
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical determination of two-photon absorption in biologically relevant pterin derivatives
Thomas
Malcomson
 *a and
Martin J.
Paterson
b
*a and
Martin J.
Paterson
b
aDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: t.malcomson@lancaster.ac.uk
bSchool of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: m.j.paterson@hw.ac.uk
First published on 24th September 2020
Abstract
Given the prevalence of fluorescence spectroscopy in biological systems, and the prevalence of pterin derivatives throughout biological systems, presented here is an assessment of the two-photon absorption spectroscopy as it applies to a range of the most commonly studied pterin derivatives. QR-CAMB3LYP//ccpVTZ calculations suggest that the use of two-photon spectroscopic methods would enable a more capable differentiation between closely related derivatives in comparison to the one-photon spectra, which show minimal qualitative deviation. Study of short tail derivatives shows that, in most cases, two-photon accessible states solely involve the π* LUMO as the particle orbital, with biopterin, neopterin, and 6-(hydroxymethyl)pterin presenting exceptional potential for targetting. Investigation of derivatives in which the tail contains an aromatic ring resulted in the observation of a series of two-photon accessible states involving charge transfer from the tail to the pterin moiety, the cross sections of which are highly dependent on the adoption of a planar geometry. The observation of these states presents a novel method for tracking the substitution of biologically important molecules such as folic acid and 5-methenyltetrahydrofolylpolyglutamate.
1. Introduction
The pterin family of molecules are defined as low weight heterocycles derived from the substitution of a pteridine ring with a amino (–NH2) and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups at the C2 and C4 ring positions, respectively. Although both nitrogen and oxygen centres are possible targets for nucleophilic attack, the central pterin moiety (Fig. 1) is more commonly functionalised through the addition of substituent groups to the C6 position, located on the pyrazine ring.1
O) groups at the C2 and C4 ring positions, respectively. Although both nitrogen and oxygen centres are possible targets for nucleophilic attack, the central pterin moiety (Fig. 1) is more commonly functionalised through the addition of substituent groups to the C6 position, located on the pyrazine ring.1
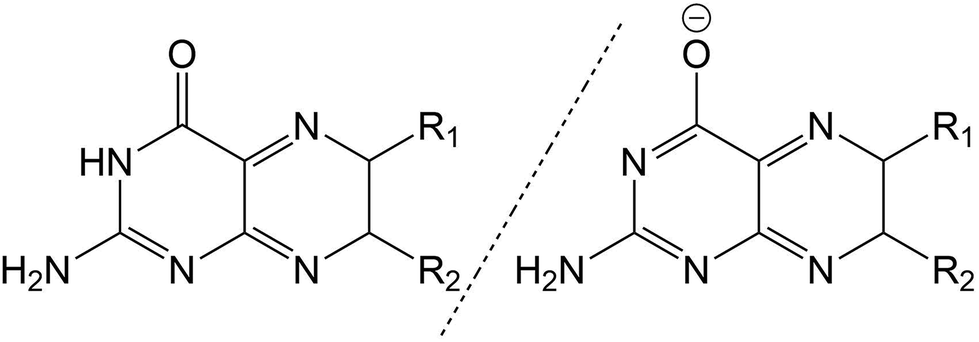 | ||
| Fig. 1 Structure of pterin moiety in its acidic (left) and basic (right) states. | ||
Behaving as weak acids in aqueous solution, pterins form a dominant equilibrium at pH > 5 consisting of the acidic (amino) and basic (phenolate) form (Fig. 1)2 with the pKa of this equilibrium, centred around the protonation state of the N3 proton, is ca. 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 for the derivatives shown in Table 1. The pKa of other pterin-based protons, specifically those located on the N1 amino group, are <2.2
3 for the derivatives shown in Table 1. The pKa of other pterin-based protons, specifically those located on the N1 amino group, are <2.2
| State | Acid | Base | ||
|---|---|---|---|---|
| Energy | σ TP | Energy | σ TP | |
| 1 | 3.8466 | 0.000 | 3.6378 | 0.000 |
| 2 | 4.3353 | 0.058 | 3.7888 | 0.077 |
| 3 | 4.7731 | 0.001 | 4.2833 | 0.000 |
| 4 | 4.9175 | 0.000 | 4.6724 | 0.026 |
| 5 | 5.0735 | 0.164 | 4.7912 | 0.002 |
| 6 | 5.4911 | 0.006 | 4.9873 | 0.003 |
| 7 | 6.0768 | 0.006 | 5.3212 | 0.286 |
| 8 | 6.2832 | 0.008 | 5.6911 | 0.007 |
| 9 | 6.3615 | 0.260 | 5.8665 | 0.010 |
| 10 | 6.5497 | 0.332 | 5.9080 | 0.002 |
Throughout this work, for the ease of discussion, the acidic and basic forms of each molecule, when appropriate, will be marked by a subscript letter (PTa and PTb representing the acidic and basic forms of the unsubstituted pterin moiety, respectively).
Pterins, and their derivatives, are found throughout biology. While their oxidised states have been found extensive medical use, acting as markers for a range of conditions, including: vitiligo,4 cardiovascular diseases,5,6 along with activation of immune responses and synthesis of neurotransmitters,7,8 pterins are also found naturally within human biology; folic acid (vit B9) acts as a coenzyme during reactions involved in the synthesis of both purine and pyridine DNA bases.9 Biopterin (BPT) has been found taking the role of a coenzyme in hydroxylation reactions in the metabolism of both amino acids and nitric oxide10,11 while neopterin (NPT), a BPT metabolite, is synthesised predominantly in activated macrophages with high levels of NPT found in response to infections caused by viruses, parasites and intracellular bacteria.12–17 Despite the role of 5,10-methenyltetrahydrofolylpolyglutamate, a derivative of folic acid (FA), in DNA repair as a light-harvesting antenna in DNA photolyases,18–20 oxidised pterins have also been shown to cause photo-activated DNA damage when exposed to UV light.21–25
The ability of pterins to produce singlet oxygen (O2(1Δg), denoted as 1O2 for simplicity)26,27 has garnered particular interest due to its role in photodynamic therapy (PDT).28,29 This metastable state of molecular oxygen, produced primarily through photosensitisation, is significantly more reactive than the triplet ground state (O2(3∑−g)). The pterin-induced production of 1O2 is brought about due to ready access of the pterin molecules to a triplet excited state through intersystem crossing,30,31 enabling a spin crossover with the 3O2 state of molecular oxygen in place of the commonly observed pterin phosphorescence.26,32
However, the ideal wavelength of light needed to achieve the production of singlet oxygen is in the region of 350 nm which has a very poor tissue penetration value as, due to scattering from the cell nuclei, mitochondria, the Golgi apparatus and the cellular surface itself,33,34 the optical penetration of biological tissue is low, usually measured in millimetres.35 Tissue penetration varies greatly depending on the tissue type and the wavelength used, with longer wavelengths appearing to penetrate to a greater degree, with Stolik et al. reporting a depth of 4.23 ± 0.03 mm.35
While 1O2 has been shown to be a dominant mediator of phototoxic effects, it is a short lived species (<200 ns in vitro).36–38 As a result of this, the diffusion of 1O2 is limited to short distances (≈1 μm),39–41 limiting the cytotoxicity to immediate area around the production location. The low diffusion of 1O2 through a biological medium does, however, limit its use as a photoactivated target due to the tissue depth penetration of the UV light required to excite the pterin molecules.42,43 This limitation can be reduced through with the use of near IR light (λ = 600–100 nm) via two-photon absorption (TPA) which, in addition to the increased tissue penetration of higher wavelength light, increases the spatial resolution of photo-activation.
