
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl dechlorination and defluorination with an organic super-photoreductant†
Felix
Glaser
 ,
Christopher B.
Larsen
,
Christoph
Kerzig
and
Oliver S.
Wenger
*
,
Christopher B.
Larsen
,
Christoph
Kerzig
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 26th June 2020
Abstract
Direct excitation of the commercially available super-electron donor tetrakis(dimethylamino)ethylene (TDAE) with light-emitting diodes at 440 or 390 nm provides a stoichiometric reductant that is able to reduce aryl chlorides and fluorides. The method is very simple and requires only TDAE, substrate, and solvent at room temperature. The photoactive excited state of TDAE has a lifetime of 17.3 ns in cyclohexane at room temperature and an oxidation potential of ca. −3.4 V vs. SCE. This makes TDAE one of the strongest photoreductants able to operate on the basis of single excitation with visible photons. Direct substrate activation occurs in benzene, but acetone is reduced by photoexcited TDAE and substrate reduction takes place by a previously unexplored solvent radical anion mechanism. Our work shows that solvent can have a leveling effect on the photochemically available redox power, reminiscent of the pH-leveling effect that solvent has in acid–base chemistry.
Introduction
Photochemistry has become remarkably popular, and numerous classes of chemical reactions can now be driven by visible light.1,2 Recently, there has been significant interest in developing photosensitizers that are able to provide very high reducing power,3–16 in order to perform ever more thermodynamically challenging reactions.17–19 At the same time, new multi-photon excitation concepts that rely on the pooling of two visible photons to access highly reducing intermediates have been developed.20–32 Whilst these and related concepts are very elegant, the need for multiple excitations can lead to complications due to counter-productive photoinduced side reactions, especially when several electron transfer steps are involved.33 Direct (single) photoexcitation remains the most straightforward way to obtain highly reducing species. To circumvent the abovementioned challenges associated with biphotonic excitation, the concept of electro-photocatalysis34 has been revitalized in order to achieve very high reducing power upon monophotonic excitation of electrochemically generated radical anions.35,36 Specific examples include dicyanoanthracene radical anion providing a potential of −3.2 V vs. SCE in its excited-state,37 and an excited naphthalene monoimide radical anion with a potential of −3.3 V vs. SCE.38 These concepts are elegant, but many radical anions have very short excited-state lifetimes,39 and catalyst-substrate preorganization may be needed because bimolecular diffusion is slow compared to excited-state deactivation.20,40 Therefore, it seemed worthwhile to explore how far the limits of molecular photoreductants can be pushed in terms of redox power.Reductive dehalogenations can be used to assess the reducing power of a photoactive compound. The scope of early investigations was limited to activated bromides or chlorides such as benzylic or α-carbonyl halides,41–43 but now there is an increasing body of literature on reductive dehalogenation of unactivated aryl and alkyl bromides,44–48 as well as unactivated aryl chlorides.6,49–55 Alkyl chlorides as well as aryl fluorides have remained very challenging targets.53,56 With UVC excitation aryl fluorides were dehalogenated successfully,57,58 but longer wavelength excitation would be much preferable because UVC radiation is usually very damaging, significantly limiting functional group tolerance. Furthermore, visible LEDs are safer and easier to handle than UV light sources.
