DOI:
10.1039/C9PY01533G
(Paper)
Polym. Chem., 2020,
11, 53-60
Carboxylate, nitrate, sulfonate, and phosphate catalysts for living radical polymerization via oxygen–iodine halogen bonding catalysis†
Received
10th October 2019
, Accepted 14th November 2019
First published on 26th November 2019
Abstract
Four families of oxyanions, i.e., carboxylate, nitrate, phosphate, and sulfonate, were studied as novel catalysts in living (or reversible deactivation) radical polymerization via oxygen–iodine halogen bonding catalysis. Oxyanions with sodium and tetraalkylammonium counter-cations exhibited good catalytic activities and high solubilities in hydrophilic and hydrophobic monomers. These oxyanion catalysts were amenable for methyl methacrylate, functional methacrylates, styrene, and acrylonitrile, and also afforded block copolymers with low dispersities. The catalytic activities of the oxyanions were also theoretically studied using density functional theory (DFT) calculation. The studied four families of oxyanions are abundant in natural and synthetic compounds. Non-toxic natural carboxylates were successfully used to synthesize well-defined biocompatible polymers. The low cost, low toxicity, and accessibility for a range of polymer designs are attractive features for practical use.
Introduction
Halogen bonding is a noncovalent attractive interaction between an electron-accepting halogen (X) and an electron-donating group (B). Halogen bonding generates a directional 180° interaction (R–X⋯B), and the strength of halogen bonding increases for heavier halogens, i.e., R–I > R–Br > R–Cl ≫ R–F. The B group can be anions and electron-donating neutral species such as halide anions and oxygen- and nitrogen-containing species.1,2 A variety of halogen bonding donor–acceptor pairs have been found and applied in crystal engineering3–5 and molecular recognition.6–8 Recently, solution-phase halogen-bonding-promoted organocatalysis has attracted emerging attention in organic synthesis and supramolecular chemistry.9–11
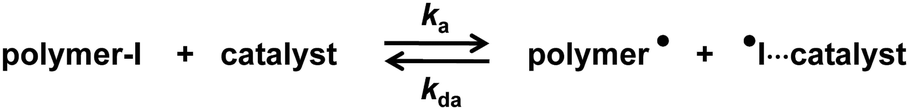
Living radical polymerization, also termed reversible-deactivation radical polymerization, is an effective method to synthesize well-defined polymers with predictable molecular weights and narrow molecular weight distributions.12–17 Our research group developed an organocatalyzed living radical polymerization using alkyl iodide (R–I) as an initiator and an organic molecule as a catalyst.18–26 Mechanistically, the dormant polymer–iodide (polymer–I) and the catalyst are supposed to form a halogen-bonding complex (polymer–I⋯catalyst). The complex subsequently reversibly generates the propagating radical (polymer˙) (Scheme 1). We term this polymerization reversible complexation mediated polymerization (RCMP). RCMP is attractive because no special capping agents or metals are required while it is amenable for a wide range of monomers and polymer structures. Several effective catalysts such as iodide (I−) and pseudo-halide anions have been utilized in RCMP.19,21 Recently, we reported that pyridine N-oxide (C5H5N+–O−) derivatives worked as efficient catalysts.27 The use of oxygen-centered anions (O−) opened a new avenue in catalyst development in RCMP. However, the rigid aromatic structure and multi-step synthesis of pyridine N-oxide derivatives limited practical applications.
 |
| | Scheme 1 Reversible activation in RCMP. | |
An oxyanion is generally described as a polyatomic anion with the chemical formula [ExOy]z−, where E is an element and O is oxygen. E can be various elements and bonds to one or more oxygen atoms to form functional oxyanions such as carboxylate (E = C, x = 1, y = 2, and z = 1), nitrate (E = N, x = 1, y = 3, and z = 1), sulfonate (E = S, x = 1, y = 3, and z = 1 or 2) and phosphate (E = P, x = 1, y = 4, and z = 1, 2 or 3). Oxyanions are widely witnessed in natural and synthetic compounds such as enzymes, lipids, drugs, and surfactants for biomedical and industrial applications.28–30 While the halogen bonding of oxyanions such as carboxylates and sulfates has been used in crystal engineering, little attention has been paid to their use in halogen bonding catalysis (catalytic reactions). Poli et al. recently reported the use of several oxyanions (or oxygen-centered anions) including carbonates, bicarbonates, sulphates, bisulphates, and nitrates as catalysts in atom transfer radical polymerization with ethyl α-bromophenylacetate as an initiator. While a comprehensive and elegant study was performed, the weak halogen bonding interaction between alkyl bromide and oxyanions led to relatively low monomer conversions (up to 37.5% conversion) and relatively large polydispersity indexes (Đ ≥ 1.34), where Đ is Mw/Mn and Mn and Mw are the number-average and weight-average molecular weights, respectively.31
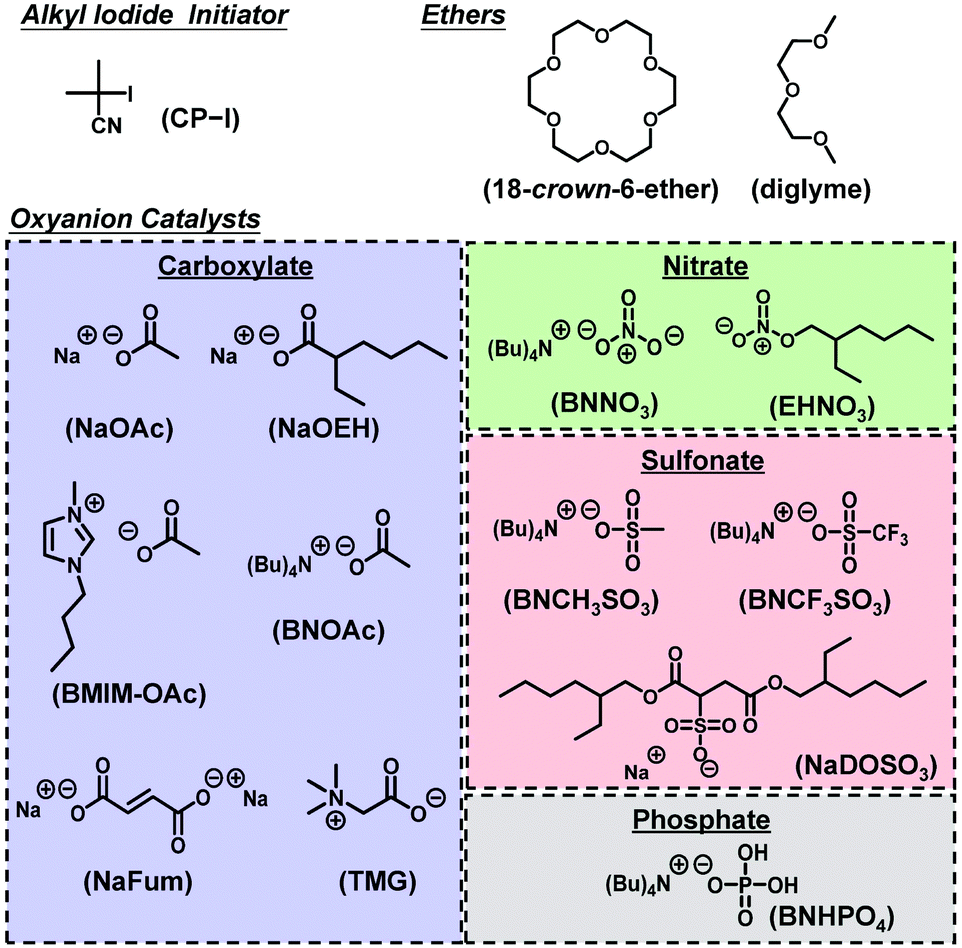
Herein, we report the use of carboxylates, nitrates, sulfonates, and a phosphate as RCMP oxyanion catalysts combined with an alkyl iodide initiator (not an alkyl bromide initiator). Unlike pyridine N-oxide derivatives, these oxyanion families enable a diverse molecular design because of their simple structures and abundance in natural, biological, and synthetic chemistries. These oxyanion catalysts were amenable for a broad range of monomers including methyl methacrylate, functional methacrylates, styrene, and acrylonitrile, which is advantageous over the previous pyridine N-oxide catalysts. Such wide scopes in catalyst structures and amenable monomers are attractive features. The catalytic activities of the oxyanion catalysts were also theoretically studied via density functional theory (DFT) calculation. Fig. 1 shows the studied alkyl iodide initiator, oxyanion catalysts, and ethers used to dissolve the catalysts. Block copolymers with hydrophobic and hydrophilic segments were also successfully synthesized using the oxyanion catalysts.
