
Designing expanded bipyridinium as redox and optical probes for DNA†
Emanuela
Trovato
 a,
Maria Letizia
Di Pietro
*b,
Antonino
Giannetto
b,
Gregory
Dupeyre
c,
Philippe P.
Lainé
*c,
Francesco
Nastasi
b,
Fausto
Puntoriero
*b and
Sebastiano
Campagna
b
a,
Maria Letizia
Di Pietro
*b,
Antonino
Giannetto
b,
Gregory
Dupeyre
c,
Philippe P.
Lainé
*c,
Francesco
Nastasi
b,
Fausto
Puntoriero
*b and
Sebastiano
Campagna
b
aChromaleont S.r.l. at Università degli Studi di Messina, Polo Annunziata, Viale Annunziata, 98168 Messina, Italy
bDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali – ChiBioFarAm – Università di Messina, Viale F. Stagno d'Alcontres 31, 98166 Messina, Italy. E-mail: fpuntoriero@unime.it
cUniversité de Paris, ITODYS, CNRS, UMR 7086, 15 rue J-A de Baïf, F-75013 Paris, France
First published on 30th December 2019
Abstract
We report on the light-switch behaviour of two head-to-tail expanded bipyridinium species as a function of their interaction with calf thymus DNA and polynucleotides. In particular, both DNA and polynucleotides containing exclusively adenine or guanine moieties quench the luminescence of the fused expanded bipyridinium species. This behaviour has been rationalized demonstrating that a reductive photoinduced electron transfer process takes place involving both adenine or guanine moieties. The charge separated state so produced recombines in the tens of picoseconds. These results could help in designing new organic substrates for application in DNA probing technology and lab on chip-based sensing systems.
Introduction
The use of intercalating species such as polypyridine metal complexes based on ruthenium, osmium, iridium, platinum,1 or organic molecules,2 may be a good strategy for DNA-probing because of their advantages such as chemical stability and reversibility of the redox processes or luminescence-switching. Recently researchers reported the use of dipyridophenazine (DPPZ) Os(II) or Ru(II) complexes as optical and redox probes for real-time methods such as real-time-PCR or PCR-free assay.3 However, it is difficult to employ these species as electrochemical probes – or combine optical and redox responses – since their relatively high oxidation potential can destroy the species immobilized on the electrode (because of guanine and adenine oxidation). Beside metal complexes, organic species can be hold as optical probes.2b,cIn this vein, pyridinium and bipyridinium salts are among the functional species the most widely used due to their appealing electron-withdrawing and redox features.4 In fact, among other implementations, these properties make pyridinium entities suitable to be integrated (i) in photochemical molecular devices (PMDs) to manage photoinduced electron transfer (PET) processes,5 as is the case for artificial multicomponent systems developed for the conversion of solar energy,5,6 (ii) within supramolecular systems designed as multielectron carriers, such as molecular batteries,7 or (iii), as redox-active stations in the field of light-powered molecular machines.8 In addition to their electrophoric behaviour, certain (bi)pyridinium salts have also recently attracted interest as chromophoric and luminophoric subunits.4n,p,9
As regards translational implementations, several years ago we studied the interaction between calf-thymus DNA, on the one hand, and both a tetraaryl-pyridinium species, namely the 1N-pyridyl-2,4,6-triphenyl-pyridinium (1) and its fused derivative 9-phenyl-benzo[c]benzo[1,2]quinolizino[3,4,5,6-ija]-[1,6]-naphthyridinium (2), on the other hand.10 It was found that the mono-cationic branched compound (1), which does not intercalate into double-stranded DNA, was switched to an intercalating agent upon irradiation with UV light (λ = 320 nm) in the presence of dioxygen. This photochemical bis-cyclization (pericondensation) was indeed leading to the formation of the fused species (2) which, conversely to the branched parent, could intercalate into the DNA double helix. Overall, this is the process we referred to as “in situ DNA intercalation on demand”. Here we study the interaction with DNA of dicationic species belonging to the family of head-to-tail (i.e. 1,4-) expanded bipyridinium compounds, in both their branched and fused forms i.e.3 and 4, respectively (Scheme 1).4o From a redox point of view, these two species have shown a completely different behaviour with respect to the previous ones, while their luminescent excited state remains at relatively high energy.4o In particular, they can be reduced at lower potential with respect to the corresponding monocationic species, so allowing the access to more oxidant excited state.
 | ||
| Scheme 1 Molecular structures with labels and synthetic relationship/filiation of here investigated branched (3) and fused (4) species. | ||
We report on the optical and luminescence properties (including ultrafast spectroscopy) of the two molecular species 3 and 4, shown in Scheme 1, as a function of their interaction with calf thymus DNA and polynucleotides of different compositions.
