
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the self-assembly and shape transformation of poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-PDLLA) polymersomes by tailoring solvent-polymer interactions†
Imke A. B.
Pijpers
a,
Fenghua
Meng
b,
Jan C. M.
van Hest
*a and
Loai K. E. A.
Abdelmohsen
 *a
*a
aDepartment of Bio-Organic Chemistry, Institute of Complex Molecular Systems (ICMS), Eindhoven University of Technology, Het Kranenveld 14, 5600 MB Eindhoven, The Netherlands. E-mail: j.c.m.v.hest@tue.nl; l.k.e.a.abdemohsen@tue.nl
bBiomedical Polymers Laboratory, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, PR China
First published on 10th September 2019
Abstract
The assembly of amphiphilic block copolymers in well-defined polymer nanoparticles is an intricate interplay between polymer composition, method of assembly and the properties of the organic solvent used to dissolve the polymer building blocks. Remarkably, the role of organic solvent composition has been much less studied in comparison to the other two parameters. Here we have systematically investigated the effect of organic solvent composition, namely, the ratio between tetrahydrofuran and dioxane, on the self-assembly of PEG-PDLLA copolymers into polymer vesicles and their subsequent shape change in different topologies. Uncovering such physical basis unlocks new opportunities for the development of complex nanoparticles without the need of extended polymer engineering processes.
Introduction
The molecular engineering of polymeric architectures from amphiphilic copolymers has been documented since the findings of Eisenberg.1 Despite the large body of research currently done on the subject, achieving control over the morphology of such supramolecular structures has proven to remain challenging.2–6 Although we have much more detailed understanding of the morphological features on the nanoscale, it is observed that apparently small changes in chemical composition (such as polymer chain length, polydispersity, composition, blending etc.) and fabrication methodology have significant consequences for the resulting particle morphology.7–14 External factors, such as temperature, pH and osmotic pressure contribute to the dynamicity of the system and, indeed, to the final supramolecular structure.15–20 With this in mind, researchers from diverse backgrounds have sought to unravel the physicochemical basis for copolymer self-assembly in order to enhance our fundamental understanding of these systems. Often, however, only the relationship between the polymeric composition and the resulting assembly is considered. Remarkably, the (organic) solvent that is crucial in the formation and subsequent manipulation (such as resizing/shape change) of the assemblies is an underexposed element in the entire structure formation. Commonly, the choice of solvent is based on its miscibility with water, and its ability to molecularly dissolve the copolymeric material. In this paper, we show that the composition of the organic solvent ensures the creation of a multitude of vesicular topologies (both during formation and manipulation), whereby small variations can have major consequences. A better understanding of this effect increases the reproducibility of the formation process and gives access to a wider range of structures.The self-assembly of polymersomes can be performed in a wide variety of manners, one of which is the solvent switch methodology, which involves dissolving a copolymer into a ‘good’ solvent followed by slow addition of a ‘poor’ solvent.16,21 Increasing the amount of the poor solvent in the mixture leads to hydrophobic membrane-formation – as a result of copolymer reorganization. The outcome of such solvent switch methodology largely depends on the respective properties of the organic solvent and its interaction with the polymer building blocks. In general, good solvents promote material-solvent interactions, wherein polymers can be molecularly dissolved. On another note, non-solvents, or poor solvents, stimulate polymer–polymer (hydrophobic) interactions where polymer chains are folded into each other and become self-knotted.22,23 Indeed, the solvation and de-solvation of polymers and the way they influence polymer aggregation play a significant role in driving the self-assembly process toward the formation of bilayered membrane or micellar structures, for instance.24 Moreover, solvents impact the way the polymers are packed in the self-assembled structures, which in turn affects the way in which they undergo subsequent shape change process.25–28