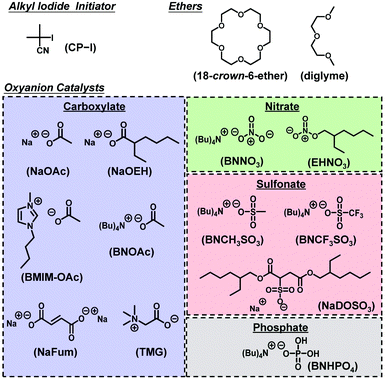 |
| | Fig. 1 Structures of the alkyl iodide, ethers, and oxyanion catalysts used in this work. | |
Results and discussion
Experimental proof of the generation of R˙ from R–I
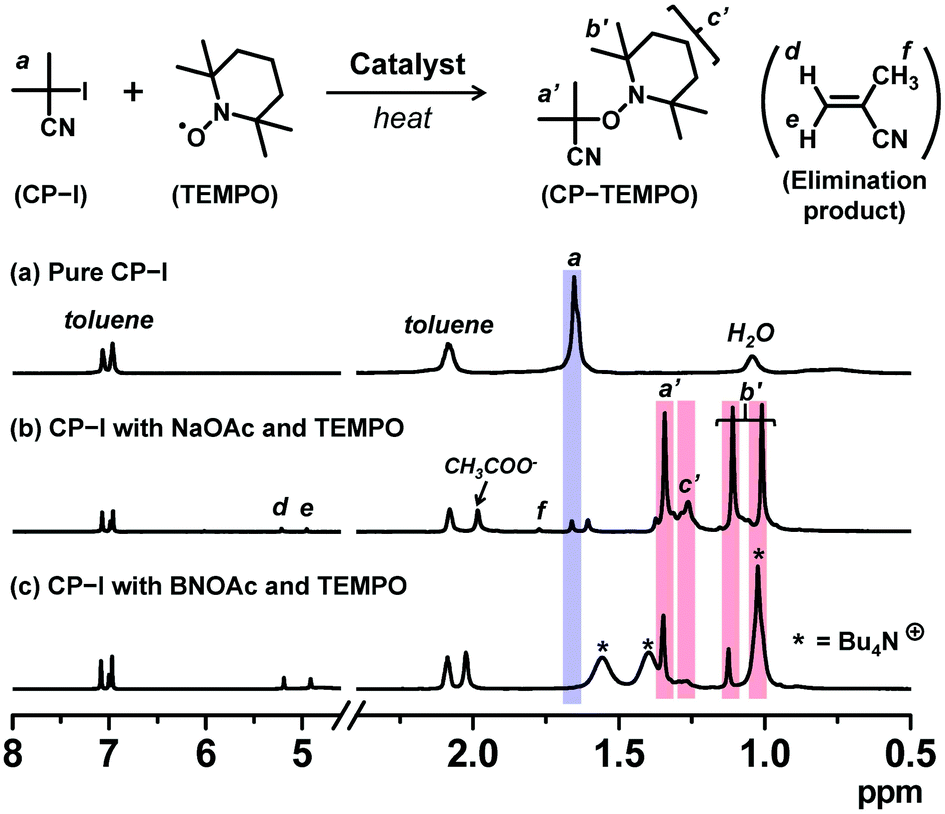
A radical trap experiment19,32 was carried out to prove the generation of an alkyl radical (R˙) from alkyl iodide using oxyanions. We heated 2-iodo-2-methylpropionitrile (CP-I (Fig. 1), 40 mM) (alkyl iodide), sodium acetate (NaOAc, 80 mM) or tetrabutylammonium acetate (BNOAc, 40 mM) (catalyst), and 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) (80 mM) (radical trap) in a mixture of toluene-d8 (90%) and acetonitrile-d3 (10%) at 70 °C for 8 h. We used 18-crown-6-ether (80 mM) to dissolve NaOAc. BNOAc is a metal-free organic acetate and was highly soluble in the studied solvent without the crown ether. The mixture of toluene-d8 (dielectric constant ε = 2.4) and acetonitrile-d3 (ε = 37.5) is a model of methyl methacrylate (MMA) medium (ε = 7.9). If CP-I generates the CP radical (CP˙) in the presence of the acetates, CP˙ is trapped by TEMPO to form CP-TEMPO. Fig. 2 shows the 1H NMR spectra of pure CP-I and the reaction mixtures after 8 h. The signals (peaks a′, b′ and c′) of CP-TEMPO were observed after the 8 h reaction (Fig. 2b and c), proving the radical generation from CP-I via the catalytic work of the acetates. The conversion of CP-I into CP-TEMPO was 92% and 49% with NaOAc and BNOAc, respectively. The acetate anions are weak bases, and the basicity can bring about the elimination of HI from CP-I as a side reaction. In the case of NaOAc, only 3% of the elimination product (methacrylonitrile) was generated in 8 h. On the other hand, BNOAc promoted the elimination and gave methacrylonitrile with 51% conversion in 8 h. The results clearly demonstrate the generation of R˙ from R-I with both acetate catalysts and also the occurrence of a rather significant side reaction (elimination) in the case of BNOAc, suggesting the importance of the choice of the counter cations. NaOAc efficiently catalyzed the radical generation with negligible elimination and should be more suitable for the polymerization catalyst. The radical trap experiment was also conducted for other oxyanions, confirming the radical generation from CP-I (Fig. S1, ESI†).