Results and discussion
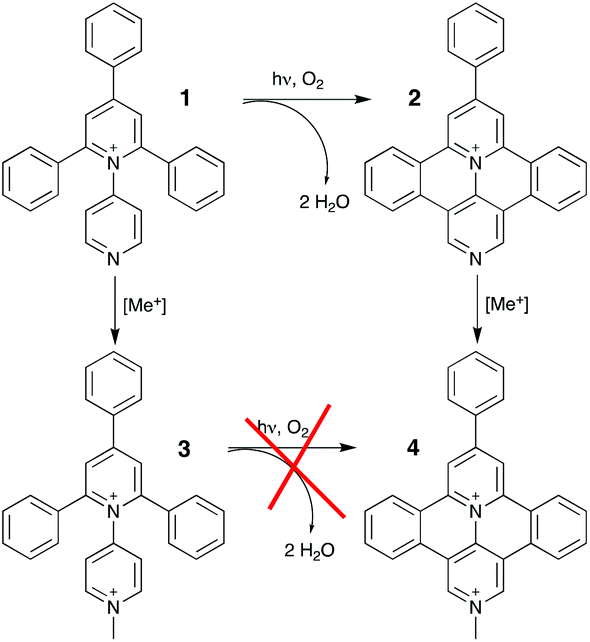
The absorption spectrum of 3 in aqueous solution, see Fig. 1, exhibits an intense band in the 300–400 nm region that, on the basis of molar absorption coefficients and band shapes, can be assigned to the lowest energy spin-allowed π–π* transitions. The spectrum recorded for 4, in the same experimental conditions, is more structured and red-shifted due to the enhanced π-delocalization over the fused polycyclic skeleton of this species (along with existence of a vibronic effect/contribution)4o as compared to 3.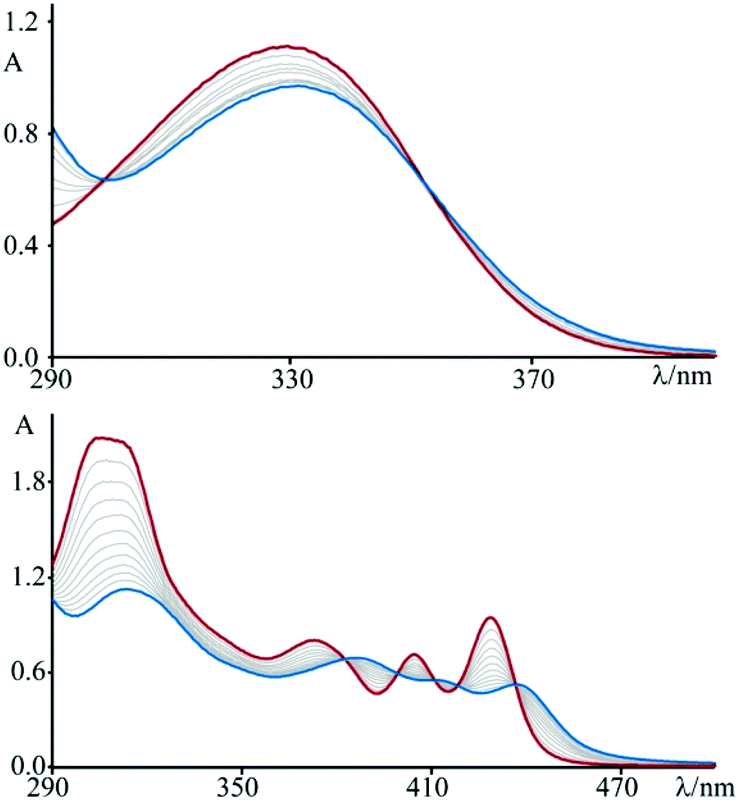 | ||
| Fig. 1 Spectrophotometric titration of (top) 3 (3.5 × 10−5 M) and (bottom) 4 (4.7 × 10–5 M) with DNA at T = 298 K and pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M); starting traces in red and last traces in blue. | ||
As a first attempt to investigate the interaction of these compounds with polynucleotides, spectrophotometric titrations of 3 and 4 in the presence of incrementally increasing amounts of DNA have been performed (see Fig. 1). In both cases, but to sharply different extents, the spectral changes are characterized by hypochromism and a shift to the red of the maxima (bathochromism), as well as isosbestic points. This observation, by far more pronounced for 4 than for 3, is indicative of a specific interaction between the fused dicationic species and DNA, which is most likely intercalation.11 Moreover, these variations are completely reversible upon addition of an excess (about 1 M) of sodium chloride, as a confirmation of the non-covalent character of the interaction process. In fact, the salt dependency of the non-covalent interaction of a cationic species with DNA12 is well recognized as an effect of the polyelectrolyte theory described by Manning and Record.13
To better characterize the interaction of the two pyridinium species with DNA, the melting temperature of DNA in the presence of 3 and 4 has been measured. Intercalation of a compound between the base pairs of the biopolymer stabilizes, in fact, the double helix, thus increasing the temperature at which it melts. The rise of the thermal denaturation temperature of the DNA in the presence of 3 (ΔTm = 6.5 ± 0.5 K) and 4 (ΔTm = 14.3 ± 0.5 K) at a 3 × 10−3 M ionic strength value (1 × 10−3 M phosphate buffer at pH 7 and 2 × 10–3 M NaCl) shows that both species stabilize the double helix similarly to what happens in the presence of the well-known reference DNA intercalator ethidium bromide (ΔTm = 10.1 ± 0.5 K),14 and much more than [Pt(en)(py)2]2+ (ΔTm = 2.7 ± 0.5 K), which is unable to intercalate.
