DOI:
10.1039/C9PY01335K
(Paper)
Polym. Chem., 2020,
11, 560-567
Facile terminal functionalization of peptides by protease-catalyzed chemoenzymatic polymerization toward synthesis of polymeric architectures consisting of peptides†
Received
5th September 2019
, Accepted 1st October 2019
First published on 30th October 2019
Abstract
Polypeptides are used as building blocks that assemble into polymeric hierarchical architectures with various functionalities based on their amino acid sequences. Chemoenzymatic polymerization using a protease as a catalyst allows us to synthesize peptides with various primary structures in an environmentally benign way. In this work, we performed papain-catalyzed polymerization in the presence of terminal-modifying agents to synthesize peptides modified with a functional group at their N-terminus. Various peptides with a reactive acrylamide group at the N-terminus were synthesized in a one-pot chemoenzymatic reaction with the side groups of the amino acid residues left intact. Acrylamide-modified poly(L-alanine) was used as a macromonomer in a radical copolymerization with N-isopropylacrylamide to give a graft copolymer consisting of poly(L-alanine) side chains.
Introduction
Polypeptides consisting of proteinogenic amino acids, which naturally exist as proteins, are fascinating biobased polymeric materials. The versatility of amino acid sequences expands the application of polypeptides, including as commodities, cosmetics, medicines, and biomedical materials. The physical properties of polypeptides are associated with their higher-order structures formed by self-assembly of the polypeptide backbone via inter-/intramolecular interactions such as hydrogen bonding and hydrophobic effects regulated by specific amino acid sequences. To take advantage of this unique advantage of polypeptides, much effort has been made to assemble peptide building blocks into polymeric architectures for functionalization.
Polypeptide-based building blocks are synthesized by various methods, including solid-phase peptide synthesis (SPPS),1 ring-opening polymerization of amino acid N-carboxyanhydrides (NCAs),2,3 and biosynthesis using microbial hosts.4,5 The amino acid sequences of polypeptides can be easily regulated by SPPS and biosynthesis; however, tedious synthetic protocols make it difficult to produce the desired polypeptides in sufficient yield and purity and on a large scale. In addition, the ring-opening polymerization of NCAs has been widely utilized for the synthesis of polypeptides that are well-defined in terms of molecular weight and polydispersity. This method allows us to synthesize block copolymers consisting of two or more different polypeptide building blocks. Block copolymers based on polypeptides are widely used to construct well-defined molecular assemblies such as polymer micelles and hydrogels, mainly for drug delivery, regenerative medicines, and tissue engineering.6–11 Polypeptide fragments have also been assembled into special-shaped polymers such as graft copolymers and star-shaped polymers.12–14
Recently, chemoenzymatic polymerization using a protease as a catalyst has emerged as another method for synthesizing peptides.15,16 Chemoenzymatic polymerization employs kinetically controlled tandem aminolysis reactions of amino acid derivatives in an aqueous buffer solution, which offers atom-economical, environmentally benign synthesis of polypeptides under mild conditions.15,17 The protease-catalyzed reaction provides stereo- and regioselectivity for the formation of peptide bonds. We have developed various types of peptides with homo-/random sequences by chemoenzymatic polymerization.16,18 Furthermore, poor substrate specificity for unnatural amino acids can be mitigated by modification with natural ones that show good affinity for proteases, resulting in successful introduction of unnatural structures such as nonproteinogenic amino acids,19,20 nylon units,21,22 telechelic structures,23,24 and aromatics25 in the peptide backbones. Peptides with a star-shaped structure were also synthesized using multifunctional substrates.26,27
Previously, we designed artificial polypeptide materials that mimic the higher-order structures of natural structural proteins by conjugating oligomeric peptide fragments. Specific amino acid motifs of structural proteins that govern their physical properties were derived from the sequence of structural proteins for this purpose. For example, we synthesized a β-sheet crystalline motif and glycine-rich amorphous motif of spider silk proteins28 and a repetitive motif of elastin29 that confers elasticity to polypeptides by chemoenzymatic polymerization. The peptide fragments were further conjugated into multiblock polypeptides, which showed specific secondary structures similar to those of natural proteins. Such peptide fragments can also be conjugated with synthetic polymers for biomimetic materials, as reported in some studies.30,31 The conjugation of peptide fragments has been performed by direct condensation, which requires relatively harsh conditions, in previous works. In this work, several types of peptides containing a reactive functional group that can be used for facile conjugation to construct special polymeric architectures were synthesized by papain-catalyzed polymerization. A terminal-modifying agent, which is expected to be involved in the polymerization reaction, was successfully introduced into a reactive functional group at the N-terminus of the peptides. The functional group was then used for postpolymerization to construct polymeric architectures such as graft copolymers.
Experimental
Materials
Papain was purchased from FUJIFILM Wako Pure Chemical Industries, Ltd (Osaka, Japan) and used as received. The activity was approximately 0.5 U g−1, where one unit is defined as the amount of enzyme needed to hydrolyze 1 mmol of N-benzoyl-DL-arginine p-nitroanilide per minute at pH 7.5 and 25 °C. Amino acid esters were purchased from Watanabe Chemical Industries, Ltd (Hiroshima, Japan) and used as received. N-Isopropylacrylamide (NIPAm) purchased from FUJIFILM Wako Pure Chemical Industries, Ltd was purified by recrystallization from hexane and stored at −4 °C until use. N,N-Dimethylformamide and triethylamine were dried over 4 Å molecular sieves and stored under nitrogen. All other reagents were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan) and used as received without any purification unless otherwise noted.
Analysis
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Varian NMR System 500 (Varian Medical Systems, Palo Alto, CA) at 25 °C and at a frequency of 500 MHz. Dimethylsulfoxide-d6 (DMSO-d6) with trifluoroacetic acid-d (TFA-d) (5/1 in volume) was used as the solvent for the polypeptides with tetramethylsilane as an internal standard. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis was conducted using an ultrafleXtreme MALDI-TOF spectrophotometer (Bruker Daltonics, Billerica, MA) operating in reflection mode at an accelerating voltage of 15 kV. The sample was dissolved in water/acetonitrile (0.8 mg mL−1) containing 0.1% TFA, mixed with a solution of α-cyano-4-hydroxycinnamic acid (CHCA) in water/acetonitrile (10 mg mL−1), and deposited on an MTP 384 ground steel BC target plate. Infrared (IR) spectra of the samples were recorded by an IRPrestige-21 Fourier transform infrared spectrophotometer (Shimadzu Corporation, Kyoto, Japan) with a MIRacle A single reflection ATR unit using a Ge prism. Gel permeation chromatography (GPC) was performed on a JASCO HPLC system (PU-2086, DG2080-54, AS-2057, CO-2065; JASCO, Tokyo, Japan) with a Shodex KD-804 column (Showa Denko K. K., Tokyo, Japan) and a UV detector (UV-2075). Measurements were carried out on polypeptide samples (2 mg mL−1) eluted with NMP containing 10 mM lithium bromide at a flow rate of 1.0 mL min−1. The number- (Mn) and weight-average molecular weights (Mw) and the molecular weight distribution (Mw/Mn) were estimated using polystyrene standards of the following molecular weights: 1.32 × 103, 3.25 × 103, 1.01 × 104, 2.85 × 104, 6.60 × 104, and 1.56 × 105. Circular dichroism (CD) spectroscopic analysis was conducted using a Jasco J-820 CD spectropolarimeter (JASCO, Tokyo, Japan). Measurements were conducted on polymer solutions (0.05 w/v%) in Milli-Q water using a quartz cuvette with a 1 mm path length. Each spectrum represents the average of 10 scans from 190 to 290 nm with a resolution of 1 nm obtained at 200 nm min−1 with a bandwidth of 1 nm.