We were curious whether the thermodynamically most challenging aryl chloride, alkyl chloride, and aryl fluoride reductions would be possible using direct excitation with visible instead of UV light, multi-photon processes, or electro-photocatalytic settings. When taking the reduction potential of chlorobenzene as a benchmark (−2.78 V vs. SCE in DMF)59 and assuming the availability of an excited state with an energy (E00) of 2.82 eV (corresponding to a blue photon, λ = 440 nm), a redox potential of 0.04 V vs. SCE or more negative is required for the photosensitizer in its ground state. Many organic super electron donors fulfill that requirement,60 but until very recently they could not be used catalytically.61 Therefore the compound tetrakis(dimethylamino)ethylene (TDAE) struck our attention, because it is commercially available. Whilst the photochemistry of TDAE has received some prior attention,62–64 reductive dehalogenations were not considered and its potential as an excited-state reductant has never been assessed quantitatively. Other, non-commercially available organic super electron donors have already been used for a variety of UVA-initiated reductions.50,65–67 Recently, the in situ generation of an organic super-electron donor permitted reductive dehalogenation of aryl chlorides and fluorobenzene with blue light, and this was exploited for borylation reactions.53
Results and discussion
TDAE has long been known to exhibit chemi- and photoluminescence,68 manifesting in a broad emission band with an onset at ca. 425 nm and a maximum at 490 nm in n-decane.69,70 We have been able to reproduce the previously reported emission spectrum using cyclohexane as solvent (ESI page S5†), and we measured a luminescence lifetime of 17.3 ns at room temperature (ESI page S6†), in line with a prior study.70 This excited-state lifetime is sufficiently long for bimolecular diffusional encounters between photoexcited TDAE and substrate molecules.Dehalogenation of aryl chlorides
We started our photochemical studies with the reduction of 2-chloro-4-fluorobenzonitrile (1) to 4-fluorobenzonitrile, using 1.25 eq. of TDAE in acetone-d6. After 2 hours of irradiation at 440 nm in a sealed NMR-tube at room temperature, a product yield of 50% (Table 1, entry 1) was obtained based on 19F-NMR spectroscopy with 1-fluoropentane as internal standard.|
|
|||||
|---|---|---|---|---|---|
| Entry | Eq. of TDAE | Solvent | [1]/mM | Time/h | Yield (conv.)/% |
| a Samples prepared in a glovebox and irradiated in sealed NMR tubes with an LED light source at room temperature. Yields determined by 19F-NMR analysis using 1-fluoropentane as internal standard. b Experiment performed in the dark. | |||||
| 1 | 1.25 | Acetone-d6 | 150 | 2 | 50 (66) |
| 2 | 1.25 | Acetone-d6 | 100 | 6 | 68 (99) |
| 3 | 2.5 | Acetone-d6 | 100 | 16 | 70 (100) |
| 4 | 1.25 | Acetone-d6 | 100 | 20 | 0b |
| 5 | 0 | Acetone-d6 | 100 | 20 | 0 |
| 6 | 1.25 | Acetonitrile-d3 | 100 | 7 | 65 (93) |
| 7 | 1.25 | Benzene-d6 | 50 | 7 | 67 (100) |
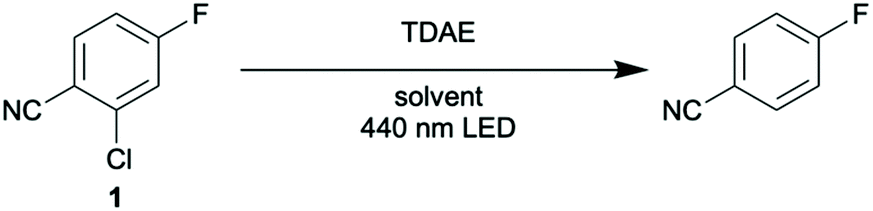
Full conversion and improvement to 68% yield was possible by lowering the substrate concentration from 150 to 100 mM and extending the irradiation period from 2 to 6 hours (entry 2). Increasing the amount of TDAE from 1.25 to 2.5 eq. combined with extending the irradiation time to 16 hours did not further improve the yield of 4-fluorobenzonitrile (entry 3). Reference experiments performed in the dark (entry 4) or under photo-irradiation in absence of TDAE (entry 5) yielded no product at all.
The change from acetone-d6 to CD3CN does not have a strong influence on the product yield (entry 6), and in benzene-d6 the reaction proceeds similarly well (entry 7). In the latter case the substrate concentration had to be limited to 50 mM, as the photochemical oxidation of TDAE results in the formation of an insoluble salt over time (see below), which can render photo-irradiation ineffective. This precipitate forms more readily in benzene than in acetone or acetonitrile, hence the need for lower substrate concentrations in benzene.
Because of the air-sensitivity of TDAE, we prepared all reaction mixtures in a glovebox, but this is not strictly necessary. When instead keeping the TDAE bottle outside the glovebox and preparing the reaction mixtures under an inert atmosphere, similar yields and conversions were obtained by using a quantity of TDAE which formally corresponds to 2 (rather than 1.25) equivalents, to compensate for (partial) decomposition of the TDAE.