Despite this promising avenue of investigation, coupled with the pterin molecule acting as a design precursor for a number of fluorescent DNA analogues44,45 and the otherwise extensive detail to which the photochemistry of these molecules has been investigated,26,27,30–32,46 studies of the potential two-photon activation of pterin derivatives is poorly explored. In response to this, we present here a study of the susceptibility of a series of well studied, biochemically relevant pterin derivatives to two-photon activation through the use of quadratic response (QR) density functional theory (DFT) methodologies. The use of these methodologies for the accurate determination of TPA spectra has been well established across a range of molecules of both biological and photochemical importance.47–53
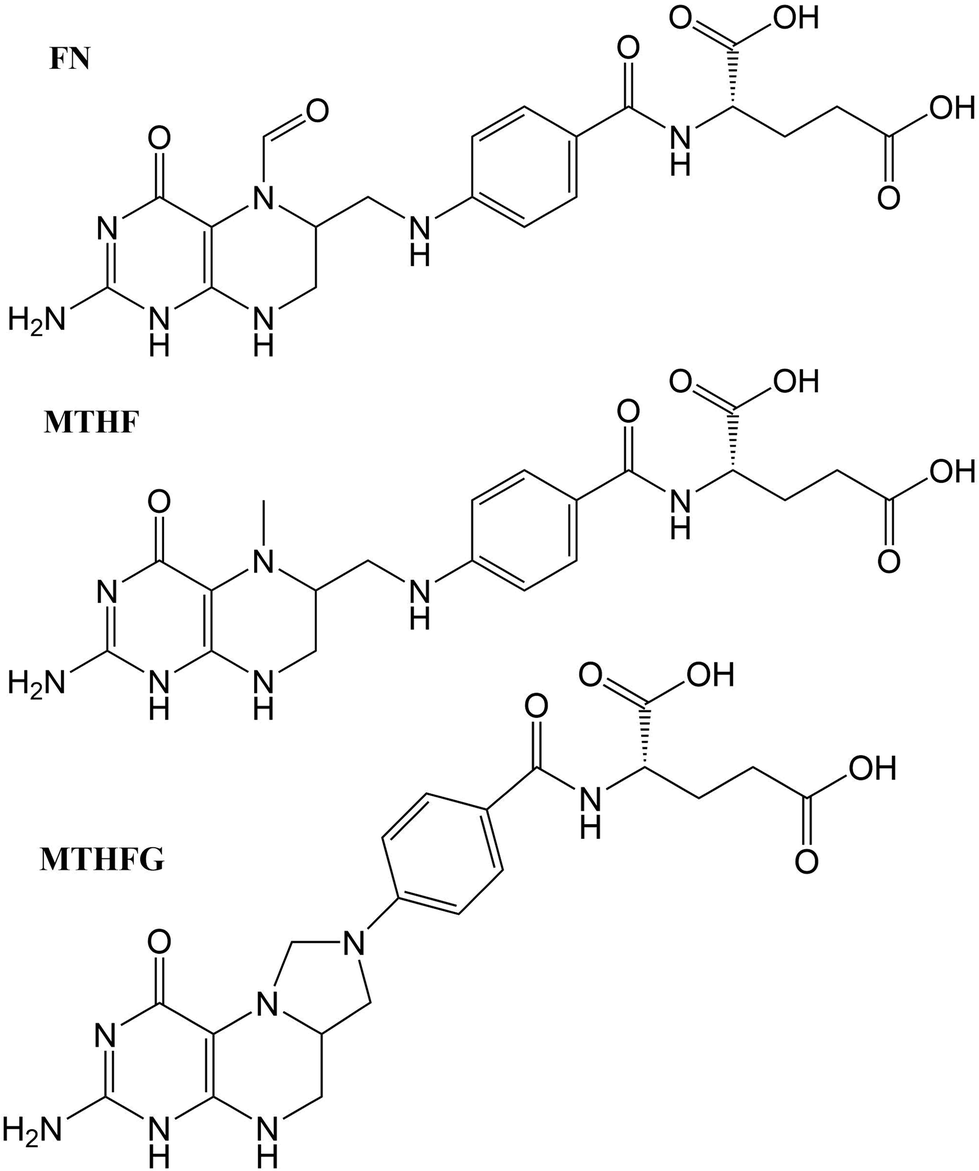
The structures studied here are presented in three groups: the first involves small functional group alterations to the core pterin moiety at the C6 (Table 1); the second involves the inclusion of extended side chains (Fig. 2) centred around the folic acid structure (FA); the final grouping of structures (Fig. 3), centred around 5-methenyltetrahydrofolylpolyglutamate (MTHFG), denote a series of biological, pterin derived, cofactors. These groups are selected to provide a thorough representation of the TPA viability of the pterin family of molecules throughout their biological function.
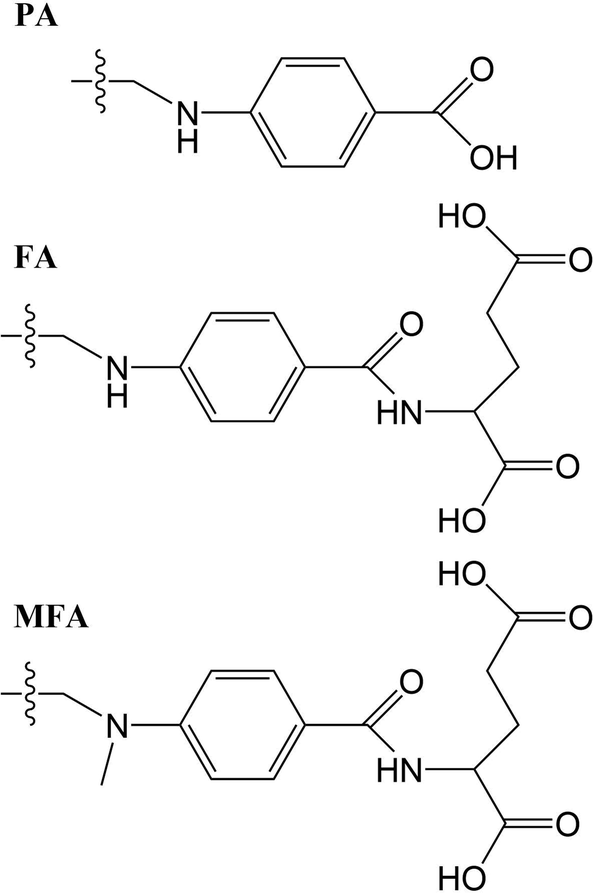 | ||
| Fig. 2 Extended R-groups for pterin derivatives. | ||
 | ||
| Fig. 3 One-photon absorption spectra of pterin derivatives in their acidic state. | ||
The successful determination of the TPA spectra for these derivatives opens up a number of avenues for further investigation, from TPA access to the triplet states of these derivatives and their potential for 1O2 production, to the ability to track the presence of specific derivatives despite the one-photon absorption (OPA) spectra of these structures showing little qualitative variation.
2. Theoretical methods
Geometry optimisations and TD-DFT calculations were carried out with the Gaussian16 software package.54,55 Optimisations utilised the B3LYP functional56–60 with Grimme's D3 dispersion61 and Becke–Johnson dampening62–65 along with the 6-311G** basis set66 with the determination of structural minima conducted through frequency analysis and noted by the presence of only positive curvature; the use of this model chemistry has been well established for giving appropriate geometry determinations for molecules of this type.17,67–71 TD-DFT data was determined using a series of DFT functionals, namely: CAM-B3LYP,72 B3LYP,56–60 BP8673,74 and PBE0,75 each with the Dunning cc-pVTZ basis.76 CAM-B3LYP values are reported here due to their agreement with the UV-Vis spectra of folic acid,77 as well as its robust performance on similar organic molecules.78,79 The use of CAM-B3LYP for the QR-DFT calculations has been well established in providing high accuracy values for similar systems.47–49,80–83Two-photon cross sections (σTP), as defined through QR-DFT implemented in Dalton,84 are determined by:
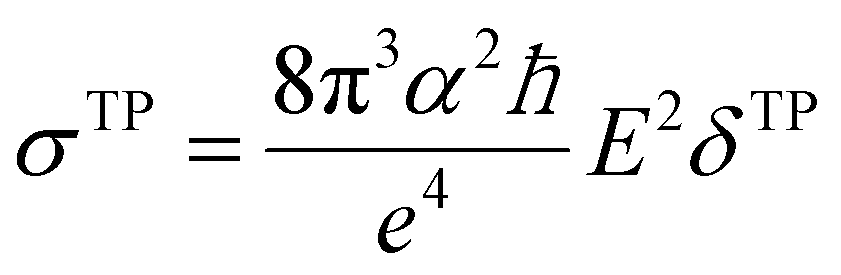 | (1) |
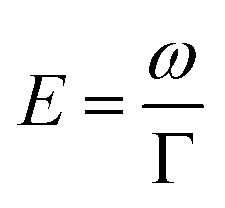 | (2) |
| δTP = FδF + GδG + HδH | (3) |
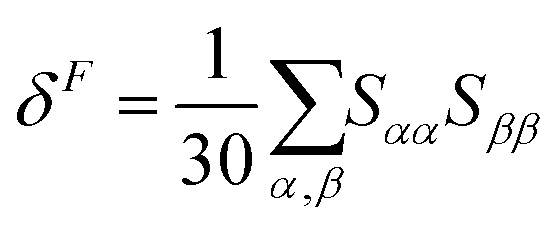 | (4) |
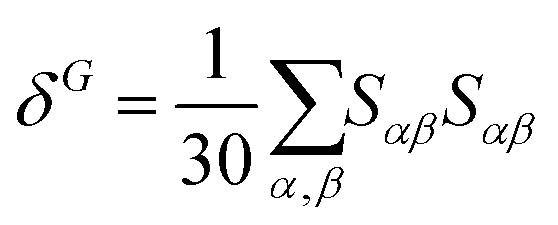 | (5) |