In our group, we have experience with the self-assembly and the shape transformation of various copolymers, one of which is poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-PDLLA). By applying the above mentioned solvent switch methodology, such copolymers assemble into spherical polymersomes (Fig. 1). Upon their exposure to hypertonic conditions, these spherical structures can undergo an osmotic-induced shape transformation process into prolates or oblates, depending on the chemical composition of the copolymer used during the initial assembly.29 Resizing such polymersomes was shown to be feasible by repeated extrusion – a process aiming to yield smaller sized nanoparticles.30
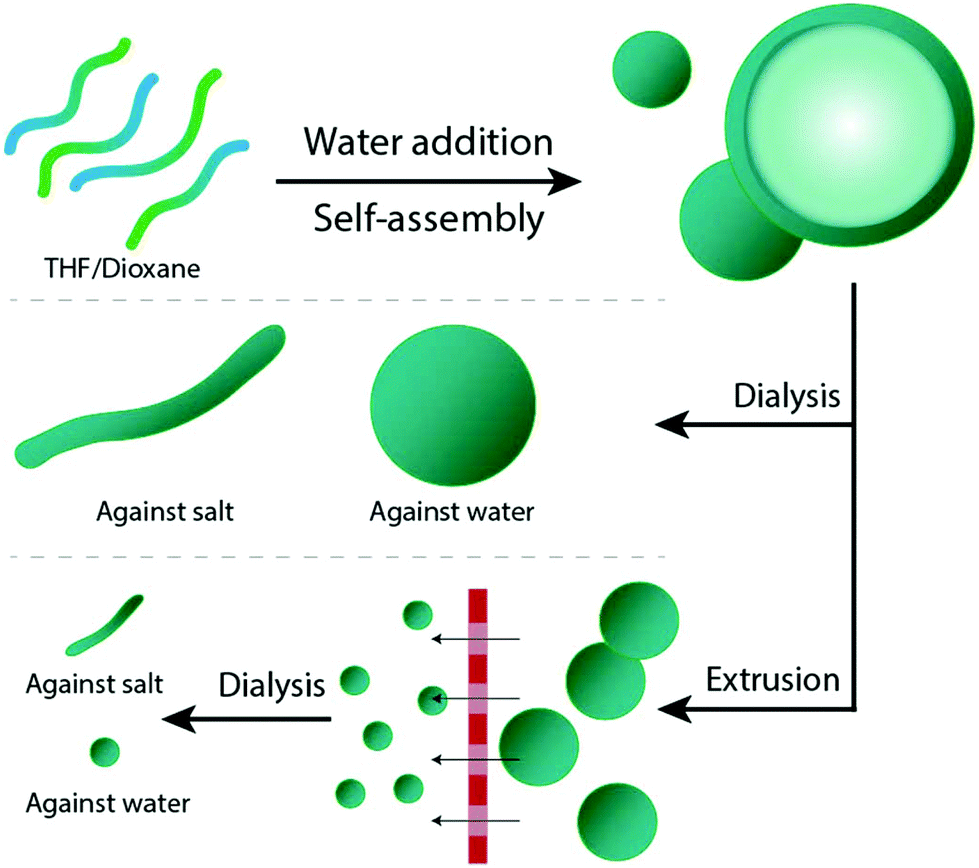 | ||
| Fig. 1 Schematic describing the self-assembly of PEG-PDLLA polymers into polymersomes and subsequent morphology control. First, polymers are assembled into polymersomes. Then, particles are either dialyzed against water to yield spherical particles or salt to induce shape transformation into tubes; or particles are first extruded and then dialyzed to form spherical polymersomes or tubes. | ||
Although the changes in the chemical composition of PEG-PDLLA affect the self-assembly and shape transformation, it is evident that the choice of solvent is crucial to bring reproducibility to this system. Herein, we provide an insight to our initial hypothesis regarding the influence of the solvent composition on the self-assembly of PEG-PDLLA polymersomes and subsequent shape transformation process – a study that aims to enhance our fundamental understanding in order to bring control and reproducibility to these systems, broadening our accessibility to new structures.
Results and discussion
A mixture of tetrahydrofuran (THF) and dioxane (in different ratios) is typically used as solvent for PEG-PDLLA copolymers, and as a plasticizer for the resulting bilayered membrane.19,29 In order to study how the composition of the organic solvent affects the behaviour of PEG22-PDLLA45 during the self-assembly and shape transformation processes, we systematically varied the ratio of THF and dioxane during the initial polymersome formation. Thereafter, the results of both the self-assembly process and shape transformation were investigated.First, PEG22-PDLLA45 with Đ ≤ 1.2 copolymer was synthesized following a previously published method.19 In short, ring-opening polymerization (ROP) was carried out with D,L-lactide, using methoxy PEG-OH as macroinitiator and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as organocatalyst. The resulting chain length and Đ were determined by 1H-NMR and Gel Permeation Chromatography (GPC), respectively (Fig. S1–3†). Previously, such copolymers were shown to form spherical polymersomes, and upon their exposure to hypertonic conditions, elongated tubes could be formed. Initial polymersome formation was performed by dissolving the polymers in a mixture of THF and dioxane with a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), followed by slow addition of an equal volume of water to the mixture. Subsequently, the organic solvent was removed via dialysis against MilliQ at 4 °C. Inverting the ratio of THF:dioxane to 1:4 during the self-assembly process resulted in unreproducible polymersome formation and major formation of aggregates – even after removing organic solvents, which was expected to force membrane rearrangement (Fig. 2).