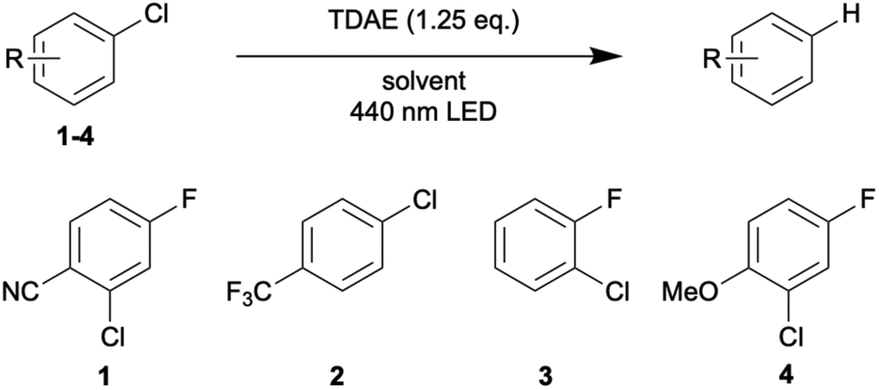
With these optimized conditions, we began to explore the scope of the reaction, always focusing on fluorinated substrates to permit quantitative analysis by 19F-NMR spectroscopy in presence of 1-fluoropentane as internal standard (Table 2). The photodriven hydrodechlorination of aryl chlorides was studied on four substrates with different types of substituents ranging from electron withdrawing (1, 2) to electron donating (4). Nearly all of these reactions proceed well and give product yields about 70%. The only outlier is substrate 2 in benzene, which seems to undergo radical polymerization under these conditions. A relatively clear trend concerning the necessary reaction time to reach full conversion emerges from Table 2: the photoreaction is slowest in acetone and fastest in benzene.
|
|
||||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| a Reaction conditions: 1.25 eq. TDAE; in acetone and acetonitrile: 100 mM substrate; in benzene: 50 mM substrate. Samples prepared in a glovebox and irradiated in sealed NMR tubes with an LED at room temperature. Yields determined by 19F-NMR analysis using 1-fluoropentane as internal standard. b 59 (64)%, 2 h. c 49 (54)%, 2 h. | ||||
| Acetone | 68 (99)%, 6 h | 60 (100)%, 7.5 h | 83 (96)%, 8 h | 79 (89)%, 6 hb |
| Acetonitrile | 65 (93)%, 7 h | 64 (100)%, 5 h | 67 (92)%, 8 h | 83 (87)%, 6 hc |
| Benzene | 70 (99)%, 2.5 h | 53 (100)%, 1.5 h | 77 (99)%, 1.5 h | 69 (92)%, 2 h |
| Benzene, 390 nm | 67 (100)%, 0.5 h | 38 (100)%, 0.5 h | 86 (99)%, 0.5 h | 67 (100)%, 0.33 h |
Dehalogenation of alkyl chlorides and bromides
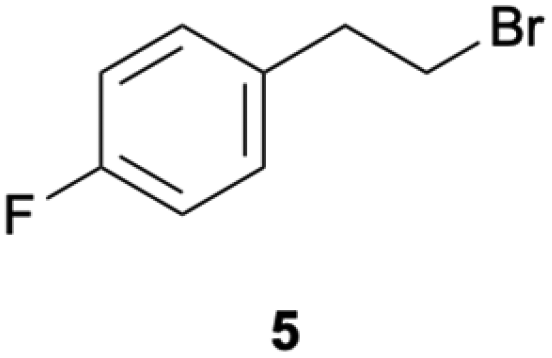
Given these encouraging results with aryl chlorides, it seemed worthwhile to investigate whether the reductive dehalogenation of an alkyl bromide (5) and an alkyl chloride (6) is also feasible with the same method (Table 3). The chosen substrates contain p-fluorobenzene units to lower volatility and to allow for 19F-NMR spectroscopy, but their chloro- and bromo-substitution is at an aliphatic position. With yields near 60% in comparable reaction times, the reductive debromination of 5 in acetone and acetonitrile proceeds nearly equally well as the reductive dehalogenation of the aryl chlorides 1–4. In benzene, the dimerization product 8 forms preferentially over the hydrodehalogenation product 7, and a change in irradiation wavelength from 440 to 390 nm amplifies this effect.|
|
||||
|---|---|---|---|---|
| Substrate | Solvent | 7 | 8 | |
| a Reaction conditions: 1.25 eq. TDAE; in acetone and acetonitrile: 100 mM substrate; in benzene: 50 mM substrate. Samples prepared in a glovebox and irradiated in sealed NMR tubes with an LED at room temperature. Yields determined by 19F-NMR analysis using 1-fluoropentane as internal standard. b 390 nm LED. | ||||
|
Acetone | 63 | — | (93)%, 6 h |
| Acetonitrile | 60 | — | (97)%, 6 h | |
| Benzene | 23 | 36 | (100)%, 2 h | |