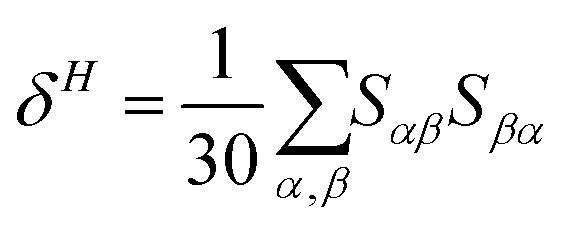 | (6) |
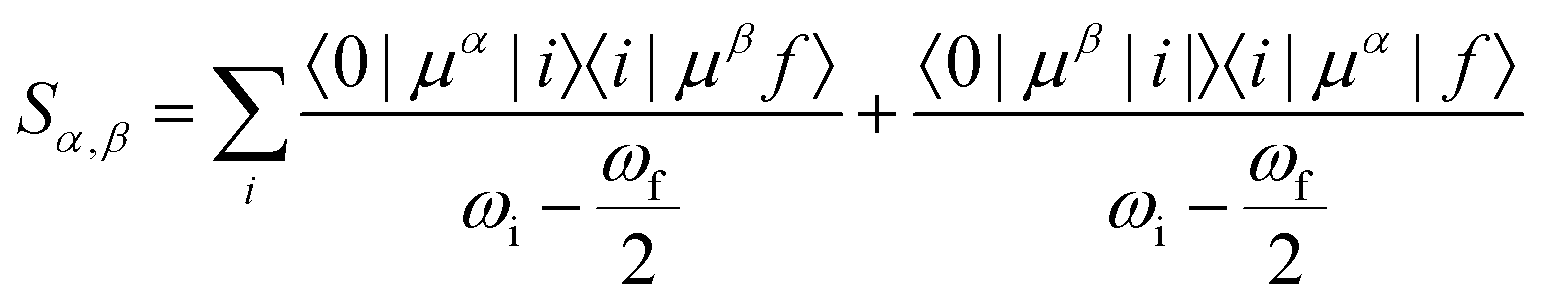 | (7) |
3. Results and discussion
3.1. Acid vs. base susceptibility for one- and two-photon absorption
Across pterins involving the addition of small, non-aromatic substituents, a single dominant (defined here as a state with a cross section >1.0 GM) can be observed in the acid form; this state can be seen moving to a lower energy in the basic form, a trend contrary to that observed in the the OPA spectra (Fig. 4 and 5). When considering the OPA spectra of the base forms of small-tail derivatives the dominant transitions are to S2 and S7 for the peaks at approx. 340 nm and 240 nm, respectively. In the two-photon spectra, these states remain accessible, with cross-sections for S2 (S1 for NPTb) ranging from 0.1–1.3 GM while S7 is seen to possess more substantial cross-section values, ranging from 4.1–10.7 GM. In contrast to the base spectra, the spectra for the acidic form (Table 4) is less predictable. While S2 is still accessible for most derivatives (with the exception of FPTa & HPTa), the state corresponding to the higher energy peak is more variable, with dominant transitions to states S5, S8, S9, or S10 observed, depending on the derivative. However, similar to what is observed in the base form, states showing strong oscillator strengths in the OPA spectra remain accessible in the TPA spectra with ranges for S2 from 0.19–1.1 GM and, with the exception of FPTa, a range of 4.6–9.5 GM for states found in the region of the higher energy OPA peak. This maintained susceptibility when moving from one- to two-photon absorption allows for the continued photo-reactivity of each pterin derivative to be monitored in a similar manner to those of modern fluorescent spectroscopy methods, while also utilising of the benefits of TPA spectroscopy, including: greater spectroscopic resolution and tissue depth penetration, as well as reduced potential for unnecessary tissue damage when targetting the higher energy peak, which would otherwise necessitate the use of a high energy UV light source.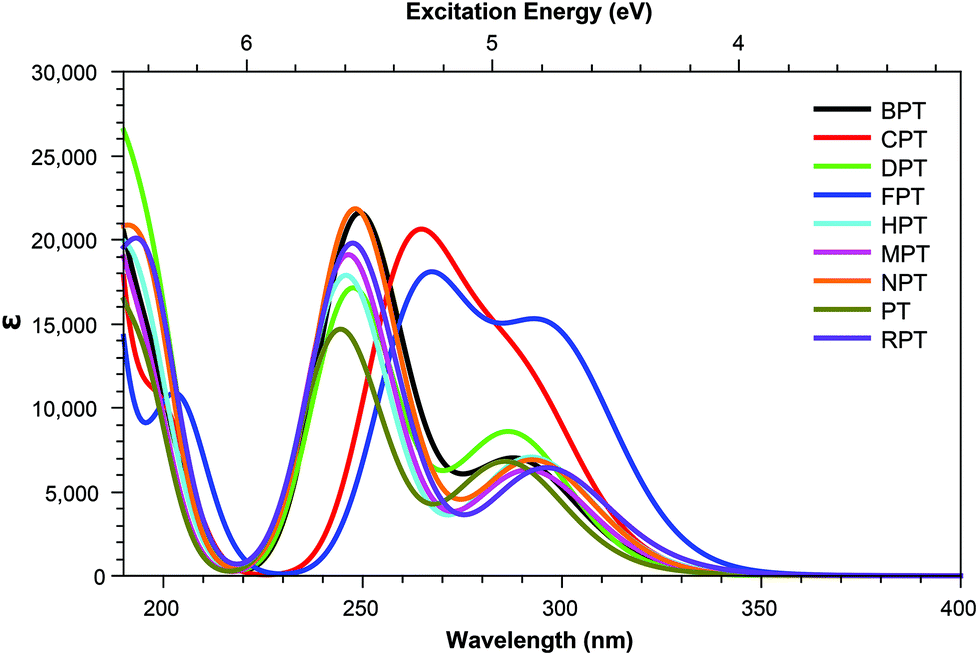 | ||
| Fig. 4 One-photon absorption spectra of pterin derivatives in their acidic state. | ||
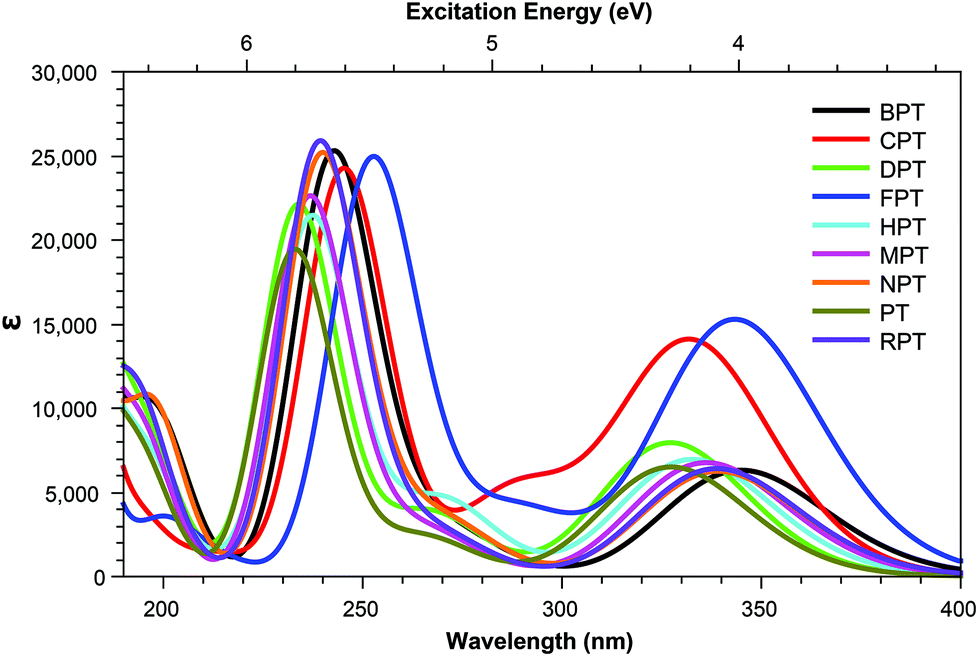 | ||
| Fig. 5 One-photon absorption spectra of pterin derivatives in their base form. | ||
The inclusion of substituents containing an aromatic group, such as those highlighted in Fig. 2, results in similar trends to those of the smaller, non-aromatic, substituents in that large cross-section values relating to the higher energy peak of the OPA spectra (Fig. 8 and 9) remaining accessible in both acid and base forms, while the high values observed in lower states are quenching when adopting the basic form (Tables 5 and 6).
Overall, despite the structures with tails inclusive of aromatic groups showing greater similarity between the acid and base forms than their small tail counterparts, the TPA spectra mimics the properties of the OPA spectra closely with similar shifts in state energies. The TPA differs from the OPA spectra, however, in the emergence of additional accessible states upon adoption of the base form of a number of derivatives; this, adding additional possibilities for the identification of the form present in vitro through the use of fluorescent spectroscopy.