Furthermore, intercalation of a species into DNA base pairs causes lengthening and stiffening of the helix which result in an increase of the DNA viscosity. Fig. 2 shows the viscosity enhancement induced on rod-like DNA solutions by increasing amounts of 3 and 4 at a 1.1 × 10−2 M ionic strength value, as well as of ethidium bromide and [Pt(en)(py)2]2+ for comparison purposes. As expected,15 the non-intercalator complex [Pt(en)(py)2]2+ does not produce any significant increase in the viscosity.
 | ||
| Fig. 2 Viscometric titration of DNA (6.0 × 10−4 M) with 3 (blue circles), 4 (orange squares), ethidium bromide (green triangles), and [Pt(en)(py)2]2+ (purple circles) at T = 298 K and pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 1.0 × 10−2 M). η = reduced viscosity of the DNA solution in the presence of compound, η0 = reduced viscosity of the DNA solution in the absence of compound, and r = [compound]bound/[DNA]tot. | ||
At a 2.2 × 10−2 M ionic strength value (phosphate buffer, 1 × 10–3 M; NaCl, 2.1 × 10−2 M) the resulting binding constant value, KB (obtained through the McGhee von Hippel equation),16 is about 7 (±0.7) × 104 M−1 for 3 and 1.31 (±0.1) × 106 M−1 for 4, respectively; it thus follows that the species 4 intercalates into DNA with a greater affinity than 3. This is not at all surprising, since the planar aromatic surface able to intercalate is obviously more extended in the case of the species 4, in perfect agreement with the results of the viscometric titrations shown in Fig. 2. It is interesting to note that whereas 3 shows a branched structure as 1, its dicationic nature allows for its intercalation into DNA (while 1 does not).10 The effect of the charges influences the strength of the interaction also in the case of 4, with respect to the analogous 2. In fact, the KB moves from 105 order for 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 to 106 order for 4.
10 to 106 order for 4.
As far as the luminescence properties are concerned, at room temperature and in buffered solution at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 1.0 × 10−2 M), compound 4 exhibits a structured luminescence at 439 nm, with an excited state lifetime of 4.5 ns, while 3 shows a broad emission band centred at 452 nm (τ = 500 ps).