| 16 | 62 | (100)%, 0.5 hb | ||

|
Acetone | 30 | — | (34)%, 20 h |
| Acetonitrile | 44 | — | (60)%, 20 h | |
| Benzene | 31 | 10 | (97)%, 6 h | |
| 23 | 48 | (98)%, 0.5 hb | ||
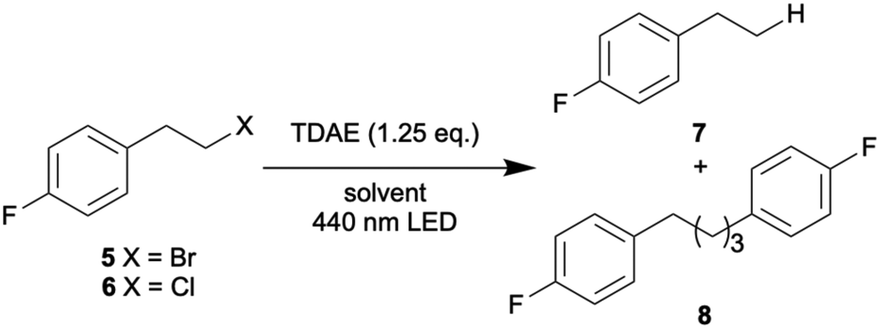
Furthermore, irradiation at 390 nm drastically shortens the reaction time to full conversion in benzene, likely due to the higher extinction coefficient of TDAE at 390 nm compared to 440 nm (ESI page S5†). The alkyl chloride 6 reacts considerably less efficiently with hydrodechlorination yields of only 30–44% in acetone and acetonitrile (Table 3), and the conversion is incomplete even after long irradiation times (20 hours). Full conversion is achievable in benzene, particularly under 390 nm irradiation, but the dimerization product 8 again dominates in that solvent. Higher radical concentrations reached under these conditions likely gear the reaction towards dimerization.71 Several side products were detectable by 19F-NMR spectroscopy (ESI page S31†), but their identification is beyond our scope.
Dehalogenation of aryl fluorides
The dechlorination reaction performed with the trifluoromethylated substrate 2 provided some evidence for possible C–F bond activation, resulting in the abovementioned polymerization and lower hydrodechlorination yields in benzene (Table 2). This observation motivated the investigation of reductive defluorination of the aryl fluoride substrates 9 and 11 (Table 4). 1,2-Difluorobenzene 9 is converted to fluorobenzene 10 in good yields (65–83%), and the reaction is considerably faster in benzene than in acetone (44 h vs. 25 h), in line with the observations made for the dehalogenation of substrates 1–6. Optimal conditions involve the use of 390 nm irradiation, leading to essentially complete conversion and 83% yield of 10 in 2.5 hours. Such efficient reductive defluorination under photochemical conditions is exceptional.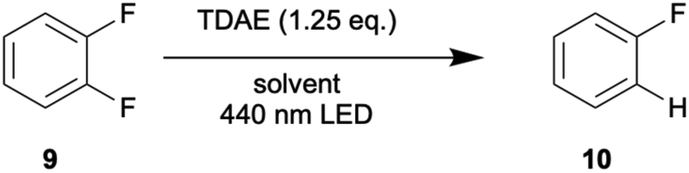
|
|
|||
|---|---|---|---|
| 12 | 10 | ||
| a Reaction conditions: TDAE; in acetone: 100 mM substrate; in benzene: 50 mM substrate. Samples prepared in a glovebox and irradiated in sealed NMR tubes with an LED at room temperature. Yields determined by 19F-NMR analysis using 1-fluoropentane as internal standard. b 3.0 eq. TDAE used. c 390 nm LED. d 3.25 eq. TDAE used. | |||
| Acetoned | 39 | 6 | (60)%, 44 h |
| 54 | 10 | (88)%, 44 hc | |
| Benzene | 37 | 20 | (84)%, 45 h |
| 18 | 40 | (95)%, 12 hc | |

1,3,5-Trifluorobenzene 11 converts to a mixture of 1,3-difluorobenzene 12 and fluorobenzene 10. Expectedly, the reaction is fastest in benzene under 390 nm irradiation, providing essentially complete conversion within 12 hours. The combined yield of 12 and 10 amounts to 58%, and the formation of significant amounts of benzene (resulting from complete defluorination of 11) seems plausible but is unfortunately not directly detectable when using benzene-d6 as solvent. Fluoride anions escape detection because they precipitate from benzene.