3.2. Effects of non-aromatic substituents
Tables 3 shows that, regardless of whether in their acidic or basic form, the unsubstituted pterin molecule (PT) exhibits no significant two-photon cross section (σTP) with the highest value of 0.332 GM for S10 of PTa, lying at 6.55 eV. Despite this low potential, the addition of a substituent at the C6 position results in a 10-fold increase in the cross sections for the majority of substituents shown across Tables 2 and 4; the only structure that does not present an accessible state with a cross section of greater than 1 GM is FPTa. In contrast to the OPA spectra (Fig. 4 and 5), which show a significant amount of overlap in terms of bright regions, the TPA spectra shows enhanced resolution with accessible states separating; this would suggest that the use of TPA spectroscopy could allow for the identification of a specific derivative even when other derivatives are present. An example of this can be seen in the NPTa derivative which, despite showing a near equivalent OPA spectra to most other derivatives (Fig. 4), shows a TPA accessible state at 5.42 eV for which the only other accessible states in the region is the 5.32 eV S6 state of RPTa and the 5.30 eV S7 state of DPTb, both of which present a cross section of less than half the size (4.78 GM (Table 2) and 4.18 GM (Table 4), respectively). A similar pattern can be observed with the S7 state of BPTb at 5.05 eV and S8 state of HPTb at 5.61 eV which presents the highest cross-section of all small-tail derivatives at 39.60 GM.| Abbrv. | Name | R1 | R2 |
|---|---|---|---|
| a R1 groups for pterin derivatives with extended side chains are shown in Fig. 2. b Structures for derivatives involving biological alterations to the central pterin moiety are shown in Fig. 3. | |||
| PT | Pterin | –H | H |
| BPT | Biopterin | –(CHOH)2CH3 | H |
| CPT | 6-Carboxypterin | –COOH | H |
| DPT | 6,7-Dimethylpterin | –CH3 | –CH3 |
| FPT | 6-Formylpterin | –CHO | H |
| HPT | 6-(Hydroxymethyl)pterin | –CH2OH | H |
| MPT | 6-Methylpterin | –CH3 | H |
| NPT | Neopterin | –(CHOH)2CH2OH | H |
| RPT | Rhamnopterin | –(CHOH)3CH3 | H |
| PA | Pteroic acid | H | |
| FA | Folic acid | H | |
| MFA | 10-Methylfolic acid | H | |
| FN | Folinic acid | H | |
| MTHF | L-5-Methyltetrahydrofolate | H | |
| MTHFG | 5,10-Methenyltetrahydrofolyl-polyglutamate | H | |
| State | BPT | CPT | DPT | FPT | HPT | MPT | NPT | RPT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | |
| 1 | 3.89 | 0.007 | 3.80 | 0.000 | 3.99 | 0.002 | 3.46 | 0.000 | 3.85 | 0.000 | 3.87 | 0.001 | 4.00 | 0.014 | 3.82 | 0.009 |
| 2 | 4.29 | 0.646 | 4.28 | 0.186 | 4.32 | 0.616 | 3.80 | 0.000 | 4.24 | 0.057 | 4.25 | 0.489 | 4.24 | 0.497 | 4.19 | 0.723 |
| 3 | 4.75 | 0.031 | 4.39 | 0.002 | 4.85 | 0.037 | 4.16 | 0.343 | 4.83 | 0.002 | 4.83 | 0.024 | 4.92 | 0.120 | 4.83 | 0.115 |
| 4 | 4.95 | 0.099 | 4.72 | 0.613 | 5.00 | 1.070 | 4.38 | 0.002 | 4.96 | 0.002 | 4.95 | 0.024 | 5.00 | 1.150 | 4.96 | 0.190 |
| 5 | 4.98 | 3.010 | 4.85 | 0.134 | 5.01 | 1.220 | 4.67 | 0.660 | 5.05 | 0.128 | 5.03 | 2.030 | 5.03 | 0.037 | 5.02 | 0.869 |
| 6 | 5.47 | 0.181 | 4.94 | 0.015 | 5.52 | 0.049 | 4.76 | 0.002 | 5.46 | 0.071 | 5.48 | 0.680 | 5.42 | 10.900 | 5.32 | 4.780 |
| 7 | 6.06 | 0.307 | 5.41 | 0.050 | 6.13 | 0.060 | 5.22 | 0.027 | 6.11 | 0.099 | 6.14 | 0.060 | 5.50 | 0.663 | 5.50 | 0.122 |
| 8 | 6.22 | 6.630 | 5.77 | 0.076 | 6.28 | 9.460 | 5.58 | 0.036 | 6.24 | 8.920 | 6.30 | 0.321 | 6.10 | 0.101 | 6.11 | 0.830 |
| 9 | 6.32 | 4.640 | 6.09 | 0.091 | 6.38 | 0.598 | 5.74 | 0.114 | 6.29 | 0.273 | 6.34 | 4.700 | 6.31 | 4.650 | 6.21 | 3.000 |
| 10 | 6.42 | 0.282 | 6.21 | 9.490 | 6.43 | 5.140 | 6.07 | 0.659 | 6.44 | 0.723 | 6.46 | 5.990 | 6.35 | 2.740 | 6.25 | 3.370 |
| State | BPT | CPT | DPT | FPT | HPT | MPT | NPT | RPT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | E | σ TP | |
| 1 | 3.57 | 0.424 | 3.45 | 0.001 | 3.75 | 0.116 | 3.30 | 0.001 | 3.64 | 0.003 | 3.60 | 0.022 | 3.65 | 1.070 | 3.64 | 0.134 |
| 2 | 3.61 | 0.859 | 3.73 | 0.268 | 3.79 | 1.330 | 3.61 | 0.327 | 3.73 | 0.107 | 3.69 | 1.340 | 3.68 | 0.157 | 3.66 | 1.280 |
| 3 | 4.26 | 0.026 | 4.10 | 0.000 | 4.37 | 0.009 | 3.83 | 0.001 | 4.30 | 0.001 | 4.26 | 0.011 | 4.36 | 0.013 | 4.31 | 0.007 |
| 4 | 4.56 | 0.701 | 4.28 | 0.326 | 4.64 | 0.882 | 4.06 | 0.001 | 4.59 | 0.232 | 4.65 | 0.703 | 4.60 | 0.772 | 4.62 | 0.758 |
| 5 | 4.76 | 0.071 | 4.31 | 0.029 | 4.85 | 0.026 | 4.28 | 0.110 | 4.78 | 0.003 | 4.83 | 0.532 | 4.82 | 0.062 | 4.81 | 0.055 |
| 6 | 4.90 | 0.021 | 4.81 | 0.123 | 5.07 | 0.040 | 4.29 | 2.730 | 4.96 | 0.042 | 4.97 | 0.032 | 4.95 | 0.014 | 4.96 | 0.058 |
| 7 | 5.11 | 4.590 | 5.05 | 10.700 | 5.30 | 4.180 | 4.77 | 0.139 | 5.22 | 9.770 | 5.24 | 4.440 | 5.17 | 4.030 | 5.18 | 5.440 |
| 8 | 5.71 | 0.566 | 5.28 | 0.016 | 5.63 | 0.848 | 4.91 | 0.001 | 5.61 | 39.600 | 5.67 | 0.078 | 5.78 | 0.200 | 5.74 | 0.110 |
| 9 | 5.85 | 0.178 | 5.39 | 0.048 | 5.69 | 0.064 | 5.30 | 0.040 | 5.66 | 0.068 | 5.90 | 0.209 | 5.89 | 0.214 | 5.75 | 20.200 |
| 10 | 5.92 | 0.100 | 5.44 | 0.055 | 5.87 | 0.151 | 5.43 | 0.102 | 5.85 | 0.048 | 5.94 | 0.035 | 5.91 | 1.800 | 5.93 | 0.104 |
With the exception of FPT, which consists of a predominantly HOMO → LUMO+1 transition, the dominant state for each derivative (Tables 2 and 4) involve the same π–π* transition (Fig. 6); the presence of this state represents a transition that, while energetically effected by the tail, remains accessible.
