
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The generation of stabilized supramolecular nanorods from star-shaped polyglutamates†
O.
Zagorodko
 ,
V. J.
Nebot‡
* and
M. J.
Vicent
*
,
V. J.
Nebot‡
* and
M. J.
Vicent
*
Polymer Therapeutics Lab. Prince Felipe Research Center, Av. Eduardo Primo Yúfera 3, 46012 Valencia, Spain. E-mail: vnebot@pts-polypeptides.com; mjvicent@cipf.es
First published on 8th January 2020
Abstract
We developed a new strategy of polyglutamate nanorod preparation based on supramolecular polymers stabilized with hydrophobic drugs. Using this strategy, we prepared a family of star-shaped polyglutamates (star-PGAs) with benzenetricarboxamide (BTA)-based cores of different hydrophobicity. We then studied the self-assembly of the resulting polymers in aqueous solutions containing a physiological level of salt using fluorescence spectroscopy, small-angle X-ray scattering (SAXS), and transmission electron microscopy (TEM). We discovered that star-PGAs behave as classical polyelectrolytes in very dilute solutions; however, compounds with hydrophobic cores assembled into one dimensional-nanorods upon an increase in concentration due to supramolecular interactions in the core. Small hydrophobic drugs, such as doxorubicin and irinotecan, stabilized the nanorods and inhibited their disassembly at concentrations below the critical aggregation concentration (CAC). We anticipate that this simple nanorod preparation strategy from star-PGAs will enable the development of new nanomedicines with unique biodistribution profiles and biological activity.
Introduction
Self-assembly is a unique yet ubiquitous phenomenon found in nature that is widely used in applied sciences.1 Biological macromolecules can self-organize into a plethora of intricate structures with specific functionalities emerging from the supramolecular complexes rather than the individual components. The propensity to mimic the complexity of biological systems has stimulated a focus on the study of synthetic polymers and their aqueous self-assembly.2–4 Synthetic polypeptides, or polyaminoacids, are the class of polymers most closely related to biomacromolecules, consisting of the same building blocks as natural proteins yet keeping a high degree of structural versatility. These characteristics make polypeptides perfectly suitable for advanced drug delivery.5–8 Among the described biocompatible polypeptides, poly-L-glutamates (PGA) display low immunogenicity, high biodegradability, and controlled biodistribution profiles, making them a focus for research.9,10Many studies have demonstrated that physicochemical properties strongly influence the in vivo behavior of particles.8 The role of charge, size, and surface decoration have been described in detail; however, the role of shape has remained mostly unstudied. Recently, Zhao et al. demonstrated that mesoporous silica rods exhibit longer circulation times and higher bioavailability than spherical analogs.11 Van Pomeren et al. also established that the shape of gold nanoparticles determined biodistribution in zebrafish embryos.12 Aqueous shaping of soft-matter nanoparticles and polyelectrolytes, in particular, remains a complicated issue due to multiple repulsive interactions, though non-spherical polyelectrolyte particles can open new avenues in drug delivery studies.
In search of new morphologies, many studies have incorporated different structural elements, such as hydrophobic blocks, into the polymer to generate unique self-assembly properties.13–15 One especially interesting strategy is the incorporation of supramolecular motifs that allow hierarchical levels of self-assembly. Benzenetricarboxamide (BTA) is one of the most well-studied motifs employed for aqueous supramolecular polymerization.16,17 Following the covalent attachment of sufficient hydrophobic domains to BTA and further shielding with hydrophilic groups, the obtained molecules assemble in water into fibers or rods due to directional non-covalent interactions. While vast numbers of studies of different BTA-based molecules (including polypept(o)ids) have been carried out, the majority of the molecules able to self-assemble were uncharged.18 Some studies attempted to combine BTA-cores with poly-amino acid derivatives; however, BTA played the role of a multifunctional core rather than supramolecular motif in all cases.18–20 Besenius pioneered a method of charge screening for small BTA-based molecules bearing ionizable groups with low molecular weight salts, achieving control for up to nine charges for Newkome dendron-decorated molecules.21 We hypothesized that replacing Newkome dendrons with small PGA chains should allow the formation of stable structures that can self-assemble in water and other physiological solutions.