To extend the applicability of our new method beyond hydrodehalogenation reactions, we investigated the dimerization of substrate 13 on a 0.5 mmol reaction scale in benzene-d6 (Scheme 1). The benzylic radical formed after reductive dechlorination was expected to be particularly stable, and indeed 79% of the dimer 14 were successfully isolated after irradiation at 440 nm for 0.5 hours.
 | ||
| Scheme 1 Preparative-scale photo-dimerization mediated by TDAE. | ||
Mechanistic studies
With the exception of substrate 2 which seems to undergo polymerization (see above), our substrate scope studies indicate that full conversion and higher yields are consistently achievable more rapidly in benzene than in acetone, particularly when using 390 nm instead of 440 nm irradiation. TDAE has an absorption band tailing more strongly into the blue spectral range in acetone than in benzene (ESI page S5†), and this is most likely the reason why excitation at shorter wavelength is preferable in benzene. However, even when irradiating at 440 nm, the photoreactions are typically a factor of 2–3 faster in benzene even though at this wavelength TDAE absorbs far more strongly in acetone, and the TDAE/substrate concentrations were typically higher in acetone. Evidently, benzene is inherently a far better solvent for these reactions than acetone, and there is a simple explanation with important consequences for this, as discussed in the following.In its electronic ground state, TDAE has an oxidation potential (Eox) of −0.78 V vs. SCE in CH3CN.72 Based on a luminescence experiment in frozen 2-methyl-THF (2-MTHF) at 77 K we determine an energy (E00) of 2.6 eV for the emissive excited state (ESI page S6†). Consequently, a potential (*Eox) of ca. −3.4 V vs. SCE can be estimated for the singlet excited state, which is an exceptionally high value in comparison to most known photoreductants.3–16 However, acetone is reduced at a potential of −2.84 V vs. SCE,73 whereas benzene reduction necessitates a potential of −3.42 V vs. SCE,74 hence photoexcited TDAE is readily able to reduce acetone, whereas benzene reduction should be much more challenging. Indeed, the fluorescence of TDAE in cyclohexane is quenched by acetone with essentially diffusion-limited kinetics (Fig. 1), whilst benzene induces practically no detectable quenching of the short-lived (τ0 = 17.3 ns) excited state of TDAE under identical conditions (inset in Fig. 1). This observation leads us to the mechanistic proposal in Scheme 2, in which photoexcited TDAE undergoes electron transfer to the acetone solvent (right half) to produce acetone radical anion, followed by onward electron transfer to the substrate. By contrast, direct electron transfer from photoexcited TDAE to the substrates occurs in benzene (left half of Scheme 2), because that solvent cannot be reduced efficiently by TDAE. Stern–Volmer experiments in cyclohexane confirm that chloro- and fluoro-benzenes quench the photoactive excited state of TDAE with essentially diffusion-limited kinetics (Table 5). With 1-chlorohexane the quenching rate constant is roughly an order of magnitude lower, and the photoinduced electron transfer step to that substrate now becomes rate-limiting. In principle, the direct quenching pathway is also viable in acetone, but due to the much higher concentration of the solvent (ca. 13.5 M) compared to the substrate (0.1 M) direct substrate reduction is unimportant in acetone. Consequently, acetone effectively levels the available reducing power at −2.84 V vs. SCE, corresponding to the oxidation potential of its ketyl radical anion form, whereas the full reducing power of photoexcited TDAE (Eox) of ca. −3.4 V vs. SCE (see above) is available in benzene. This can explain why the photochemical reactions discussed above proceed considerably more efficiently in benzene than in acetone. After initial formation of the aryl halogenide radical anions, C–X bond cleavage is expectable based on prior studies of hydrodehalogenation reactions. For aryl chlorides and aryl fluorides, this bond cleavage typically occurs in a separate step after the initial electron transfer.59,75 The irradiation times needed for comparable conversions are considerably shorter for aryl chlorides than for aryl fluorides (Tables 2 and 4), but the initial photoinduced electron transfer step is similarly fast in both types of substrates (Table 5). Thus, it seems that C–F bond cleavage becomes the rate-determining step for the aryl fluorides, and furthermore it is possible that unproductive recombination between the aryl halogenide radical anion and TDAE radical cation limits the progress of the reaction with these substrates.59 Once an aryl radical has been liberated through C–X bond cleavage, it is expected to be highly reactive, and the necessary H-atom equivalent to access the final hydrodehalogenation product is likely provided by TDAE˙+. Oxidation of tertiary amines commonly leads to radical cations, which are potent donors of H-atom equivalents either via direct H-atom transfer or via consecutive proton and electron transfer steps.76,77 These onward oxidations of TDAE˙+ are likely responsible for the formation of an insoluble salt over the course of the reaction.