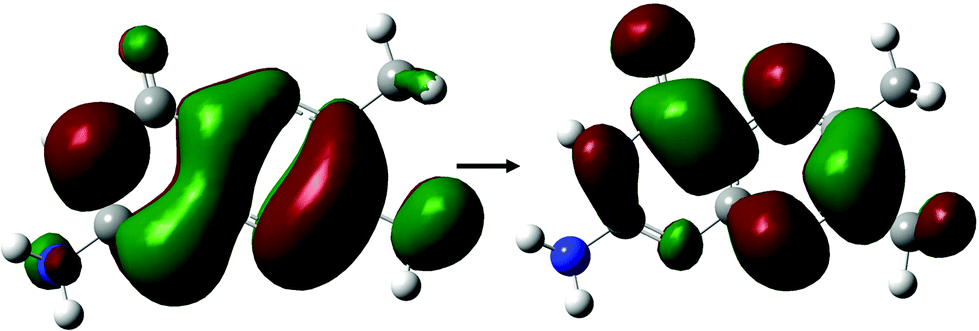 | ||
| Fig. 6 Dominant orbital transition describing the two-photon accessible state showing high cross sections across small tail pterin derivatives, as shown for the DPTa structure. | ||
3.3. Effect of aromatic substituents
Derivatives including an aromatic moiety in the tail (Fig. 2) show similar spectral features to that of the small tail derivatives; showing a small number of accessible states with high cross sectional values spread throughout a manifold of states that show low to moderate accessibility. The differences between these derivatives and those containing small tails becomes evident when assessing the OPA spectra (Fig. 8 and 9), in which the stereotypical shoulder shown at the 300–350 nm range remains present and at a similar intensity to that of the small tail derivatives while the larger peak seen at ≈250 nm presents a significant increase in intensity.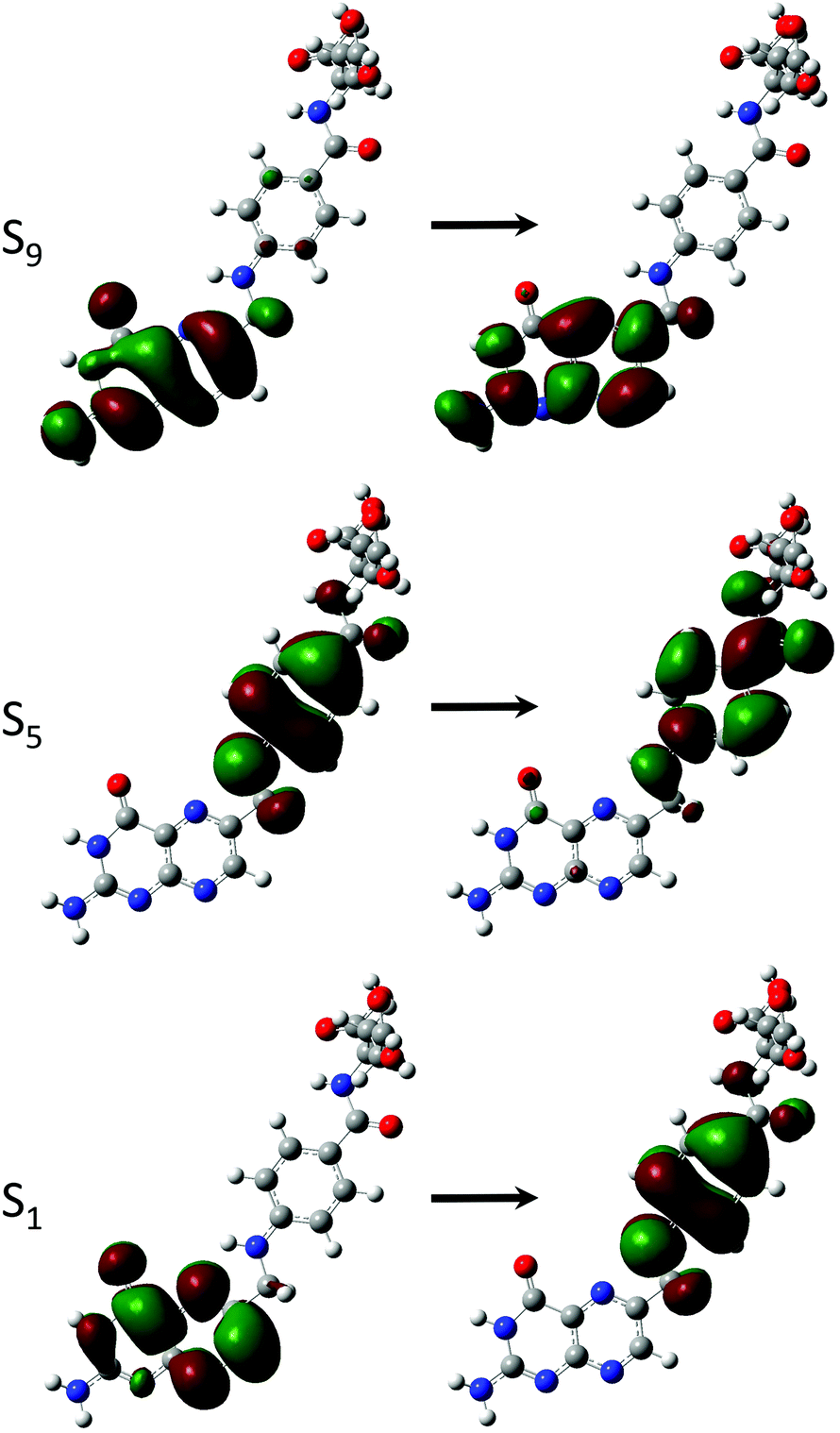 | ||
| Fig. 7 Dominant orbital transitions describing the two-photon accessible states showing high cross sections in FAa as a representation of those excitations that are two-photon accessible within derivatives containing an aromatic ring in the tail. | ||
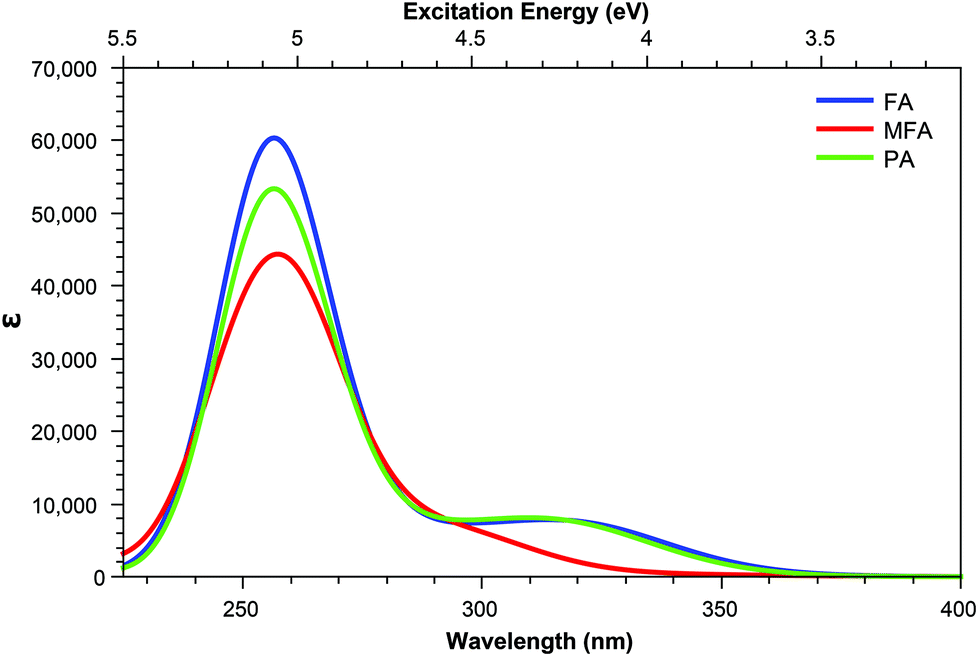 | ||
| Fig. 8 One-photon absorption spectra of the acid form of pterin derivatives with extended tails. | ||
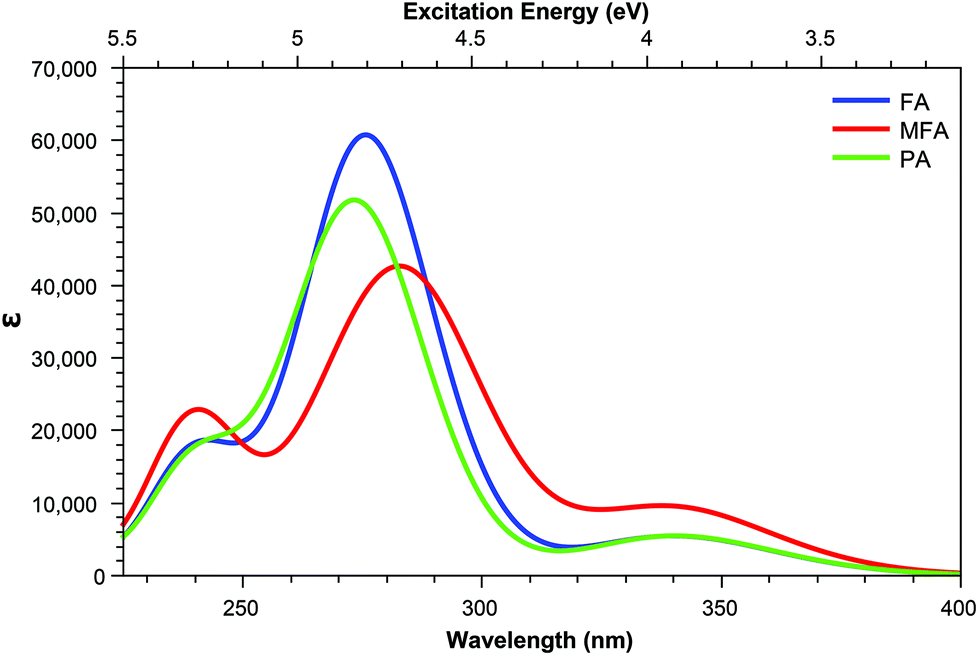 | ||
| Fig. 9 One-photon absorption spectra of the base form of pterin derivatives with extended tails. | ||
In contrast, the TPA spectra appears to consist of states which can be characterised in one of three ways: charge transfer states, in which density moves from the aromatic ring of the tail to the pterin moiety; or π–π* states isolated to either the pterin moiety or the tail; representations of these states are shown in Fig. 7.