BTA-based supramolecular polymers, as well as polyelectrolyte rod-like or worm-like micelles, suffer from disassembly upon dilution that significantly limits their applicability in drug delivery. Micelles require crosslinking for system stabilization, and hydrophobic drugs, such as paclitaxel (PTX) or doxorubicin (Dox), have been used to non-covalently stabilize micelles.22 BTA-based supramolecular polymers usually lack functional groups, and to the best of our knowledge, their crosslinking or stabilization has yet to be reported.
In this study, we synthesized the first compound from the class of supramolecular PGAs based on BTA cores (Fig. 1A) that self-assembles into one dimensional (1D) aggregates both in pure water and in the presence of salts and bears a sufficient amount of charged functional groups for further modification. In the second part of this study, we developed a method of supramolecular stabilization of rod-like aggregates with flat hydrophobic drugs, such as Dox and irinotecan (Iri) (which play a dual role as both active agent and particle stabilizer). The obtained particles display stability upon dilution for at least 48 hours.
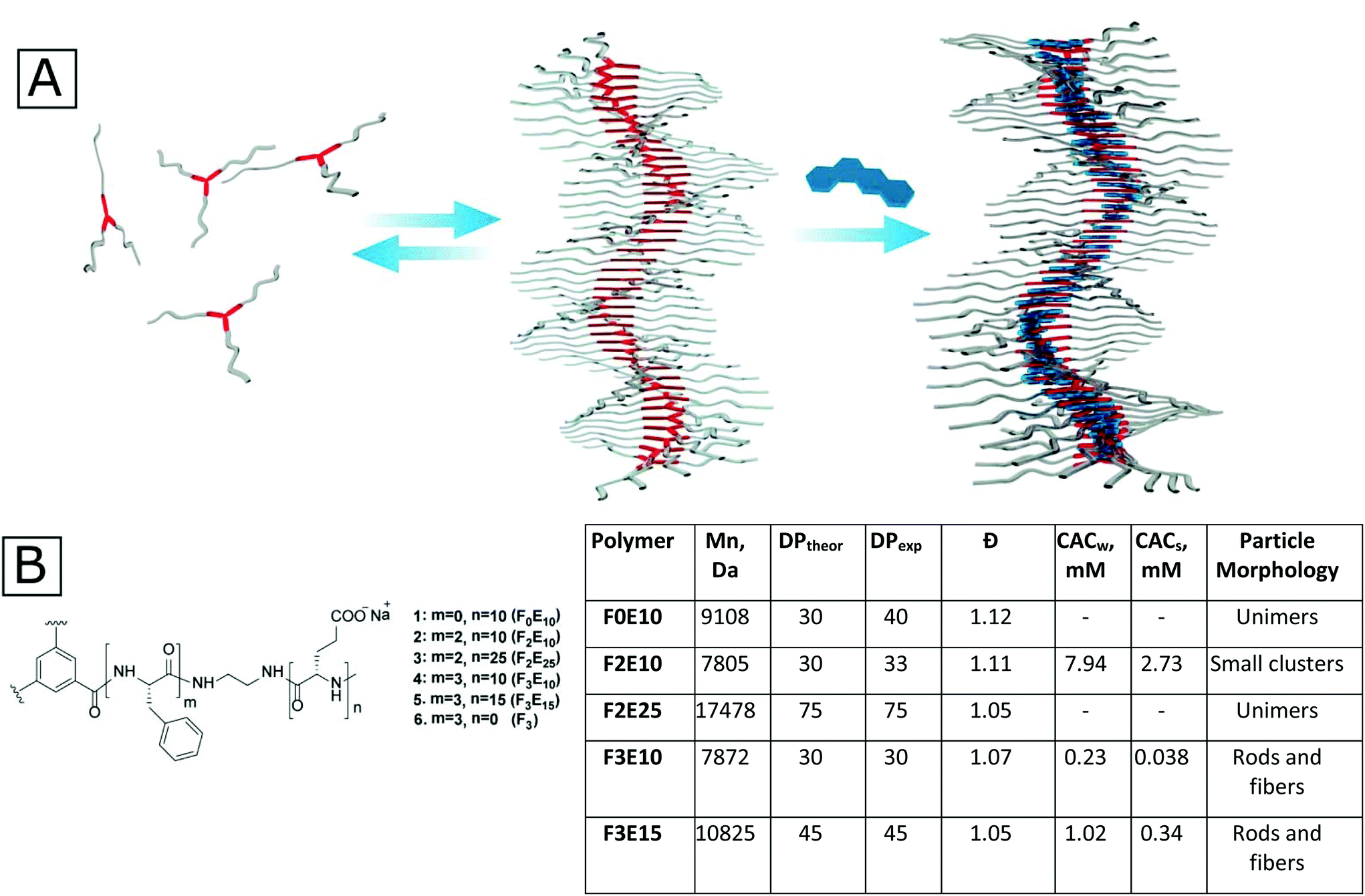 | ||
| Fig. 1 (A) Suggested general scheme of self-assembling benzenetricarboxamide (BTA)-based polyglutamates (PGAs) and their further stabilization with flat hydrophobic drugs. (B) General structure of the polymers used in this study and their main physico-chemical characteristics. Mn, DPexp, and polydispersity (Đ) determined by GPC for benzyl protected polymers. CAC determined by fluorimetry. | ||
Results and discussion
As a basis for our star-PGAs, we chose previously described hexa- and nona-phenylalanine-BTA modified with small ethylenediamine spacer that was used as an initiator to prepare short PGAs with 10–25 glutamic units per arm (total 30–75 units per molecule). We prepared an initiator for star-PGAs bearing no hydrophobic core, as previously described by Duro-Castano et al.23 Therefore, by employing ring-opening polymerization of N-carboxyanhydrides (NCA-ROP) of γ-benzyl glutamic acid with further deprotection, we synthesized water-soluble star-PGAs (F0E10, F2E10–25, and F3E10–15) as sodium salts (Fig. 1B) (see ESI for synthetic details and Scheme 1S†). NMR analyses allowed us to assess identity and appropriate purity of the polymers, confirming the experimental degree of polymerization in good agreement with theoretical values and in line with the molecular weight determined via GPC that confirmed very low Đ for the obtained polymers (1.06–1.12) (Fig. 1C, Fig. S1†).BTA-based molecules assemble into 1D supramolecular polymers through the formation of a directional C3-symmetric network of non-covalent bonds.17 In aqueous solutions, this process is coupled with the formation of a hydrophobic microenvironment that can be probed with solvatochromic dyes such as Nile Red (NR). According to previous reports, NR in water displays emission at 654 nm with very low fluorescence intensity irrespective of the salt concentration. In the presence of hydrophobic domains in solution, NR changes its conformation accompanied by an increase in intensity and a hypsochromic shift.24 As a first step, we performed fluorimetry experiments to determine the critical aggregation concentration (CAC) of F2E10, F3E10, and F3E15 in pure water and 120 mM NaCl solution (Fig. 1B and Fig. 2A). We performed all measurements at pH 7.4, adjusted, if necessary, with 0.1 M NaOH. We observed a substantial increase in NR peak intensity for all three compounds and the hypsochromic shift of the peak maximum to 633, 630, and 629–630 nm for F2E10, F3E15, and F3E10, respectively, confirming the self-assembly of the compounds with the formation of hydrophobic microenvironments. F3E10 displayed a CAC 30-times lower than F2E10 and five-times lower than F3E15 (Fig. 1B), which emphasizes the significantly stronger tendency of nona-phenylalanine-based cores to self-assemble. The hypsochromic shift of NR directly correlates with the hydrophobicity of the environment; therefore, we conclude that compound F3E10, which possesses a more hydrophobic core and a lower polymerization degree, not only aggregates more strongly but also contains an altered inner structure of the self-assembled particles. In a 120 mM solution of NaCl, we observed a similar trend for self-assembly, with CAC values decreasing for all compounds by three- to six-times due to the partial screening of Coulomb repulsions. The CAC value of F3E10 decreased the most, as this compound exhibits a lower spatial charge density than F2E10 and fewer negative charges than F3E15.
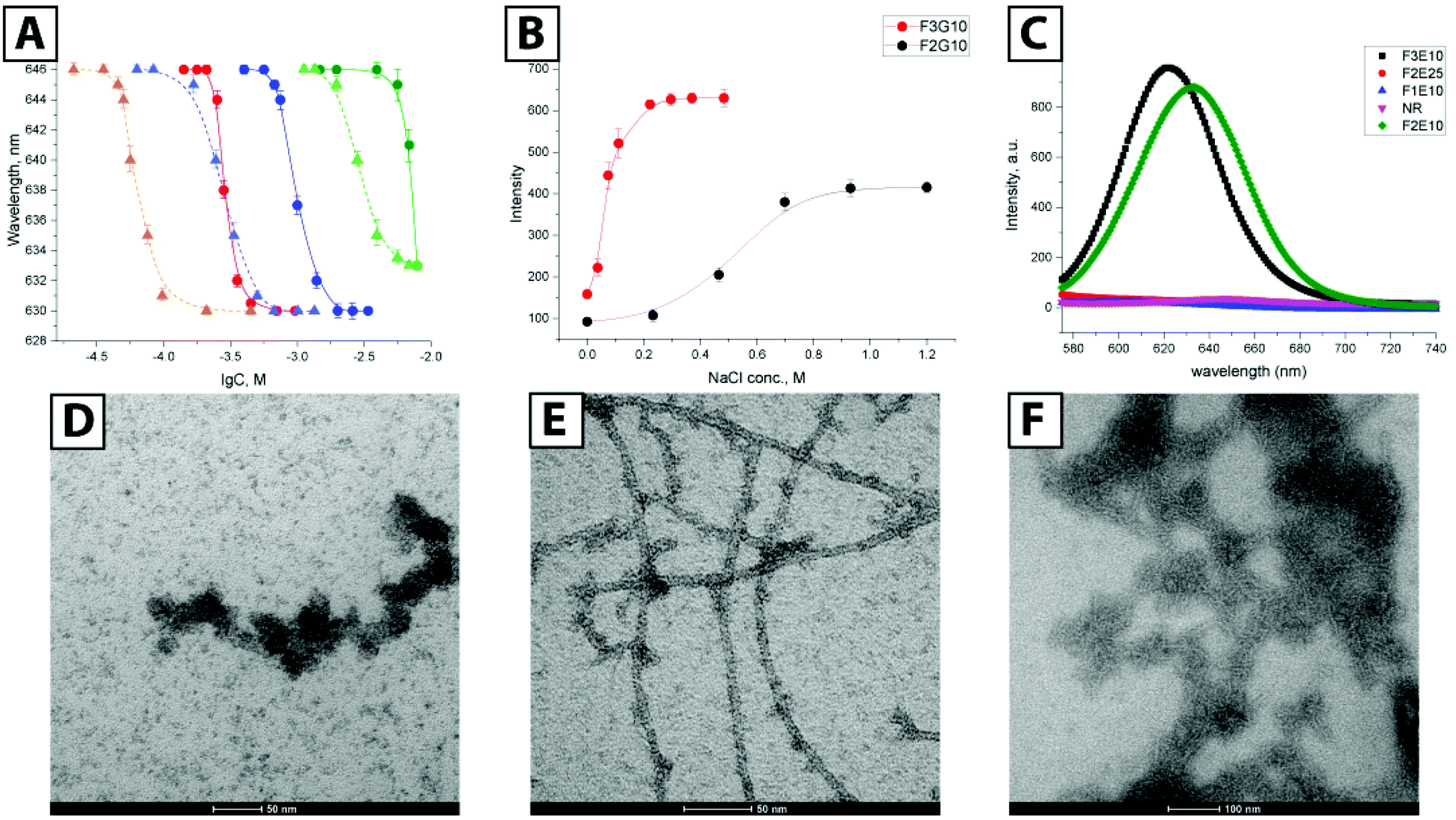 | ||