:1 (v/v), followed by slow addition of an equal volume of water to the mixture. Subsequently, the organic solvent was removed via dialysis against MilliQ at 4 °C. Inverting the ratio of THF:dioxane to 1:4 during the self-assembly process resulted in unreproducible polymersome formation and major formation of aggregates – even after removing organic solvents, which was expected to force membrane rearrangement (Fig. 2).
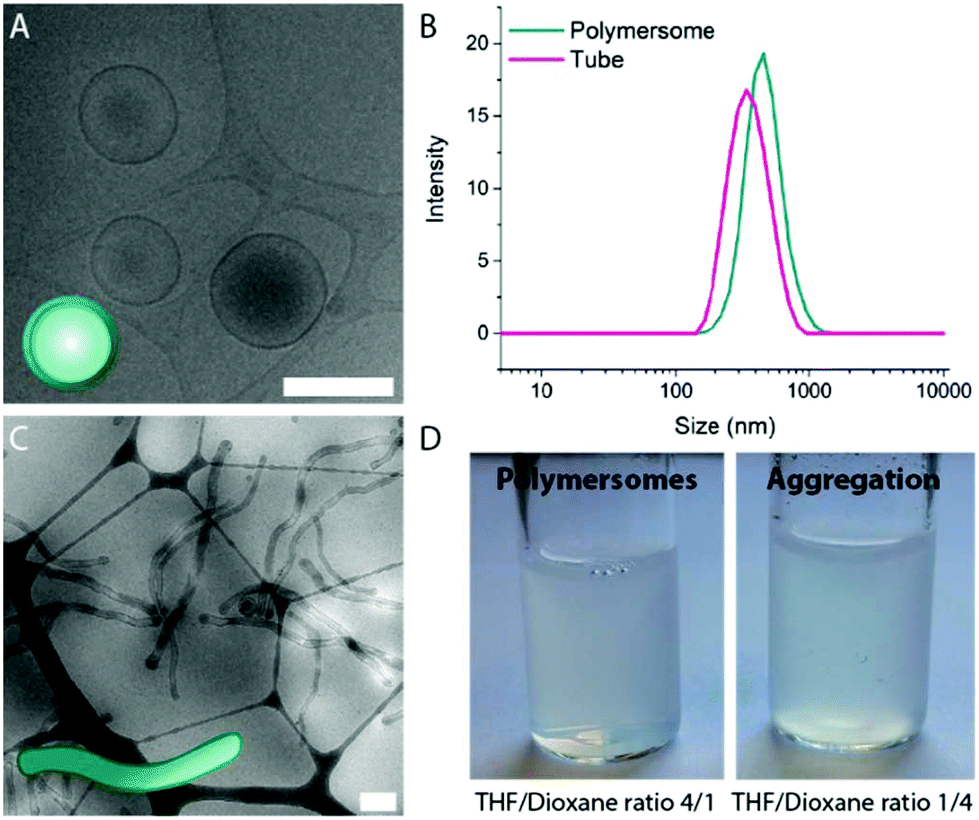 | ||
| Fig. 2 Cryo-TEM images of spherical polymersomes (A) and tubular polymersomes (C). (B) Hydrodynamic size of polymersomes and tubes measured by DLS. Please note the difference in size in polymersomes and tubes which is a result of the volume reduction following the shape transformation process. (D) Formation of polymersomes (left) and aggregation (right) with varying THF:dioxane ratio. Scale bars are 500 nm. | ||
This result indicates that indeed THF is crucial to the formation of stable PEG-PDLLA polymersomes, as the increased polarity of THF increases repulsive (electrostatic) interactions between polymer chains, which is necessary for the formation of ordered polymeric membranes with well-defined organization. As such, undefined polymer aggregates were formed by increasing polymer–polymer charge interactions – a result of decreasing the amount of THF during the self-assembly in the initial polymer solution.
