
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and complex self-assembly of amphiphilic block copolymers with a branched hydrophobic poly(2-oxazoline) into multicompartment micelles, pseudo-vesicles and yolk/shell nanoparticles†
Davy
Daubian
 a,
Jens
Gaitzsch
*ab and
Wolfgang
Meier
*a
a,
Jens
Gaitzsch
*ab and
Wolfgang
Meier
*a
aDepartment of Physical Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058 Basel, Switzerland. E-mail: gaitzsch@ipfdd.de; wolfgang.meier@unibas.ch
bLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Strasse 6, 01069 Dresden, Germany
First published on 23rd December 2019
Abstract
We report on the synthesis and self-assembly of poly(ethylene oxide)-block-poly(2-(3-ethylheptyl)-2-oxazoline) (PEO-b-PEHOx), a new amphiphilic diblock copolymer obtained via microwave-assisted polymerization of EHOx using a new nosylated PEO macroinitiator. The kinetics of the polymerization in different solvents was crucial to optimize the synthesis and revealed a controlled, yet fast polymerization of the AB diblock copolymer. Differential scanning calorimetry proved that PEO-b-PEHOx shows glass transition temperatures below room temperature, making it suitable for a wide range of self-assembly methods, especially under mild and solvent-free conditions. Self-assembly of PEO-b-PEHOx was then performed using film rehydration and solvent switch. In both cases, we were able to show the formation of various complex structures (multi-compartment micelles (MCMs), pseudo-vesicles and yolk/shell nanoparticles) by light scattering, TEM and Cryo-TEM. Our results show that PEO-b-PEHOx is a potent new AB diblock copolymer due to its fast synthesis and unique self-assembly behavior.
Introduction
In recent years, amphiphilic block copolymers have attracted a considerable amount of attention thanks to their ability to self-organize into various complex structures.1–3 These structures formed by amphiphilic block copolymers can mimic complex structures found in nature (e.g. lipid bilayers) while exhibiting increased chemical and mechanical stability, which makes them interesting for different bio-inspired applications, like drug delivery and nanoreactors.4–10 Most investigated structures come from AB diblock copolymers (where A is a hydrophilic block and B is a hydrophobic block) or ABA triblock copolymers. Amphiphilic diblock copolymers have been the focus of numerous studies, like PSS-b-PAA,11 PEO-b-PCL,12 PEO-b-P(d)MCL,13 PEO-b-P(d)MCL,14 and PEO-b-PDMS,15 notably to form vesicles. Combining two different diblock copolymers AB and AC also leads to new exotic structures like multi-compartment micelles (MCMs),16 vesicles with a patterned surface17,18 or yolk/shell nanoparticles.19,20 Most of these studies use hydrophobic blocks with a short side chain to ensure mobility. There are only a few studies investigating AB diblock copolymers including a B block with a long side chain.21–23 This could lead to novel interactions during the self-assembly. The aim of this work is to shed some light on the self-assembly of such polymers and an AB diblock copolymer was designed accordingly.Poly(2-oxazolines) have been extensively used to form amphiphilic block copolymers thanks to their biocompatibility and versatility.24–26 The water solubility of poly(2-oxazolines) can be precisely tailored by the length of the side chain as they become completely water insoluble with at least 4 carbons.27 Moreover, their mechanical properties, notably their glass transition temperature (Tg) and crystallinity, can be modified significantly by branching their side chain in the right position.28 Previously, Kempe et al. synthesized the first hydrophobic poly(2-oxazoline) with a Tg of −6 °C, namely poly(2-(3-ethylheptyl)-2-oxazoline (PEHOx).29 This polymer benefits from a purely hydrocarbon side chain of nine carbon atoms, providing sufficient hydrophobicity and branching to suppress crystallinity. PEHOx is thus perfectly suitable as a hydrophobic block in an amphiphilic block copolymer but has only been used so far to form polymer micelles to achieve a higher drug loading capacity.30 The side chain of PEHOx with its ethyl branch and long length will generate additional hydrophobic–hydrophobic interactions. It will also prevent compact order or strong entanglement of the formed AB diblock copolymers in a membrane, potentially enabling more complex self-assembled structures. Combined with its backbone structure close to a peptide, these properties make PEHOx a compelling hydrophobic block to lead to new insights into self-assembled polymer nanoparticles.
In order to achieve a straightforward one-pot synthesis of our AB diblock copolymer, modified poly(ethylene oxide) (PEO) was used as a macroinitiator. A tosylated version has already been used as a macroinitiator for the polymerization of 2-oxazolines, but has significant disadvantages due to the low reactivity of tosylates.31 We aimed to optimize this method by replacing tosylates with the more reactive nosylate.32
Herein, we report the one-pot synthesis of new poly(ethylene oxide)-block-poly(2-(3-ethylheptyl)-2-oxazoline (PEO-b-PEHOx) amphiphilic AB diblock copolymers using microwave-assisted polymerization of EHOx on a poly(ethylene oxide)-nosylate (PEO-Nos) macroinitiator. Using the obtained library of PEO-b-PEHOx, we then examined their thermal properties by differential scanning calorimetry (DSC) comparing it to the homopolymer PEHOx. Aqueous self-assembly of these diblock copolymers was examined by two different techniques: film rehydration and solvent switch. We studied the self-assemblies in depth by dynamic and static light scattering (DLS/SLS), transmission electron microscopy (TEM) and cryogenic transmission electron microscopy (Cryo-TEM). The combined analysis proved the formation of a variety of potent self-assembled structures like multi-compartment micelles (MCMs) and pseudo-vesicles, amongst others. The new polymer was set to be a showcase example to elaborate which experiments are necessary to prove the existence of vesicles, and notably the importance of Cryo-TEM insights into those complex structures.
Experimental section
Materials
Glassware for polymerization was dried overnight at 120 °C prior to use. Isopropanol, sulfolane, 2-ethylhexyl bromide, n-butyllithium, N,N,N′,N′-tetramethylenediamine (TMEDA), triethylamine, p-nitrobenzenesulfonyl chloride, anhydrous chlorobenzene, 2-methyl-2-oxazoline, barium oxide, methyl p-toluenesulfonate, poly(ethylene oxide) monomethyl ether (PEO, 2000 g mol−1) and poly(ethylene oxide) monomethyl ether tosylate (PEO-Tos, 2000 g mol−1) were obtained from Sigma-Aldrich (CH) and used as received. 2-Methyl-2-oxazoline was distilled over barium oxide (BaO) and stored under argon. Poly(ethylene oxide) monomethyl ether (PEO, 2000 g mol−1) was dissolved in water and then lyophilized. Sulfolane was dried over CaH2 for 24 h under reduced pressure at 40 °C, distilled, and stored in a glovebox. The Milli-Q water (18.2 MΩ cm) used was obtained from a Purelab Option-R 7/15 system (ELGA LabWater, UK). Anhydrous dichloromethane (DCM), tetrahydrofuran (THF) and acetonitrile were obtained from an inert solvent purification system PureSolv MD 5 (Inert, USA).Nuclear magnetic resonance spectroscopy (NMR)
1H NMR spectra were recorded at 295 K in CDCl3 (0.05% tetramethylsilane) on a Bruker Avance III NMR spectrometer (400.13 MHz). To buffer any possible acidity, CDCl3 was saturated with K2CO3. The instrument was equipped with a direct observe 5 mm BBFO smart probe. The experiments were performed at 295 K and the temperature was calibrated using a methanol standard showing accuracy within ±0.2 K. Spectra were processed with MestReNova software, and chemical shifts were reported in ppm.Gel permeation chromatography (GPC)
GPC traces were analysed and recorded using WinGPC Unichrom software (v 8.20 build 8251, PSS polymer, Germany). Traces of the diblock copolymers or of the reaction mixtures were recorded using an Agilent based system composed of a 1200 series pump and autosampler. The GPC system was equipped with a series of linear-S SDV columns (pre-column (5 cm), three analytical columns (30 cm), all 5 μm particles and 0.8 cm in diameter, PSS polymer, Germany), followed by a Variable Wavelength Detector (VWD) (1100 series) and a Refractive Index Detector (RI) (1100 series). Detectors and columns were kept at 35 °C. CHCl3, stabilized with EtOH, was used as the eluent at a flow rate of 1 mL min−1. The system was calibrated against narrowly distributed polystyrene (PS) standards.Microwave-assisted synthesis