| Fig. 2 (A) Dependence of NR wavelength emission maximum on the concentration of F3E10, F3E15, and F2E10 in water (red, blue, and green circles, respectively) and 120 mM NaCl (orange, blue, and green triangles, respectively). (B) Fluorescent salt titration of F3E10 and F2E10 at 0.2 mM at pH 7.4, data as mean ± SD (n = 3). (C) Fluorescence spectra for F3E10, F2E25, F0E10, F2E10, and Nile Red (NR). Transmission electron microscopy images of F3E10 in water at pH 7.4 at 1.0 mM, stained with uranyl acetate (D), at 3.5 mM stained with uranyl acetate (E), and 1.0 mM stained with phosphotungstic acid (F). | ||
We further demonstrated this effect with an ionic strength variation experiment (Fig. 2B). Keeping concentrations of F2E10 and F3E10 at 0.2 mM in water at pH 7.4 and varying salt concentration from 0 to 1.2 M, we determined CAC values of 0.73 M and 0.068 M, respectively. F3E10 reached an intensity plateau at much lower salt concentration and higher intensity value that F2E10; in fact, complete charge screening can be reached in solutions with high salt concentration, which forces compounds to self-assembly even at lower concentrations. To confirm that the NR signal does not arise from alternative mechanisms, we synthesized compounds F0E10 and F2E25. Control compound F0E10 is a star-PGA without a hydrophobic core that we used to demonstrate that specific properties of supramolecular PGAs do not arise from topological or polyelectrolyte issues. Compound F2E25 contains a hydrophobic core with two phenylalanines and 25 glutamic acid units per arm, that we used to eliminate the possibility of assembly due to the flexibility of the core and formation of hydrophobic domains within a single molecule. As expected, NR fluorescence intensity quenched rapidly to zero in both water and salt solution in the solutions of F0E10 and F2E25 irrespective of concentration (up to 70 mg ml−1) and ionic strength (from 5 mM to 1 M), thus excluding the alternative mechanisms (Fig. 2C).
Next, we performed transmission electron microscopy (TEM) studies of compound F3E10 at concentrations of 1.0 and 3.5 mM with uranyl acetate staining. At concentrations above the CAC (1.0 mM), we found short flexible nanofilaments with a diameter of around 15 nm and a length varying from 40 to 150 nm (Fig. 2D). Finally, at high concentrations (3.5 mM), we observed very long fibers with lengths up to micrometers (Fig. 2E).
When we evaluated phosphotungstic acid staining, we observed straight nanorods; however, the contrast of the images was very low (Fig. 2F). We performed Cryo-TEM for solutions at near the CAC value to confirm particle shape. We observed nanorods with a length from 50 to 150 nm and a diameter of around 7 nm (Fig. 3A). In contrast with TEM with uranyl staining where particles appeared as flexible filaments, cryo-TEM images showed rod-like particles, without apparent bending. The appearance of artifacts with uranyl acetate may be explained by the fact that, at concentrations around the CAC, only a portion of the molecules in solution become aggregated into rods while the majority of molecules exist as unimers. Uranyl acetate interacts with nanorods and, in parallel, complexate the surrounding unimers, thus forming thick, seemingly flexible aggregates. A supporting argument for this assumption is the presence of more defined long fibers at concentrations above the CAC, where the majority of the molecules becomes aggregated, leaving only minor artifacts visible (Fig. 2E). Even though not directly suitable for the study of polyanions like F3E10 at concentrations around the CAC, uranyl staining can be useful for systems with a high degree of aggregation.
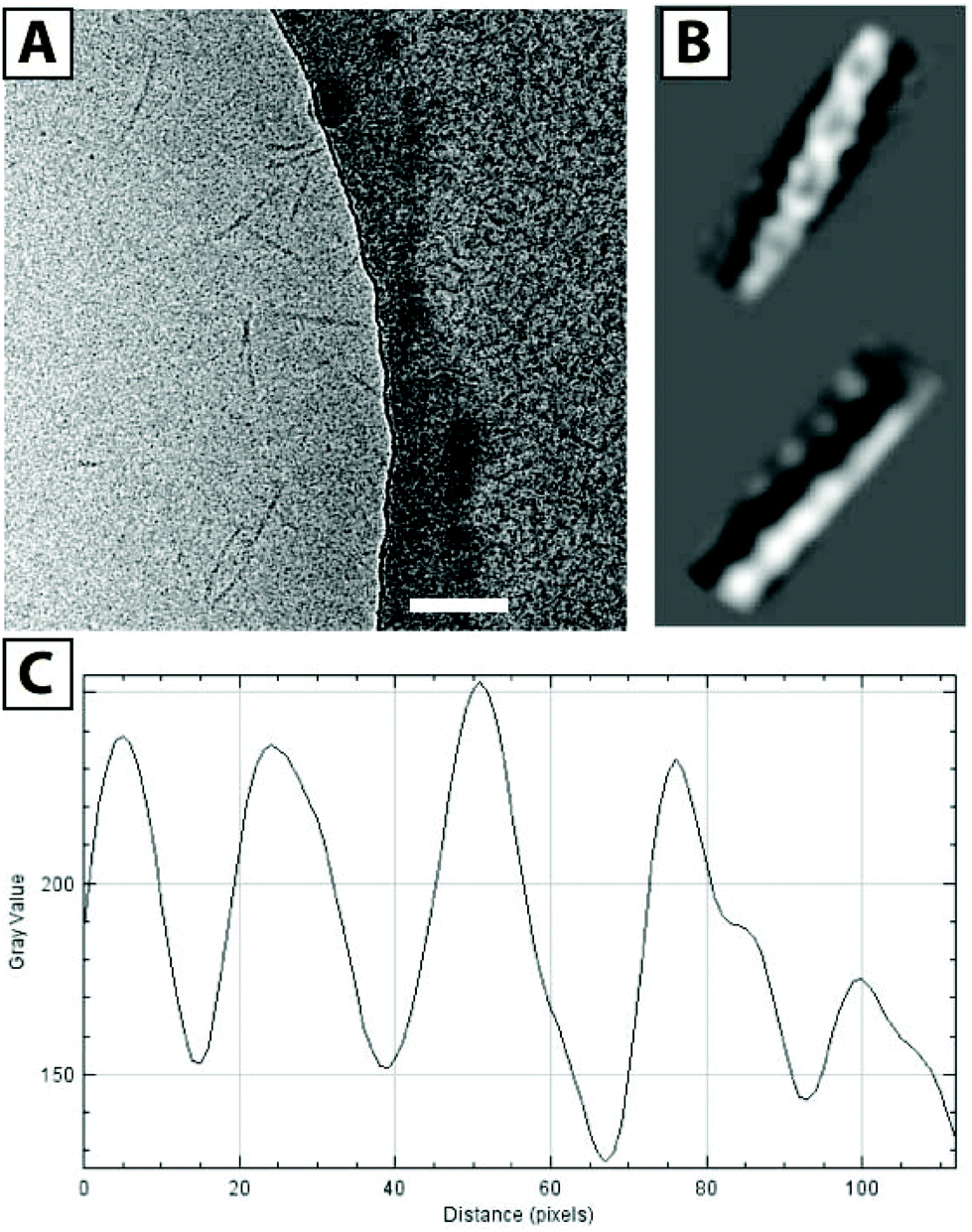 | ||
| Fig. 3 Cryo-TEM image of F3E10 at 0.14 mM in 120 mM NaCl (A, scale bar 100 nm), reconstructed images (B), and corresponding plot profile (C). | ||
We also acquired cryo-TEM images of F3E10 at 0.14 mM in 120 mM NaCl (Fig. 3A), and their reconstruction demonstrated a periodic structure for the nanorods (Fig. 3B). Fascinatingly, these images resembled a double helix, a concept lacking an adequate explanation within the framework of what we currently know regarding the self-assembly of BTA-based molecules. This periodic structure may arise from unusual self-assembly mechanisms involving interactions of several fibers and the assembly of individual fibers in a spiral manner. The distance between the two groove maxima in nanorods is 4.6–4.9 nm (Fig. 3C), the diameter of nanorods is around 5 nm, and the maximum distance between the centers of the fibers is 2.8 nm. PGA chains in 0.12 M NaCl display partial collapse, and the diameter of individual fibers is smaller than the theoretically calculated diameter of the molecule with fully extended chains. To better understand this unexpected twisted architecture, we conducted deeper fluorescence and small-angle X-ray scattering (SAXS) experiments.