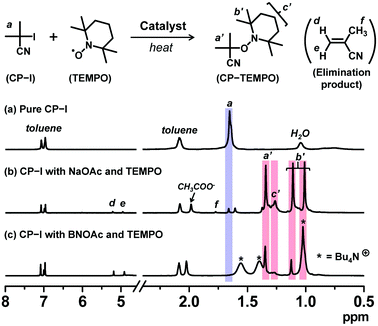 |
| | Fig. 2
1H NMR spectra (in the range of 0.5–8.0 ppm) of (a) pure CP-I, (b) the solution of CP-I (40 mM), NaOAc (80 mM), 18-crown-6-ether (80 mM), and TEMPO (80 mM) heated at 70 °C for 8 h, and (c) the solution of CP-I (40 mM), BNOAc (40 mM), and TEMPO (80 mM) heated at 70 °C for 8 h. The solvent was a mixture of toluene-d8 and acetonitrile-d3 (v/v = 9/1). | |
Polymerizations of MMA with carboxylate catalysts
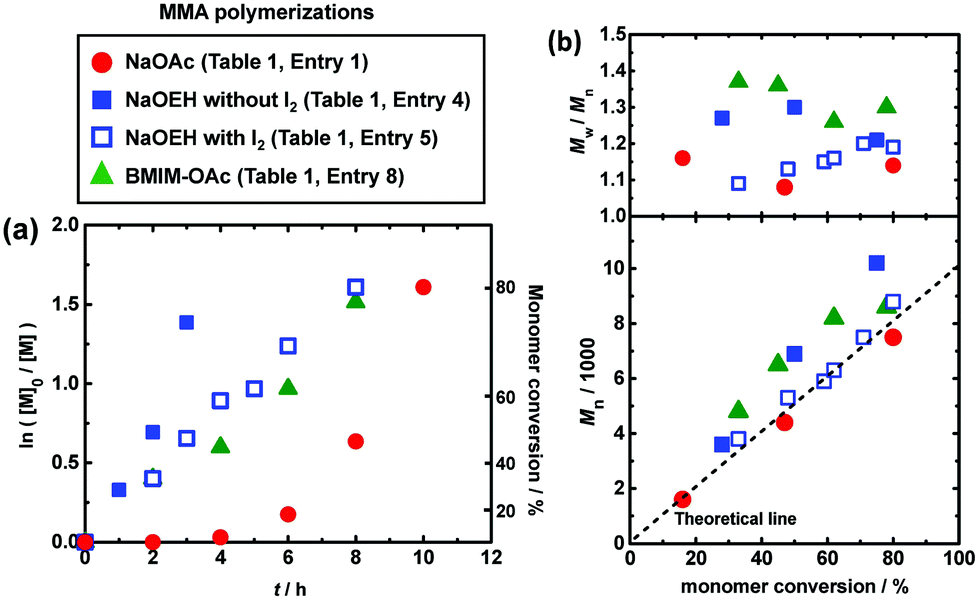
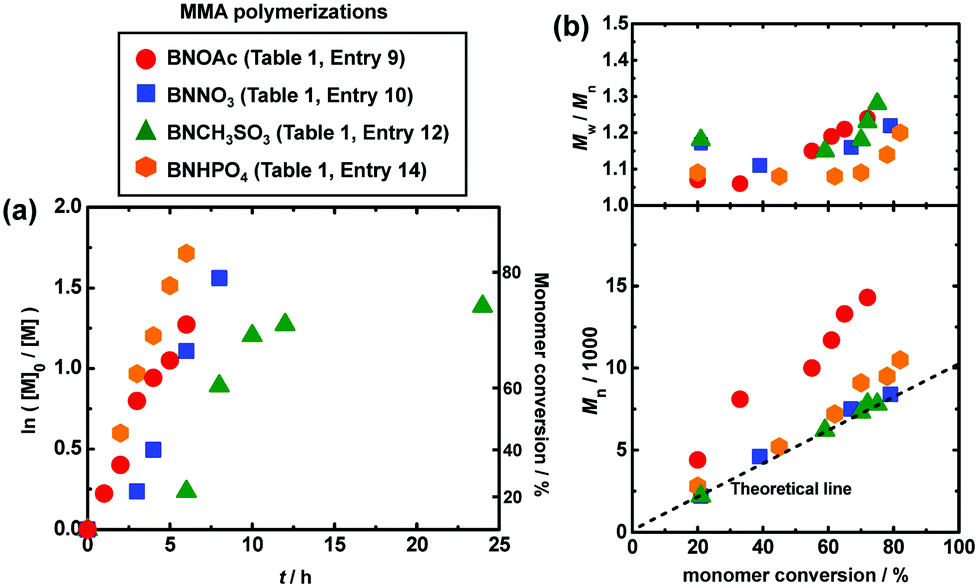
We studied the bulk polymerizations of MMA using an R-I initiator and oxyanion catalysts, i.e., carboxylates, nitrates, sulfonates, and a phosphate with a wide range of different molecular structures (Fig. 1). Fig. 3, 4 (circle), and Table 1 (entries 1 and 3–9) show the polymerization results using carboxylate catalysts. We heated a mixture of MMA (8 M), CP-I (80 mM) as an alkyl iodide initiator, NaOAc (80 mM) as a catalyst, and 18-crown-6-ether (80 mM) to dissolve the catalyst at 70 °C (Fig. 3 (circle) and Table 1 (entry 1)). An induction period (Fig. 3a (circle)) was observed in the first 4 h due to a slow dissolution of NaOAc in MMA. (NaOAc was similarly or slightly better dissolved in a toluene-d8/acetonitrile-d3 (v/v = 9/1) mixture in the radical trap experiment mentioned above.) Subsequently, the polymerization proceeded up to 80% monomer conversion in 10 h. The Mn value was in agreement with the theoretical value (Mn,theo), and the Đ value was below 1.2 from an early stage of polymerization. No polymerization occurred without the addition of NaOAc or 18-crown-6-ether (Table 1 (entries 2 and 3)), meaning that the NaOAc dissolved by the crown ether worked as a catalyst.
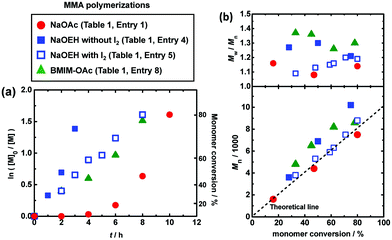 |
| | Fig. 3 Plots of (a) ln([M]0/[M]) vs. t and (b) Mn and Mw/Mnvs. conversion for the MMA/CP-I/catalyst/I2 systems (70 °C): [MMA]0 = 8 M; [CP-I]0 = 80 mM; [catalyst]0 = 80 mM; [I2]0 = 0 or 5 mM. 18-crown-6-ether (80 mM) was added to NaOAc and NaOEH systems. The symbols are indicated in the figure. | |
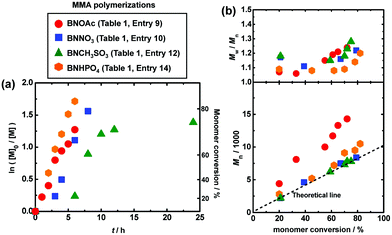 |
| | Fig. 4 Plots of (a) ln([M]0/[M]) vs. t and (b) Mn and Mw/Mnvs. conversion for the MMA/CP-I/catalyst/I2 systems (70 °C): [MMA]0 = 8 M; [CP-I]0 = 80 mM; [catalyst]0 = 80 mM; [I2]0 = 5 mM. The symbols are indicated in the figure. | |
Table 1 Polymerizations of MMA with oxyanion catalysts
| Entry |
Target DPa |
Catalyst |
[MMA]0/[CP-I]0/[catalyst]0/[18-crown-6-ether]0/[I2]0 (mM) |
T (°C) |
T (h) |
Conv. (%) |
M
n (Mn,theo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b) b) |
Đ
|
|
Target degree of polymerization at 100% monomer conversion (calculated by [MMA]0/[CP-I]0).
Theoretical Mn calculated with [MMA]0, [CP-I]0, and monomer conversion.