Synthesis of N-acryloyl-L-alanine ethyl ester (1)
To a flask equipped with an addition funnel and a stirring bar were added L-alanine ethyl ester hydrochloride (4.61 g, 30.0 mmol), triethylamine (9.2 mL, 66.0 mmol) and chloroform (50 mL) at 0 °C under nitrogen. To this solution, a solution of acryloyl chloride (2.42 mL, 30.0 mmol) in chloroform (20 mL) was added dropwise over 30 min, and the resulting mixture was stirred at 0 °C for 2 h. After the mixture was allowed to warm to 25 °C, the solution was washed with water twice and brine. The organic layer was dried with Na2SO4 and concentrated by a rotary evaporator. The crude product was dried in vacuo and purified by silica gel column chromatography eluted with ethyl acetate to yield a pale-yellow solid. The yield was 2.90 g (56%). IR (neat, cm−1): 3263, 2960, 1746, 1643, 1544, 1272, 1200, 1152, 1033. 1H NMR (CDCl3, ppm, 25 °C): δ 6.82 (s, 2H), 4.55 (m, 2H), 4.18 (q, J = 6.9 Hz, 4H), 2.56 (m, 4H), 1.69 (m, 2H), 1.57 (m, 4H), 1.27 (t, J = 6.9 Hz, 6H), 0.94 (d, J = 6.3 Hz, 12H). 13C NMR (CDCl3, ppm, 25 °C): δ 173.36, 172.13, 61.16, 50.96, 40.93, 31.74, 24.73, 22.74, 21.67, 14.01.
Synthesis of N-propargyl-L-alanine ethyl ester (2)
A terminal modifier containing an alkyne group was synthesized by a modified protocol according a previously reported work.32 Anhydrous lithium hydroxide (1.32 g, 55 mmol), molecular sieves (4 Å, 18.8 g), and N,N-dimethylformamide (150 mL) were placed in a flask equipped with a stirring bar, and the mixture was stirred at 25 °C for 30 min under nitrogen. Propargyl bromide (2.23 mL, 30 mmol) and L-alanine ethyl ester hydrochloride (3.84 g, 25 mmol) were added to this dispersion, and the mixture was stirred at 25 °C for 12 h. After the reaction, the inorganic salt and molecular sieves were removed by filtration. Ethyl acetate was added, and the organic solution was washed with water and brine. The organic layer was dried with MgSO4 and concentrated by a rotary evaporator. The crude product was dried in vacuo and purified by silica gel column chromatography eluted with hexane/ethyl acetate (1/1) to yield a pale-yellow liquid. The yield was 1.33 g (34%). The spectroscopic data were identical to the reported values.
Chemoenzymatic polymerization of L-alanine ethyl ester with acrylamide derivative 1 using papain
To a glass tube equipped with a stirring bar were added L-alanine ethyl ester hydrochloride (0.922 g, 6.0 mmol), 1 (0.514 g, 3.0 mmol), phosphate buffer (2.0 mL, 1.0 M, pH 8.0), and methanol (1.0 mL), and the mixture was stirred at 40 °C until all the substrates were completely dissolved. A solution of papain (0.30 g) in phosphate buffer (2.0 mL) was added to this solution in one portion. The final concentrations of alanine and papain were 1.0 M and 50 mg mL−1, respectively. The mixture was stirred at 800 rpm and 40 °C for 2 h. After cooling to 25 °C, the precipitate was collected by centrifuging at 9000 rpm for 15 min at 4 °C. The crude was washed with deionized water and water/methanol (1/1 in volume) and lyophilized to provide the acrylamide-modified poly(L-alanine) (acryl-polyAla) as a white solid. The yield was 0.189 g (23.5%). The chemoenzymatic polymerization of glycine ethyl ester hydrochloride, L-glutamic acid diethyl ester hydrochloride, L-tyrosine ethyl ester hydrochloride, or Nε-Boc-protected L-lysine methyl ester hydrochloride with 1 using papain was also carried out under the same conditions. All the polymerizations were triplicated to determine the standard deviation.
Chemoenzymatic polymerization of L-alanine ethyl ester with alkyne derivative 2 using papain
To a glass tube equipped with a stirring bar were added L-alanine ethyl ester hydrochloride (0.154 g, 1.0 mmol), 2 (0.031 g, 0.2 mmol), phosphate buffer (0.33 mL, 1.0 M, pH 8.0), and methanol (0.17 mL), and the mixture was stirred at 40 °C until all the substrates were completely dissolved. A solution of papain (0.050 g) in phosphate buffer (0.33 mL) was added to this solution in one portion. The final concentrations of alanine and papain were 1.0 M and 50 mg mL−1, respectively. The mixture was stirred at 800 rpm and 40 °C for 2 h. After cooling to 25 °C, the precipitate was collected by centrifuging at 9000 rpm for 15 min at 4 °C. The crude product was washed twice with deionized water and lyophilized to provide the alkyne-modified poly(L-alanine) as a white solid. The yield was 0.017 g (17.7%).
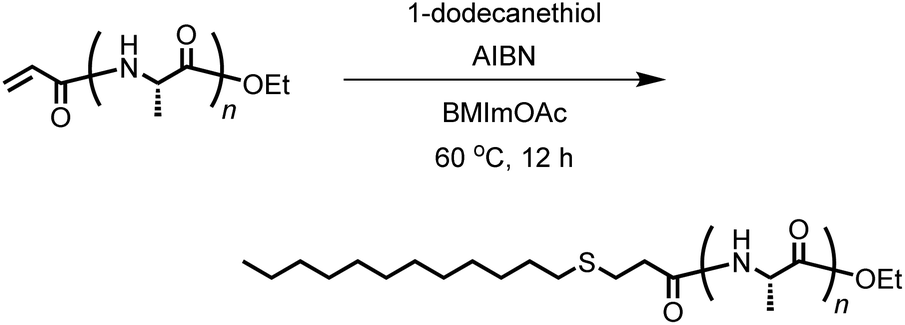
Thiol–ene reaction for conjugation between acryl-polyAla and 1-dodecanethiol
Acryl-polyAla (0.05 g, 0.1 mmol of acrylamide moiety), 1-dodecanethiol (24 μL, 0.1 mmol), and 1-butyl-3-methylimidazolium acetate (BMImOAc, 2.0 mL) were added to a flask equipped with a stir bar and stopcock, and the mixture was sonicated until all the solid was dissolved. After 2,2′-azobis(isobutyronitrile) (AIBN, 0.016 g, 0.1 mmol) was added, the solution was deoxygenated by purging with nitrogen. The mixture was stirred at 60 °C for 12 h under nitrogen. After cooling to 25 °C, the solution was poured into water. The precipitate was collected by centrifugation at 9000 rpm for 15 min at 4 °C, washed twice with water, and lyophilized to provide the product. The yield was 0.057 g (81%).