Systematic variation of the THF and dioxane ratios in the initial polymer solution, and accordingly, in the final assembled-polymersome solution resulted in clear differences in the formation of spherical vesicles. High amounts of THF resulted in the self-assembly of spherical polymersomes, while addition of increasing volumes of dioxane induced aggregation in the resulted structures (Table 1). Contrarily, the presence and quantity of dioxane in the polymer mixture proved to influence the shape transformation of the spherical particles (Fig. 3 and S4B, 5†). At the first instance, it was clear that the size of the formed spherical polymersomes was bigger at higher dioxane ratios (Fig. S4†). Moreover, the length of the resulting tubes increases at a higher dioxane to THF ratio. Additionally, aggregates were observed when no THF was present in the system, confirming the importance of THF in modulating the stability of the assembled membrane. From cryo-TEM image analysis, it is apparent that the mean aspect ratio of the tubular structures increased with increasing dioxane content (Fig. 3F and S6†).
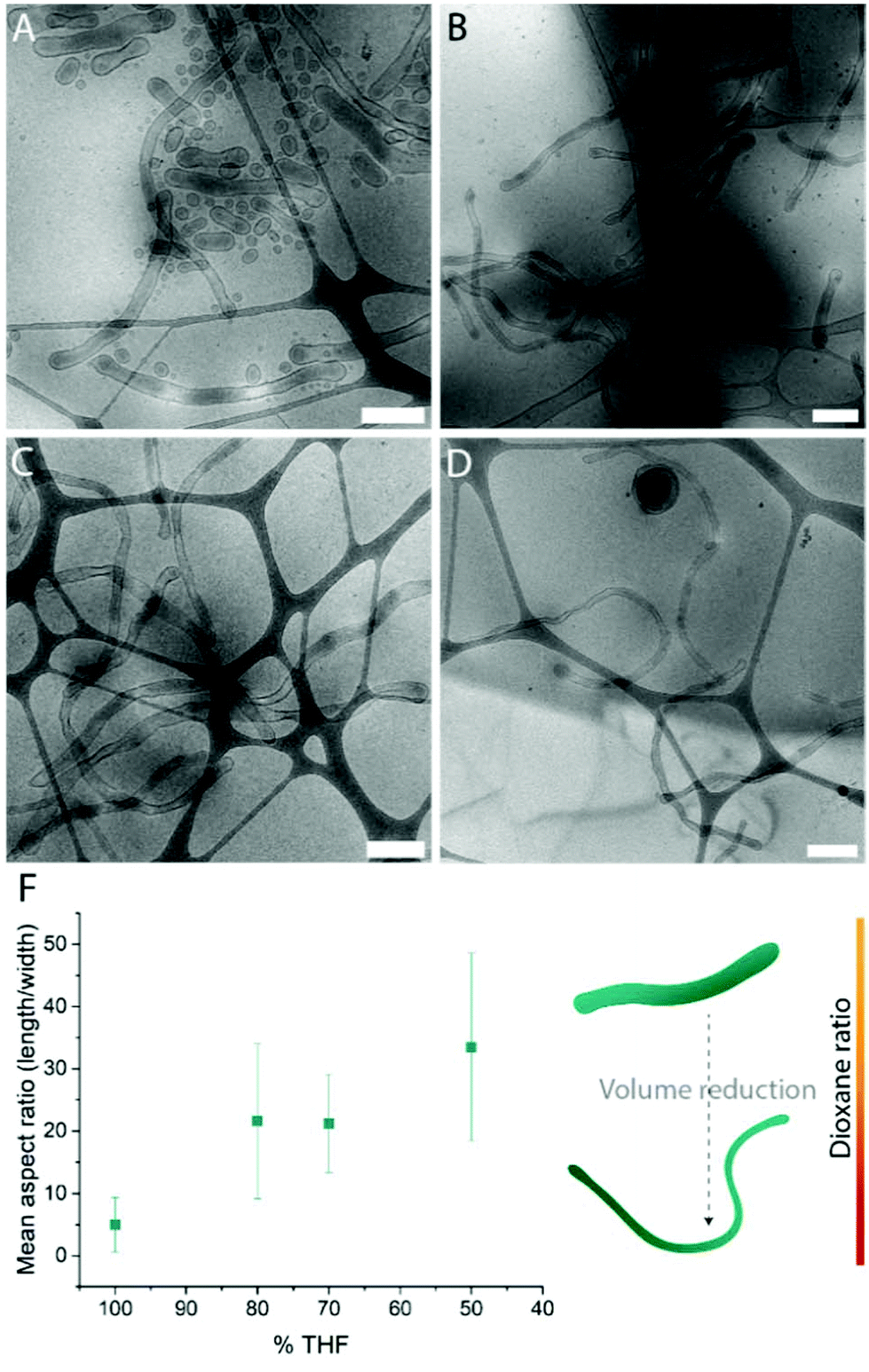 | ||
| Fig. 3 Cryo-TEM images of tubes formed with THF:dioxane ratio (v/v%) (A) 100:0, (B) 80:20, (C) 70:30 and (D) 50:50. Mean aspect ratio measured from image analysis (F). Scale bars are 500 nm. | ||
:dioxane solvent ratio during polymersome formation and shape transformation
| Solvent composition scheme | |||||
|---|---|---|---|---|---|
| Ratio solvents | Initial morphology | Structure after shape transformation | Mean aspect ratio | ||
| Dioxane | Tetrahydrofuran | ||||
|
0 | 10 | Spheres | Tubes + prolates | 4.99 |
| 2 | 8 | Spheres | Tubes | 21.60 | |
| 3 | 7 | Spheres | Tubes | 21.22 | |
| 5 | 5 | Spheres | Tubes | 33.49 | |
| 7 | 3 | Aggregation | Aggregation | ||
| 8 | 2 | Aggregation | Aggregation | ||
| 10 | 0 | Aggregation | Aggregation | ||
Interestingly, the presence of nested vesicles, closed stomatocytes, became increasingly apparent at higher THF ratios (Fig. S5†). Experimental details are given in Table 1.