The addition of 10 wt% 1-butanol (MMA/1-butanol = 9/1 (w/w)).
|
| 1 |
100 |
NaOAc |
8000/80/80/80/0 |
70 |
10 |
80 |
7500 (8000) |
1.14 |
| 2 |
100 |
None |
8000/80/0/80/0 |
70 |
10 |
No polymerization |
| 3 |
100 |
NaOAc |
8000/80/80/0/0 |
70 |
10 |
No polymerization |
| 4 |
100 |
NaOEH |
8000/80/80/80/0 |
70 |
3 |
75 |
10000 (7500) |
1.21 |
| 5 |
100 |
NaOEH |
8000/80/80/80/5 |
70 |
8 |
80 |
8800 (8000) |
1.19 |
| 6 |
200 |
NaOEH |
8000/40/40/40/5 |
70 |
8 |
80 |
21000 (16000) |
1.15 |
| 7 |
400 |
NaOEH |
8000/20/20/20/5 |
70 |
24 |
81 |
30000 (32000) |
1.38 |
| 8 |
100 |
BMIM-OAc |
8000/80/80/0/0c |
70 |
8 |
78 |
8600 (7800) |
1.30 |
| 9 |
100 |
BNOAc |
8000/80/80/0/5 |
70 |
6 |
72 |
14300 (7200) |
1.24 |
| 10 |
100 |
BNNO3 |
8000/80/80/0/5 |
70 |
8 |
79 |
8400 (7900) |
1.22 |
| 11 |
100 |
EHNO3 |
8000/80/80/0/0 |
70 |
24 |
No polymerization |
| 12 |
100 |
BNCH3SO3 |
8000/80/80/0/5 |
70 |
24 |
75 |
7800 (7500) |
1.28 |
| 13 |
100 |
BNCF3SO3 |
8000/80/80/0/5 |
70 |
24 |
No polymerization |
| 14 |
100 |
BNHPO4 |
8000/80/80/0/5 |
70 |
6 |
82 |
10500 (8200) |
1.20 |
For a better solubility of the catalyst, we used sodium 2-ethylhexanoate (NaOEH) bearing a 2-ethylhexyl group together with 18-crown-6-ether (Fig. 3 (filled square) and Table 1 (entry 4)). NaOEH brought no induction period because of the good solubility and hence led to faster polymerization, reaching 75% monomer conversion in 3 h. The Mn value linearly increased with the monomer conversion but deviated from the Mn,theo value, probably because NaOEH is a highly active catalyst to rapidly generate a large amount of alkyl radicals from alkyl iodides, where the termination among short oligomer radicals significantly took place at an early stage of polymerization (until a sufficient amount of deactivator is accumulated). The generated low-molar-mass terminated species were not counted as polymers in the GPC analysis. In order to suppress the termination, we added a small amount of deactivator, i.e., molecular iodine (I2), so that the generated alkyl radical can smoothly be capped with iodide to generate the dormant species. With the addition of I2 (5 mM), the Mn value was in agreement with the Mn,theo value and the Đ value was below 1.2 from an early stage of polymerization (Fig. 3 (open square) and Table 1 (entry 5)). The high catalytic activity of NaOEH also allowed the synthesis of higher molecular weight polymers with Mn = 21000–30000 and Đ = 1.15–1.38 (Table 1 (entries 6 and 7)). These results demonstrate the effectiveness of the NaOEH catalyst.
As completely metal-free (sodium-free) catalysts, we also used 1-butyl-3-methylimidazolium acetate (BMIM-OAc) (Fig. 3 (triangle) and Table 1 (entry 8)) and BNOAc (Fig. 4 (circle) and Table 1 (entry 9)) and obtained polymers with Đ = 1.24–1.30. 1-Butanol (10 wt%) was added to dissolve BMIM-OAc in MMA. Both catalysts are sodium-free and did not require the crown ether for the dissolution. However, the Mn value deviated from the Mn,theo value. Unlike NaOEH, the deviation was observed even in the presence of I2 (deactivator) for both catalysts, probably because the deviation is attributed to the HI elimination (side reaction) from CP-I (and short polymer–I), as was observed in the radical trapping experiment in the case of BNOAc (Fig. 2c). In general, a higher basicity (a higher nucleophilicity) leads to stronger halogen bonding and is beneficial for promoting halogen bonding catalysis. However, too high basicity leads to significant elimination as a competitive side reaction. While the ion pair (carboxylate anion and counter cation) is fully dissociated in aqueous solutions, it tends to associate in organic solutions. In the MMA medium, because of this association, the basicity may differ for Na and organic carboxylates. A possible reason why the elimination was insignificant and significant for Na and organic carboxylates, respectively, would be their different basicities in MMA, although the exact mechanism is unclear at the moment.
Nitrate, sulfonate and phosphate catalysts
A nitrate has a similarity to a carboxylate in the isoelectronic structures of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+–O− and OC–O−. However, a nitrate (NO3−) is more conjugated than a carboxylate (CO2−) (Fig. 1). Because of the delocalization of the electron, a nitrate is less basic and may suppress the elimination. Fig. 4 (square) and Table 1 (entry 10) show the result using tetrabutylammonium nitrate (BNNO3) as a catalyst. BNNO3 is organic and is also highly soluble in MMA. Unlike BNOAc (carboxylate), BNNO3 (nitrate) led to a good agreement of Mn with Mn,theo, suggesting the suppression of the elimination. BNNO3 is a bimolecular pair of the cationic BN+ and anionic NO3− ions. An electrically neutral single molecular nitrate ester, i.e., 2-ethylhexyl nitrate (EHNO3), showed a weak catalytic ability and resulted in no polymerization after 24 h (Table 1 (entry 11)).
N+–O− and OC–O−. However, a nitrate (NO3−) is more conjugated than a carboxylate (CO2−) (Fig. 1). Because of the delocalization of the electron, a nitrate is less basic and may suppress the elimination. Fig. 4 (square) and Table 1 (entry 10) show the result using tetrabutylammonium nitrate (BNNO3) as a catalyst. BNNO3 is organic and is also highly soluble in MMA. Unlike BNOAc (carboxylate), BNNO3 (nitrate) led to a good agreement of Mn with Mn,theo, suggesting the suppression of the elimination. BNNO3 is a bimolecular pair of the cationic BN+ and anionic NO3− ions. An electrically neutral single molecular nitrate ester, i.e., 2-ethylhexyl nitrate (EHNO3), showed a weak catalytic ability and resulted in no polymerization after 24 h (Table 1 (entry 11)).
A sulfonate (RSO3−) anion is another EO3− type anion structurally similar to NO3−. Similar to BNNO3, tetrabutylammonium methanesulfonate (BNCH3SO3) worked as an efficient catalyst and afforded a good agreement of Mn with Mn,theo and low Đ values (≤1.28) (Fig. 4 (triangle) and Table 1 (entry 12)), suggesting a minor occurrence of the elimination. In contrast to the successful polymerization with methyl sulfonate (BNCH3SO3), no polymerization took place with trifluoromethanesulfonate (tetrabutylammonium trifluoromethanesulfonate (BNCF3SO3)) (Table 1 (entry 13)). The electron-withdrawing trifluoromethyl group decreases the basicity (nucleophilicity) of SO3− and hence suppresses the halogen bonding catalysis, giving no polymerization.
A phosphate (PO4−) anion forms an (RO)2PO2− structure. The resonance of PO2− is similar to that of a carboxylate (CO2−) and hence the basicity of PO4− is relatively high. Therefore, the polymerization with tetrabutylammonium phosphate monobasic (BNHPO4) led to a marked deviation of Mn from Mn,theo due to the elimination (Fig. 4 (hexagon) and Table 1 (entry 14)).
