearing polymer-grafted NaGdF4 nanoparticles. The random copolymer of bisphosphonic ester (BPE)-P(OEGA-co-DBM) was first synthesized through reversible addition–fragmentation chain transfer (RAFT) copolymerization of oligo(ethylene glycol)methyl ether acrylate (OEGA) and DBM-based monomers using a BPE-terminated RAFT agent. The resulting polymers were subjected to deprotection with the formation of bisphosphonic acid (BPA) terminals, allowing for the attachment of the as-synthesized BPA-P(OEGA-co-DBM) chains onto the surface of paramagnetic NaGdF4 nanoparticles through the ligand exchange reaction. Azide moieties could be readily incorporated into the hybrid nanoparticles by the coupling reaction between the highly reactive DBM moieties and amine derivatives. Intriguingly, the coupling reaction was characterized by a unique fluorescence turn-on even in aqueous media, which subsequently enabled the fluorescence imaging applications of the resulting hybrid nanoparticle. Furthermore, a single-chain antibody (scFv), which is specifically used for the active conformation of the GPIIb/IIIa integrin, was successfully attached onto the nanoparticles by a strain-promoted copper-free “click” reaction, allowing the targeting of activated platelets in acute thrombosis. The hybrid nanoparticles prepared through this new surface functionalization protocol possessed not only high colloidal stability under physiological conditions but also potential MR/optical imaging capacity. The thrombosis targeting capacity of the hybrid nanoparticle-based probe was then demonstrated by exploiting DBM conjugation-induced fluorescence in living cells.
- This article is part of the themed collection: Polymer Chemistry Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