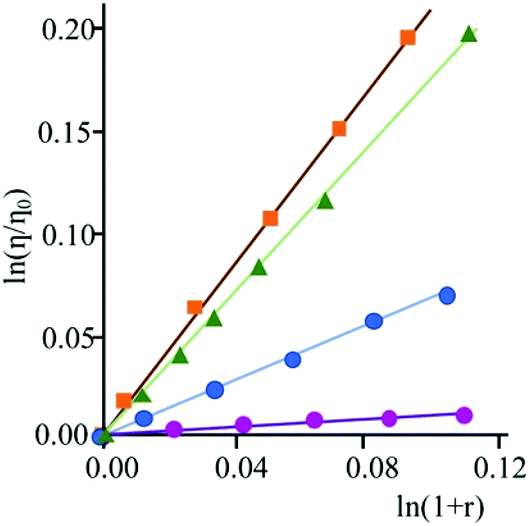
Upon addition of DNA, no significant change in the luminescence of the species 3 is observed until the achievement of a high [DNA]/[3] ratio (about 40), for which a very small decrease in emission intensity is detected. This behaviour is assumed to be a consequence of the low affinity of this species towards nucleic acids. On the contrary, addition of DNA aliquots to a solution of 4 produces noteworthy changes of the emission intensity (see Fig. 3). The luminescence, in fact, decreases sharply with increasing nucleic acid concentration, up to a total quenching when the DNA concentration becomes about six-fold that of the bipyridinium species. The species 4 then turns out to be not only sensitive to the presence of the biopolymer, but even to behave as a genuine DNA-sensitive ON/OFF light-switch.
 | ||
| Fig. 3 Emission spectral changes of 3 (top) and 4 (bottom) with DNA at T = 298 K and pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M); λexc = 345 nm and 383 nm for 3 and 4, respectively. | ||
In order to rationalize the effect of DNA on the luminescence properties of 4, experiments with synthetic polynucleotides [poly(dA-dT)2] and [poly(dG-dC)2] have been performed (see ESI – Fig. S1 and S2†). In both cases a decrease in the luminescence intensity has been observed, until its complete quenching at a [polynucleotide]/[compound] ratio of 6.8 in the case of [poly(dA-dT)2] and of 5.3 for [poly(dG-dC)2].
This behaviour is presumably due to a photo-induced electron transfer process involving guanine or adenine as donors, and the excited state of 4 as the acceptor, in line with usual findings.3d,17
The driving force associated with these reductive quenching processes can be estimated using the following equation:
 | (1) |
Noteworthy, whereas in the case of 2 only a decrease of the luminescence intensity was found after intercalation,10 in this case the luminescence of 4 is completely quenched. This behaviour could be rationalized taking into account the higher oxidant power of 4 with respect to 2 (E00 for 2 is 2.81 eV while Ered is −0.84 V, 410 mV higher with respect to 4 in the same experimental conditions).
To get further insights into photochemical processes at work, transient absorption experiments have been performed. Fig. 4 shows the pump–probe transient absorption spectra (TAS) of 4 in buffered solution. The initial transient spectrum is characterized by a negative contribution at high energy and a broad absorption centred at around 580 nm. By comparison with the absorption and emission spectra of 4, the prominent negative signal below 450 nm can be attributed to the bleaching of the ground state absorption, whereas the structured band in the range of 460–510 nm is ascribed to a stimulated emission contribution. The ground state is recovered, without spectral changes, with an experimental time constant of about 5 ns, in agreement with emission lifetime.
 | ||
| Fig. 4 Pump–probe transient absorption spectra (TAS) of 4 in buffered solution at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M). | ||
Fig. 5–7 and Fig. S3† gather the TAS of 4 in the presence of DNA, [poly(dG-dC)2] and [poly(dA-dT)2] at a 2.2 × 10−2 M ionic strength value.
 | ||
| Fig. 5 TAS of 4 at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M) in the presence of an excess of DNA; λexc = 400 nm. Top panel report the spectral changes in the early time after excitation, middle panels and bottom panel the evolutions in the following 40 ps. In the inset of the top panel is reported an enlargement of the 420–480 nm spectral region. | ||
 | ||
| Fig. 6 TAS of 4 at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M) in the presence of an excess of [poly(dG-dC)2]; λexc = 400 nm. In the upper panel are reported the spectral changes in the first ps after excitation, in the lower panel the evolutions in the following 11 ps. | ||
 | ||
| Fig. 7 TAS of 4 at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M) in the presence of an excess of [poly(dA-dT)2]; λexc = 400 nm. In the upper panel are reported the spectral changes in the first ps after excitation, in the lower panel the evolutions in the following 21 ps. | ||
As can be observed, in all cases the initial TAS appear to be qualitatively similar. They are characterized by a bleaching at wavelengths lower than 440 nm and intense transient absorption band that extends from 450 nm to 750 nm. On comparing with the absorption spectrum of the ground state, the bleaching at wavelengths lower than 440 nm is attributed to the disappearance of the lowest-energy absorption band.