Of note, we failed to observe any aggregated or assembled particles for F2E10, even in solutions with concentration far above the CAC. Considering the lower hypsochromic shift obtained for F2E10, we hypothesize that this compound forms tiny aggregates incorporating only a few molecules.
We performed SAXS experiments to determine the structure of nanorods; at low concentration, all compounds behaved as typical polyelectrolyte solutions displaying practically identical scattering curves at a concentration of 0.5 mM in pure water (Fig. 4A). These curves fit well with regards to the polyelectrolyte model.25 Such SAXS profiles are typical for solutions of charged particles, with a dip at low q-values and curve bending in medium q-range, which varies slightly with molecular weight and concentration. For similar 0.5 mM solutions containing 120 mM NaCl, we observed almost identical curves for both F0E10 and F2E10, providing scattering profiles typical for unimolecular solutions (Fig. 4B). Both compounds behave as dilute polyelectrolyte solutions with repulsive interactions screened by salt ions, resulting in the linear range in low q-values.26 We found a completely different curve for F3E10 characterized by a power law −1 dependence, typical for cylindrical objects.
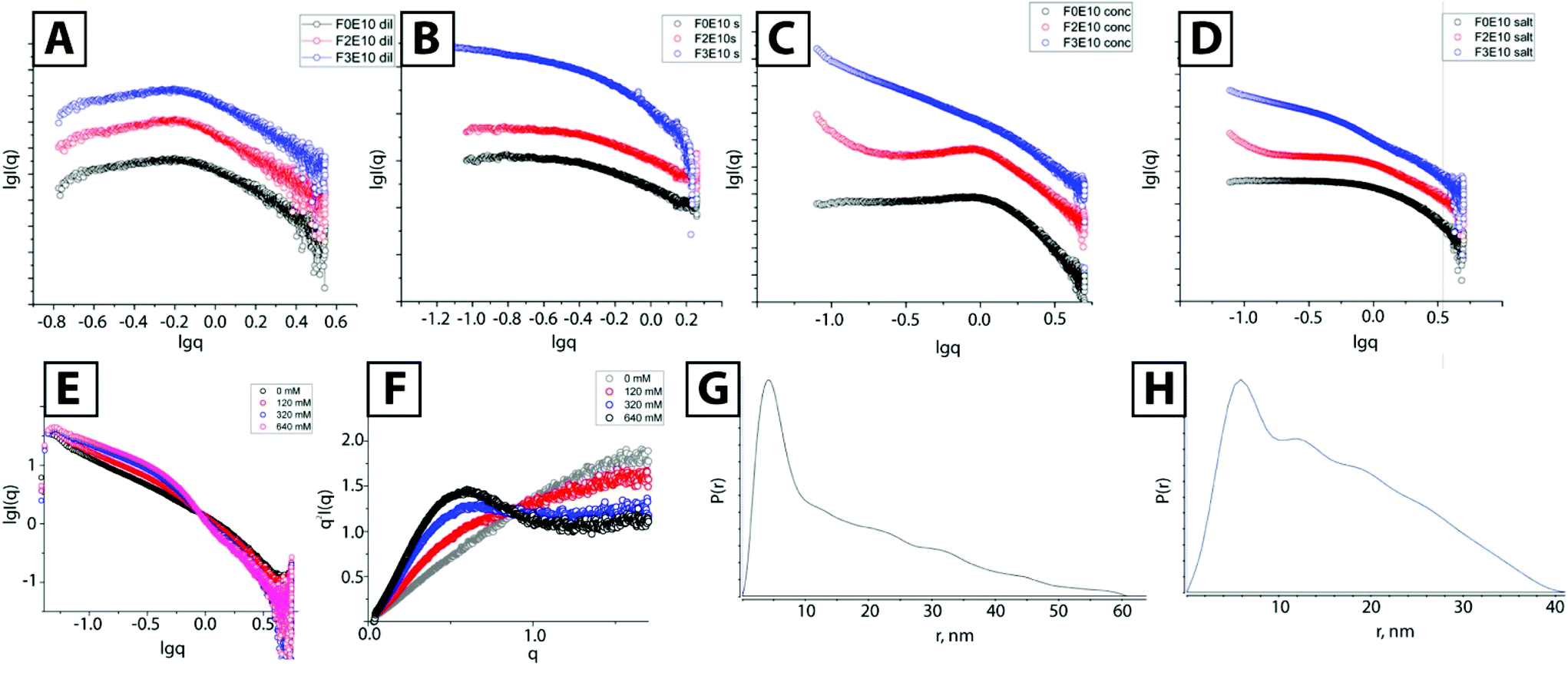 | ||
| Fig. 4 SAXS profiles of F0E10, F2E10, and F3E10 at 0.5 mM and 3.5 mM in water (A and C) and 120 mM NaCl (B and D, respectively). SAXS curves (E) and Kratky plots (F) for F3E10 at 3.5 mM and 0–640 mM NaCl concentration. P(r)-distribution function for F3E10 (G) and F3 (H). | ||
We also performed SAXS studies at higher concentrations to study the possible self-assembly of F2E10 and confirm the formation of long fibers for F3E10 (concentration 3.5 mM, which is below the CAC for F2E10 but far above the CAC for F3E10 in water and 120 mM salt solution). At higher concentrations in solutions of varying concentrations of salt (from 0 to 640 mM), we discovered substantially different SAXS curves for compounds F3E10, F0E10, and F2E10. In pure water (Fig. 4C), differences in the appearance of the curves were already apparent. We found a less pronounced dip in the low q-region of the curve for F0E10 than for diluted compound, and peak maximum shifted to the higher q-value.
This alteration occurs as charged particles at higher concentrations remain at smaller distances between each other in solution. The curves acquired for F2E10 appeared similar to the curves for F0E10 in water, with the uphill section in the low q-range shifted to q = 1 nm−1. In 120 mM NaCl solution, we observed similar profiles for F0E10 and F2E10 in high and medium q-range with a power law dependence of 0, corresponding to a unimolecular solution. Small uphill sections at low q-values on the curve corresponding to F2E10 may be related to a clustering effect. Curves obtained for compound F3E10 in the presence and the absence of salt possess −1 power law dependence in low to medium q-range, which corresponds to cylindrical particles, and −4 at high q-range, which corresponds to the scattering of individual molecules. At 120 mM salt solution, the curve possesses a more defined S-shape that can be explained in terms of overlapping the two curves – polyelectrolyte repulsive scattering and rod scattering (Fig. 4D). Of note, a Guinier approximation is not valid for these systems as the aggregation distorts low-q regions. Therefore, we employed a modified Guinier approximation for rod-like particles and determined that a radius of the cross-section of the rods is around 2.3 nm.