For the acid form (Table 5) shows that, for both PAa and FAa, the S1 state situated at 3.87 and 3.84 eV, respectively, is significantly more accessible than its OPA counterpart with respect to the rest of their corresponding spectra; this state is, notably, quenched upon adopting the base form (Table 6). Unlike the S1 state, the second TPA accessible region, represented by S5 of each derivative in the acid form remains relatively intact in the base form with an average shift from 4.80 eV to 4.40 eV (noting that the TPA accessible S5 state of the acid form (Table 5) migrates to the S4 position when in the base form (Table 6)).
| State | PA | FA | MFA | |||
|---|---|---|---|---|---|---|
| E | σ TP | E | σ TP | E | σ TP | |
| 1 | 3.87 | 51.100 | 3.84 | 60.600 | 3.58 | 0.078 |
| 2 | 3.95 | 0.040 | 3.95 | 0.027 | 4.02 | 0.086 |
| 3 | 4.22 | 9.930 | 4.22 | 10.200 | 4.20 | 0.977 |
| 4 | 4.69 | 6.830 | 4.67 | 0.987 | 4.54 | 0.825 |
| 5 | 4.85 | 41.900 | 4.82 | 55.700 | 4.74 | 23.100 |
| 6 | 4.89 | 1.090 | 4.86 | 13.900 | 4.81 | 5.770 |
| 7 | 4.90 | 6.360 | 4.89 | 0.944 | 4.89 | 0.012 |
| 8 | 4.99 | 0.197 | 4.99 | 0.268 | 4.99 | 2.800 |
| 9 | 5.09 | 10.000 | 5.08 | 12.900 | 5.09 | 1.680 |
| 10 | 5.43 | 0.000 | 5.25 | 3.320 | 5.23 | 0.950 |
| State | PA | FA | MFA | |||
|---|---|---|---|---|---|---|
| E | σ TP | E | σ TP | E | σ TP | |
| 1 | 3.64 | 0.765 | 3.64 | 0.822 | 3.56 | 0.125 |
| 2 | 3.66 | 0.193 | 3.66 | 0.206 | 3.65 | 3.020 |
| 3 | 4.32 | 0.005 | 4.32 | 0.006 | 4.22 | 0.237 |
| 4 | 4.48 | 16.700 | 4.47 | 34.000 | 4.31 | 64.000 |
| 5 | 4.54 | 6.220 | 4.54 | 8.490 | 4.49 | 3.500 |
| 6 | 4.69 | 1.390 | 4.59 | 1.880 | 4.57 | 3.860 |
| 7 | 4.76 | 5.890 | 4.77 | 3.360 | 4.61 | 7.790 |
| 8 | 4.78 | 0.078 | 4.78 | 0.091 | 4.72 | 0.131 |
| 9 | 4.92 | 0.027 | 4.92 | 0.036 | 4.87 | 1.200 |
| 10 | 5.16 | 2.560 | 5.15 | 3.380 | 5.10 | 3.660 |
The S1 state of these derivatives, which show a large cross sectional value for both PAa and FAa (Table 5), is characterised by a charge transfer from the π HOMO, isolated on the pterin moiety, to the π* LUMO on the aromatic ring of the tail, as shown in Fig. 7. The methylation of the bridging nitrogen in MFA (Fig. 2), which causes the structure to fold so that the phenyl ring of the tail is orientated perpendicular to the plane of the pterin moiety, may explain the near zero cross section observed for this derivative through the breaking of any π overlap that would aid in the transfer of electron density. The reduction in cross section as these derivatives move to their base form can be explained in a similar manner, in which the change in electronic structure of the pterin moiety, including the development of a formal negative charge on the oxygen, would reduce the ability of this moiety to accommodate charge from the tail. In contrast, the higher energy state (S5 in the acid form; S4 in the base form) is characterised by a π–π* transition isolated to the aromatic ring located in the tail which explains the observation that, while the cross sections of the states to change from the acid to base form, the state remains accessible across both forms.
3.4. Oxidised derivatives
The derivatives achieved through oxidation of the pterin moiety, shown in Fig. 3, present a significantly blue shifted spectra compared to structures in which the pterin moiety is left intact, with the additional feature of increased intensity in the shoulder region. The TPA spectra of these structures (Table 7) show a shared accessible state slightly red shifted in comparison to the lower energy peak of the OPA spectra. A second, TPA accessible, state can be seen for MTHFG at 5.76 eV with a cross sectional value of 22.40 GM; access to the MTHFG 4.74 eV S1 state can be achieved with a cross sectional value of 12.80 GM.| State | FNA | MTHF | MTHFG | |||
|---|---|---|---|---|---|---|
| E | σ TP | E | σ TP | E | σ TP | |
| 1 | 4.73 | 9.220 | 4.73 | 5.680 | 4.74 | 12.800 |
| 2 | 4.84 | 0.575 | 4.74 | 6.310 | 4.76 | 3.330 |
| 3 | 4.88 | 3.020 | 4.82 | 27.200 | 4.91 | 23.000 |
| 4 | 4.95 | 19.500 | 5.02 | 1.240 | 4.92 | 0.570 |
| 5 | 5.21 | 2.900 | 5.25 | 4.190 | 5.09 | 4.870 |
| 6 | 5.24 | 1.730 | 5.26 | 5.340 | 5.17 | 7.310 |
| 7 | 5.27 | 1.600 | 5.28 | 3.100 | 5.25 | 1.790 |
| 8 | 5.51 | 0.374 | 5.74 | 0.498 | 5.76 | 22.400 |
| 9 | 5.60 | 4.390 | 5.86 | 3.130 | 5.82 | 3.310 |
| 10 | 5.88 | 1.340 | 5.94 | 3.750 | 5.85 | 0.028 |
| 11 | 5.92 | 0.175 | 6.00 | 6.640 | 5.97 | 0.173 |
| 12 | 6.01 | 1.030 | 6.02 | 0.012 | 6.05 | 0.159 |
| 13 | 6.01 | 0.011 | 6.06 | 0.113 | 6.11 | 0.782 |
| 14 | 6.07 | 0.079 | 6.13 | 1.090 | 6.18 | 16.400 |
| 15 | 6.12 | 23.200 | 6.16 | 2.370 | 6.27 | 4.120 |
| 16 | 6.16 | 7.360 | 6.22 | 7.850 | 6.33 | 1.830 |
| 17 | 6.25 | 10.100 | 6.33 | 2.870 | 6.36 | 0.262 |
| 18 | 6.29 | 1.010 | 6.36 | 0.084 | 6.41 | 1.180 |
| 19 | 6.34 | 7.400 | 6.47 | 6.050 | 6.44 | 2.210 |
| 20 | 6.46 | 5.200 | 6.53 | 4.100 | 6.52 | 9.670 |
| 21 | 6.46 | 2.300 | 6.57 | 2.280 | 6.53 | 4.410 |
| 22 | 6.52 | 12.600 | 6.57 | 6.940 | 6.61 | 1.450 |
| 23 | 6.61 | 0.817 | 6.68 | 18.900 | 6.63 | 1.160 |
| 24 | 6.68 | 2.290 | 6.71 | 5.680 | 6.73 | 2.400 |
| 25 | 6.73 | 4.490 | 6.73 | 2.980 | 6.78 | 27.200 |
| 26 | 6.75 | 2.490 | 6.76 | 41.100 | 6.80 | 4.940 |
| 27 | 6.78 | 13.200 | 6.81 | 9.400 | 6.82 | 4.260 |
| 28 | 6.84 | 0.972 | 6.86 | 4.900 | 6.83 | 2.830 |
| 29 | 6.86 | 1.300 | 6.92 | 1.280 | 6.86 | 0.421 |
| 30 | 6.92 | 0.757 | 6.95 | 2.460 | 6.90 | 12.000 |
In a similar manner to the pterins involving tails which contain an aromatic moiety, the oxidised derivatives studied here present a bright π–π* state that is shared across the derivatives (Table 7). This state can be found at S4 for FNA (E = 4.95 eV; σTP = 19.5 GM) and S3 for MTHF and MTHFG (E = 4.82 eV; σTP = 27.2 GM and E = 4.91 eV; σTP = 23.0 GM, respectively). However, while this is the only bright state on the TPA spectra of FNA and MTHF that overlaps with the lower band of the OPA spectra, this is not the case with MTHFG. The cyclisation between the tail and the oxidised pterin moiety results in the structure of MTHFG adopting a more planar geometry which, as seen with the acidic form of both PAa and FAa (Table 5), results in additional bright states. The first of these states is S1 (E = 4.74 eV; σTP = 12.8 GM), which is characterised as a π–π* transition from a hole orbital with density on the substituted ring of the oxidised pterin moiety, the 5-membered ring linking it to the tail, and the aromatic ring of the tail itself, to a particle π* orbital isolated on the oxidised pterin moiety. The second of these states is S8 (E = 5.76 eV; σTP = 22.4 GM) which is characterised by a charge transfer from a π hole orbital isolated on the oxidised pterin moiety to a particle π* orbital on the aromatic ring of the tail (Fig. 3; bottom); the charge transfer nature of this transition makes the high cross section associated to it dependent on the planarity of the geometry compared to the twisted geometry of MTHF in an analogous manner as seen when comparing the acid forms of FAa and MFAa (Table 5).