Contrary to what was observed in TAS matrix of 4 alone (see Fig. S3, panel A†), in the presence of the polynucleotides the stimulated emission band is no longer observed and, furthermore, the recovery of the ground state takes place in very short times.
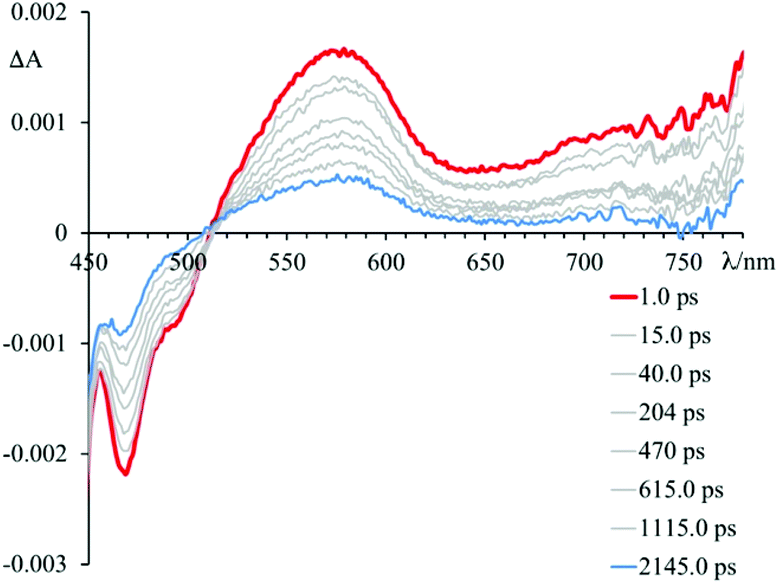
In particular, the initial spectrum of the species 4 in the presence of an excess of DNA (see Fig. 5, top panel) evolves in approximately 1 ps towards a new species characterized by a contribution in absorption at about 450 nm, which appears simultaneously with an apparent recovery of the bleaching at 435 nm and to a decrease of the transient absorption at 560 nm. An increased absorption at wavelengths longer than 700 nm also appears. This transient species decays, at least biphasically, to the ground state in less than 50 ps.
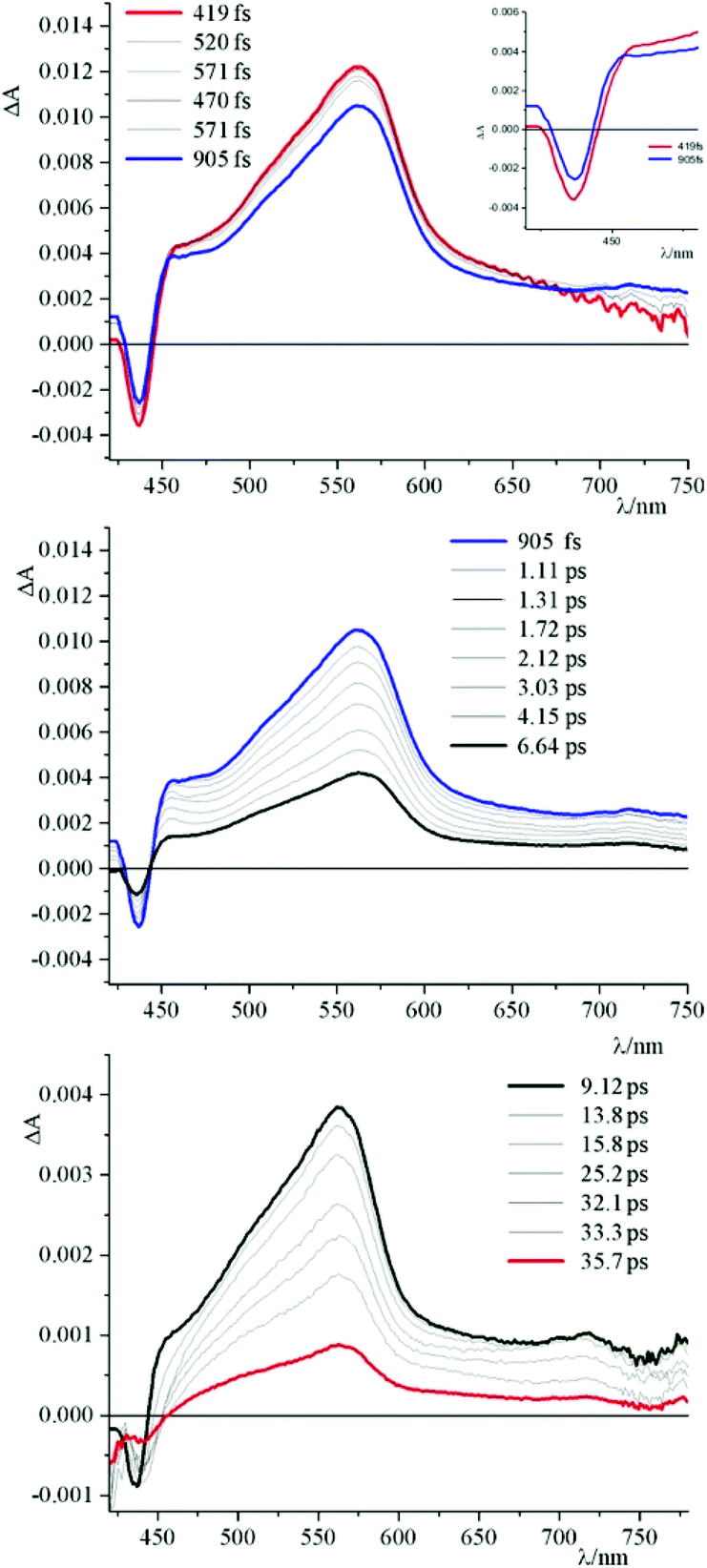