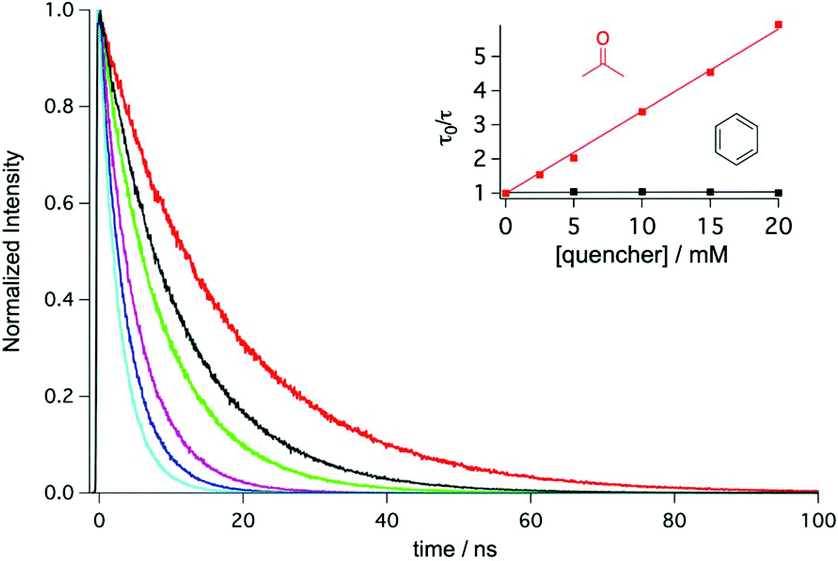 | ||
| Fig. 1 Emission decays of TDAE (10 mM) in neat de-aerated cyclohexane (red trace) and in de-aerated cyclohexane with different concentrations of acetone (2.5, 5, 10, 15, 20 mM) detected at 470 nm (λexc = 405 nm). The inset displays the Stern–Volmer plots obtained from the TDAE luminescence lifetime quenching by acetone (red trace) and benzene (black trace) in cyclohexane. | ||
 | ||
| Scheme 2 Proposed mechanisms for the light-driven dehalogenation of aryl halides. | ||
| Quencher | k Q/1010 M−1 s−1 |
|---|---|
| a Details given in ESI pages S6–S9.† | |
| Chlorobenzene | 1.43 |
| 1-Chloro-2-fluorobenzene (3) | 1.52 |
| 1,2-Difluorobenzene (9) | 1.45 |
| Fluorobenzene (10) | 1.04 |
| 1-Chlorohexane | 0.13 |
| Acetone | 1.42 |
| Benzene | <0.002 |
| Toluene | <0.002 |
Conclusions
In summary, photoexcitation of an organic super-electron donor provides extraordinarily high reducing power, enabling efficient reductive dehalogenation of aryl chlorides and aryl fluorides under irradiation with blue (440 nm) and violet/UVA light (390 nm). The excited-state oxidation potential of TDAE (−3.4 V vs. SCE) compares very favorably with the strongest known photoreductants operating on the basis of single excitation with visible photons (Fig. 2).3–16,78,79 Furthermore, the reducing power of photoexcited TDAE is close to that achievable via electrochemical generation of organic radical anions and their subsequent photo-excitation (green arrows in Fig. 2).37,38 Most of the currently known multi-photon excitation-based processes do not provide competitive reducing powers.20 Our method is not catalytic, but it is operationally simple, merely requiring excitation of a commercial reagent without additives, electrodes or high excitation densities. The key point is the very low oxidation potential of the electronic ground state of TDAE. In situ generation of an organic super electron donor recently gave access to an excited-state oxidation potential as low as −3.5 V vs. SCE,53 but for most other (non-commercial) organic super electron donors the relevant excited-state energies are unknown, hence their *Eox values cannot be estimated. It might be interesting to explore this substance class further from a more photophysical and photochemical viewpoint, with particular focus on their excited-state properties. The above-mentioned in situ generated super electron donor is ca. 0.1 V more reducing than photoexcited TDAE, but the latter has an excited-state lifetime roughly 3 times as long.53 Both donors are able to activate a similar substrate scope. Unfortunately, common sacrificial electron donors are unable to regenerate TDAE from TDAE˙+, and therefore TDAE cannot be used in catalytic fashion.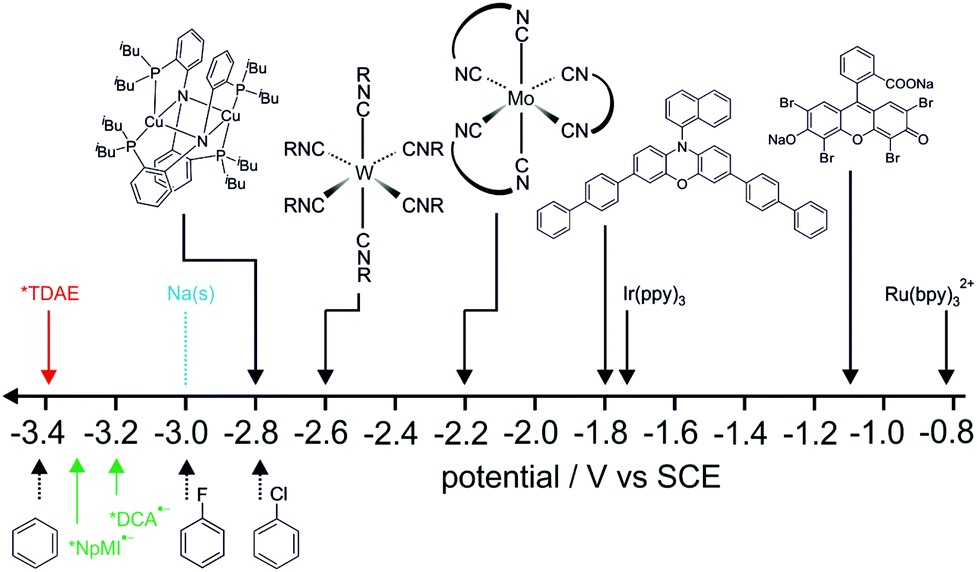 | ||
| Fig. 2 Overview of some of the most potent excited-state photoreductants operating on the basis of single excitation with visible photons known to date.3,5,7,78,79 See ref. 80 and 5, 12 for chemical structures of the isocyanide ligands. Chloro- and fluorobenzene reduction potentials from ref. 59 and 81, benzene reduction potential from ref. 74. *NpMI˙− = photoexcited naphthalene monoimide radical anion, *DCA˙− = photoexcited dicyanoanthracene radical anion (both generated with electro-photocatalytic settings).37,38 | ||