In addition to the TPA accessibility of the low energy shoulder of the OPA spectra (Fig. 10), all three structures studied also show significant accessibility for the high energy OPA peak. Each structure presents a constantly accessible π–π* state isolated to the tail such as that seen in S5 of FAa (Fig. 7); this state remains at ≈6.80 eV across the structures. In comparison, the second accessible state of this band more structurally specific; S22 of FNA (E = 6.52 eV; σTP = 12.6 GM) shows a π–π* isolated to the head in which the particle orbital presents significant Rydberg character, whereas S23 (E = 6.68 eV; σTP = 18.9 GM) in MTHF and S30 (E = 6.90 eV; σTP = 12.0 GM) in MTHFG present charge transfer from tail to head and π–π* character isolated to the head, respectively.
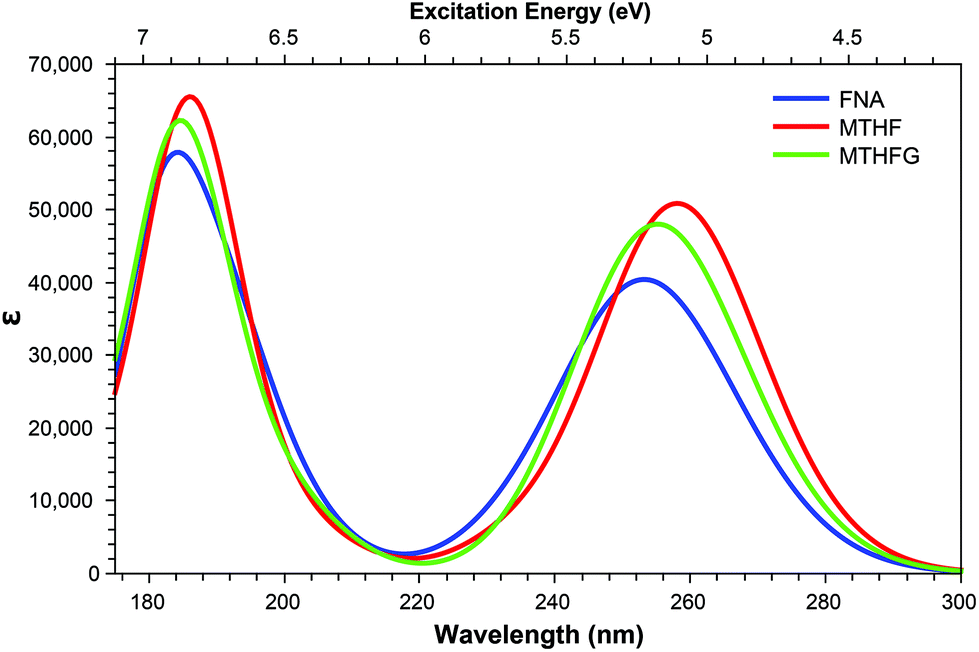 | ||
| Fig. 10 One-photon absorption spectra of structures derived from modifications to the pterin moiety. | ||
Of particular interest in the comparison of the OPA and TPA spectra is the emergence of TPA accessible states in both FNA and MTHFG which lie in the dark region between the OPA spectra (≈6.0 eV; Fig. 10) in the form of a charge transfer state in which density shifts from the pterin moiety to the aromatic ring of the tail; these states are S15 in FNA (E = 6.12 eV; σTP = 23.2 GM) and S14 in MTHFG (E = 6.18 eV; σTP = 16.4 GM). While these states present similar character to that of S3 in MTHF (E = 4.82 eV; σTP = 27.2 GM), these states are not accessible in the OPA spectra.
The accessibility of these two states for MTHFG (S1 & S8) is a particularly promising find as, in comparison to the OPA spectra (Fig. 10) which shows minimal differentiation between MTHFG and MTHF, the TPA spectra does enable the differentiation of these structures. This presents a novel route for investigating the activity of the methylenetetrahydrofolate reductase enzyme (MTHFR)85–87 which catalyses the reduction and decyclisation of MTHFG to MTHF, a cofactor in the conversion of homocysteine to methionine via the methionine synthase enzyme, a process vital for DNA reproduction as part of the cysteine cycle.88,89
4. Conclusion and outlook
Analysis of the structures presented here reveals a trend in the accessibility of secondary states (defined here as those that are of significantly smaller cross sectional value than the dominant TPA peaks for a given structure); this trend shows that, while these states have a near zero value for a large number of states when looking to the small tail derivatives (Tables 2 and 4), the cross sectional values of these states increases with structural complexity. The derivatives containing an aromatic element in the tail (Tables 5 and 6) show an increased number of secondary states with a cross sectional value >1 GM while, when considering the oxidised derivatives (Table 7), the majority of secondary states present a >1 GM cross section.Due to the lack of symmetry present throughout the pterin systems, the parity rules which act to limit the accessible states of symmetric systems do not apply to the structures studied here; this has the effect of, theoretically, rendering the entire singlet manifold assessable, with the cross section of a given state determined predominantly by the overlap of the particle and hole orbitals.
Across the pterin structures, TPA accessible states can be isolated into three different groups of π–π* transitions: those where both the hole and particle orbitals are located on the pterin moiety; those where both are orbitals are isolated to the tail; and those involving charge transfer from the tail to the pterin moiety. The later two types of transitions are only present in derivatives containing an aromatic ring in the tail; derivates without an aromatic ring present TPA accessible states that are depicted solely by transitions from various hole orbitals to the same particle orbital (with the exception of FPT, this particle orbital is shown to be the LUMO). The analysis of the TPA spectra, when compared to its OPA counterpart, has shown the potential for targetting specific derivatives despite the minimal qualitative variation evident in the OPA spectra; this is particularly relevant for NPTa, BPTb, and HPTb.
Of particular interest are the third category of states, those involving charge transfer from tail to pterin moiety where the nature of these states means that access to them is heavily dependent on the molecular geometry. This geometry dependence enables photochemical investigation of methylation and substitution events such as the conversion of FA to MFA, or the ring opening of MTHFG to form MTHF; the utilisation of TPA in these situations presents the possibility for structural differentiation that is not observable in the OPA spectra, due to the density of accessible states, potentially lending new insight into these biologically important processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. P. thanks the EPSRC for funding through Platform Grant EP/P001459/1 and Programme Grant EP/T021675, while T. M. thanks the The High End Computing facility at Lancaster University.References
- M. Vignoni, N. Walalawela, S. M. Bonesi, A. Greer and A. H. Thomas, Mol. Pharm., 2018, 15, 798 CrossRef CAS.
- A. Albert, Biochem. J., 1953, 54, 646 CrossRef CAS.
- C. Lorente and A. H. Thomas, Acc. Chem. Res., 2006, 39, 395 CrossRef CAS.
- H. Rokos, W. D. Beazlwy and K. U. Schallreuter, Biochem. Biophys. Res. Commun., 2002, 292, 805 CrossRef CAS.
- M. Pacileo, P. Cirillo, S. De Rosa, G. Ucci, G. Petrillo, S. Musto D'Amore, L. Sasso, P. Maietta, R. Spangnuolo and M. Chiariello, Monaldi Arch. Chest Dis., 2007, 68, 68 Search PubMed.
- A. Csordas, D. Fuchs, A. H. Frangieh, G. Reibnegger, B. Stähli, M. Cahenzly, F. Nietlispach, W. Maier, F. Maisano, R. K. Binder, C. Liebetreu, W. Kim, C. Hamm and T. F. Lüscher, IJC Metab Endocr, 2016, 10, 7 CrossRef.
- C. Murr, B. Widner, B. Wirleitner and D. Fuchs, Curr. Drug Metab., 2002, 3, 175 CrossRef CAS.
- C. A. Firth, A. D. Laing, S. K. Baird, J. Pearson and S. P. Gieseg, Clin. Biochem., 2008, 41, 1078 CrossRef CAS.
- R. L. Blakley, The Biochemistry of Folic Acid, North-Holland Publishing Co., 1969 Search PubMed.
- C. A. Nichol, G. K. Smith and D. S. Duch, Annu. Rev. Biochem., 1985, 54, 729 CrossRef CAS.
- J. M. Hevel and M. A. Marletta, Biochemistry, 1992, 31, 7160 CrossRef CAS.
- D. Fuchs, A. Hausen, G. Reibnegger, E. R. Werner, M. P. Dierich and H. Wachter, Immunol. Today, 1988, 9, 150 CrossRef CAS.
- C. Chandrashekar, K. Balaji, P. H. Vasudev, G. M. Panachiyil and T. Babu, Int. J. Pediatr. Adolesc. Med., 2019, 6, 151 CrossRef.
- S. Geisler, S. D. Lytton, N. L. Toan, T. H. Nghia, N. M. Nam, H. V. Hung, N. T. Son, D. T. Anh, H. T. Tuyen, T. V. Tien, D. Quyet, H. V. Tong, N. X. Hoan, L. H. Song, S. R. Pallerla, J. M. Gostner, D. Fuchs and T. P. Velavan, Int. J. Infect. Dis., 2020, 91, 162 CrossRef CAS.
- S. E. Dibakou, A. Souza, L. Boundenga, L. Givalois, S. Mercier-Delarue, F. Simon, F. Prugnolle, E. Huchard and M. J. E. Charpentier, Int. J. Parasitol. Parasites Wildl., 2020, 11, 198 CrossRef.