Microwave polymerization was conducted on a Biotage Initiator System (Biotage, Sweden) equipped with Robot Eight. The microwave synthesizer was operated at a constant set temperature (mentioned in the Results and discussion) and monitored by using an infrared (IR) sensor.Differential scanning calorimetry (DSC)
Thermal transitions were measured using 10 mg of polymer for each measurement on a DSC 214 Polyma (Netzsch GmbH, Austria) under a nitrogen atmosphere from −120 °C to 190 °C with a heating and cooling rate of 40 K min−1. The DSC curves shown correspond to the third heating curve.Transmission electron microscopy (TEM)
5 mL of solution containing self-assembled polymers (0.2 w/w %) was left adsorbing on a Formvar-coated and glow discharged 200 mesh copper grid and blotted off after 1 min. A drop of 5 μL of water was placed on the grid and blotted off immediately. The action was repeated two times. This procedure was repeated with 5 μL of 2% aqueous uranyl acetate, where the solution was left for 10 s in the second step, ensuring sufficient staining of the assemblies. The prepared grids were left drying in air for at least 10 min before imaging them at an acceleration voltage of 80 kV on a Philipps CM100 (Netherlands) transmission electron microscope.Static and dynamic light scattering (SLS/DLS)
SLS and DLS experiments were performed on a light scattering spectrometer (LS instruments, Switzerland), equipped with a He–Ne 21 mW laser (λ = 632.8 nm) at scattering angles from 30° to 150° at 25 °C. All samples were diluted in order to suppress multiple scattering. For samples with a radius smaller than 100 nm that satisfy the Rayleigh-Gans-Debye (RGD) scattering model, the radius of gyration (Rg) was obtained from the SLS data using Zimm plots. For samples with a radius larger than 100 nm, Rg was obtained from the SLS data using MIE scattering models (MiePlot, UK). The intensity versus angle curve of samples was fit using Mie scattering models for η = 1.35 and 5% polydispersity. Rg is then calculated using the obtained R and the formula for a spherical structure: Rg2 = (3/5)R2. In the case of DLS, second order cumulant analysis of the data for various angles was performed to obtain the hydrodynamic radius (Rh).Determination of the refractive index increment
SLS experiments required the refractive index increment value, dn/dc, of the analyte in the respective solvent. It was obtained using an automatic refractometer Reichert AR7 series (Reichert, USA) at 25 °C. After being calibrated with Milli-Q water (18.2 MΩ cm), the self-assemblies of PEO-b-PEHOx were analysed and dn/dc = 0.15 was obtained (see Fig. S6 in the ESI†).Cryogenic transmission electron microscopy (Cryo-TEM)
A 4 μL aliquot of a sample was adsorbed onto a holey carbon-coated grid (Lacey, Tedpella, USA), blotted off with Whatman 1 filter paper and vitrified into liquid ethane at −178 °C using a Leica GP plunger (Leica, Austria). Frozen grids were transferred onto a Talos electron microscope (FEI, USA) using a Gatan 626 cryo-holder. Electron micrographs were recorded at an accelerating voltage of 200 kV and a nominal magnification of 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×, using a low-dose system (20 e− Å−2) by maintaining the sample at low temperature. Micrographs were recorded on a CETA camera. Images were then processed using ImageJ (NIH, USA) to measure notably the micelle sizes and membrane thicknesses.
000×, using a low-dose system (20 e− Å−2) by maintaining the sample at low temperature. Micrographs were recorded on a CETA camera. Images were then processed using ImageJ (NIH, USA) to measure notably the micelle sizes and membrane thicknesses.
Synthesis of 2-(3-ethylheptyl)-2-oxazoline (EHOx)
The monomer was prepared by using the synthetic procedure described by Kempe et al.29 Briefly, TMEDA (10.32 mL, 69 mmol, 1 eq.) was dissolved in 300 mL THF at −78 °C under argon. N-Butyllithium (2.5 M in hexane, 26 mL, 65 mmol, 0.96 eq.) was then added and after 60 min of stirring, 2-methyl-2-oxazoline (5.8 mL, 68 mmol, 1 eq.) was added. The stirring was continued for 2 hours at −78 °C and was concluded by the addition of 2-ethylhexyl bromide (10.13 mL, 57 mmol, 0.83 eq.). The solution was allowed to warm to room temperature overnight which was then terminated after 25 hours with 150 mL of methanol and the solvents were evaporated under reduced pressure. The residue was dissolved in 200 mL of a biphasic solution 1:1 CHCl3/NaHCO3 (sat). The aqueous phase was extracted twice with 75 mL of chloroform. The combined organic phases were washed with water and brine. After drying over MgSO4, the solvent was removed under reduced pressure, and the crude product was purified by distillation. The purified product was confirmed to be EHOx by 1H NMR. It was then stored under an argon atmosphere. 1H NMR (400 MHz, CDCl3, 295 K, δ, ppm): 0.86 (m, 6H, CH3), 1.27 (m, 9H, CH(CH2CH3)–CH2CH2CH2CH3), 1.60 (m, 2H, NCOCH2CH2), 2.25 (t, J = 8.2 Hz, 2H, NCOCH2CH2), 3.82 (t, J = 9.5 Hz, 2H, CNCH2), 4.2 (t, J = 9.4 Hz, 2H, COCH2).
Homopolymerization of EHOx
We used the procedure from Kempe et al.29 Briefly, a microwave vial was prepared containing methyl p-toluenesulfonate, EHOx and acetonitrile. The monomer concentration was adjusted to 2 M and a monomer-to-initiator ratio of 60 was used. The microwave vial was then heated to 140 °C for 30 min to reach full monomer conversion. To remove the solvent and residual components, the polymerization mixture was placed into a dialysis membrane (regenerated cellulose, MWCO 3.5 kDa, RC6, Spectra Por, USA) and dialyzed against THF for 3 days (the solvent was exchanged 6 times). 1H NMR (400 MHz, CDCl3, 295 K, δ, ppm): 0.86 (m, 6H, CH3), 1.24 (m, 9H, CH(CH2CH3)–CH2CH2CH2CH3), 1.54 (m, 2H, N(COCH2CH2)), 2.24 ppm (m, 2H, N(COCH2CH2)), 3.43 ppm (m, 4H, N(COCH2CH2)-CH2CH2). ĐM (GPC) = 1.18. Mn = 11800 Da.
Synthesis of the PEO-Nos macroinitiator
Poly(ethylene oxide) monomethyl ether (PEO, 2000 g mol−1) (4 g, 2 mmol, 1 eq.) was dissolved in 80 mL dry DCM and chilled to 0 °C. Triethylamine (2.8 mL, 20 mmol, 10 eq.) and p-nitrobenzenesulfonyl chloride (4.4362 g, 20 mmol, 10 eq.) were then added. The reaction was allowed to stir for 10 h at 0 °C under an argon atmosphere. The reaction mixture was concentrated under vacuum and the high excess of unreacted reagents was then extracted 3 times with 400 mL of isopropanol at RT exploiting the poor solubility of PEO at RT in isopropanol. It was then placed into a dialysis membrane (regenerated cellulose, MWCO 3.5 kDa, RC6, Spectra Por, USA) and dialyzed against acetonitrile for 2 days (the solution was exchanged 4 times, at least 8 hours in between exchanges). The purified product was stored under an argon atmosphere and was stable over 12 months, as confirmed by 1H NMR. The resulting ω-nosylation (96%) was determined by integrating the peak from the terminal group of PEO at 3.38 ppm and the peak of the methylene group next to nosylate at 4.32 ppm (see Fig. S1 in the ESI†). 1H NMR (400 MHz, CDCl3, 295 K, δ, ppm): 3.38 (s, 3H, –OCH2CH2O–CH3), 3.65 (m, 180H, OCH2CH2O), 4.32 (t, J = 4.6 Hz, 2H, Ar–O–CH2–), 8.15 (m, 2H, Ar–NO2), 8.41 (m, 2H, Ar–NO2).Synthesis of PEO-b-PEHOx diblock copolymers
PEO-b-PEHOx was synthesized via cationic ring-opening polymerization of EHOx in a microwave reactor using PEO-Nos as a macroinitiator. In a glovebox, a stock solution containing the initiator PEO-Nos, monomer EHOx, and the solvent (sulfolane, acetonitrile or chlorobenzene, see in the main paper) was prepared. The monomer concentration was set to 1 M for all experiments. A monomer-to-initiator ratio of 100 was used for the synthesis of PEO45-b-PEHOx8–57 and a monomer-to-initiator of 200 for PEO45-b-PEHOx95–171. The desired volume of the solution was transferred into microwave vials. The vials were sealed in a glovebox under an argon atmosphere prior to the transfer into the microwave reactor.To study the kinetics of EHOx polymerization at 140 °C, reactions were performed three times. The kinetics were monitored by 1H NMR and GPC analysis of the crude polymerization mixture. The final PEO-b-PEHOx polymers were purified by dialyzing (regenerated cellulose, MWCO 3.5 kDa, RC6, Spectra Por, USA) the polymerization mixture against THF for 3 days (the solvent was exchanged 6 times, at least 8 hours in between exchanges). The block ratio was determined by integrating the PEO backbone peak at 3.60 ppm and peaks of the PEHOx side chain at 1.24 and 0.86 ppm. 1H NMR (400 MHz, CDCl3, 295 K, δ, ppm): 0.86 (m, 6H, CH3), 1.24 (m, 9H, CH(CH2CH3)–CH2CH2CH2CH3), 1.54 (m, 2H, N(COCH2CH2)), 2.24 (m, 2H, N(COCH2CH2)), 3.38 (s, 3H, –OCH2CH2O–CH3), 3.43 (m, 4H, N(COCH2CH2)–CH2CH2), 3.65 (m, 180H, OCH2CH2O).