As a whole, all the four families of oxyanions (carboxylate, nitrate, sulfonate, and phosphate) were effective for halogen bonding catalysis and induced polymerization. The four families of oxyanions are abundant in natural and synthetic compounds with biological, enzymatic, and amphipathic functions. The low cost, low toxicity, and accessibility for broad molecular structures are attractive features for practical use and future catalyst development. The basicity increases in the order of a sulfonate CH3SO3− (pKb = 15.92) < a nitrate NO3− (15.30) < a phosphate PO4− (11.88) < a carboxylate AcO− (9.24), where the pKb values are those in aqueous solutions and can be viewed for qualitative comparison in the organic MMA medium. As mentioned, higher basicity (higher nucleophilicity) promotes the halogen bonding catalysis. However, the carboxylate and phosphate are too basic and brought about the elimination, losing the livingness in the polymerization, in the case of the organic (BN) counter cation. Importantly, the carboxylate with the Na counter cation (NaOEH) significantly suppressed the elimination (as observed in the radical trap experiment and will be demonstrated in the chain-end analysis shown below), leading to a good control of polymerization. In summary, among the studied oxyanions, sodium carboxylate (NaOEH) and BN nitrate (BNNO3) are particularly useful catalysts with respect to their high polymerization rate and good livingness (regarding Mn and Đ values).
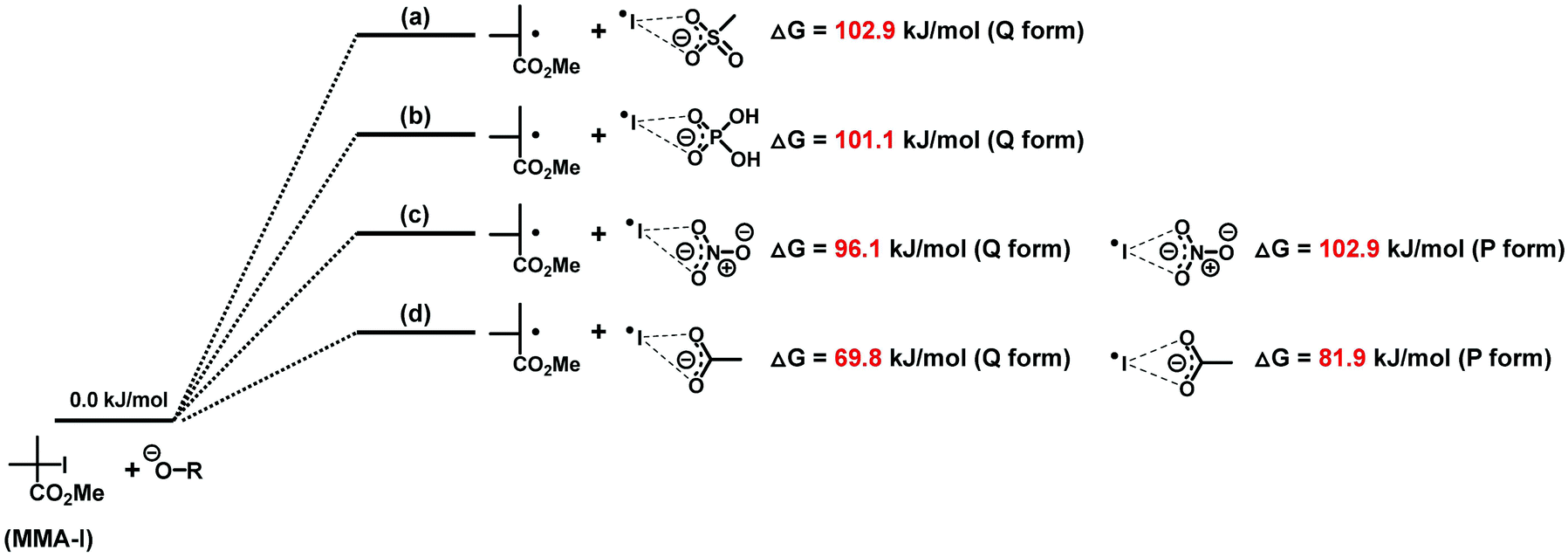
Density functional theory (DFT) calculation was performed to support the experimental results. Methyl 2-iodo-2-methylpropionate (MMA-I (Scheme 2)) was studied as a unimer model of poly(methyl methacrylate)-iodide (PMMA-I). We calculated the Gibbs free energy change (ΔG) from MMA-I and oxyanion (reactants) to the corresponding radicals (products) (Scheme 2). The ΔG value increased in the order of acetate (69.8 kJ mol−1) < nitrate (96.1 kJ mol−1) < dihydrogen phosphate (101.1 kJ mol−1) < methanesulfonate (102.9 kJ mol−1) in the bifurcated asymmetric form (Q form). These ΔG values are smaller than that for the most effective pyridine oxide catalyst (116.1 kJ mol−1) previously reported,27 suggesting the higher catalytic abilities of the studied oxyanions. Acetate has the lowest ΔG value (hence the highest catalytic activity), which is consistent with the strongest basicity among the four types of oxyanions. The ΔG value was relatively small for nitrate. These results support the experimentally observed effectiveness of Na carboxylate (NaOEH) and BN nitrate (BNNO3) as catalysts. Phosphate has a strong basicity (the second strongest basicity among the four oxyanions) but showed the second-largest ΔG value, probably because the basicity is not always proportional to the nucleophilicity that actually influences the halogen-bonding catalysis. The use of methanesulfonate (BNCH3SO3) led to a relatively slow polymerization compared to the other three catalysts (Fig. 4), which is consistent with the calculation result showing the largest ΔG value. Not only the bifurcated asymmetric form (Q form) but also the symmetric form (P form) was observed for acetate (ΔG = 81.9 kJ mol−1) and nitrate (ΔG = 102.9 kJ mol−1). However, the Q form gave smaller ΔG values than the P form, and hence the Q form process is more favourable. The P form was not found for dihydrogen phosphate and methanesulfonate. The mono-coordinate halogen-bonding (R form) of R–O−⋯I˙ was not found in the DFT calculation, as also reported in the literature.33,34
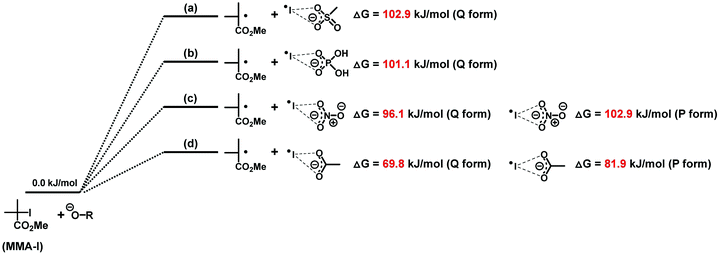 |
| | Scheme 2 Gibbs free energy change in the reaction of MMA-I with (a) methanesulfonate, (b) dihydrogen phosphate, (c) nitrate and (d) acetate. | |
Increase in the polymerization rate
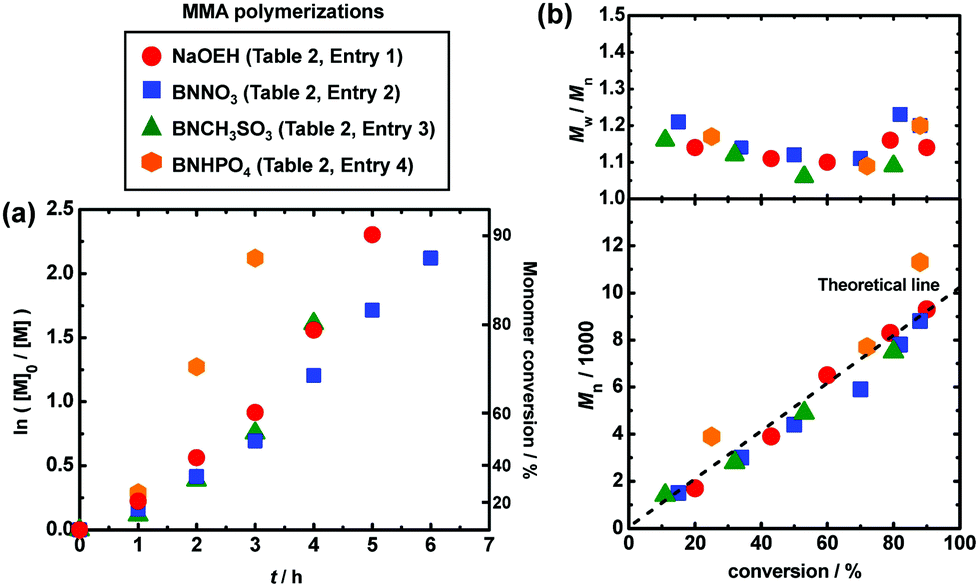
In order to suppress the elimination (side reaction), we decreased the polymerization temperature from 70 °C to 60 °C. The slowed polymerization caused by a decreased temperature was overcome by the addition of a small amount of an azo initiator, i.e., 2,2′-azobis(2,4-dimethylvaleronitrile) (V65). Azo initiators are often used to decrease the deactivator concentration and hence effectively increase the polymerization rate in other living radical polymerizations.35,36 As Fig. 5 and Table 2 (entries 1–4) show, the addition of V65 (10 mM) maintained the high polymerization rate or even increased the polymerization rate at a lower temperature of 60 °C, reaching 80–90% monomer conversions in 3–6 h, for the studied four families of oxyanions (NaOEH, BNNO3, BNCH3SO3 and BNHPO4). Importantly, BNHPO4 afforded a good agreement of Mn with Mn,theo at 60 °C (Fig. 5 (hexagon)), successfully suppressing the elimination that was significant at the higher temperature of 70 °C (Fig. 4 (hexagon)).