We performed a more detailed study of F3E10 at a higher salt concentration (0–640 mM) and observed a smooth transition with an isosbestic point at q = 0.89 nm−1. We observed an increase in intensity at the low to middle q-range due to an increased fraction of aggregated fibers and a slight decrease in the slope at the high q-range corresponding to chain collapse. Kratky plots (Fig. 4F) demonstrated the appearance of a hyperbolic peak with a maximum at 0.48 nm−1 with increasing intensity typical for the formation of ordered or frozen structures in solution.
We additionally proved the presence of supramolecular fibers using a small-angle neutron scattering (SANS) study of F3E10 in water and 120 mM NaCl (see ESI Fig. S2†). While this system scatters weakly, the results were sufficient to confirm the results of the SAXS experiments. We observed −1 power law dependence typical for rod-like particles and salt-induced aggregation. At this concentration, the length of the particles is higher than the resolution of the instrument, and Guinier approximation is not applicable to determine the absolute length.
Finally, we extracted the P(r) pair distribution function for the F3G10 solution in 120 mM NaCl to eliminate the interference of chain repulsion (Fig. 4G). The function is typical for rod-like particles with a sharp maximum, corresponding to rod diameter and Dmax slightly above 50 nm. Of particular interest, we note the appearance of small periodic oscillations in the P(r) function. Burian and Amenitsch performed dummy atom modeling for SAXS scattering for helical and twisted structures and established that P(r) functions of these structures have similar oscillations with a distance between the maxima corresponding to the length of the full twist.27 In our case, the distance between the maxima is 10–11 nm, which correlates well with the cryo-TEM data.
As a control experiment, we prepared compound F3 in the form of a water-soluble hydrochloride salt; this compound comprises the nona-phenylalanine-BTA without PGA. We assumed that the self-assembly of the core should be similar to the assembly of the corresponding polymer. The P(r) function for this compound also displayed decaying oscillations typical for periodic structures with the distance between the grooves of 9.7 nm. However, taking into account the complexity of the system, we cannot consider P(r) function appearance as final proof for the helical structure of the particles.
PGA can adopt different solution conformations depending on the degree of ionization, while pH represents a critical parameter for controlled supramolecular polymerization.28,29 We performed circular dichroism studies of F3E10 at pH 7.5 (Fig. SI1†) and observed that PGA adopts a random coil conformation. From these data, we suggest the oligophenylalanine core and not the PGA chains as the main driving force behind self-assembly.
By combining SAXS and cryo-TEM data, we suggest that fibers assemble as depicted in Fig. 1A. We provide a more detailed characterization of this hypothetical model in the ESI.† Existing models for the self-assembly of BTA-based molecules include a twist of the ring, as shown in Fig. S8A,† with the centers of the BTA cores situated on the z-axis. Repulsions between corresponding donors and acceptors are balanced by the formed hydrogen bonds. This model is sufficient to explain the appearance of chirality in the systems and explains the majority of BTA-based low molecular weight systems well. We hypothesized that our systems assembled into helical structure according to the molecule twist mechanism, as shown in Fig. 1A and Fig. S8B.† We hypothesized that π–π stacking interactions are more energetically favorable in the case of offset stacking than for sandwich stacking. Therefore, the system of directional hydrogen and aromatic stacking interactions will not be disrupted. However, in the center of the molecule, BTA rings will be shifted one against the other, so that interactions will occur through amide–π stacking and π–π stacking. Considering the distance between the groove maxima as 11.5 ± 1.5 nm (from cryo-TEM data), which corresponds to a full 360-degree twist, and considering the distance between molecules in the assembly as 0.3 nm, we estimate the number of molecules per full twist as 38 ± 5 units. That provides a twist angle of 9.47 ± 1.43 degrees. Considering that the maximum distance from the z-axis to the chain center is around 1.4 nm, we determine that the shift of each molecule in the XY plane is equal to around 1.5 Å. It is not completely clear how two fibers interact with each other as it is highly improbable that PGA chains can somehow be packed into a double-helical structure without repulsions. Additionally, the distance between fiber centers, as determined from cryo-TEM, is 1.4 nm, which is not enough to reach an overlap of phenylalanine aromatic rings from two separate molecules. We suggest that due to the break of C3 symmetry, an exposed hydrophobic region appears on the single fiber, which then interacts with a similar region on the other fiber thus forming a double helix. However, we do highlight the requirement for further experiments to confirm the proposed model.
Both supramolecular and polymeric self-assembling systems suffer from low stability upon dilution and require additional stabilization. One possible stabilization strategy is the use of hydrophobic molecules to stabilize the whole structure by the formation of additional non-covalent bonds. We employed the common anthracycline chemotherapeutic drug Dox as it is both water-soluble and strongly hydrophobic.
We prepared a series of solutions with F3E10/Dox ratios of 50/1, 25/1, 10/1, and 5/1 (mol/mol) and a polymer concentration of 1.0 mM. By TEM (Fig. 5A–D), we observed the presence of flexible filaments and small nanorods of 20–40 nm in length in low Dox solutions. Upon increasing Dox content, we observed an increase in the fiber lengths to 50–120 nm for the 10/1 ratio. For the 5/1 ratio of PGA/Dox, we observed the formation of long fibers from 100 nm to a few hundred nanometers. The rod diameter in all the cases was maintained at around 10 nm. We selected the 10/1 ratio for subsequent stability studies.