Self-assembly
The self-assembly of PEO-b-PEHOx diblock copolymers was performed using two different techniques: film rehydration and solvent switch. In all experiments, the final concentration of the self-assembled polymer was 0.2 w/w %.Results and discussion
Synthesis of PEO-b-PEHOx polymers
We started by selecting the best initiator and tested tosylated (Tos) and nosylated (Nos) PEO as macroinitiators for the synthesis of PEO-b-PEHOx diblock copolymers. Tosylated macroinitiators were already used for the polymerization of 2-oxazolines13,33,34 but small molecule nosylates were shown to be advantageous over triflates and tosylates for polymerization of various 2-oxazolines.32 Nosylates are more stable compared to triflates and result in faster initiation compared to tosylates. While the PEO-Tos macroinitiator was commercially available, PEO-Nos was not reported previously and synthesized by adapting a procedure described for tosylation of PEO.13 The reaction proceeded smoothly at 0 °C for 10 h, resulting in 100% of nosylation of PEO. Purification of crude PEO-Nos via extraction in isopropanol and dialysis in acetonitrile resulted in a product of 96% purity (see Fig. S1 in the ESI†). The residual 4% corresponded to non-nosylated PEO. Partial cleavage of the nosyl group during purification occurred because of its high reactivity and sensitivity towards protic solvents such as isopropanol and water traces in acetonitrile. Aprotic purification through precipitation in diethyl ether and using column chromatography was also conducted but proved to be ineffective as impurities were still present.PEO-b-PEOHx polymers were synthesized via microwave-assisted polymerization of EHOx on a PEO macroinitiator (Fig. 1). Using microwave-assisted polymerization, we were able to work with small volumes (as low as 0.5 mL), which was an advantage over conventional heating. Small volumes allowed for a kinetic study screening different parameters in triplicate while keeping the amount of reagents used to a bare minimum. It was especially advantageous considering that both the macroinitiator and monomer were self-synthesized. On top of that, the temperature was monitored precisely using an infrared sensor and kept constant by the microwave input. The synthesis was optimized by varying the macroinitiator (PEO-Tos or PEO-Nos), solvent, and temperature.
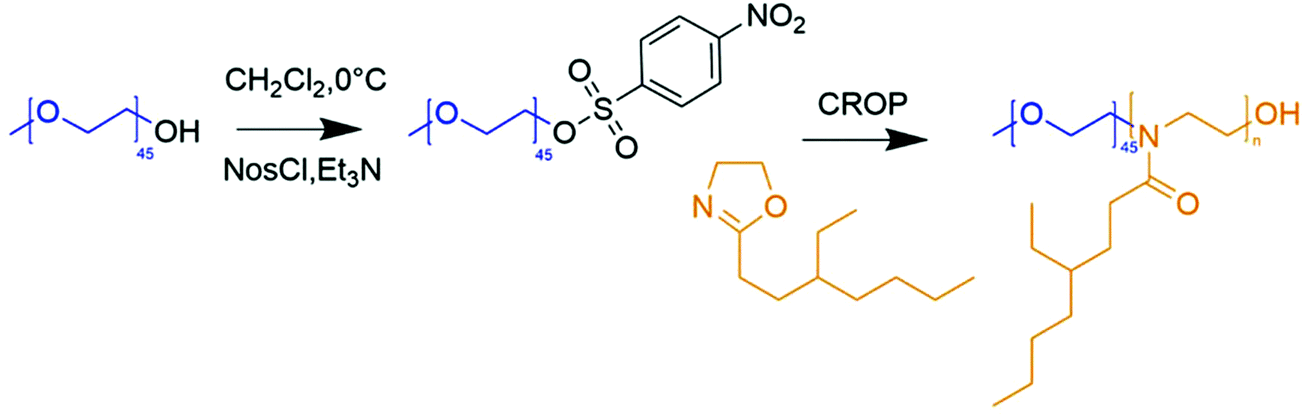 | ||
| Fig. 1 Schematic representation of the synthesis of poly(ethylene oxide)-nosylate followed by the synthesis of poly(ethylene oxide)-b-poly(2-(3-ethylheptyl)-2-oxazoline (PEO-b-PEHOx). | ||
In line with previous reports, the PEO-Tos macroinitiator resulted in side reactions, leading to the broadening of PEO-Tos elugram at various temperatures (see Fig. S2 in the ESI†). PEO-Nos, on the other hand, did lead to polymerization (see Fig. S3 in the ESI†). Therefore, all further experiments were performed using PEO-Nos as a macroinitiator and a constant monomer-to-initiator ratio of 100.
We then investigated three different solvents adequate for the CROP of 2-oxazolines: sulfolane, acetonitrile and chlorobenzene as they showed to solubilize the macroinitiator PEO-Nos and the monomer EHOx. The correct choice of solvent is of utmost importance as it has to fully solubilize the initiator, the monomer and its growing polymer while having a high solvency power for cations (i.e., high Hildebrand solubility parameters) which is favourable for the CROP of 2-oxazolines.35 Sulfolane was shown to accelerate the CROP of 2-oxazolines,36 but was not suited for our system as PEO-b-PEHOx is not soluble in sulfolane. With a growing polymer and the already dense sulfolane, the polymerization mixture becomes very viscous to the point of obtaining two different phases, which leads to an uncontrolled polymerization (see Fig. S4 in the ESI†). Acetonitrile and chlorobenzene were compared in a kinetic study. We monitored the monomer conversion by 1H NMR and stopped the polymerisation after 30 min as it was already shown that it reaches 80% in chlorobenzene as compared to the 52% in acetonitrile when polymerizing at 140 °C (Fig. 2B/C), proving that chlorobenzene is the better solvent for this system. Similarly to sulfolane, growing PEO-b-PEHOx decreases in solubility in acetonitrile. After 7 min (10 units of EHOx), the reactive solution becomes turbid, resulting in less control over the reaction from that point onwards as shown by the sharp increase in the dispersity. The change in solubility is due to the isomerization of the cyclic imino ether structure (EHOx) to a tertiary amide structure (PEHOx). The first order kinetic plot (Fig. 2A) illustrates this lack of control even further, revealing two different trends: a late initiation with a steady but irregular increase in concentration until 20 min where the polymerization reaches its limit and is slowed down dramatically. For chlorobenzene, the linear increase in ln([M]0/[M]t) with time demonstrates a constant concentration of propagating species, indicative of a living polymerization of the 2-oxazolines. In all further experiments, chlorobenzene was then chosen as the solvent for polymerization of EHOx. Assuming a 100% initiator efficiency, the polymerization rate (kp) was determined from the slope of the graph. With a value of 60 ± 2 L mol−1 s−1, the rate is lower than the homopolymerization rate of EHOx in acetonitrile using methyl tosylate as an initiator (106 ± 2 L mol−1 s−1).29 This decrease most probably comes from the use of a PEO-modified macroinitiator and its decreased mobility compared to a small molecule initiator like methyl tosylate. This can also be explained by the lower Hildebrand solubility parameter of chlorobenzene (9.5) compared to acetonitrile (11.9).36
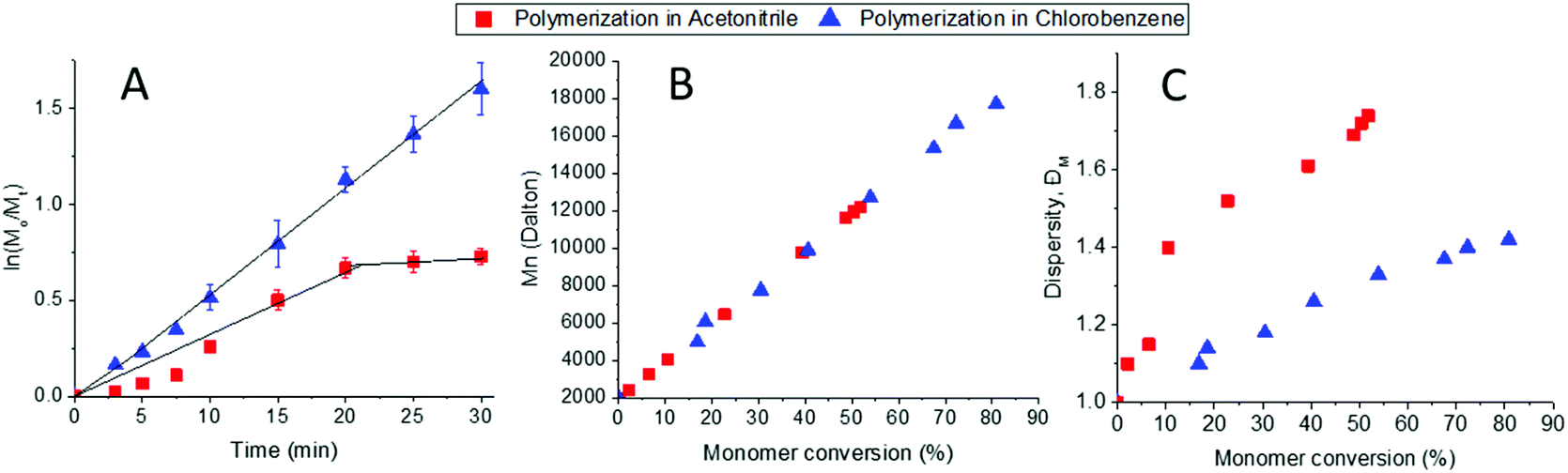 | ||
| Fig. 2 Microwave-assisted polymerization kinetics of EHOx in acetonitrile and chlorobenzene at 140 °C using PEO-Nos as a macroinitiator. A – First-order kinetic plot. B – Mn values against monomer conversion. C – ĐM against monomer conversion. | ||
The used temperature (140 °C) was also carefully chosen. The temperatures investigated varied from 80 to 150 °C. For temperatures lower than 140 °C, there is an increased amount of side reactions like homopolymerization while 150 °C leads to a broadening of the peak (see Fig. S3 in the ESI†). Hence, all temperatures other than 140 °C proved to be inadequate for this system.