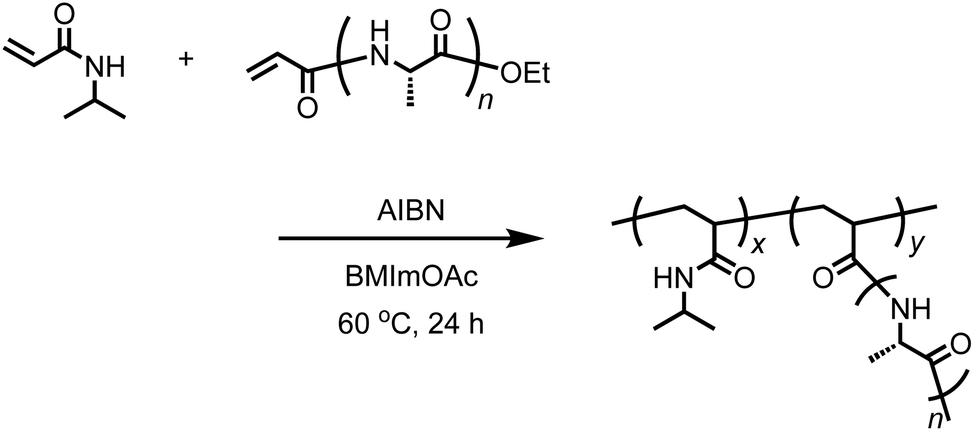
Free-radical copolymerization of acryl-polyAla with N-isopropylacrylamide (NIPAm)
Acryl-polyAla (0.05 g, 0.1 mmol of acrylamide moiety), NIPAm (0.554 g, 4.9 mmol), and BMImOAc (2.0 mL) were added to a flask equipped with a stir bar and stopcock, and the mixture was sonicated until all the solid was dissolved. After AIBN (0.016 g, 0.1 mmol) was added, the solution was deoxygenated by purging with nitrogen. The resulting solution was then stirred at 60 °C for 24 h under nitrogen. After cooling to 25 °C, the mixture was poured into cold water. An insoluble precipitate was removed by filtration at 0 °C. The filtrate was then heated to 80 °C, and the precipitate was collected by filtration. The filtrate was washed with hot water several times and dried under vacuum to obtain the graft polymer poly(NIPAm-g-Ala) as a white solid. The yield was 0.1632 g (27%). Poly(N-isopropylacrylamide) [poly(NIPAm)] as a control was also synthesized by radical polymerization using AIBN in BMImOAc under the same conditions.
Turbidity measurement for the solution of poly(NIPAm-g-Ala)
Poly(NIPAm-g-Ala) was dissolved in Milli-Q water (0.05 w/v%) at 20 °C. The solution was placed into a quartz cuvette with a 1 mm path length. The transmittance of the solution at 600 nm was obtained by a V-750 UV-visible spectrophotometer (JASCO, Tokyo, Japan) every 2 °C during a heating and cooling process. The sample was heated from 20 to 50 °C at a heating rate of 1 °C min−1, maintained at 50 °C for 10 min, and cooled down to 20 °C at a cooling rate of 1 °C min−1. The data points were fit by a sigmoid function, and cloud points were determined from a threshold of the fitting curve. As a control, the same measurement was carried out with a solution of poly(NIPAm) (0.05 w/v%).
Results and discussion
Chemoenzymatic polymerization
The protease-catalyzed polymerization of amino acid esters can afford a broad spectrum of peptide sequences. Papain is a cysteine protease with relatively broad substrate specificity and has been used for the chemoenzymatic polymerization of various amino acid esters to date. Not only natural amino acids but also unnatural amino acids can be introduced into peptides via papain-catalyzed polymerization.19–21 Therefore, papain was used as a catalyst for the chemoenzymatic polymerization in this study. We selected an acryloyl group as a terminal functional group because it undergoes Michael addition reactions with thiol or amine groups and radical polymerization, enabling the peptides to be utilized as a building block for constructing special polymer architectures. A terminal-modifying agent containing the acryloyl group (1) was synthesized from acryl chloride and L-alanine ethyl ester (Ala-OEt). Introduction of the Ala unit in the terminal-modifying agent is important for efficient recognition by proteases. For the one-pot synthesis of terminal-functionalized peptides, the papain-catalyzed polymerization of various amino acid esters was carried out at 40 °C in the presence of 1 in 1 M sodium phosphate buffer (Scheme 1). A small amount of methanol (20 vol%) was added to the reaction mixture to ensure the complete dissolution of the monomer and 1. We tried Ala-OEt, glycine ethyl ester (Gly-OEt), diethyl L-glutamate [Glu-(OEt)2], L-tyrosine ethyl ester (Tyr-OEt), and Nε-Boc-protected L-lysine methyl ester [Lys(Boc)-OMe] as amino acid monomers to examine the versatility of this polymerization. The results of the papain-catalyzed polymerization are summarized in Table 1. First, the polymerization of Ala-OEt was carried out at various Ala-OEt/1 feed ratios (Runs 1–4). A white precipitate was gradually obtained within 2 h under all conditions, and the yield of the precipitate was 15.1–31.5%. The yield slightly decreased when the amount of 1 in the feed increased because of the lower affinity of 1 than Ala-OEt for papain. In the chemoenzymatic polymerization, the aminolysis reaction is kinetically facilitated by the use of weakly activated ester derivatives of amino acids. The aminolysis reaction competes with hydrolysis reaction which decreases the molecular weight, especially at the latter stage of polymerization.19,20 The use of 1 with a lower affinity to papain probably retarded the copolymerization with Ala-OEt, resulting in lower yields compared to the homopolymerization of Ala-OEt.33,34 Using a suitable protease optimized by screening proteases including artificially modified ones can improve the yield and molecular weight of the resulting peptides.
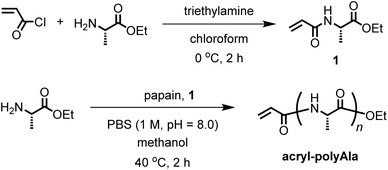 |
| | Scheme 1 Synthesis of an acrylamide terminal-modifying agent (1) and terminal-functionalized poly(L-alanine) via papain-catalyzed chemoenzymatic polymerization in the presence of 1 as a terminal modifier. | |
Table 1 Papain-catalyzed polymerization of amino acid esters in the presence of acrylamide derivative 1a
| Run |
Monomer |
[M]/[1] (mmol mmol−1) |
Yieldb (%) |
Terminal modification ratec (%) |
|
Polymerization was carried out using monomer (HCl salt, 1 M), acrylamide terminal modifier 1 (0.1–1 M) and papain (50 mg mL−1) in phosphate buffer (1 M, pH 8.0) and methanol at 40 °C for 2 h.
Precipitate was collected by centrifugation, washed with water, and lyophilized.