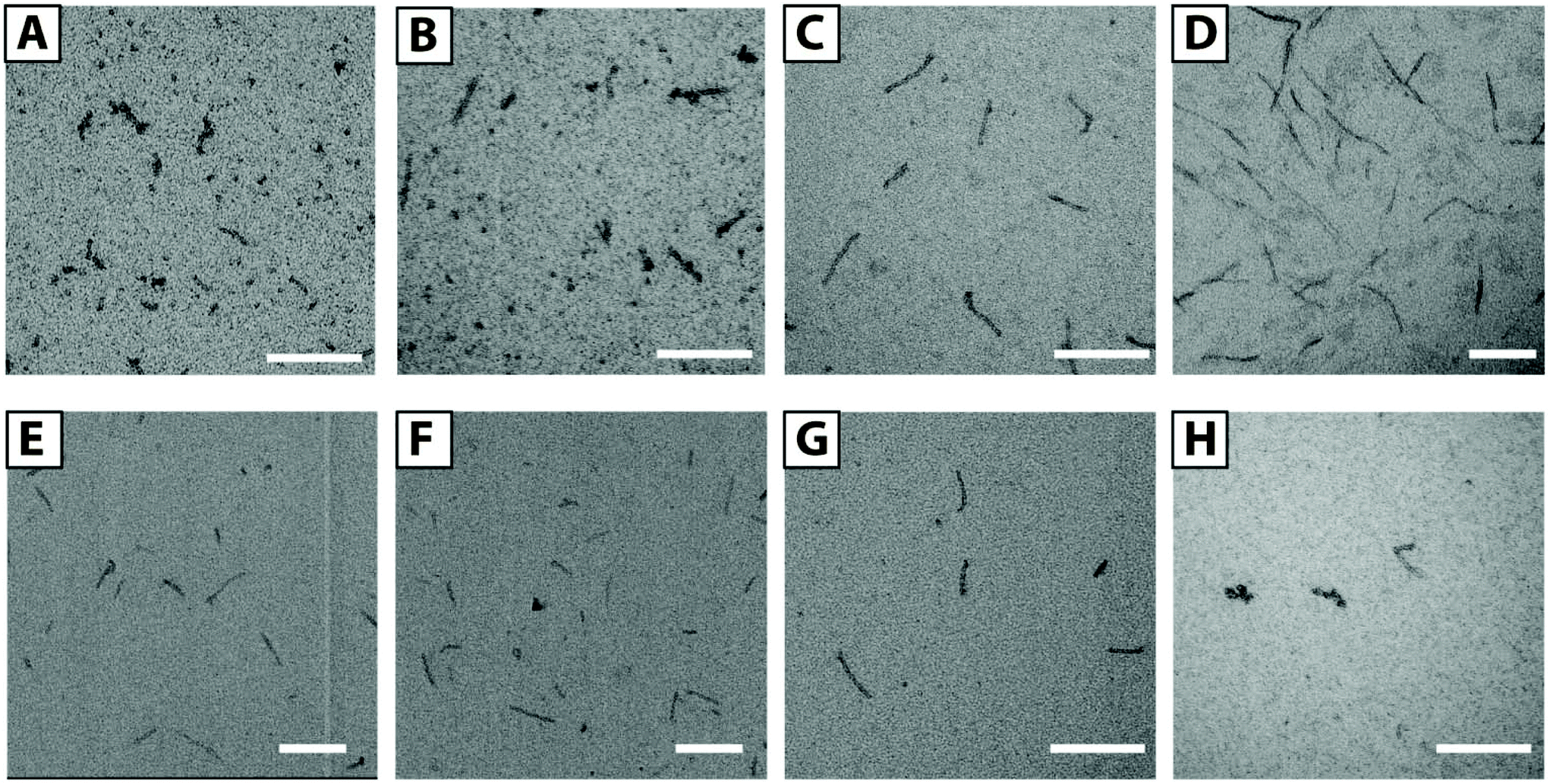 | ||
Fig. 5 TEM images of F3E10/Dox complexes with molar ratio 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (A), 20:1 (B), 10:1 (C), 5:1 (D). Images of 10:1 complex, diluted 10 times below CAC of F3E10 after 24 h (E) and 48 h (F). F3E10/Iri (G) and F3E10/doxycycline (H) with molar ratio 10:1. Scale bar 200 nm. :1 (A), 20:1 (B), 10:1 (C), 5:1 (D). Images of 10:1 complex, diluted 10 times below CAC of F3E10 after 24 h (E) and 48 h (F). F3E10/Iri (G) and F3E10/doxycycline (H) with molar ratio 10:1. Scale bar 200 nm. | ||
To determine the influence of pH on self-assembly, we prepared F3E10/Dox complexes (10/1 mol/mol) in a pH range from 5.5 to 8.5 (see ESI, Fig. S4†). At low pH, we observed the formation of longer nanofibers, reaching 1 μm in length, as a consequence of a lower degree of glutamic residue ionization and lower chain repulsions. At pH 6.5–7.5, we observed similar nanorods of around 100 nm in length. At pH 8.5, we observed shorter rods (30–50 nm); however, it remains unclear if this occurs due to higher PGA chain repulsions or partial Dox degradation.
These studies demonstrated that the obtained particles remained stable during dialysis for five days, and we failed to observe any Dox loss (as measured by UV spectroscopy) (see ESI, Fig. S3†). We additionally studied how particles respond to dilution below their CAC values for non-stabilized F3E10 in 120 mM NaCl solution using TEM. We prepared a 1.0 mM solution of F3E10/Dox at a ratio of 10/1 in 120 mM NaCl, diluted it to 0.014 mM, and then studied both solutions after 24 and 48 hours with TEM (Fig. 5E and F). In all cases, we observed the maintenance of a similar nanorod size even after 48 hours. A control experiment with non-stabilized F3E10 demonstrated the absence of assembled particles (see ESI, Fig. S4†). Dox loading efficiency was determined as 96 ± 2% (see ESI† for details).
Next, we tried to ascertain whether other flat hydrophobic but water-soluble molecules can also stabilize the structure in the same way. To this end, we performed a series of TEM experiments for solutions containing different amounts of NR, doxycycline, or Iri and performed TEM studies. Excluding NR, we used all compounds as hydrochloride salts that were added to the F3E10 solutions at pH 7.4 and in similar molar ratios of 5/1, 10/1, and 20/1. For F3E10 mixtures with Iri, we observed nanorods similar to those containing Dox (Fig. 5G, Fig S4 and S6†), but shorter and more polydisperse in length, ranging from 30 to 200 nm. When we employed doxycycline at ratios of 5/1, 10/1, and 20/1, we only detected a few short nanorods of 50–70 nm (Fig. 5H) independently of the content of doxycycline. In all cases, we observed a significantly lower number of observed particles for F3E10/doxycycline than for F3E10/Dox and F3E10/Iri. Finally, NR failed to support the formation of nanorods, and the dye precipitated in a few days.
To determine the stabilization mechanism involved, we selected the F3E10/Dox complex at a ratio of 10:1, given the suitable size range of the particles and high sensitivity of Dox to different environments. As a first step, we studied how star-PGA without a hydrophobic core interacted with Dox by mixing Dox hydrochloride with a solution of F3E10 and F0E10 at pH 7.4 at a ratio of 10:1 star-PGA to Dox. We observed that the addition of Dox to solutions of F0E10 results in immediate cloudiness with rapid precipitation. In solutions of F3E10, light cloudiness appeared immediately, but which subsequently completely disappeared in a few seconds (see images in ESI, Fig. S5†). In the case of F0E10, the precipitate formed most probably due to point-to-point Dox-induced aggregation. In the case of F3E10 complexes, Dox forms some point-to-point interactions as a kinetic product, but either intercalates in the core or diffuse deeper into the molecule at later times.
Studies have also shown that Dox forms nanorods by itself, which can then further co-assemble with PGA forming rod-like particles that comprise an inner Dox core and outer PGA shells.30 To exclude this possibility, we performed cryo-TEM for F3E10/Dox at 0.014 mM in 120 mM NaCl (Fig. 6A) and found identical nanorods to those not stabilized with Dox. Image reconstruction provided evidence of a similar helical structure; this fact suggests that the particles represent supramolecular polymers stabilized with Dox rather than Dox nanorods stabilized with PGA. We note that we discovered a particle length and diameter identical to that obtained by TEM with uranyl acetate staining, confirming our suggestion that this technique may be of use for studies of highly aggregated systems.