Indeed, both solvents play their respective part in the self-assembly process; their individual properties, such as dielectric constant and solubility parameter, have significant consequences for how PEG and PDLLA chains are folded in the membrane. The dielectric constant (ε) can be used as an indication of solvents’ polarity, with values for THF and dioxane being ε = 7.6 and ε = 2.3, respectively. The higher polarity of THF results in increased repulsive interactions between the hydrophilic chains of the membrane. Dioxane, having a lower polarity, leads to weaker repulsive interactions.31 As a consequence, the ratio between both THF/dioxane in the solvent mixture determines the polymer conformation (or stretching), and therefore membrane architecture.22 In general, solvent-induced repulsive (electrostatic) interactions among polymeric chains of copolymeric assemblies originate predominantly from electrostatic repulsion between hydrophilic polymer chains,31 and possibly from adsorption of negative ions onto the hydrophobic chains.32 The effect of various solvents with distinct dielectric constants on the resulting vesicle morphology was established by Yu and Eisenberg, where varying amounts of DMF, THF and dioxane yielded vesicles, spheres or rods with various sizes.33 These results were explained to be a consequence of a combination of effects that involve interactions between the solvent used and the copolymers, which in turn affects both polymer folding in solution and the repulsion of chains in the membrane of the resulting assembly. Moreover, the organic solvent composition influences the dimensions of the polymer during self-assembly (i.e. packing parameter) and the critical water content necessary to initiate self-assembly. Our hypothesis states that based on the ε values, an increase in THF content should lead to the self-assembly of stable structures. On the other hand, dioxane can modulate the rigidity of the polymer chains in the membrane, allowing subsequent shape transformation process to occur. From our research into vesicular shape changes of polymersomes, we understand that both membrane flexibility and vesicle volume reduction are deciding factors in whether a spherical polymersome undergoes shape transformation. Originally, the shape change of PEG22-PDLLA45 spherical polymersomes was induced by dialyzing flexible spherical polymersomes (in 50% organic solvent v/v) against salt solution. Indeed, organic solvent directly contributes towards the degree by which the self-assembled membrane is plasticized, facilitating efflux of solvents from the polymersomes’ lumen upon their exposure to hypertonic conditions. Eqn 1 describes the membrane's bending energy (Eb) in terms of its bending rigidity k.