Degree of terminal functionalization with acrylamide moiety was determined by 1H NMR spectroscopy.
|
| 1 |
Ala-OEt |
1/0.1 |
31.5 ± 2 |
18 ± 2 |
| 2 |
Ala-OEt |
1/0.3 |
26.8 ± 3 |
68 ± 1 |
| 3 |
Ala-OEt |
1/0.5 |
23.2 ± 3 |
78 ± 4 |
| 4 |
Ala-OEt |
1/1 |
15.1 ± 2 |
>99 |
| 5 |
Gly-OEt |
1/1 |
0 |
— |
| 6 |
Glu-(OEt)2 |
1/1 |
13.8 ± 4 |
>99 |
| 7 |
Tyr-OEt |
1/1 |
7.9 ± 1 |
>99 |
| 8 |
Lys(Boc)-OMe |
1/1 |
55.3 ± 2 |
>99 |
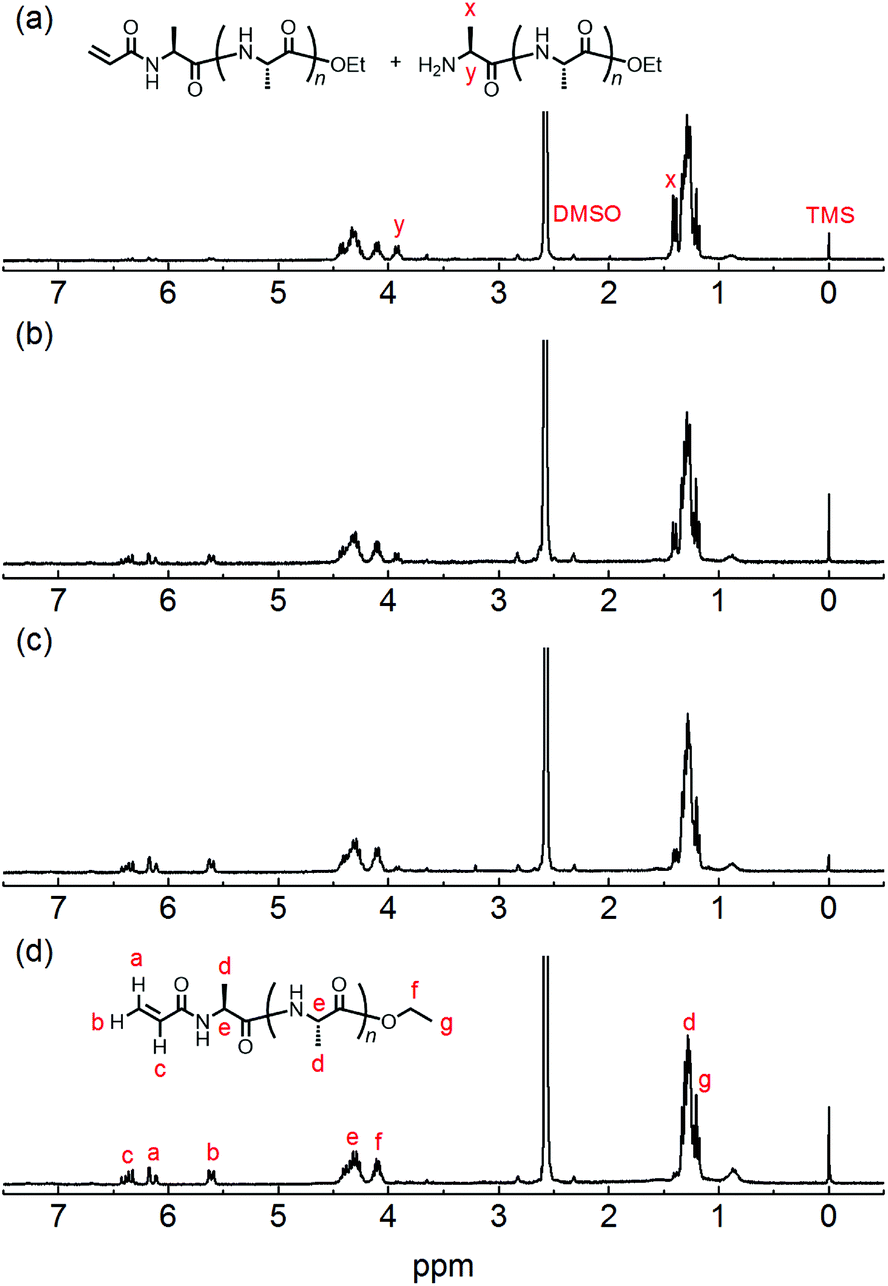
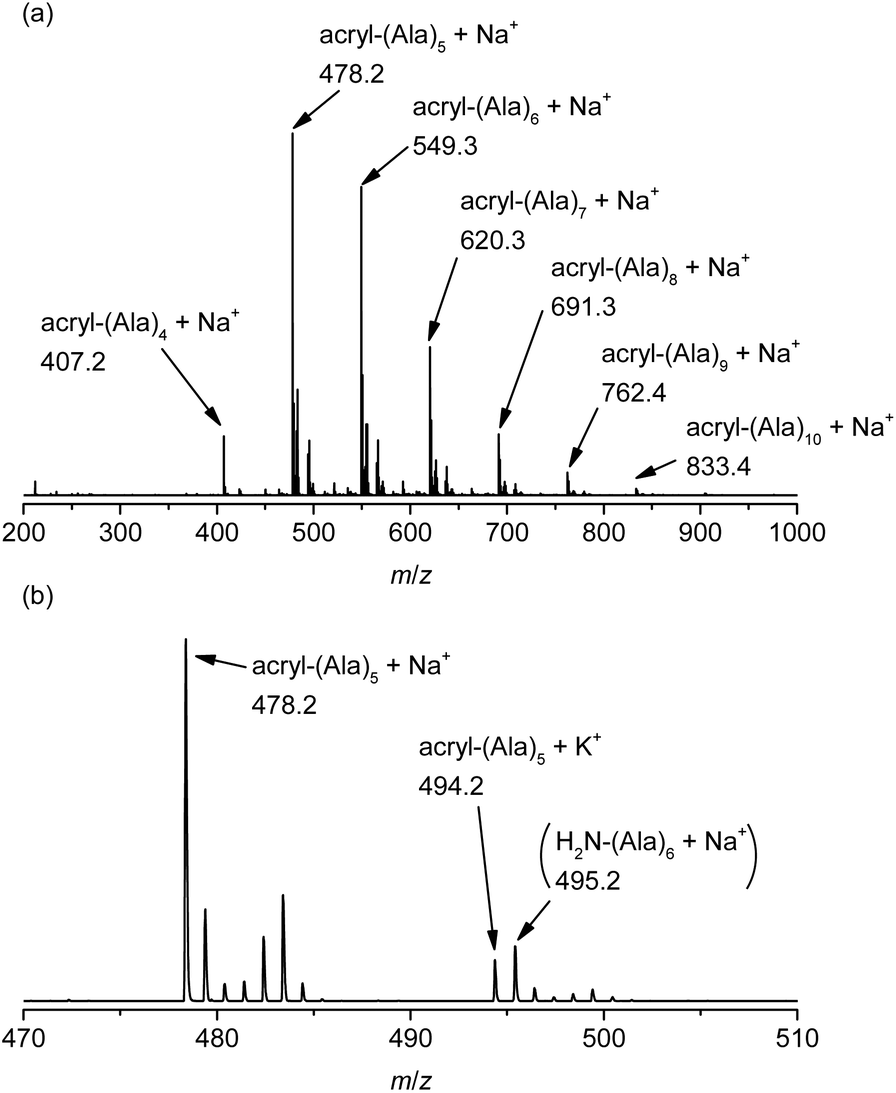
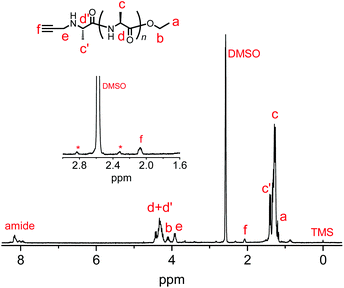
The chemical structure of the obtained products was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) and 1H NMR spectroscopy. Fig. 1 shows the 1H NMR spectra of poly(L-alanine) (polyAla) obtained by chemoenzymatic polymerization at different Ala-OEt/1 feed ratios (Run 1–4). In addition to the signals of the polyAla main chain, new signals derived from the vinyl protons of the acrylamide group (assigned as a, b, and c in Fig. 1) appear at 5.5–6.5 ppm in all the spectra. The terminal modification rate with the acrylamide group was calculated by comparing the integral ratio of the vinyl protons with the methylene protons of the C-terminal ester group appearing at 4.2 ppm (assigned as f in Fig. 1) and is listed in Table 1. In the case of Ala-OEt/1 = 1/0.1, the vinyl signals are small, and the terminal modification rate was 20%. Increasing the amount of 1 in the feed composition increased the terminal modification rate, and almost complete modification was achieved at a feed ratio of Ala-OEt/1 = 1/1. The MALDI-TOF MS spectrum of the obtained polyAla revealed that a series of peaks with an m/z interval corresponding to the Ala monomer unit (71.0 m/z) appeared, as shown in Fig. 2 (Run 4 in Table 1). The molecular mass of each peak was identical to polyAla containing an acrylamide unit and an ethyl ester unit at the N- and C-termini, respectively, confirming the synthesis of acrylamide-terminated polyAla (acryl-polyAla) via chemoenzymatic polymerization. When the feed ratio of Ala-OEt/1 was in the range from 1/0.1 to 1/0.5, another series of major peaks appeared in addition to the peaks of acryl-polyAla (Fig. S3†). The molecular mass of these peaks corresponds to polyAla with a free amine N-terminus and an ethyl ester C-terminus, indicating that the modification with acrylamide was insufficient under these conditions. The peak intensity of free amine-terminated polyAla increased with a decrease in the feed amount of 1, corresponding to the results of 1H NMR spectroscopy. The peak corresponding to polyAla with a free amine N-terminus was overlapping with the potassium ion adduct of the acrylamide-terminated polyAla as shown in Fig. 2b, indicating that there is a possibility that the product was still contaminated with a small amount of polyAla with a free amine N-terminus. However, judging from the integral ratio of 1H NMR spectra, the terminal defect without the acrylamide moiety is almost negligible for the product prepared at the feed ratio of Ala-OEt/1 = 1/1. The molecular weights detected in the MALDI-TOF MS spectra were in the range between 400 to 1000 Da regardless of the Ala-OEt/1 composition. PolyAla