The optimized conditions, i.e. PEO-Nos in chlorobenzene and 140 °C, were used to synthesize several PEO-b-PEHOx, characterized by NMR and GPC. GPC showed a complete shift, indicating no left-over macroinitiator, and 1H NMR allowed assigning all peaks of PEO-PEHOx, thus proving its formation (Fig. 3). Table 1 comprises the library of synthesized PEO-b-PEHOx diblock copolymers with their respective average molecular weight Mn, dispersity ĐM and hydrophilic weight fraction, f. In order to achieve a longer B block, we increased the monomer-to-initiator ratio to 200. To obtain a phase diagram of the self-assemblies formed with PEO-b-PEHOx, an extensive spectrum of different f was essential to obtain a holistic phase diagram of the self-assemblies formed with PEO-b-PEHOx: from the most hydrophilic PEO45-b-PEHOx8 (f = 57%) to the most hydrophobic PEO45-b-PEHOx171 (f = 6%).
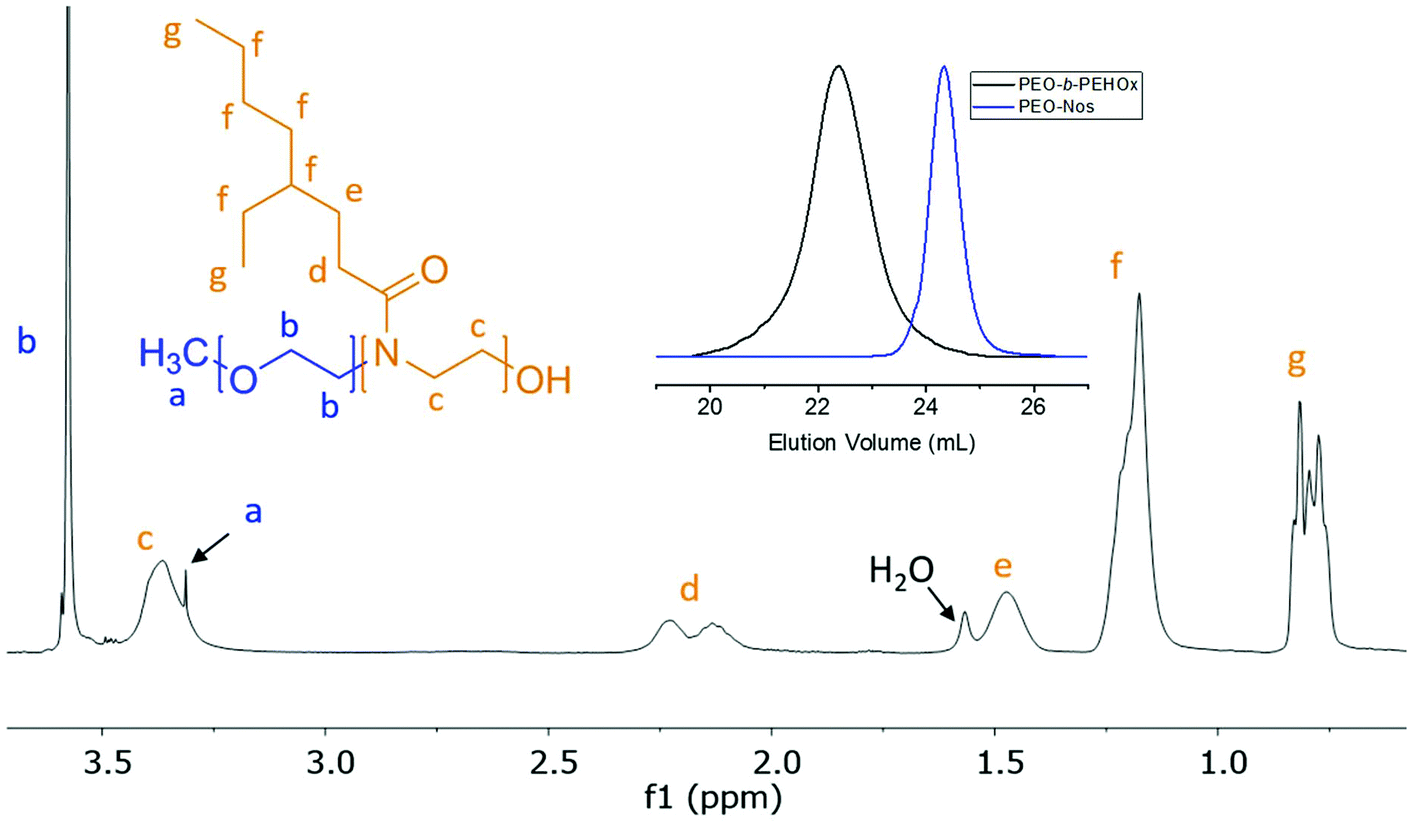 | ||
| Fig. 3 Representative NMR (1H, 400 MHz, CDCl3) of PEO40-b-PEHOx46. All peaks have been assigned to the chemical structure of the polymer with representative GPC traces (CHCl3) of PEO40-b-PEHOx46 and the macroinitiator PEO-Nos. | ||
| Polymers |
M
na [Da] |
Đ
Mb |
fc [%] |
|---|---|---|---|
| a Obtained from 1H NMR. b Obtained by GPC. c Calculated by the equation f = (Mn of PEO)/(Mn(PEO) + Mn(PEHOx)). | |||
| PEO45-b-PEHOx8 | 3500 | 1.14 | 56 |
| PEO45-b-PEHOx26 | 6000 | 1.20 | 28 |
| PEO45-b-PEHOx40 | 9900 | 1.27 | 20 |
| PEO45-b-PEHOx57 | 13200 |
1.32 | 15 |
| PEO45-b-PEHOx95 | 21000 |
1.35 | 10 |
| PEO45-b-PEHOx128 | 27600 |
1.37 | 7 |
| PEO45-b-PEHOx151 | 32200 |
1.4 | 6 |
| PEO45-b-PEHOx171 | 36200 |
1.41 | 6 |
| PEHOx60 | 11800 |
1.18 | — |
Determination of the glass transition temperature (Tg)
The thermal properties were then assessed by differential scanning calorimetry (DSC) of the homopolymer PEHOx as well as of the diblock copolymers PEO45-b-PEHOx8 and PEO45-b-PEHOx171, the smallest and longest of the library (Fig. 4; for the DSC curves of the rest of the diblock copolymers, see Fig. S5 in the ESI†). The thermal properties of the homopolymer PEO were obtained from the literature.37 A glass transition temperature (Tg) below room temperature is key for self-assembly with biological actives under mild conditions that do not harm the enzyme activity for example.38 Being in a flexible and rubbery state above the Tg enables the amphiphilic block copolymers to reorganize readily in response to environmental changes and attain particular structures through self-assembly.39,40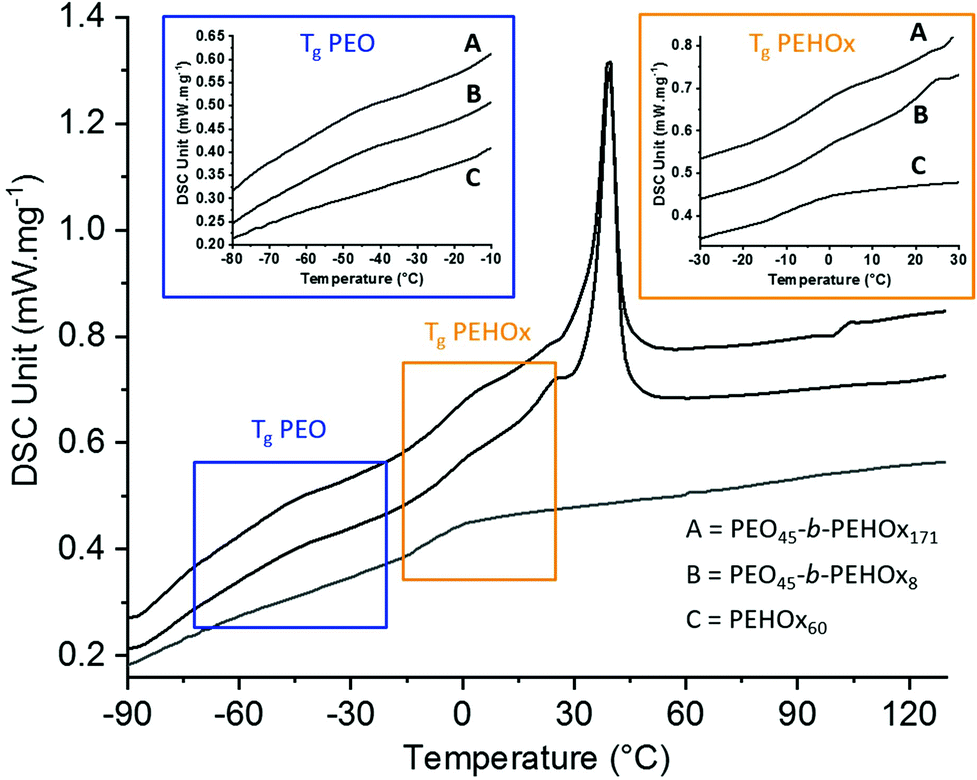 | ||
| Fig. 4 DSC measurements of the diblock copolymers PEO45-b-PEHOx8 and PEO45-b-PEHOx171 and homopolymer PEHOx60, highlighting the regions of the glass transition temperatures, in blue for PEO and in orange for PEHOx. | ||
The homopolymer PEHOx revealed a Tg of −7 °C and no melting point which is coherent with the value (−6 °C) from Kempe et al.29 and the amorphous behaviour of this polymer. Interestingly, every diblock copolymer PEO-b-PEHOx revealed two glass transition temperatures and a melting peak. The broad melting peak of PEO starts at about 15 °C and passes its maximum around 45 °C. PEO45-b-PEHOx8 and PEO45-b-PEHOx171 both exhibit two Tg values with the first one in the range of −40 °C (PEO block) and a second one in the range of 3 °C (PEHOx block). No significant difference in the glass transitions temperatures was observed with increasing block length of the EHOx. The fact