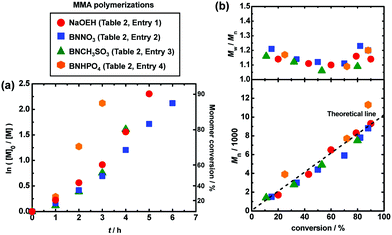 |
| | Fig. 5 Plots of (a) ln([M]0/[M]) vs. t and (b) Mn and Mw/Mnvs. conversion for the MMA/CP-I/catalyst/V65/I2 systems (60 °C): [MMA]0 = 8 M; [CP-I]0 = 80 mM; [catalyst]0 = 80 mM; [V65]0 = 10 mM; [I2]0 = 5 mM. The symbols are indicated in the figure. | |
Table 2 Polymerizations of MMA with V65
| Entry |
Target DPa |
Catalyst |
[MMA]0/[CP-I]0/[catalyst]0/[V65]0/[I2]0 (mM) |
T (°C) |
t (h) |
Conv. (%) |
M
n (Mn,theob) |
Đ
|
|
Target degree of polymerization at 100% monomer conversion (calculated by [MMA]0/[CP-I]0).
Theoretical Mn calculated with [MMA]0, [CP-I]0, and monomer conversion.
The addition of 18-crown-6-ether (80 mM).
|
| 1 |
100 |
NaOEH |
8000/80/80/10/5c |
60 |
5 |
90 |
9300 (9000) |
1.14 |
| 2 |
100 |
BNNO3 |
8000/80/80/10/5 |
60 |
6 |
88 |
8800 (8800) |
1.20 |
| 3 |
100 |
BNCH3SO3 |
8000/80/80/10/5 |
60 |
4 |
80 |
7600 (8000) |
1.09 |
| 4 |
100 |
BNHPO4 |
8000/80/80/10/5 |
60 |
3 |
88 |
11300 (8800) |
1.20 |
| 5 |
100 |
NaFum |
8000/80/40/10/2c |
60 |
4 |
89 |
8400 (8900) |
1.19 |
| 6 |
100 |
TMG |
8000/80/80/10/2 |
60 |
6 |
90 |
9100 (9000) |
1.24 |
| 7 |
100 |
NaDOSO3 |
8000/80/80/10/0c |
60 |
16 |
78 |
8400 (7800) |
1.28 |
Naturally existing oxyanions as catalysts
Naturally existing oxyanions were also used as catalysts for the polymerization of MMA. Sodium fumarate (NaFum) and trimethylglycine (TMG, also known as glycine betaine) are naturally existing and non-toxic carboxylates that are used as food additives.37,38
NaFum showed a relatively slow polymerization rate in the absence of V65 (Table S1, ESI†). With a small amount of V65 (10 mM), NaFum and TMG yielded polymers with high monomer conversions (≥89%) and low Đ values (≤1.24) in 6 h (Table 2 (entries 5 and 6)). Sodium dioctyl sulfosuccinate (NaDOSO3) is a commercial laxative.39 This alkyl sulfonate drug also worked as an efficient catalyst (Table 2 (entry 7)).
Chain-end fidelity
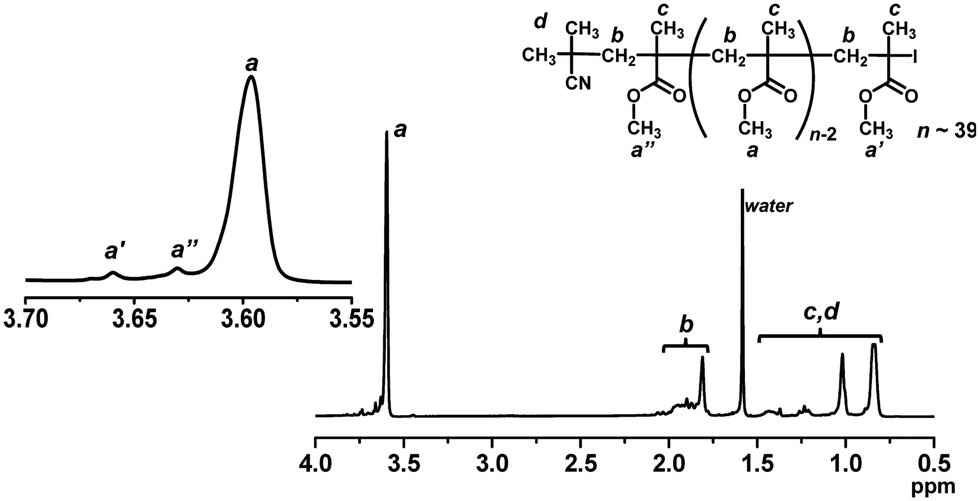
We studied the iodide chain-end fidelity (livingness) of PMMA synthesized with the NaOEH catalyst. PMMA obtained after 2 h (monomer conversion = 33%) in Fig. 3 (open square) was purified through reprecipitation from hexane (non-solvent) and subsequently by preparative GPC (Mn = 4000 and Đ = 1.14 after purification) and was analyzed by high-resolution 1H NMR (500 MHz) (Fig. 6). The signals of the methoxyl protons (a, a′, and a′′) at the side chain appeared at 3.58–3.67 ppm. The main peak at 3.58–3.62 ppm was assigned to the repeating MMA units (a) in the middle of the chain. The downfield-shifted peak at 3.65–3.67 ppm was assigned to the ω-terminal chain-end unit (a′) adjacent to the chain-end iodide. The peak at 3.62–3.65 ppm was assigned to the α-terminal chain-end unit (a′′ adjacent to the initiating 2-methylpropionitrile (CP) group). From the 1H NMR peak area and the Mn value (= 4000) determined by GPC, the fraction of the iodine chain end was calculated to be 94%, meaning a high iodide-chain-end fidelity. A small amount of dead polymer should be generated via radical–radical termination, because this polymerization is a radical polymerization. A minor elimination may also occur, as observed in the radical trap experiment (3% elimination product at 70 °C for 8 h). This result clearly shows the good livingness (high iodide-chain-end fidelity) with the NaOEH catalyst. The obtained (purified) PMMA was used as a macroinitiator in the block copolymerizations shown below.