obtained under the same condition without 1 also showed a similar molecular weight distribution as previously reported.34 This indicates that the molecular weight obtained in the polymerization was mainly limited by precipitation of the product. However, a slight shift to lower molecular weight in the MALDI-TOF MS spectra was observed at a higher feed amount of 1. Therefore, 1 was assumed to function as an end-capper to decrease the molecular weight during the polymerization proceeding in a step-growth fashion.16 So far, the molecular weight obtained by the chemoenzymatic polymerization in aqueous media has reached up to approximately 2000 Da depending on the amino acid residues that characterize the reactivity of monomers and the solubility of resulting polypeptides in water.29 The use of water-free conditions such as solution polymerization in an organic solvent35 or bulk polymerization36 can improve the molecular weight of the obtained polypeptides, although the greenness of the chemoenzymatic synthesis is deteriorated.
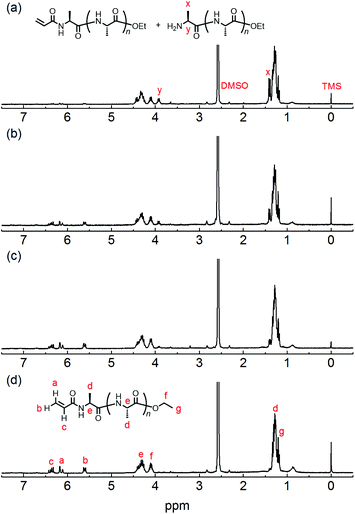 |
| | Fig. 1
1H NMR spectra of poly(L-alanine)s with an acrylamide moiety at the N-terminus synthesized via papain-catalyzed polymerization at different Ala-OEt/1 feed ratios: (a) Ala-OEt/1 = 1/0.1, (b) Ala-OEt/1 = 1/0.3, (c) Ala-OEt/1 = 1/0.5, and (d) Ala-OEt/1 = 1/1. | |
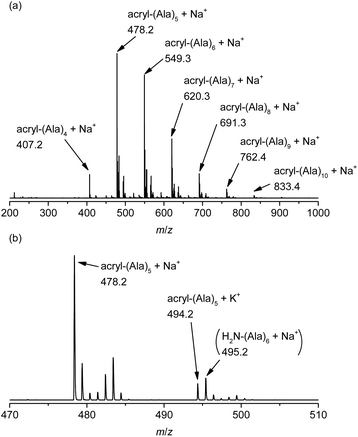 |
| | Fig. 2 (a) MALDI-TOF MS spectrum of poly(L-alanine)s with an acrylamide moiety at the N-terminus synthesized via papain-catalyzed polymerization at an Ala-OEt/1 feed ratio of 1/1 and (b) the expanded spectrum from 470 to 510 m/z. | |
The papain-catalyzed polymerization in the presence of 1 was also applied to other amino acid esters. All the polymerization reactions afforded a white precipitate except for Gly-OEt. This result is probably because of the low affinity of Gly compared to Ala for papain.33 Poly(ethyl L-glutamate) (polyGlu), poly(L-tyrosine) (polyTyr), and poly(Nε-Boc-L-lysine) (polyLys) were successfully obtained with yields of 13.8, 7.9, and 55.3%, respectively. The yield of polyLys was relatively high compared to the other amino acid monomers. This is due to the high hydrophobic structure of Nε-Boc-protected Lys which facilitates the precipitation of resulting polyLys. The chemical structures of the obtained peptides, which were characterized by 1H NMR spectroscopy and MALDI-TOF mass spectrometry, revealed that all the peptides possess an acrylamide group at the N-terminus (Fig. S4–S9†). Notably, the Tyr ester can also be polymerized via chemoenzymatic polymerization; by contrast, protection of the phenolic hydroxy group is unavoidable for synthesizing the well-defined terminal-functionalized polyTyr by conventional polymerization methods such as ring-opening polymerization of NCAs.37–39 There are very few examples that employed unprotected Lys NCA derivative for the polymerization.40 A series of major peaks in the MALDI-TOF MS spectrum were assigned to polyTyr with an acrylamide terminus, confirming that the N-terminus of polyTyr was completely modified with the acrylamide group via one-pot chemoenzymatic synthesis without any protection of the side groups.