Herein we focused on hydrodehalogenation, but in principle it should be possible to intercept the aryl radicals by pyrroles to form C–C bonds,26 or by other trapping reagents to effect borylation,53 phosphorylation or sulfide formation.28 The dimerization experiment in Scheme 1 suggests that bimolecular reactions are within reach.
Our mechanistic studies clearly point to different reaction mechanisms in different solvents. Acetone is reduced by photoexcited TDAE and mediates electron transfer to the substrate via its radical anion form, whereas benzene is not reduced and direct electron transfer to the substrate must occur (Scheme 2). As the search for increasingly potent excited-state reductants continues, the solvent-radical-anion-mediated mechanism observed herein for acetone will likely become increasingly important.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swiss National Science Foundation through grant 200021_178760 is acknowledged. C. K. is grateful to the Research Fund of the University of Basel for a Novartis University of Basel Excellence Scholarship for Life Sciences.Notes and references
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- K. Li, Q. Y. Wan, C. Yang, X. Y. Chang, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2018, 57, 14129–14133 CrossRef CAS PubMed.
- L. A. Büldt and O. S. Wenger, Angew. Chem., Int. Ed., 2017, 56, 5676–5682 CrossRef PubMed.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. D. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 2030–2031 CrossRef CAS PubMed.
- F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed.
- I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS PubMed.
- M. Marchini, G. Bergamini, P. G. Cozzi, P. Ceroni and V. Balzani, Angew. Chem., Int. Ed., 2017, 56, 12820–12821 CrossRef CAS PubMed.
- J. H. Shon, S. Sittel and T. S. Teets, ACS Catal., 2019, 9, 8646–8658 CrossRef CAS.
- P. Herr, F. Glaser, L. A. Büldt, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 14394–14402 CrossRef CAS PubMed.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milian-Medina, L. Luer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- B. L. Buss, C.-H. Lim and G. M. Miyake, Angew. Chem., 2020, 59, 3209–3217 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- M. Brasholz, Angew. Chem., Int. Ed., 2017, 56, 10280–10281 CrossRef CAS PubMed.
- B. K. Qiao and Z. Y. Jiang, ChemPhotoChem, 2018, 2, 703–714 CrossRef CAS.
- M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2019, 58, 1869–1874 CrossRef PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- M. Majek, U. Faltermeier, B. Dick, R. Perez-Ruiz and A. Jacobi von Wangelin, Chem. – Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- C. G. López-Calixto, M. Liras, V. A. de la Peña O'Shea and R. Pérez-Ruiz, Appl. Catal., B, 2018, 237, 18–23 CrossRef.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- L. Zeng, T. Liu, C. He, D. Y. Shi, F. L. Zhang and C. Y. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- C. Kerzig and M. Goez, Chem. Sci., 2016, 7, 3862–3868 RSC.