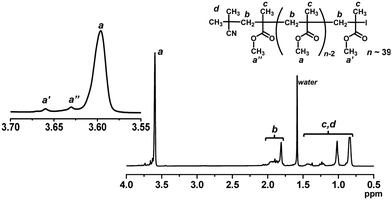 |
| | Fig. 6
1H NMR spectrum (CDCl3) of PMMA-I obtained with NaOEH (Fig. 3 (open square) for 2 h) (Mn = 4000 and Đ = 1.14 after purification). | |
Polymerizations of functional methacrylates, styrene and acrylonitrile
The monomer scope encompassed functional methacrylates, styrene, and acrylonitrile. Table 3 (entries 1–8) summarizes the polymerizations of butyl methacrylate (BMA), benzyl methacrylate (BzMA), 2-methoxyethyl methacrylate (MEMA), 2-(dimethylamino)ethyl methacrylate (DMAEMA), di(ethylene glycol) methyl ether methacrylate (DEGMA), and poly(ethylene glycol)methyl ether methacrylate (PEGMA) using the NaOEH, NaFum and TMG carboxylate catalysts. Low-dispersity (Đ = 1.14–1.24) polymers were obtained for the hydrophobic BMA, BzMA, and MEMA monomers with 70–80% monomer conversions at 70 °C for 4–12 h. A lower polymerization temperature (50 °C) was used for the hydrophilic DMAEMA, DEGMA and PEGMA monomers in order to suppress the HI elimination from the polymer chain end. The HI elimination is promoted under polar conditions. For DMAEMA, DEGMA and PEGMA, V65 was added to increase the polymerization rate at the lower temperature (50 °C), yielding low-dispersity (Đ = 1.28–1.40) polymers. Poly(di(ethylene glycol) methyl ether methacrylate) and poly(poly(ethylene glycol)methyl ether methacrylate) are biocompatible and may be used in biomedical materials.40 The use of naturally existing non-toxic NaFum and TMG as the polymerization catalysts offers an attractive route to synthesize well-defined biomedically useful polymers without the remaining toxic catalyst residues. Besides methacrylates, styrene and acrylonitrile were also successfully used. Table 3 (entries 9–14) shows the polymerizations of styrene and acrylonitrile using NaOEH (carboxylate), BNNO3 (nitrate), and BNCH3SO3 (sulfonate) as catalysts, yielding low-dispersity (Đ = 1.31–1.41) polymers. These results demonstrate that the oxyanion catalysts are amenable for various monomer families and functional monomers.
Table 3 Polymerizations of methacrylates, styrene, and acrylonitrile with oxyanion catalysts
| Entry |
Monomer |
Catalyst |
Azo initiator |
Solvent |
[Monomer]0/[CP-I]0/[catalyst]0/[Azo initiator]0 (mM) |
T (°C) |
t (h) |
Conv. (%) |
M
na (Mn,theob) |
Đ
|
|
PMMA-calibrated THF-GPC values for entries 1–3. Polystyrene-calibrated THF-GPC values for entries 9–11. PMMA-calibrated DMF-GPC values for entries 4–8 and 12–14.
Theoretical Mn calculated with [monomer]0, [CP-I]0, and monomer conversion.
The addition of diglyme (25 wt%). NaOEH was dissolved in this solution polymerization.
The addition of ethylene carbonate (EC) (50 wt%). NaOEH was dissolved in this solution polymerization (entry 12).
The addition of 18-crown-6-ether (80 mM).
The addition of I2 (2 mM).
|
| 1 |
BMA |
NaOEH |
None |
None |
8000/80/80/0e,f |
70 |
12 |
80 |
15000 (14000) |
1.14 |
| 2 |
BzMA |
NaOEH |
None |
None |
8000/80/80/0e |
70 |
8 |
70 |
14000 (12000) |
1.24 |
| 3 |
MEMA |
NaOEH |
None |
None |
8000/80/80/0e |
70 |
4 |
75 |
15000 (12000) |
1.24 |
| 4 |
DMAEMA |
NaOEH |
V65 |
None |
8000/80/80/20e |
50 |
5 |
55 |
8100 (8600) |
1.34 |
| 5 |
DEGMA |
NaFum |
V65 |
None |
8000/80/40/40e,f |
50 |
4 |
97 |
17000 (18000) |
1.39 |
| 6 |
DEGMA |
TMG |
V65 |
None |
8000/80/40/40f |
50 |
4 |
81 |
15000 (15000) |
1.34 |
| 7 |
PEGMA |
NaOEH |
V65 |
None |
8000/80/80/10e |
50 |
8 |
60 |
24000 (18000) |
1.40 |
| 8 |
PEGMA |
NaFum |
V65 |
None |
8000/80/40/40e,f |
50 |
4 |
67 |
17000 (20000) |
1.28 |
| 9 |
Styrene |
NaOEH |
AIBN |
Diglymec |
8000/80/80/30 |
80 |
7 |
70 |
7000 (7300) |
1.41 |
| 10 |
Styrene |
BNNO3 |
AIBN |
None |
8000/80/80/30 |
80 |
4 |
80 |
8400 (8300) |
1.37 |
| 11 |
Styrene |
BNCH3SO3 |
AIBN |
None |
8000/80/80/40 |
80 |
5 |
89 |
8700 (9200) |
1.31 |
| 12 |
Acrylonitrile |
NaOEH |
AIBN |
ECd |
8000/80/160/10 |
75 |
4 |
54 |
5000 (3000) |
1.40 |
| 13 |
Acrylonitrile |
BNNO3 |
AIBN |
ECd |
8000/80/160/10 |
75 |
2 |
57 |
8000 (3500) |
1.34 |
| 14 |
Acrylonitrile |
BNCH3SO3 |
AIBN |
ECd |
8000/80/160/10 |
75 |
3 |
90 |
12000 (4800) |
1.39 |
Block copolymerization
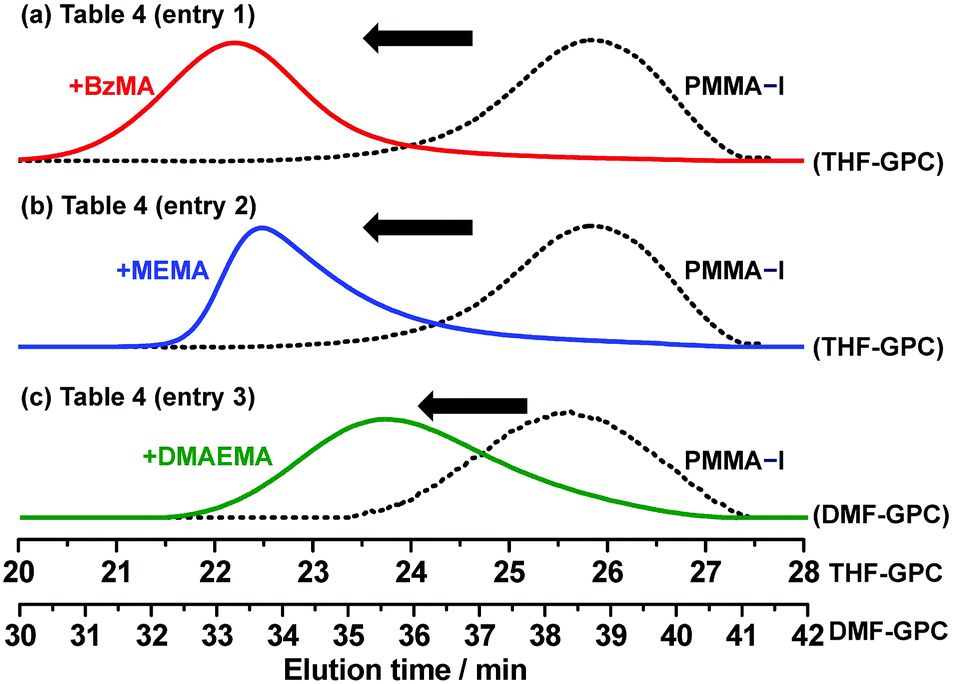
Exploiting the living character, block polymerizations were performed using the NaOEH catalyst. We used the purified PMMA-iodide (PMMA-I) (Mn = 4000 and Đ = 1.14) shown in Fig. 6 as a macroinitiator. Diglyme was used as the solvent to dissolve NaOEH. The block polymerizations with BzMA, MEMA, and DMAEMA gave well-defined hydrophobic–hydrophobic and hydrophobic–hydrophilic block copolymers with low polydispersity (Mn = 11000–19000 and Đ = 1.37–1.42) (Table 4). Fig. 7 shows the GPC chromatograms before and after the block polymerization. In all cases, a large fraction of the macroinitiator extended to the block copolymers, demonstrating the high block efficiency. The results show the accessibility for various block copolymers.