that we have two independent visible Tg's confirms a phase separation between PEO and PEHOx. In comparison with the Tg's of the homopolymers (PEO: −73 °C and PEHOx: −7 °C), the increased values obtained for PEO-b-PEHOx can be explained by a decrease in flexibility of the polymer chains within the segments of the diblock copolymer. Nonetheless, the values remain well below room temperature and thus enable a self-assembly process at room temperature with solvent-free techniques like film rehydration, which is advantageous for incorporation of biological actives in the future.
Self-assembly of PEO-b-PEHOx
We studied the self-assembly of PEO-b-PEHOx polymers in aqueous solution using two different techniques: film rehydration (FR) and solvent switch (SS). The final concentration of the self-assembled polymer was always set to 0.2 w/w %. For film rehydration, the polymer solutions were stirred extensively for one week as a stirring time of only one day only resulted in a precipitated polymer. For solvent switch, the self-assemblies were formed much faster as they were completely formed at the end of the dialysis step after 2 days. Further stirring did not yield any change in the self-assemblies (see Fig. S6 in the ESI†). All self-assemblies were then characterized extensively using SLS/DLS, TEM and Cryo-TEM. PEO-b-PEHOx151 and PEO-b-PEHOx171 were not investigated as they did not yield any self-assembly structures. With a hydrophilic weight fraction of 6% for those two diblock copolymers, they reached the limit in terms of hydrophobicity and cannot self-assemble.Light scattering was conducted as the first step to assess the form factor ρ, which is the ratio of the radius of gyration (Rg) to the hydrodynamic radius (Rh). With dynamic light scattering (DLS), Rh can be obtained while static light scattering (SLS) allows obtaining Rg. The form factor ρ is a structure property that reflects the radial density distribution of the particle. Typical values for ρ are ρ = 0.78 for solid spheres and ρ = 1.0 for hollow spheres with an infinitely thin shell. The latter is viewed typical for vesicles.41,42 The Rg, Rh and ρ for both self-assembly techniques are shown in Table 2. For most polymers and both self-assembly techniques ρ was close to 0.78 which thus indicated solid-filled particles as a common morphology but in different sizes. Among those, three results stood out: FR of PEO45-b-PEHOx26 and SS of PEO45-b-PEHOx95 showed values of ρ = 0.93 and ρ = 0.94 respectively, which indicated a hollow sphere with a thin shell, while SS of PEO45-b-PEHOx128 with ρ = 0.88 indicated a hollow sphere with a thicker shell.
| Film rehydration (FR) | Solvent switch (SS) | |||||
|---|---|---|---|---|---|---|
| Diblock copolymers | R h [nm] | R g [nm] | ρ = Rg/Rh | R h [nm] | R g[nm] | ρ = Rg/Rh |
| PEO45-b-PEHOx8 | 19 ± 3 | 15 | 0.79 | 13 ± 2 | 10 | 0.77 |
| PEO45-b-PEHOx26 | 67 ± 20 | 62 | 0.93 | 21 ± 4 | 16 | 0.80 |
| PEO45-b-PEHOx40 | 80 ± 8 | 61 | 0.76 | 77 ± 14 | 58 | 0.75 |
| PEO45-b-PEHOx57 | 91 ± 12 | 73 | 0.80 | 93 ± 16 | 69 | 0.74 |
| PEO45-b-PEHOx95 | 139 ± 19 | 112 | 0.81 | 132 ± 6 | 124 | 0.94 |
| PEO45-b-PEHOx128 | 178 ± 21 | 120 | 0.67 | 101 ± 19 | 89 | 0.88 |
TEM and Cryo-TEM images of all samples showed the various PEO-b-PEHOx self-assemblies obtained by FR and SS with increasing EHOx length (Fig. 5). The nature of the self-assemblies formed was confirmed by combining the results from these images and the light scattering data.