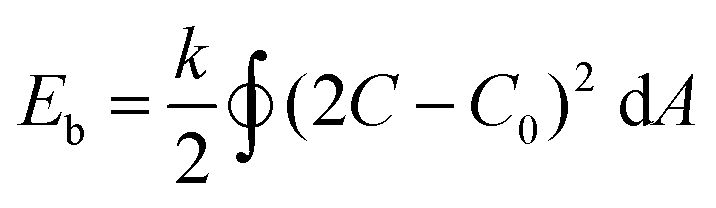 | (1) |
In order to confirm our hypothesis on the effect of both THF and dioxane on PDLLA-based polymersomes, we set out to study their extrusion (i.e. mechanical resizing), and the consecutive shape transformation of the resulted structures in presence of different THF:dioxane ratios. In general, extrusion of polymeric particles require high degree of membrane flexibility. Such extrusion process results in the formation of small polymersomes (ca. 100 nm), which possess higher surface curvature when compared to their counterparts (ca. 400 nm) (Fig. S7†). Such size control over PEG-PDLLA polymersomes was previously realized by repeated extrusion of ca. 400 nm polymersomes, which are suspended in a higher content of organic solvent to water (66/33 v/v%). This solvent condition ensured membrane flexibility as well as permeability to allow efflux of solvent from the polymersomes lumen during their shape transformation into tubular morphologies. Without dioxane present in the solution, ±400 nm polymersomes were successfully extruded into ±150 nm polymersomes (Fig. 4A and S8, 9†); however, no shape transformation could be observed when osmotic shock was applied. These results highlight the role dioxane plays in reducing k and Eb, so that the shape transformation of high surface curvature structures can take place.
 | ||
| Fig. 4 Cryo-TEM images depicting the effect of increasing the concentration of dioxane on the shape transformation of extruded polymersomes (ca. 150 nm) Respective THF:dioxane ratios (v/v%) are: (A) 100:0, (B) 80:20, (C) 70:30, (D) 50:50 and (E) 30:70. The mean aspect ratio calculated from image analysis (F) depicts the increase in length-to-width variation with increasing dioxane ratios. Scale bars are 200 nm. | ||
Higher aspect ratio particles could be obtained by further increasing the concentration of dioxane to a final THF:dioxane ratio of 30:70 (Fig. 4 and S8–10†), elongating spherical nanoparticles into prolates and eventually tubes – demonstrating the role dioxane plays in plasticizing PDLLA membrane. At a mixture of THF and dioxane with a ratio 1:4, the shape transformation of the extruded polymersomes, in most cases, yielded a mixture of polymersomes and aggregates – again, highlighting the role of THF in modulating the polymer-solvent interactions and yielding a well-ordered membrane. Table 2 displays the results of the extrusion of polymersomes and their shape transformation with varying solvent composition.
:dioxane ratios in extrusion of polymersomes and subsequent shape transformation
| Solvent composition scheme | |||||
|---|---|---|---|---|---|
| Ratio solvents | Initial morphology | Structure after shape transformation | Mean aspect ratio | ||
| Dioxane | Tetrahydrofuran | ||||
|
0 | 10 | Spheres | Spheres | 0.99 |
| 2 | 8 | Spheres | Spheres + prolates | 1.33 | |
| 3 | 7 | Spheres | Spheres + prolates | 1.82 | |
| 5 | 5 | Spheres | Tubes | 2.19 | |
| 7 | 3 | Spheres | Tubes | 4.89 | |
| 8 | 2 | Aggregation | Aggregation | ||
| 10 | 0 | Aggregation | Aggregation | ||
From these experiments it is clear that the properties of the solvent are of utmost importance in modulating not only membrane formation but also the bending rigidity, either allowing or impeding shape transformation. Furthermore, it is clear that increasing solvent polarity (by increasing the amount of THF) is a significant driving factor in tailoring repulsive interactions between PEG-PDLLA polymer chains to yield stable assemblies. On the other hand, membrane flexibility is necessary to induce shape transformation; as evident by the formation of longer tubes at higher dioxane content.