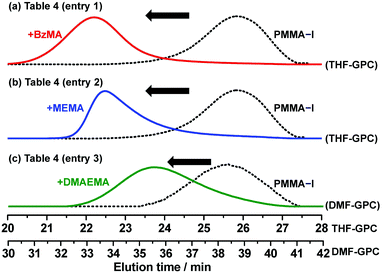 |
| | Fig. 7 GPC chromatograms before (dashed lines) and after (solid lines) the block polymerizations of (a) BzMA, (b) MEMA, and (c) DMAEMA from PMMA-I macroinitiator. The polymerization conditions are shown in Table 4 (entries 1–3). | |
Table 4 Block polymerizations from the PMMA-I macroinitiator (Mn = 4000 and Đ = 1.14)
| Entry |
Monomer |
Target DP |
[Monomer]0/[PMMA-I]0/[NaOEH]0/[V65]0a (mM) |
T (°C) |
t (h) |
Conv. (%)b |
M
nc (Mn,theo)d |
Đ
|
|
The addition of diglyme (25 wt% of diglyme and totally 75% of monomer and PMMA-I).
Calculated by 1H NMR.
PMMA-calibrated THF-GPC values for entries 1 and 2. PMMA-calibrated DMF-GPC values for entry 3.
Theoretical Mn calculated with [monomer]0, [PMMA-I]0, and monomer conversion.
|
| 1 |
BzMA |
100 |
8000/80/80/0 |
70 |
5 |
80 |
19000 (18000) |
1.37 |
| 2 |
MEMA |
100 |
8000/80/80/0 |
70 |
5 |
60 |
13000 (13000) |
1.40 |
| 3 |
DMAEMA |
100 |
8000/80/80/20 |
50 |
6 |
50 |
11000 (12000) |
1.42 |
Conclusions
The four families of oxyanions (carboxylate, nitrate, sulfonate, and phosphate) were effective for halogen bonding catalysis and induced polymerization. Among the studied catalysts, sodium carboxylate (NaOEH) and BN nitrate (BNNO3) are particularly effective with negligible side reactions (elimination), maintaining good livingness. Naturally existing non-toxic sodium fumarate (NaFum) and trimethylglycine (TMG) also successfully worked as catalysts. These catalysts were amenable for MMA, functional methacrylates, styrene, and acrylonitrile, and also successfully yielded block copolymers. The four families of oxyanions are abundant in natural and synthetic compounds. The low cost, low toxicity, and accessibility for broad molecular structures are attractive features for practical use and future catalyst development.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation (NRF) Investigatorship in Singapore (NRF-NRFI05-2019-0001) and the Academic Research Fund (AcRF) Tier 2 from the Ministry of Education in Singapore (MOE2017-T2-1-018).
Notes and references
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- J.-C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- Y. Ni, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2019, 10, 2504–2515 RSC.
- A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsujii and H. Kaji, Macromolecules, 2011, 44, 8709–8715 CrossRef CAS.
- A. Goto, A. Ohtsuki, H. Ohfuji, M. Tanishima and H. Kaji, J. Am. Chem. Soc., 2013, 135, 11131–11139 CrossRef CAS.
- A. Ohtsuki, L. Lei, M. Tanishima, A. Goto and H. Kaji, J. Am. Chem. Soc., 2015, 137, 5610–5617 CrossRef CAS.
- C.-G. Wang and A. Goto, J. Am. Chem. Soc., 2017, 139, 10551–10560 CrossRef CAS.
- C.-G. Wang, F. Hanindita and A. Goto, ACS Macro Lett., 2018, 7, 263–268 CrossRef CAS.
- J. Zheng, C.-G. Wang, Y. Yamaguchi, M. Miyamoto and A. Goto, Angew. Chem., 2018, 130, 1568–1572 CrossRef.
- C.-G. Wang, C. Chen, K. Sakakibara, Y. Tsujii and A. Goto, Angew. Chem., Int. Ed., 2018, 57, 13504–13508 CrossRef CAS.
- X. Liu, C.-G. Wang and A. Goto, Angew. Chem., Int. Ed., 2019, 58, 5598–5603 CrossRef CAS.
- C.-G. Wang, X. Y. Oh, X. Liu and A. Goto, Macromolecules, 2019, 52, 2712–2718 CrossRef CAS.
- H. Xu, C.-G. Wang, Y. Lu and A. Goto, Macromolecules, 2019, 52, 2156–2163 CrossRef CAS.
- S. D. Gangolli, P. A. van den Brandt, V. J. Feron, C. Janzowsky, J. H. Koeman, G. J. A. Speijers, B. Spiegelhalder, R. Walker and J. S. Wishnok, Eur. J. Pharmacol., Environ. Toxicol. Pharmacol. Sect., 1994, 292, 1–38 CrossRef CAS.
- K. G. Raghothama, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1999, 50, 665–693 CrossRef CAS.
- A. Czajka, G. Hazell and J. Eastoe, Langmuir, 2015, 31, 8205–8217 CrossRef CAS.
- J. Wang, J. Han, H. Peng, X. Tang, J. Zhu, R.-Z. Liao, X. Xie, Z. Xue, C. Fliedel and R. Poli, Polym. Chem., 2019, 10, 2376–2386 RSC . The data in Table 1 (entries 20–34) in this reference are related to oxyanion (oxygen-centered anion) catalysts.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
- S. Saha, S. Ganguly and G. R. Desiraju, Aust. J. Chem., 2014, 67, 1840–1848 CrossRef CAS.
- S. Tothadi, P. Sanphui and G. R. Desiraju, Cryst. Growth Des., 2014, 14, 5293–5302 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- R. K. Das, S. K. Brar and M. Verma, Pharmacol. Rep., 2016, 68, 404–414 CrossRef CAS.
- F. J. de Zwart, S. Slow, R. J. Payne, M. Lever, P. M. George, J. A. Gerrard and S. T. Chambers, Food Chem., 2003, 83, 197–204 CrossRef CAS.
- M. Donowitz and H. J. Binder, Gastroenterology, 1975, 69, 941–950 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, measurement, experimental procedures, radical trapping experiments, polymerization results, and DFT calculation. See DOI: 10.1039/c9py01533g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Chen-Gang
Wang‡
,
Chen-Gang
Wang‡