 | ||
| Fig. 5 TEM (A–F, K–P) and Cryo-TEM (G–J, Q–T) images of the self-assemblies by film rehydration (A–J) and solvent switch (K–T) of AB diblock PEO-b-PEHOx for increasing length of the B block – EHOx. Scale bars, 200 nm. Supplementary Cryo-TEM images of SS of PEO45-b-PEHOx95 (S) and PEO45-b-PEHOx128 (T) can be seen in respectively Fig. S7 and S8 in the ESI.† | ||
All of these results are too large for normal micelles. In a stretched conformation, PEO45-b-PEHOx46 is 32 nm in length, which is in theory the largest possible radius for a micelle from this polymer (see section 5b of the ESI†). With 36 nm for PEO45-b-PEHOx57 and 50 nm for PEO45-b-PEHOx95, these polymers are also too short to form micelles of the size measured by DLS and the form factor is too small for vesicles. To clarify the type of nanoparticles formed, Cryo-TEM was conducted as it can visualize them in their native environment and discriminate between solid spherical particles, vesicles or other types of nanoparticles. Surprisingly, all spheres were filled with inverse micelles, forming multi-compartment micelles (MCMs) as shown by the absence of any membrane and the presence of many compartments of the size of micelles (Fig. 5G/H/Q/R/I/J and Fig. 6A, see Fig. S11 in the ESI† for a higher resolution).43–46 It should also be noted that MCMs of PEO45-b-PEHOx46 and PEO45-b-PEHOx57 formed by SS are completely filled with monodisperse inverse micelles (Rmicelles = 12.3 ± 1.4 nm and Rmicelles = 12.3 ± 1.4 nm, respectively) compared to MCMs from PEO45-b-PEHOx46 and PEO45-b-PEHOx57 formed by FR which are more loosely filled with polydisperse inverse micelles (Rmicelles = 22.6 ± 6.3 nm). The different sizes of the inverse micelles can be explained by the random presence of a few small direct micelles in their center. Since FR does not give the PEO chains time to equilibrate the size of the micelles, a higher dispersity can be explained as observed for PEO45-b-PEHOx57. The bigger MCMs observed for the FR for PEO45-b-PEHOx95 and PEO-b-PEHOx128 are filled completely with monodisperse inverse micelles similar to the MCMs of smaller diblocks formed by SS (Rmicelles = 13.1 ± 1.3 nm and Rmicelles = 14.5 ± 1.7 nm, respectively). The core of these inverse micelles is now composed of PEO and water as a solvent. It is likely that the PEO chains are present as a coil rather than a stretched conformation. R of the PEO block is 2.8 nm as a perfect random coil. In order to fill the volume of the internal micelles, a solvent must be present either to hydrate the polymer chains or to fill the gaps between them (see section 5a of the ESI†).
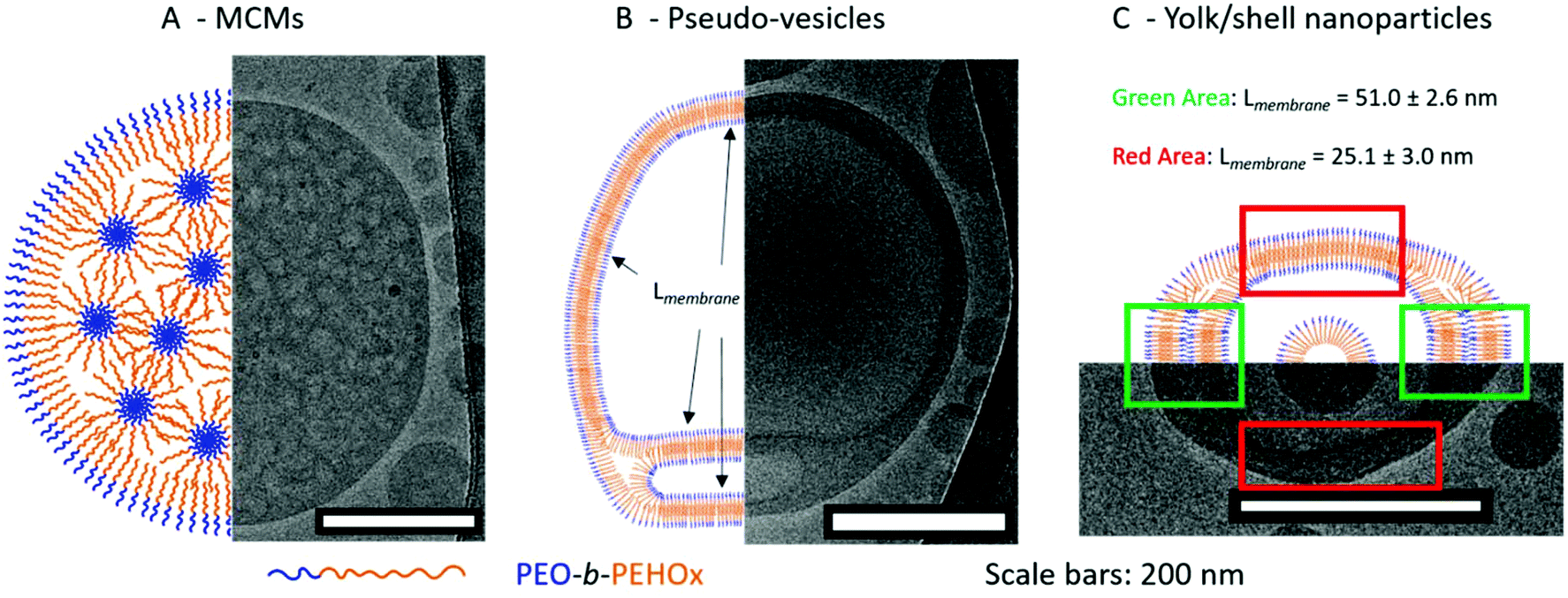 | ||
| Fig. 6 Representative cartoons with the corresponding model Cryo-TEM image of the various self-assemblies. A – Multi-compartment micelles (MCMs). B – Pseudo-vesicles. C – Yolk/shell nanoparticles. | ||
MCMs are composed of discrete and structured nanodomains within the core of the nanoparticles. This explains the form factor close to 0.78 as it is a myriad of inverse micelles that make the overall structure comparable to a solid sphere. The most common pathway to induce the formation of MCMs is either by peptides45,46 or by a terpolymer system in the presence of two mutually incompatible hydrophobic segments and one hydrophilic segment.16 For PEO-b-PEHOx, there is only one hydrophobic block and to the best of our knowledge, this is the first synthetic amphiphilic AB diblock copolymer forming MCMs. We hypothesize that the self-assembly of the MCMs is driven by the hydrophobic interactions between the branched 2-ethyl-3-heptyl side chains of the PEHOx, while the backbone of the 2-oxazolines forms discrete subdomains due to a certain incompatibility with the purely hydrocarbon side-chains. Consequently, the system contains PEO, the PEHOx main chain and the PEHOx side chains as three distinctly different contributors, which act like a terpolymer system when forming the MCMs. Moreover, those self-assembled structures are stable over months. Both TEM and Cryo-TEM pictures were taken three months after the sample self-assembled and TEM images did not change over that period (see Fig. S12 in the ESI†). Together with the fact that both film rehydration and solvent switch techniques yielded predominantly MCMs, we presume those structures to be near-equilibrium and thermodynamically favourable.
This is a pseudo-triple-layer ellipsoid structure where an irregular bilayer membrane encloses a cavity which then hosts a micelle at its core (Fig. 5P/T and 6C; see Fig. S16 in the ESI† for a higher resolution). The core has a radius of 43 ± 3.6 nm which is now in line with a stretched length of PEO45-b-PEHOx128 of 62 nm. The polymer will be present at least somewhat coiled, making a radius of 43 nm completely reasonable. The bilayer forms two discrete domains defined by a certain thickness of the membrane: a narrow one with a thickness of 25.1 ± 3.0 nm (green area in Fig. 6C) and a broad one with a thickness of 51.0 ± 2.6 nm (red area in Fig. 6C). We hypothesize that the broad bilayer is actually a double bilayer of diblock copolymers which dissociate and transition to a simple bilayer. Those compact bilayers and the transition between a simple and a double bilayer are stabilized by the hydrophobic interactions of the side chains. These yolk/shell nanoparticles present similarities to the pseudo-vesicles. The main differences are that the hydrophilic domain between bilayers remains minimal and is not an actual hollow cavity filled with water and that a micelle is encapsulated in its core. Just like for the pseudo-vesicles, an extensive comparison of all measurable membrane thicknesses of yolk/shell nanoparticles was conducted and the results are shown in Table S2.† We hypothesized that the differences observed can be explained by the dispersity of the diblock copolymers and the remaining traces of THF lingering in the hydrophobic domains due to the solvent switch method. We confirmed it by NMR (see Fig. S15 in the ESI†). Additionally, by adopting a more coiled up conformation it maximizes the hydrophobic interactions between the side chains, which enables the formation of a compact bilayer of entangled side chains.
Interestingly, in all Cryo-TEM images of MCMs, pseudo-vesicles and yolk/shell nanoparticles, the PEO corona can be visualized and measured as it forms a clear black halo around all nanoparticles (see the orange arrows in Fig. S8/S10/S13 in the ESI†). It is 2.9 ± 0.3 nm thick across all Cryo-TEM images. This length is perfectly in line with the 2.8 nm of a PEO45 as a random coil mentioned earlier (see section 5a of the ESI†).
All results were summarized in a self-assembly phase diagram (Fig. 7), to emphasize the broad range of self-assembled structures possible with this AB diblock copolymer, which could only be proven using a combination of DLS, SLS, TEM and Cryo-TEM. Both film rehydration and solvent switch yield unique yet distinct self-assembled structures with a variety which is usually seen for ABC triblock copolymers.3 This great variety reflects the unique influence of the side chain of PEHOx, as it is the first synthetic amphiphilic AB diblock copolymer to efficiently form such a variety of complex self-assemblies on its own. Further studies on the influence of THF or other solvents on the solvent switch might also elucidate how to form regular vesicles with this system.