Conclusions
The intricate interplay between dioxane and THF demonstrates that polymer swelling in cosolvents contributes immensely to the supramolecular structure of the self-assembled polymers and, in turn, morphology can be controlled. This provides us with means to control the morphology of PEG-PDLLA nanoparticles without directly adjusting the chemical composition of the copolymer, and therefore, facilitating easier techniques to control nanoparticle morphology for applications in bio-(medical) chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the NWO-NSFC Advanced Materials (project 792.001.015) and the National Natural Science Foundation of China (NSFC 51561135010).References
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- A. C. Anselmo, C. L. Modery-Pawlowski, S. Menegatti, S. Kumar, D. R. Vogus, L. L. Tian, M. Chen, T. M. Squires, A. Sen Gupta and S. Mitragotri, ACS Nano, 2014, 8, 11243–11253 CrossRef CAS.
- J. Wang, J. D. Byrne, M. E. Napier and J. M. Desimone, Small, 2011, 7, 1919–1931 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753–10758 CrossRef CAS.
- R. Salva, J. F. Le Meins, O. Sandre, A. Bruîlet, M. Schmutz, P. Guenoun and S. Lecommandoux, ACS Nano, 2013, 7, 9298–9311 CrossRef CAS.
- M. C. M. van Oers, W. S. Veldmate, J. C. M. van Hest and F. P. J. T. Rutjes, Polym. Chem., 2015, 6, 5358–5361 RSC.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- J. F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E, 2011, 34, 1–17 CrossRef.
- P. Schuetz, M. J. Greenall, J. Bent, S. Furzeland, D. Atkins, M. F. Butler, T. C. B. McLeish and D. M. A. Buzza, Soft Matter, 2011, 7, 749–759 RSC.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- I. A. B. Pijpers, L. K. E. A. Abdelmohsen, Y. Xia, S. Cao, D. S. Williams, F. Meng, J. C. M. van Hest and Z. Zhong, Adv. Ther., 2018, 1800068 CrossRef.
- Z. Li, J. Ma, N. S. Lee and K. L. Wooley, J. Am. Chem. Soc., 2011, 133, 1228–1231 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- J. D. Robertson, G. Yealland, M. Avila-Olias, L. Chierico, O. Bandmann, S. A. Renshaw and G. Battaglia, ACS Nano, 2014, 8, 4650–4661 CrossRef CAS.
- U. Borchert, U. Lipprandt, M. Bilang, A. Kimpfler, A. Rank, R. Peschka-Süss, R. Schubert, P. Lindner and S. Förster, Langmuir, 2006, 22, 5843–5847 CrossRef CAS.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. Van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H. A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS.
- P. L. Soo and A. D. I. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- E. Antoniou and P. Alexandridis, Eur. Polym. J., 2010, 46, 324–335 CrossRef CAS.
- V. V. Vasilevskaya, P. G. Khalatur and A. R. Khokhlov, Macromolecules, 2003, 36, 10103–10111 CrossRef CAS.
- D. J. Pochan, K. L. Wooley, H. Cui, S. Zhong and Z. Chen, Science, 2007, 317, 647–650 CrossRef.
- K. T. Kim, J. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. Van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS.
- C. K. Wong, A. F. Mason, M. H. Stenzel and P. Thordarson, Nat. Commun., 2017, 8, 1–10 CrossRef CAS.
- R. P. M. Lafleur, X. Lou, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2018, 9, 6199–6209 RSC.
- R. S. M. Rikken, H. Engelkamp, R. J. M. Nolte, J. C. Maan, J. C. M. van Hest, D. A. Wilson and P. C. M. Christianen, Nat. Commun., 2016, 7, 12606 CrossRef CAS.
- I. A. B. Pijpers, L. K. E. A. Abdelmohsen, D. S. Williams and J. C. M. Van Hest, ACS Macro Lett., 2017, 6, 1217–1222 CrossRef CAS.
- A. C. Wauters, I. A. B. Pijpers, A. F. Mason, D. S. Williams, J. Tel, L. K. E. A. Abdelmohsen and J. C. M. Van Hest, Biomacromolecules, 2018, 20, 177–183 CrossRef.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS.
- R. W. N. Nugroho, T. Pettersson, K. Odelius, A. Höglund and A.-C. Albertsson, Langmuir, 2013, 29, 8873–8881 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 9297, 1144–1154 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01089k |
| This journal is © The Royal Society of Chemistry 2020 |