Chemoenzymatic synthesis of alkyne-modified polyAla
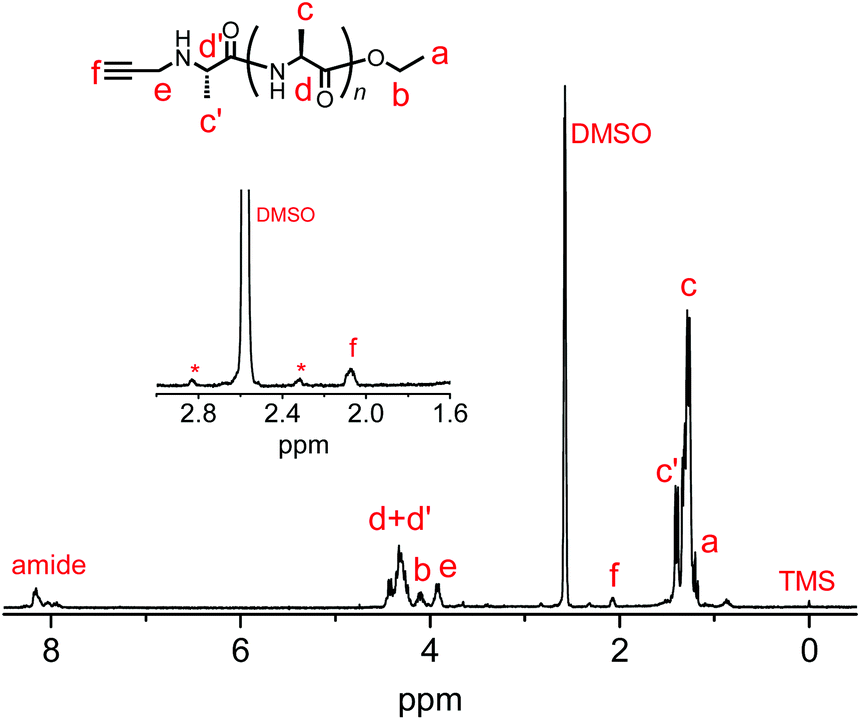
We synthesized another terminal-modifying agent, 2, containing an alkyne moiety as a functional group. Terminal modifier 2 consists of an alanine ethyl ester moiety for effective recognition by papain during chemoenzymatic polymerization. The papain-catalyzed polymerization of Ala-OEt was performed in the presence of 2 under the optimized conditions (Scheme 2). Alkyne-modified polyAla was obtained as a white precipitate in 17.7% yield. The chemical structure of alkyne-modified polyAla was analyzed by 1H NMR spectroscopy (Fig. 3). A characteristic signal assignable to a proton of the propargyl group appeared at 2.1 ppm, confirming the introduction of the alkyne moiety at the N-terminus of polyAla.
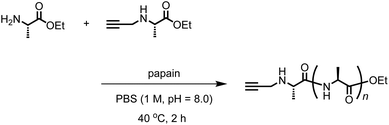 |
| | Scheme 2 One-pot chemoenzymatic synthesis of poly(L-alanine) functionalized with an alkyne moiety at the N-terminus. | |
 |
| | Fig. 3
1H NMR spectrum of poly(L-alanine) with an alkyne moiety at the N-terminus synthesized via papain-catalyzed polymerization at the Ala-OEt/2 feed ratio of 1/0.2. Asterisk shows spinning side bands of solvent peak. | |
Conjugation using the acrylamide terminal group of polyAla
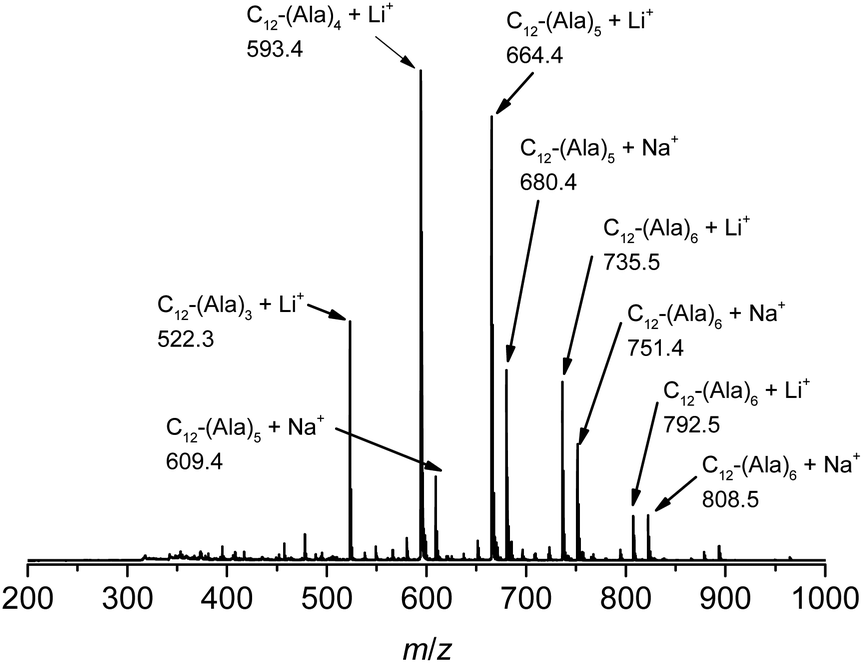
The vinyl group in the acrylamide terminal group of acryl-polyAla can be used as a conjugation site, which results in blocky structures. We attempted the radical-mediated thiol–ene addition reaction between the acrylamide moiety of acryl-polyAla and 1-dodecanethiol (Scheme 3). The reaction was performed in the presence of AIBN in 1-butyl-3-methylimidazolium acetate (BMImOAc), an ionic liquid that is known to dissolve cellulose and polypeptides. The chemical structure of the resulting polyAla conjugated with 1-dodecanethiol was characterized by 1H NMR spectroscopy and MALDI-TOF MS spectrometry (Fig. 4). The MALDI-TOF MS spectrum of the obtained polyAla showed a series of peaks with an m/z interval corresponding to the Ala monomer unit (71.0 m/z). Compared to the peaks of acryl-polyAla in Fig. 2, all the peaks shifted to higher mass values by 202.2 m/z, which corresponds to the mass of 1-dodecanethiol. This result indicates that complete conjugation with a thiol derivative was achieved via a thiol–ene reaction.
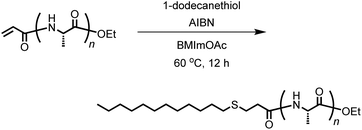 |
| | Scheme 3 Conjugation of acryl-polyAla with a thiol derivative via a radical-mediated thiol–ene reaction. | |
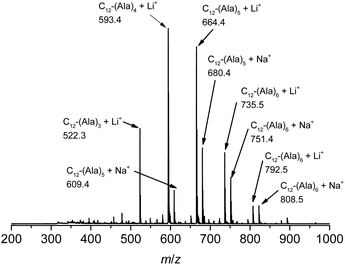 |
| | Fig. 4 MALDI-TOF MS spectrum of poly(L-alanine)s conjugated with 1-dodecanthiol at the acrylamide N-terminal. | |
Postpolymerization of acrylamide-terminated polyAla
The acrylamide moiety is a group that is polymerizable via radical or anionic polymerization. The peptides with the acrylamide group at the N-terminus synthesized in this work can be used as a macromonomer for radical polymerization, which results in the formation of graft polymers.41,42 We attempted the free radical polymerization of acryl-polyAla in the presence of AIBN as a radical initiator. To completely dissolve the acryl-polyAla macromonomer, BMImOAc was used as a reaction medium. Unfortunately, the homopolymerization of acryl-polyAla failed due to the high viscosity of the polymer solution. Therefore, N-isopropylacrylamide (NIPAm) was copolymerized with acryl-polyAla using AIBN in BMImOAc (Scheme 4). The copolymerization in the ionic liquid successfully proceeded to afford the graft copolymer poly(NIPAm-g-Ala). The number-average molecular weight (Mn) and the molecular weight dispersity (Mw/Mn) estimated by GPC were 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and 1.46, respectively. Poly(NIPAm) was also synthesized via radical polymerization in BMImOAc for comparison (Mn = 73000 Da; Mw/Mn = 1.52). The structure of poly(NIPAm-g-Ala) was confirmed by 1H NMR spectroscopy (Fig. S11†). The GPC trace poly(NIPAm-g-Ala) was unimodal and showed a shift to lower molecular weight compared to that of poly(NIPAm) prepared at the same condition (Fig. S12†). This fact indicates that the poly(NIPAm-g-Ala) assumedly possesses a branched structure which has smaller hydrodynamic radius than the linear poly(NIPAm).