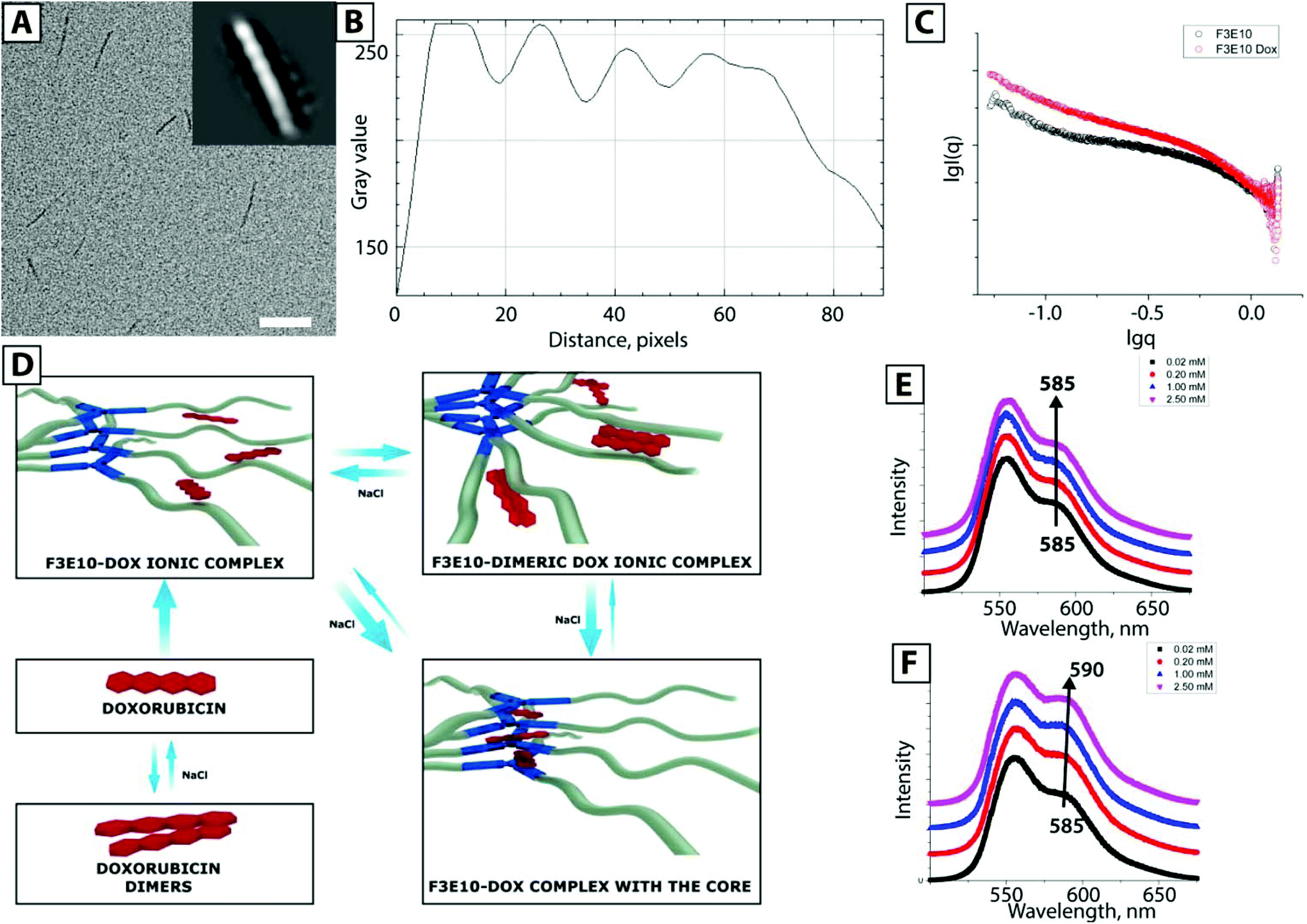 | ||
| Fig. 6 Cryo-TEM image of F3E10/Dox, particle reconstruction image (A) and corresponding plot profile (B). (C) SAXS profile for F3E10 at 1.0 mM in 0.12 M NaCl solution and in the presence of 0.1 eq. of Dox. (D) General scheme of Dox-F3E10 solution equilibrium. Fluorescence spectroscopy data for F0E10 (E) and F3E10 (F) at 0.02–2.5 mM concentration in 0.12 M NaCl in the presence of 6 μmol Dox. | ||
The behavior of Dox in polyanionic solution is quite diverse (Fig. 6D) – it forms ionic complexes with PGA chains, dimeric ionic complexes, dimers, and oligomers with itself, and also interacts with hydrophobic cores. We employed fluorescence spectroscopy to determine a more detailed mechanism of Dox interaction with F3E10. We studied solutions of F0E10 and F3E10 with concentrations ranging from 0.02 to 2.5 mM in 120 mM salt solution (Fig. 6E and F). For the control experiment with F0E10, we observed changes in the intensity of the peaks in the emission spectrum with no peak maximum shift or peak ratio change. Intensity changes occur due to the formation of dimeric ionic complexes (i.e., point-to-point interactions) at lower concentrations, which disassemble in the presence of salt or high concentration of the polymer. Upon increased polymer concentration, the equilibrium shifts towards the formation of unimeric ionic complexes, and intensity increases. For F3E10 at low concentrations, we observed an identical trend; however, at concentrations above CAC, the peak shifts from 585 to 590 nm, and we observed a decrease in the ratio between peaks at 559 nm and 590 nm. At concentrations above the CAC, Dox interacts with the core, resulting in a red shift of the 585 nm peak. Considering that the expected mechanism of self-assembly of F3-based compounds does not suppose the formation of a dense ordered structure of planar stacks, like in DNA, Dox cannot directly intercalate into the structure. Similar interactions were observed for poly-AT complexes with Dox. Unlike poly-CG nucleotides, Dox fails to intercalate into the poly-AT structure; however, Dox does interact with the poly-AT structure.31 Therefore, we can conclude the formation of an association product with the F3 core, although we cannot unambiguously define the precise orientation of Dox within the core.
Finally, we performed SAXS studies of F3E10 with Dox in 0.12 M NaCl. For the samples at 0.2 mM (prepared from 1.0 mM solutions and diluted 24 h before analysis) in the presence of Dox, we observed an increase in the intensity and a higher slope of the curve, which confirms the existence of rod-like particles and an increase in their quantity. In addition to the simple stabilization of the particles, Dox additionally stimulated their aggregation.
We repeated the synthesis of F3E10/Dox at a 10:1 ratio several times to study the reproducibility of the method (see ESI, Fig. S6†) and, encouragingly, we observed identical sizes for the particles. Finally, we note the fundamental nature of the preparation strategy for the formation of stabilized nanorods. Following the addition of highly concentrated solutions of Dox to polymer without strong stirring, the mixture consisted of very long rods of a few mm long that sometimes gathered in “clots” and uranyl-induced artifacts arising from non-assembled unimers (see ESI, Fig. S7†).
Finally, we studied drug release from F3E10/Dox (10/1 mol/mol) nanorods at pH 5.5, pH 7.4, and in the presence of cathepsin B (Fig. 7). At both pH 5.5 and pH 7.4, Dox release remained below 2% of total drug loading after 72 hours. However, we believe that this value corresponds to the free drug rather than the slow release as levels remain almost constant. In the presence of Cathepsin B, we observed significantly higher cumulative drug release, reaching 25% after 72 h. These results confirm our hypothesis of Dox localization in the nanorods cores as opposed to interacting with the PGA chains.