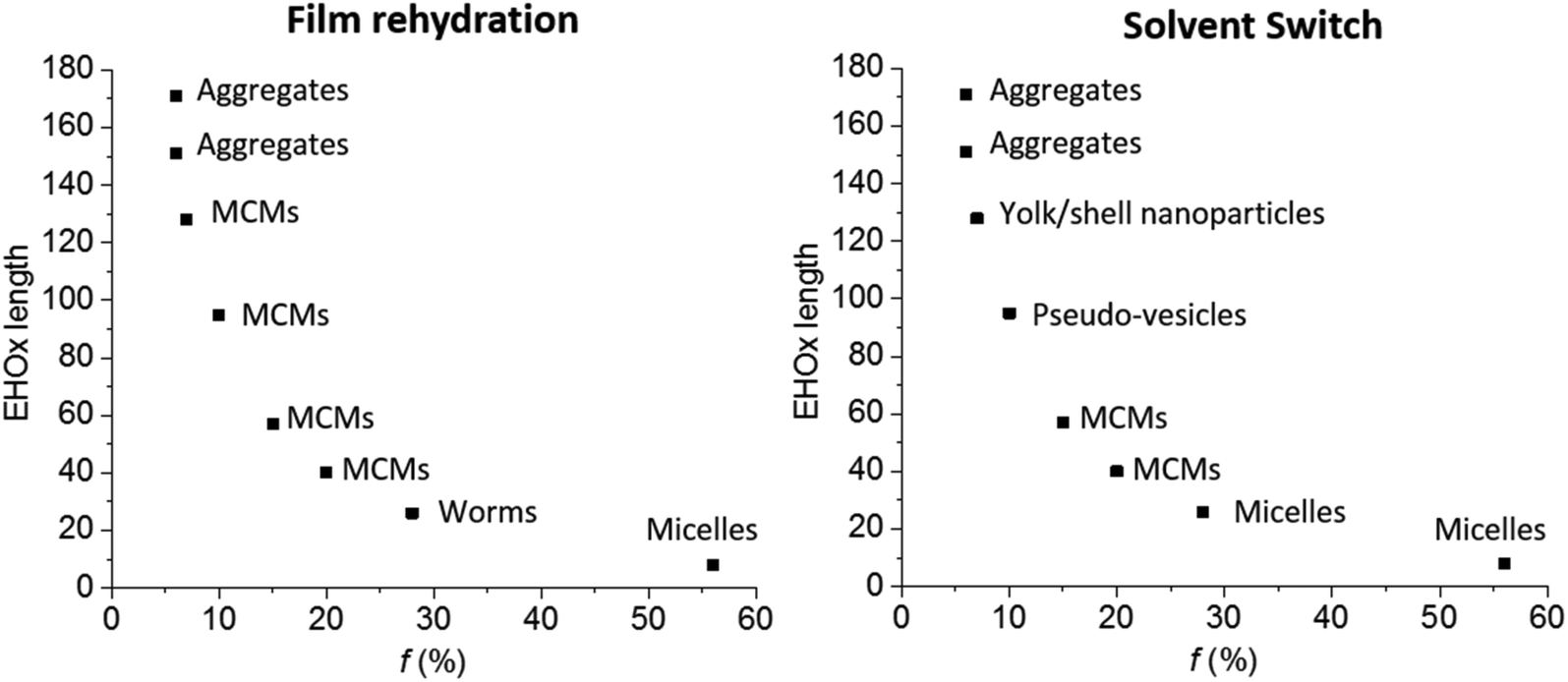 | ||
| Fig. 7 Self-assembly phase diagram of AB diblock PEO-b-PEHOx self-assemblies by film rehydration and solvent switch. MCMs = multicompartment micelles. | ||
Conclusions
We presented the synthesis of a new amphiphilic diblock copolymer PEO-b-PEHOx by cationic ring opening microwave-assisted polymerization using a previously unknown nosylate-based PEO macroinitiator. The choice of solvent proved to be as paramount as the choice of temperature, with chlorobenzene and 140 °C being the best combination. A library of AB diblock copolymers with different PEHOx was synthesized and designed for self-assembly into defined nanoparticles. DSC revealed that the resulting diblock copolymers have a Tg below 0 °C, ensuring a high flexibility of the system for the following self-assembly at room temperature. Using film rehydration and solvent switch, we showed that PEO-b-PEHOx predominantly self-assembles into multi-compartment micelles (MCMs). MCMs grow in size when the PEHOx becomes longer. As hydrophilic and hydrophobic molecules could be loaded in the same carrier, those MCMs could find applications in advanced drug delivery applications.Solvent switch of PEO-b-PEHOx with a long PEHOx block also leads to pseudo-vesicles and yolk/shell nanoparticles. As the organic solvent was trapped within the walls of those vesicle-like structures, further studies can focus on loading those distinct hydrophobic and hydrophilic domains with different catalysts in order to perform cascade reactions. All those complex structures were formed from a single AB diblock copolymer which shows the potential of hydrophobic blocks with a long side chain to obtain complex nanoparticles from a single material. Further studies will be conducted to gain deeper insights into the influence of various side chains of the hydrophobic block on the self-assembly of block copolymers. Only an in-depth study using Cryo-TEM and SLS revealed these structures and should motivate the community to look deeper if regular TEM suggests the presence of vesicles as this might not always be the case. This work will also be used as a stepping stone towards the one-pot synthesis of new self-assembling triblock amphiphilic copolymers based on this system using the sequential polymerization of oxazolines.3,31
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Swiss National Science Foundation (SNSF) especially in light of the National Centre for Competence in Research – Molecular Systems Engineering (NCCR-MSE) and J. G. thanks the Novartis-University of Basel fund for excellence in life sciences. We gratefully acknowledge the BioEM Lab, Dr Mohamed Chami and Carola Alampi for the cryogenic transmission electron microscopy measurements and useful discussions. D. D. is very grateful to Dr Samuel Lörcher and Dr Evgeniia Konishcheva for their mentorship in polymer synthesis and many fruitful discussions.References
- Y. Mai and A. Eisenberg, Self-assembly of block copolymers, Chem. Soc. Rev., 2012, 41(18), 5969–5985 RSC.
- D. E. Discher and A. Eisenberg, Polymer Vesicles., Science, 2002, 297(5583), 967–973 CrossRef CAS PubMed.
- E. Konishcheva, D. Daubian, J. Gaitzsch and W. Meier, Synthesis of Linear ABC Triblock Copolymers and Their Self-Assembly in Solution, Helv. Chim. Acta, 2018, 101(2), e1700287 CrossRef.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Bioinspired polymer vesicles and membranes for biological and medical applications, Chem. Soc. Rev., 2016, 45(2), 377–411 RSC.
- N. Uehlein, B. Otto, A. Eilingsfeld, F. Itel, W. Meier and R. Kaldenhoff, Gas-tight triblock-copolymer membranes are converted to CO2 permeable by insertion of plant aquaporins, Sci. Rep., 2012, 2, 538 CrossRef PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Polymer nanoreactors shown to produce and release antibiotics locally, Chem. Commun., 2013, 49(2), 128–130 RSC.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Polymersomes with engineered ion selective permeability as stimuli-responsive nanocompartments with preserved architecture, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Engineering Functional Polymer Capsules toward Smart Nanoreactors, Chem. Rev., 2016, 116(3), 1053–1093 CrossRef CAS PubMed.
- A. Najer, D. Wu, M. G. Nussbaumer, G. Schwertz, A. Schwab, M. C. Witschel, A. Schafer, F. Diederich, M. Rottmann, C. G. Palivan, H. P. Beck and W. Meier, An amphiphilic graft copolymer-based nanoparticle platform for reduction-responsive anticancer and antimalarial drug delivery, Nanoscale, 2016, 8(31), 14858–14869 RSC.
- J. Gaitzsch, S. Hirschi, S. Freimann, D. Fotiadis and W. Meier, Directed Insertion of Light-Activated Proteorhodopsin into Asymmetric Polymersomes from an ABC Block Copolymer, Nano Lett., 2019, 19(4), 2503–2508 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Multiple Morphologies and Characteristics of “Crew-Cut” Micelle-like Aggregates of Polystyrene-b-poly(acrylic acid) Diblock Copolymers in Aqueous Solutions, J. Am. Chem. Soc., 1996, 118(13), 3168–3181 CrossRef CAS.
- E. Konishcheva, D. Häussinger, S. Lörcher and W. Meier, Key aspects to yield low dispersity of PEO- b -PCL diblock copolymers and their mesoscale self-assembly, Eur. Polym. J., 2016, 83, 300–310 CrossRef CAS.