- S. P. Gieseg, G. Baxter-Parker and A. Lindsay, Antioxidants, 2018, 7, 80 CrossRef.
- G. Reibnegger, ChemistrySelect, 2018, 3, 10925 CrossRef CAS.
- P. Heelis, S. T. Kim, T. Okamura and A. Sancar, J. Photochem. Photobiol., B, 1993, 17, 219 CrossRef CAS.
- J. L. Johnson, S. Hamm-Alvarez, G. Payne, G. B. Sancar, K. V. Rajagopalan and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 2046 CrossRef CAS.
- J. E. Hearst, Science, 1995, 268, 1858 CrossRef CAS.
- K. Ito and S. Kawanishi, Biochemistry, 1997, 36, 1774 CrossRef CAS.
- S. Estébanez, A. H. Thomas and C. Lorente, ChemPhysChem, 2018, 19, 300 CrossRef.
- M. P. Serrano, M. Vignoni, C. Lorente, P. Vicendo, E. Oliveros and A. H. Thomas, Free Radicals Biol. Med., 2016, 96, 418 CrossRef CAS.
- M. P. Serrano, C. D. Borsarelli and A. H. Thomas, Photochem. Photobiol., 2013, 89, 1456 CrossRef CAS.
- C. Lorente, A. H. Thomas, L. S. Villata, D. Hozbor, A. Lagares and A. L. Capparelli, Pteridines, 2000, 11, 100 CAS.
- S. Y. Egorov, A. A. Krasnovsky Jr., M. E. Bashtanov, E. A. Mironov, T. A. Lyudnikova and M. S. Kritsky, Biochemistry, 1999, 64, 1325 Search PubMed.
- E. Oliveros, M. L. Dántola, M. Vignoni, A. H. Thomas and C. Lorente, Pure Appl. Chem., 2011, 83, 801 CAS.
- A. A. Buglak, T. A. Telegina, E. A. Vorotelyak and A. I. Kononov, J. Photochem. Photobiol., A, 2019, 372, 254 CrossRef CAS.
- A. H. Thomas, C. Lorente, A. L. Capparelli, C. G. Martínez, A. M. Braun and E. Oliveros, J. Photochem. Photobiol., A, 2019, 372, 254 CrossRef.
- J. W. Ledbetter Jr., W. Pfleiderer and J. H. Freisheim, Photochem. Photobiol., 1995, 62, 71 CrossRef.
- K. V. Neverov, E. A. Mironov, T. A. Lyudnikova, A. A. Krasnovsky and M. S. Kritsky, Biochemistry, 1996, 61, 1149 Search PubMed.
- A. H. Thomas, C. Lorente, A. L. Capparelli, M. R. Pokhrel, A. M. Braun and E. Oliveros, Photochem. Photobiol. Sci., 2002, 1, 421 RSC.
- J. R. Mourant, J. Frever, A. Hielscher, A. Fick, D. Shen and T. Johnson, Appl. Opt., 1998, 37, 3586 CrossRef CAS.
- J. R. Mourant, M. Canpolat, C. Brocker, O. Esponda-Ramos, T. Johnson, A. Matanock, K. Stetter and J. Freyer, J. Biomed. Opt., 2000, 5, 131 CrossRef CAS.
- S. Stolik, J. A. Delgado, A. Perez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90 CrossRef CAS.
- G. G. Kramarenko, S. G. Hummel, S. M. Martin and G. R. Buetner, Photochem. Photobiol., 2006, 82, 1634 CrossRef CAS.
- D. Havrylyuk, D. K. Heidary, L. Nease, S. Parkin and E. C. Glazer, Eur. J. Inorg. Chem., 2017, 12, 1687 CrossRef.
- E. C. Glazer, Photochem. Photobiol., 2017, 12, 1326 CrossRef.
- E. Skovsen, J. W. Snyder, J. D. Lambert and P. R. Ogilby, J. Phys. Chem. B, 2005, 109, 8570 CrossRef CAS.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707 CrossRef CAS.
- T. L. To, K. F. Medzihradszky, A. L. Burlingame, W. F. DeGrado, H. Jo and X. Shu, Bioorg. Med. Chem. Lett., 2016, 26, 3359 CrossRef CAS.
- C. Ash, M. Dubec, K. Donne and T. Bashford, Lasers Med. Sci., 2017, 32, 1909 CrossRef.
- H. Zhang, D. Salo, D. M. Kim, S. Komarov, Y. Tai and M. Y. Berezin, J. Biomed. Opt., 2016, 21, 126006 CrossRef.
- K. Datta, N. P. Johnson, G. Villani, A. H. Marcus and P. H. von Hippel, Nucleic Acids Res., 2012, 40, 1191 CrossRef CAS.
- K. Yang, S. Matsika and R. J. Stanley, J. Phys. Chem. B, 2007, 111, 10615 CrossRef CAS.
- M. L. Dántola, M. Vignoni, C. González, C. Lorente, P. Vicendo, E. Oliveros and A. H. Thomas, Free Radicals Biol. Med., 2010, 49, 1014 CrossRef.
- J. Arnberg, A. Jiménez-Banzo, M. J. Patersin, S. Nonell, J. I. Borrell, O. Christiansen and P. R. Ogilby, J. Am. Chem. Soc., 2007, 129, 5188 CrossRef.
- L. T. Bergendahl and M. J. Paterson, J. Phys. Chem. B, 2012, 116, 11818 CrossRef CAS.
- A. Graczyk, J. M. Žurek and M. J. Paterson, Photochem. Photobiol. Sci., 2014, 13, 103 RSC.
- S. E. Greenough, M. D. Hornbury, N. A. Smith, P. J. Sadler and M. J. Paterson, Photochem. Photobiol. Sci., 2014, 13, 103 RSC.
- P. Zhang, C. K. C. Chiu, H. Huang, Y. P. Y. Lam, A. Habtemariam, T. Malcomson, M. J. Paterson, G. J. Clarkson, P. B. O'Connor, H. Chao and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 14898 CrossRef CAS.
- H. Huang, S. Banerjee, K. Qiu, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stravos, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041 CrossRef CAS.
- T. Malcomson, R. McKinlay and M. J. Paterson, ChemPhotoChem, 2019, 3, 825 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, Semichem Inc., Shawnee Mission KS, 2016.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, 37, 785 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- A. D. Becke, Phys. Rev., 1988, 38, 3098 CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2007, 127, 154108 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 174104 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- P. Jaramillo, K. Coutinho and S. Canuto, Int. J. Quantum Chem., 2010, 110, 2371 CrossRef CAS.
- G. Reibnegger, Pteridines, 2015, 26, 135 CAS.
- M. Soniat and C. B. Martin, Pteridines, 2008, 19, 120 CAS.
- M. Soniat and C. B. Martin, Pteridines, 2009, 20, 124 CAS.
- M. Soniat and C. B. Martin, Pteridines, 2015, 26, 13 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 38, 3098 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. Baibarac, I. Smaranda, A. Nila and C. Serbschi, Sci. Rep., 2019, 9, 14278 CrossRef.
- A. Ali, M. I. Rafiq, Z. Zhang, J. Cao, R. Geng, B. Zhou and W. Tang, Phys. Chem. Chem. Phys., 2020, 22, 7864 RSC.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420 CrossRef CAS.
- M. J. Patersin, O. Christiansen, F. Pawłowski, P. Jørgensen, C. Hättig, T. Helgaker and P. J. Sałek, Chem. Phys., 2006, 124, 054322 Search PubMed.
- J. Arnberg, M. J. Paterson, C. B. Nielsen, M. Jørgensen, O. Christiansen and P. R. Ogilby, J. Phys. Chem. A, 2007, 111, 5756 CrossRef.
- M. Johnsen, M. J. Paterson, F. Arnberg, O. Christiansen, C. B. Nielsen, M. Jørgensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2008, 10, 1177 RSC.
- L. T. Bergendahl and M. J. Paterson, Chem. Commun., 2012, 48, 1544 RSC.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernádez, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hátig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. H. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansik, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klooper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. Rybkin, P. Salek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269 CAS.
- E. E. Trimmer, Curr. Pharm. Des., 2013, 19, 2574 CrossRef CAS.
- R. Reilly, H. McNulty, K. Pentieva, J. J. Strain and M. Ward, Proc. Nutr. Soc., 2014, 73, 47 CrossRef CAS.
- R. P. Ojha and J. G. Gurney, Leuk. Lymphoma, 2014, 55, 67 CrossRef CAS.
- V. Forster, F. van Delft, S. Baird, S. Mair, R. Skinner and C. Halsey, Cancer Chemother. Pharmacol., 2016, 78, 1093 CrossRef CAS.
- A. Banjac, T. Perisic, H. Sato, A. Seiler, S. Bannai, N. Weiss, P. Kölle, K. Tschoep, R. D. Issels, P. T. Daniel, M. Conrad and G. W. Bornkamm, Oncogene, 2008, 27, 1618 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2020 |