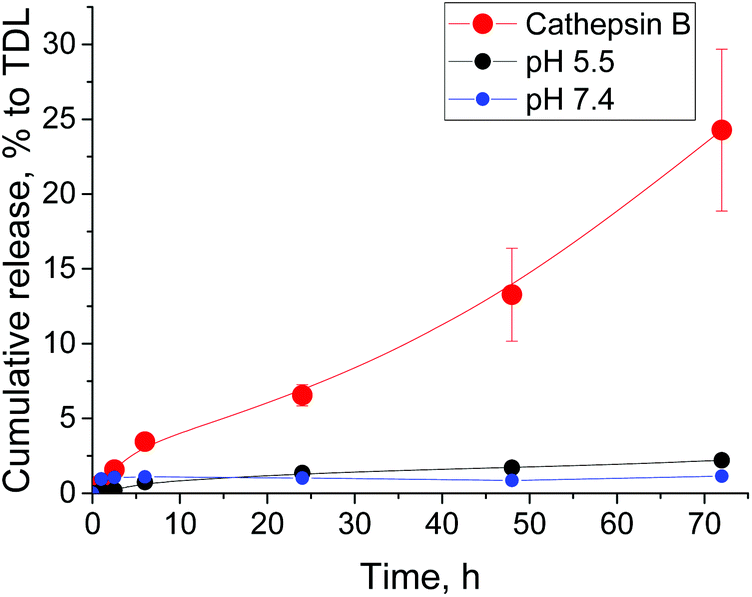 | ||
| Fig. 7 Time-dependent release of doxorubicin from F3E10/Dox (10:1) nanorods at pH 5.5, pH 7.4, and in the presence of cathepsin B. | ||
Conclusions
In conclusion, we prepared the first compound from a class of supramolecular star-polyelectrolytes combining BTA supramolecular chemistry and PGA functionality. We observed that the molecules behave as polyelectrolytes in diluted solutions but self-assemble into rod-like particles in more concentrated solutions or the presence of salts. We controlled the length of the fibers over a broad range from 40 nm to micrometers. TEM and SAXS results confirmed that F3E10 behaves as a molecular gel at higher concentrations.Finally, as these systems are prone to disassembly upon dilution and considering future applications in drug delivery technologies, we developed a method for the stabilization of nanorods with first-line chemotherapeutic agents such as Dox and Iri, most probably through intercalation in the hydrophobic core. Other hydrophobic drugs with cationic substituents could also be used through this strategy. The careful control over the preparation of the constructs allowed us to generate nanorods with a length of 50–120 nm that displayed stability upon dilution for several days. Dox incorporation not only stabilizes the nanorods but strongly pulls the equilibrium towards nanorod formation, minimizing the presence of unimers or small oligomer species within the system. We believe that these systems represent promising candidates for biomedical applications due to their unique morphological structure, expected flow alignment, and vast availability of carboxylic groups that can be easily functionalized with drugs or targeting and diagnostic moieties. Importantly, due to their inherent versatility and multivalency, these polyelectrolytes will allow the generation of a new family of targeted drug delivery technologies, representing the core constituents of combination therapeutics and allowing the application of theranostic moieties. Combination therapies represent one of the four recognized strategic directions that can foster nanomedicine translation.32 Due to fiber formation and the ability to modulate the gelation properties of these materials, polyelectrolytes could also be applied as therapies for topical/local administration after the application of an adequate design strategy. Therefore, potential applications include pathologies such as spinal cord injury after intrathecal injection or skin diseases.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge N. Mahmoudi from ISIS Neutron and Muon Source (UK) for performing SANS measurements, J. Chichon and R. Arranz from the CNB-CiB (CSIC) cryo-EM facility for the sample vitrification and data acquisition support, and Stuart P. Atkinson for English editing. Authors also acknowledge E. Solano and M. Malfois from BL-11 NCD-SWEET beamline at ALBA Synchrotron for their support with SAXS experiments. The authors would like to thank the Spanish Ministry of Economy and Competitiveness (SAF2013-44848-R, SAF2016-80427-R, PTQ-13-06465) and the European Research Council (Grant ERC-CoG-2014-648831 MyNano) for financial support. Part of the equipment employed in this work has been funded by Generalitat Valenciana and co-financed with FEDER funds (PO FEDER of Comunitat Valenciana 2014–2020).Notes and references
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154–1196 CrossRef.
- H. Feng, X. Lu, W. Wang, N. G. Kang and J. W. Mays, Polymers, 2017, 9, 494 CrossRef.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 CAS.
- S. P. Atkinson, Z. Andreu and M. J. Vicent, J. Pers. Med., 2018, 8, 6 CrossRef.
- C. Deng, J. Wu, R. Cheng, F. Meng and H. Klok, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS.
- C. M. Gonz and M. A. Sarabia-Vallejos, Polymers, 2017, 8, 551 Search PubMed.
- O. Zagorodko, J. J. Arroyo-Crespo, V. J. Nebot and M. J. Vicent, Macromol. Biosci., 2016, 17, 1600316 CrossRef.
- J. J. Arroyo-Crespo, A. Armiñán, D. Charbonnier, L. Balzano-Nogueira, F. Huertas-López, C. Martí, S. Tarazona, J. Forteza, A. Conesa and M. J. Vicent, Biomaterials, 2018, 186, 8–21 CrossRef CAS.
- C. Apfelthaler, M. Anzengruber, F. Gabor and M. Wirth, Eur. J. Pharm. Biopharm., 2017, 115, 131–139 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 4131 CrossRef PubMed.
- M. van Pomeren, W. J. G. M. Peijnenburg, R. C. Vlieg, S. J. T. van Noort and M. G. Vijver, Nanotoxicology, 2019, 13, 558–571 CrossRef CAS PubMed.
- J. Babin, D. Taton, M. Brinkmann and S. Lecommandoux, Macromolecules, 2008, 41, 1384–1392 CrossRef CAS.
- W. Agut, A. Brûlet, C. Schatz, D. Taton and S. Lecommandoux, Langmuir, 2010, 26, 10546–10554 CrossRef CAS.
- Z. Ahmad, S. Lv, Z. Tang, A. Shah and X. Chen, J. Biomater. Sci., Polym. Ed., 2016, 27, 40–54 CrossRef CAS PubMed.
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- R. Holm, M. Douverne, B. Weber, T. Bauer, A. Best, P. Ahlers, K. Koynov, P. Besenius and M. Barz, Biomacromolecules, 2019, 20, 375–388 CrossRef CAS PubMed.
- R. Holm, D. Schwiertz, B. Weber, J. Schultze, J. Kuhn, K. Koynov, U. Lächelt and M. Barz, Macromol. Biosci., 2019, 1900152, DOI:10.1002/mabi.201900152.
- A. Duro-Castano, V. J. Nebot, A. Niño-Pariente, A. Armiñán, J. J. Arroyo-Crespo, A. Paul, N. Feiner-Gracia, L. Albertazzi and M. J. Vicent, Adv. Mater., 2017, 29, 1702888 CrossRef PubMed.
- P. Ahlers, H. Frisch, R. Holm, D. Spitzer, M. Barz and P. Besenius, Macromol. Biosci., 2017, 17, 1700111 CrossRef PubMed.
- K. E. Washington, R. N. Kularatne, M. C. Biewer and M. C. Stefan, ACS Biomater. Sci. Eng., 2018, 4, 997–1004 CrossRef CAS.
- A. Duro-Castano, R. M. England, D. Razola, E. Romero, M. Oteo-Vives, M. A. Morcillo and M. J. Vicent, Mol. Pharm., 2015, 12, 3639–3649 CrossRef CAS.
- I. de Feijter, P. Besenius, L. Albertazzi, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Soft Matter, 2013, 9, 10025–10030 RSC.
- V. Yu. Borue and I. Ya. Erukhimovich, Macromolecules, 1988, 21, 3240–3249 CrossRef.
- J. B. Hayter and J. Penfold, Mol. Phys., 1981, 42, 109–118 CrossRef CAS.
- M. Burian and H. Amenitsch, IUCrJ, 2018, 5, 390–401 CrossRef CAS.
- H. Xia, H. Fu, Y. Zhang, K. Shih, Y. Ren, M. Anuganti, M. Nieh, J. Cheng and Y. Lin, J. Am. Chem. Soc., 2017, 139, 11106–11116 CrossRef CAS PubMed.
- J. Wang, H. Lu, R. Kamat, S. V. Pingali, V. S. Urban, J. Cheng and Y. Lin, J. Am. Chem. Soc., 2011, 133, 12906–12909 CrossRef CAS PubMed.
- S. Yang, F. Zhu, Q. Wang, F. Liang, X. Qu, Z. Gan and Z. Yang, J. Mater. Chem. B, 2016, 4, 7283–7292 RSC.
- M. Airoldi, G. Barone, G. Gennaro, A. M. Giuliani and M. Giustini, Biochemistry, 2014, 53, 2197–2207 CrossRef CAS PubMed.
- R. van Der Meel, E. Sulheim, Y. Shi, F. Kiessling and W. J. M. Mulder, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01442j |
| ‡ Current address: Polypeptide Therapeutics Solutions SL. Av. Benjamin Franklin 46980 Valencia (Spain). |
| This journal is © The Royal Society of Chemistry 2020 |