- M. A. Petersen, L. Yin, E. Kokkoli and M. A. Hillmyer, Synthesis and characterization of reactive PEO-PMCL polymersomes, Polym. Chem., 2010, 1(8), 1281–1290 RSC.
- J. Gaitzsch, P. C. Welsch, J. Folini, C.-A. Schoenenberger, J. C. Anderson and W. P. Meier, Revisiting monomer synthesis and radical ring opening polymerization of dimethylated MDO towards biodegradable nanoparticles for enzymes, Eur. Polym. J., 2018, 101, 113–119 CrossRef CAS.
- D. Wu, M. Spulber, F. Itel, M. Chami, T. Pfohl, C. G. Palivan and W. Meier, Effect of Molecular Parameters on the Architecture and Membrane Properties of 3D Assemblies of Amphiphilic Copolymers, Macromolecules, 2014, 47(15), 5060–5069 CrossRef CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Multicompartment Block Polymer Micelles, Macromolecules, 2012, 45(1), 2–19 CrossRef CAS.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Controlling Cellular Uptake by Surface Chemistry, Size, and Surface Topology at the Nanoscale, Small, 2009, 5(21), 2424–2432 CrossRef CAS PubMed.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Novel aspects of encapsulation and delivery using polymersomes, Curr. Opin. Pharmacol., 2014, 18, 104–111 CrossRef CAS PubMed.
- W.-M. Wan and C.-Y. Pan, Formation of Polymeric Yolk/Shell Nanomaterial by Polymerization-Induced Self-Assembly and Reorganization, Macromolecules, 2010, 43(6), 2672–2675 CrossRef CAS.
- R. Purbia and S. Paria, Yolk/shell nanoparticles: classifications, synthesis, properties, and applications, Nanoscale, 2015, 7(47), 19789–19873 RSC.
- J. Du and S. P. Armes, pH-Responsive Vesicles Based on a Hydrolytically Self-Cross-Linkable Copolymer, J. Am. Chem. Soc., 2005, 127(37), 12800–12801 CrossRef CAS PubMed.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Biomimetic pH Sensitive Polymersomes for Efficient DNA Encapsulation and Delivery, Adv. Mater., 2007, 19(23), 4238–4243 CrossRef CAS.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Self-Assembly of Amphiphilic Block Copolypeptoids – Micelles, Worms and Polymersomes, Sci. Rep., 2016, 6(1), 33491 CrossRef PubMed.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Poly(2-Oxazoline)s – Are They More Advantageous for Biomedical Applications Than Other Polymers?, Macromol. Rapid Commun., 2012, 33(19), 1648–1662 CrossRef CAS PubMed.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, The chemistry of poly(2-oxazoline)s, Eur. Polym. J., 2017, 88(Supplement C), 451–469 CrossRef CAS.
- N. Zhang, S. Huber, A. Schulz, R. Luxenhofer and R. Jordan, Cylindrical Molecular Brushes of Poly(2-oxazoline)s from 2-Isopropenyl-2-oxazoline, Macromolecules, 2009, 42(6), 2215–2221 CrossRef CAS.
- R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich, E. V. Batrakova, R. Jordan and A. V. Kabanov, Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs, Biomaterials, 2010, 31(18), 4972–4979 CrossRef CAS PubMed.
- K. Kempe, E. F. J. Rettler, R. M. Paulus, A. Kuse, R. Hoogenboom and U. S. Schubert, A systematic investigation of the effect of side chain branching on the glass transition temperature and mechanical properties of aliphatic (co-)poly(2-oxazoline)s, Polymer, 2013, 54(8), 2036–2042 CrossRef CAS.
- K. Kempe, S. Jacobs, H. M. L. Lambermont-Thijs, M. M. W. M. Fijten, R. Hoogenboom and U. S. Schubert, Rational Design of an Amorphous Poly(2-oxazoline) with a Low Glass-Transition Temperature: Monomer Synthesis, Copolymerization, and Properties, Macromolecules, 2010, 43(9), 4098–4104 CrossRef CAS.
- M. M. Lübtow, L. Keßler, A. Appelt-Menzel, T. Lorson, N. Gangloff, M. Kirsch, S. Dahms and R. Luxenhofer, More Is Sometimes Less: Curcumin and Paclitaxel Formulations Using Poly(2-oxazoline) and Poly(2-oxazine)-Based Amphiphiles Bearing Linear and Branched C9 Side Chains, Macromol. Biosci., 2018, 18(11), 1800155 CrossRef PubMed.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten, B. M. v. Lankvelt and U. S. Schubert, One-pot synthesis of 2-phenyl-2-oxazoline-containing quasi-diblock copoly(2-oxazoline)s under microwave irradiation, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(3), 416–422 CrossRef CAS.
- M. Glassner, D. R. D′hooge, J. Young Park, P. H. M. Van Steenberge, B. D. Monnery, M.-F. Reyniers and R. Hoogenboom, Systematic investigation of alkyl sulfonate initiators for the cationic ring-opening polymerization of 2-oxazolines revealing optimal combinations of monomers and initiators, Eur. Polym. J., 2015, 65(Supplement C), 298–304 CrossRef CAS.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, PEO-b-PCL-b-PMOXA Triblock Copolymers: From Synthesis to Microscale Polymersomes with Asymmetric Membrane, Macromolecules, 2017, 50(4), 1512–1520 CrossRef CAS.
- B. Brissault, C. Guis and H. Cheradame, Kinetic study of poly(ethylene oxide-b-2-methyl-2-oxazoline) diblocs synthesis from poly(ethylene oxide) macroinitiators, Eur. Polym. J., 2002, 38(2), 219–228 CrossRef CAS.
- U. Tilstam, Sulfolane: A Versatile Dipolar Aprotic Solvent, Org. Process Res. Dev., 2012, 16(7), 1273–1278 CrossRef CAS.
- M. Vergaelen, B. Verbraeken, B. D. Monnery and R. Hoogenboom, Sulfolane as Common Rate Accelerating Solvent for the Cationic Ring-Opening Polymerization of 2-Oxazolines, ACS Macro Lett., 2015, 4(8), 825–828 CrossRef CAS.
- L. M. Robeson, Polymer blends : a comprehensive review, Hanser, Munich, 2007 Search PubMed.
- V. P. Privalko and Y. S. Lipatov, Glass transition and chain flexibility of linear polymers, J. Macromol. Sci., Part B: Phys., 1974, 9(3), 551–564 CrossRef CAS.
- C. Nardin, T. Hirt, J. Leukel and W. Meier, Polymerized ABA Triblock Copolymer Vesicles, Langmuir, 2000, 16(3), 1035–1041 CrossRef CAS.
- H. Huang, B. Chung, J. Jung, H.-W. Park and T. Chang, Toroidal Micelles of Uniform Size from Diblock Copolymers, Angew. Chem., Int. Ed., 2009, 48(25), 4594–4597 CrossRef CAS PubMed.
- J. Hotz and W. Meier, Vesicle-Templated Polymer Hollow Spheres, Langmuir, 1998, 14(5), 1031–1036 CrossRef CAS.
- S. U. Egelhaaf and P. Schurtenberger, Shape Transformations in the Lecithin-Bile Salt System: From Cylinders to Vesicles, J. Phys. Chem., 1994, 98(34), 8560–8573 CrossRef CAS.
- J.-N. Marsat, F. Stahlhut, A. Laschewsky, H. v. Berlepsch and C. Böttcher, Multicompartment micelles from silicone-based triphilic block copolymers, Colloid Polym. Sci., 2013, 291(11), 2561–2567 CrossRef CAS.
- J. Babinot, E. Renard, B. Le Droumaguet, J.-M. Guigner, S. Mura, J. Nicolas, P. Couvreur and V. Langlois, Facile Synthesis of Multicompartment Micelles Based on Biocompatible Poly(3-hydroxyalkanoate), Macromol. Rapid Commun., 2013, 34(4), 362–368 CrossRef CAS PubMed.
- D. de Bruyn Ouboter, T. B. Schuster, A. Mantion and W. Meier, Hierarchical Organization of Purely Peptidic Amphiphiles into Peptide Beads, J. Phys. Chem. C, 2011, 115(30), 14583–14590 CrossRef CAS.
- S. J. Sigg, V. Postupalenko, J. T. Duskey, C. G. Palivan and W. Meier, Stimuli-Responsive Codelivery of Oligonucleotides and Drugs by Self-Assembled Peptide Nanoparticles, Biomacromolecules, 2016, 17(3), 935–945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01559k |
| This journal is © The Royal Society of Chemistry 2020 |