000 Da and 1.46, respectively. Poly(NIPAm) was also synthesized via radical polymerization in BMImOAc for comparison (Mn = 73000 Da; Mw/Mn = 1.52). The structure of poly(NIPAm-g-Ala) was confirmed by 1H NMR spectroscopy (Fig. S11†). The GPC trace poly(NIPAm-g-Ala) was unimodal and showed a shift to lower molecular weight compared to that of poly(NIPAm) prepared at the same condition (Fig. S12†). This fact indicates that the poly(NIPAm-g-Ala) assumedly possesses a branched structure which has smaller hydrodynamic radius than the linear poly(NIPAm).
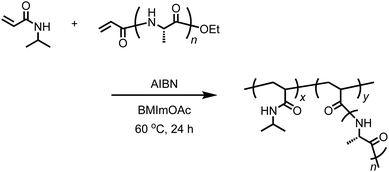 |
| | Scheme 4 Free radical copolymerization of acryl-polyAla with NIPAm. | |
CD spectroscopic analysis was performed on aqueous solutions of poly(NIPAm-g-Ala) and poly(NIPAm). The resulting CD spectrum of poly(NIPAm-g-Ala) showed a Cotton effect with a small positive peak at 215 nm and a large negative peak at 195 nm, whereas no peak appeared in the spectrum of poly(NIPAm) (Fig. S12†). This result indicates that optically active polyAla was successfully incorporated into the polyNIPAm backbone via radical copolymerization of the acryl-polyAla macromonomer. The CD spectrum of poly(NIPAm-g-Ala) was characterized as a typical random coil structure.43 Therefore, the polyAla grafts did not adopt any secondary structures under the solubilized, dispersed condition in an aqueous media, although polyAla generally tends to form β-sheet structure in natural structural proteins.44,45
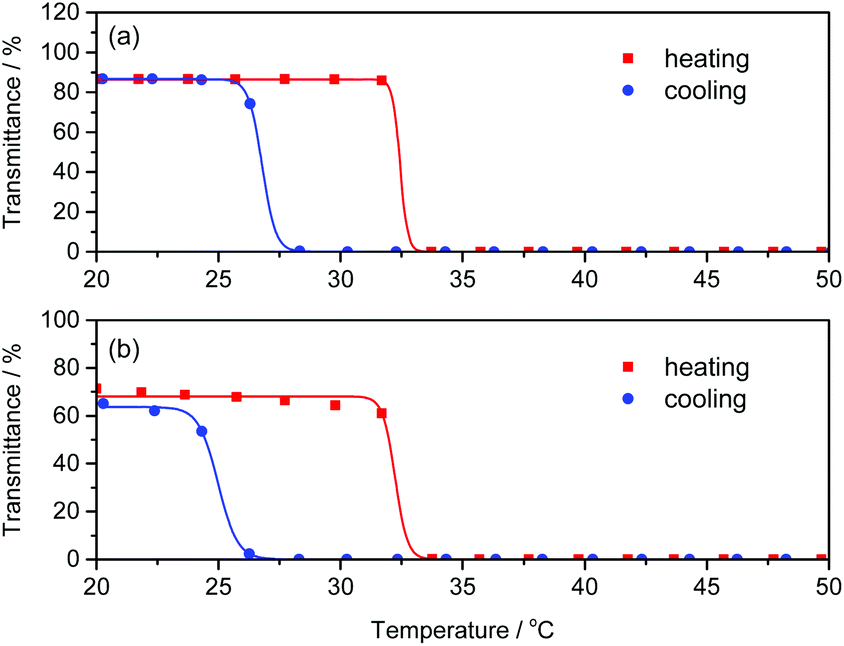
Poly(NIPAm) is known to show a lower critical solution temperature (LCST) at approximately 32 °C due to its coil-globule transition caused by dehydration of the polymer chains.46–48 We investigated the effect of polyAla grafts on the physical properties of poly(NIPAm) by measuring the turbidity of polymer solutions in water. The aqueous solutions of poly(NIPAm-g-Ala) and poly(NIPAm) were subjected to a heating and cooling cycle at a rate of 1 °C min−1, and the transmittance of the solutions was monitored, as shown in Fig. 5. A steep decrease in transmittance was observed at 32.0 and 31.8 °C for polyNIPAm and poly(NIPAm-g-Ala) in the heating process, respectively, indicating that both polymers showed a cloud point attributable to the coil-globule transition of the poly(NIPAm) chains. In the case of poly(NIPAm), a slightly lower cloud point at 26.2 °C was observed in the cooling process. This hysteresis is caused by weak crosslinking via hydrogen bonding between amide bonds in the globule state, as reported in previous studies.49,50 In contrast, poly(NIPAm-g-Ala) showed a lower cloud point at 24.2 °C than poly(NIPAm), indicating a large hysteresis. The cloud point of polymers showing an LCST highly depends on their chemical structures; however, the introduction of polyAla grafts at the poly(NIPAm) side chains only affected the cloud point in the cooling process. This fact indicates that aggregates of the polyAla fragments are assumed to function as cross-linking points in the globule states at a high temperature above the LCST. The transmittance of the poly(NIPAm-g-Ala) solution after the heating-to-cooling cycle became slightly lower than the initial transmittance, also indicating the aggregation of the polyAla fragments.
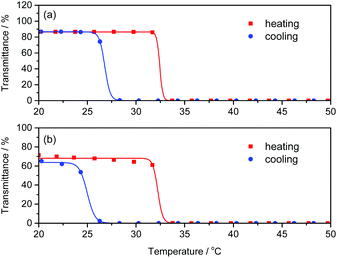 |
| | Fig. 5 Temperature dependence of transmittance at 600 nm for (a) poly(NIPAm) and (b) poly(NIPAm-g-Ala) aqueous solutions (1 wt%) in the heating (red) and cooling (blue) scan at a rate of 1 °C min−1. | |
Conclusions
In conclusion, we successfully synthesized various oligomeric peptides modified with an acrylamide functional group at the N-terminus via chemoenzymatic polymerization catalyzed by papain in the presence of an acrylamide terminal-modifying agent. The acrylamide group was introduced at the polyTyr N-terminus in a one-pot chemoenzymatic synthesis without interacting with the side groups of the Tyr residues, enabling the direct synthesis of a functional peptide with unprotected side groups. Using an alkyne-containing terminal-modifying agent can also introduce an alkyne group at the N-terminus of polyAla. This result indicates that chemoenzymatic one-pot synthesis offers a wide variety of peptide structures. The conjugation of acryl-polyAla with a thiol derivative completely proceeded at the acrylamide moiety via a radical-mediated thiol–ene reaction, which is beneficial for the facile synthesis of block copolymers based on peptides. The obtained acrylamide-modified polyAla was further utilized as a macromonomer to prepare a graft copolymer containing polyAla side chains via radical copolymerization with NIPAm. The graft copolymer poly(NIPAm-g-Ala) showed different LCST behavior from poly(NIPAm), probably due to the aggregation of the polyAla side chains. These results strongly suggest that our methodology for synthesizing terminal-functionalized peptides is a promising tool for constructing various polymeric architectures based on peptide building blocks.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by JST ERATO Grant Number JPMJER1602 and Grants-in-Aid for Young Scientists (B) (17K18361) from the Japan Society for the Promotion of Science (JSPS).