- R. Naumann, C. Kerzig and M. Goez, Chem. Sci., 2017, 8, 7510–7520 RSC.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- S. S. Shukla and J. F. Rusling, J. Phys. Chem., 1985, 89, 3353–3358 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201913767.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- N. T. La Porte, J. F. Martinez, S. Chaudhuri, S. Hedstrom, V. S. Batista and M. R. Wasielewski, Coord. Chem. Rev., 2018, 361, 98–119 CrossRef CAS.
- J. Haimerl, I. Ghosh, B. König, J. Vogelsang and J. M. Lupton, Chem. Sci., 2019, 10, 681–687 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- K. Tahara and Y. Hisaeda, Green Chem., 2011, 13, 558–561 RSC.
- T. Maji, A. Karmakar and O. Reiser, J. Org. Chem., 2011, 76, 736–739 CrossRef CAS PubMed.
- G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342–13345 CrossRef CAS PubMed.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed.
- S. O. Poelma, G. L. Burnett, E. H. Discekici, K. M. Mattson, N. J. Treat, Y. D. Luo, Z. M. Hudson, S. L. Shankel, P. G. Clark, J. W. Kramer, C. J. Hawker and J. R. de Alaniz, J. Org. Chem., 2016, 81, 7155–7160 CrossRef CAS PubMed.
- J. J. Devery, J. D. Nguyen, C. H. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS.
- L. Zhang, Z.-Q. Wu and L. Jiao, Angew. Chem., 2020, 59, 2095–2099 CrossRef CAS PubMed.
- H. L. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS PubMed.
- E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Z. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673–3676 CrossRef CAS PubMed.
- S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed.
- R. Matsubara, T. Yabuta, U. M. Idros, M. Hayashi, F. Ema, Y. Kobori and K. Sakata, J. Org. Chem., 2018, 83, 9381–9390 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef PubMed.
- S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- Q. M. Wang, M. Poznik, M. Y. Li, P. J. Walsh and J. J. Chruma, Adv. Synth. Catal., 2018, 360, 2854–2868 CrossRef CAS.
- T.-H. Ding, J.-P. Qu and Y.-B. Kang, Org. Lett., 2020, 22, 3084–3088 CrossRef CAS PubMed.
- T. Fukuyama, Y. Fujita, H. Miyoshi, I. Ryu, S. C. Kao and Y. K. Wu, Chem. Commun., 2018, 54, 5582–5585 RSC.
- D. Cao, C. Yan, P. Zhou, H. Zeng and C.-J. Li, Chem. Commun., 2019, 55, 767–770 RSC.
- L. Pause, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef CAS PubMed.
- S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454–11458 CrossRef CAS PubMed.
- T. Juspin, M. Laget, T. Terme, N. Azas and P. Vanelle, Eur. J. Med. Chem., 2010, 45, 840–845 CrossRef CAS PubMed.
- O. Amiri-Attou, T. Terme, M. Médebielle and P. Vanelle, Tetrahedron Lett., 2008, 49, 1016–1020 CrossRef CAS.
- S. Ait-Mohand, N. Takechi, M. Médebielle and W. R. Dolbier, Org. Lett., 2001, 3, 4271–4273 CrossRef CAS PubMed.
- S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474–478 CrossRef PubMed.
- E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239–2242 CrossRef CAS PubMed.
- E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934–10937 CrossRef CAS PubMed.
- H. E. Winberg, J. R. Downing and D. D. Coffman, J. Am. Chem. Soc., 1965, 87, 2054–2055 CrossRef CAS.
- A. N. Fletcher and C. A. Heller, J. Phys. Chem., 1967, 71, 1507–1518 CrossRef CAS.
- M. Hori, K. Kimura and H. Tsubomura, Spectrochim. Acta, Part A, 1968, 24, 1397–1404 CrossRef CAS.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC.
- C. Burkholder, W. R. Dolbier and M. Médebielle, J. Org. Chem., 1998, 63, 5385–5394 CrossRef CAS.
- T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, John Wiley & Sons, Chichester, West Sussex, U. K., 2015 Search PubMed.
- J. Mortensen and J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 84–85 CrossRef.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- P. J. Delaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- Y. Du, R. M. Pearson, C. H. Lim, S. M. Sartor, M. D. Ryan, H. S. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- W. Sattler, M. E. Ener, J. D. Blakemore, A. A. Rachford, P. J. LaBeaume, J. W. Thackeray, J. F. Cameron, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 10614–10617 CrossRef CAS PubMed.
- C. P. Andrieux, C. Blocman and J. M. Savéant, J. Electroanal. Chem., 1979, 105, 413–417 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0pp00127a |
| This journal is © The Royal Society of Chemistry and Owner Societies 2020 |