Notes and references
- B. Merrifield, Science, 1986, 232, 341–347 CrossRef CAS.
- G. J. M. Habraken, A. Heise and P. D. Thornton, Macromol. Rapid Commun., 2012, 33, 272–286 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- K. P. McGrath, M. J. Fournier, T. L. Mason and D. A. Tirrell, J. Am. Chem. Soc., 1992, 114, 727–733 CrossRef CAS.
- A. Harada and K. Kataoka, Polym. J., 2017, 50, 95 CrossRef.
- C. A. Machado, I. R. Smith and D. A. Savin, Macromolecules, 2019, 52, 1899–1911 CrossRef CAS.
- V. Mikhalevich, I. Craciun, M. Kyropoulou, C. G. Palivan and W. Meier, Biomacromolecules, 2017, 18, 3471–3480 CrossRef CAS.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS.
- H. Robson Marsden and A. Kros, Macromol. Biosci., 2009, 9, 939–951 CrossRef CAS PubMed.
- K. Wu, J. Yang, Č. Koňák, P. Kopečková and J. Kopeček, Macromol. Chem. Phys., 2008, 209, 467–475 CrossRef CAS.
- F. Sanda, G. Gao and T. Masuda, Macromol. Biosci., 2004, 4, 570–574 CrossRef CAS.
-
K. Tsuchiya, Y. Miyagi, T. Miyamoto, P. G. Gudeangadi and K. Numata, in Enzymatic Polymerization towards Green Polymer Chemistry, ed. S. Kobayashi, H. Uyama and J. Kadokawa, Springer, Singapore, 2019, ch. 8, pp. 233–265 Search PubMed.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 17, 1700177 CrossRef.
- J. Gimenez-Dejoz, K. Tsuchiya and K. Numata, ACS Chem. Biol., 2019, 14, 1280–1292 CrossRef CAS.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
-
K. Tsuchiya and K. Numata, in Green Polymer Chemistry: New Products, Processes, and Applications, American Chemical Society, 2018, vol. 1310, ch. 7, pp. 95–105 Search PubMed.
- K. Tsuchiya and K. Numata, Chem. Commun., 2017, 53, 7318–7321 RSC.
- K. Yazawa, J. Gimenez-Dejoz, H. Masunaga, T. Hikima and K. Numata, Polym. Chem., 2017, 8, 4172–4176 RSC.
- K. Yazawa and K. Numata, Polymers, 2016, 8, 194 CrossRef.
- K. Tsuchiya, H. Masunaga and K. Numata, Biomacromolecules, 2017, 18, 1002–1009 CrossRef CAS.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2016, 16, 1001–1008 CrossRef CAS PubMed.
- K. Tsuchiya, N. Kurokawa, J. Gimenez-Dejoz, P. G. Gudeangadi, H. Masunaga and K. Numata, Polym. J., 2019 DOI:10.1038/s41428-019-0242-z.
- J. M. Ageitos, J.-A. Chuah and K. Numata, Macromol. Biosci., 2015, 15, 990–1003 CrossRef CAS PubMed.
- J. M. Ageitos, P. J. Baker, M. Sugahara and K. Numata, Biomacromolecules, 2013, 14, 3635–3642 CrossRef CAS.
- K. Tsuchiya and K. Numata, ACS Macro Lett., 2017, 6, 103–106 CrossRef CAS.
- P. G. Gudeangadi, K. Tsuchiya, T. Sakai and K. Numata, Polym. Chem., 2018, 9, 2336–2344 RSC.
- T.-B. Yu, J. Z. Bai and Z. Guan, Angew. Chem., Int. Ed., 2009, 48, 1097–1101 CrossRef CAS.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231–5239 CrossRef CAS.
- Q. Yang, Y.-Y. Lai, W.-J. Xiao and H. Alper, Tetrahedron Lett., 2008, 49, 7334–7336 CrossRef CAS.
- J. M. Ageitos, K. Yazawa, A. Tateishi, K. Tsuchiya and K. Numata, Biomacromolecules, 2016, 17, 314–323 CrossRef CAS PubMed.
- P. J. Baker and K. Numata, Biomacromolecules, 2012, 13, 947–951 CrossRef CAS.
- I. S. Aguirre-Díaz, C. Montiel, I. Bustos-Jaimes, Y. Medina-Gonzalez, A. Tecante and M. Gimeno, RSC Adv., 2018, 8, 35936–35945 RSC.
- F. Totsingan, R. Centore and R. A. Gross, Chem. Commun., 2017, 53, 4030–4033 RSC.
- C. Rosu, R. Cueto and P. S. Russo, Langmuir, 2016, 32, 8392–8402 CrossRef CAS.
- H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef CAS.
- R. K. Scheule, F. Cardinaux, G. T. Taylor and H. A. Scheraga, Macromolecules, 1976, 9, 23–33 CrossRef CAS PubMed.
- A. Saha, T. K. Paira, M. Biswas, S. Jana, S. Banerjee and T. K. Mandal, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2313–2319 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, H. Iatrou and S. Pispas, Macromol. Rapid Commun., 2003, 24, 979–1013 CrossRef CAS.
- K. Matyjaszewski, K. L. Beers, A. Kern and S. G. Gaynor, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 823–830 CrossRef CAS.
-
J. T. Yang, C.-S. C. Wu and H. M. Martinez, in Methods in Enzymology, Academic Press, 1986, vol. 130, pp. 208–269 Search PubMed.
- C. Riekel, C. Bränden, C. Craig, C. Ferrero, F. Heidelbach and M. Müller, Int. J. Biol. Macromol., 1999, 24, 179–186 CrossRef CAS.
- J. M. Gosline, P. A. Guerette, C. S. Ortlepp and K. N. Savage, J. Exp. Biol., 1999, 202, 3295–3303 CAS.
- H. Kojima, Polym. J., 2018, 50, 411–418 CrossRef CAS.
- T. Kawaguchi, Y. Kojima, M. Osa and T. Yoshizaki, Polym. J., 2008, 40, 455 CrossRef CAS.
- M. Ito and T. Ishizone, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4832–4845 CrossRef CAS.
- H. Lai, G. Chen, P. Wu and Z. Li, Soft Matter, 2012, 8, 2662–2670 RSC.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9PY01335K |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Keiji
Numata
* and
Keiji
Numata