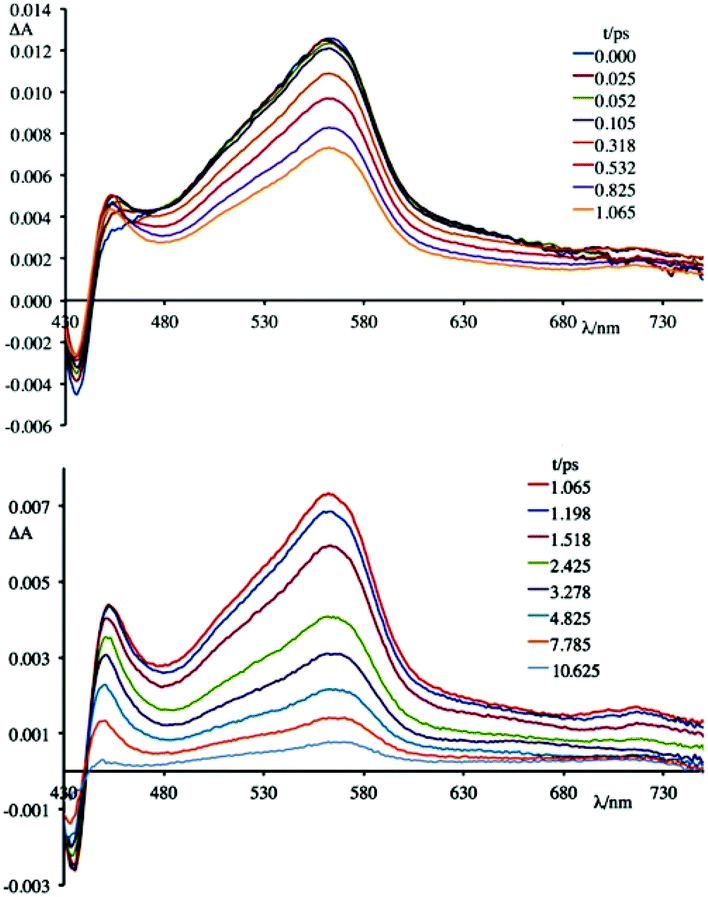
To better understand this scenario, it is worthwhile analysing in greater detail the results obtained in the presence of the synthetic polynucleotides, so as to discriminate the contributions of individual nucleobases. From a further analysis of Fig. 6 it can be seen that, in the presence of [poly(dG-dC)2], the optically formed transient species evolves in 580 fs (±80 fs) to a new type that exhibits (i) a new absorption transient band centred at 450 nm, (ii) a recovery of the bleaching at 435 nm and (iii) a decrease of the transient absorption intensity at 560 nm. This new species returns to the ground state without further significant changes in about 8.0 ± 0.5 ps. From comparison with the data in literature,19 in a first approximation, it is possible to assign the absorption band at 450 nm to the formation of the guanine radical cation.
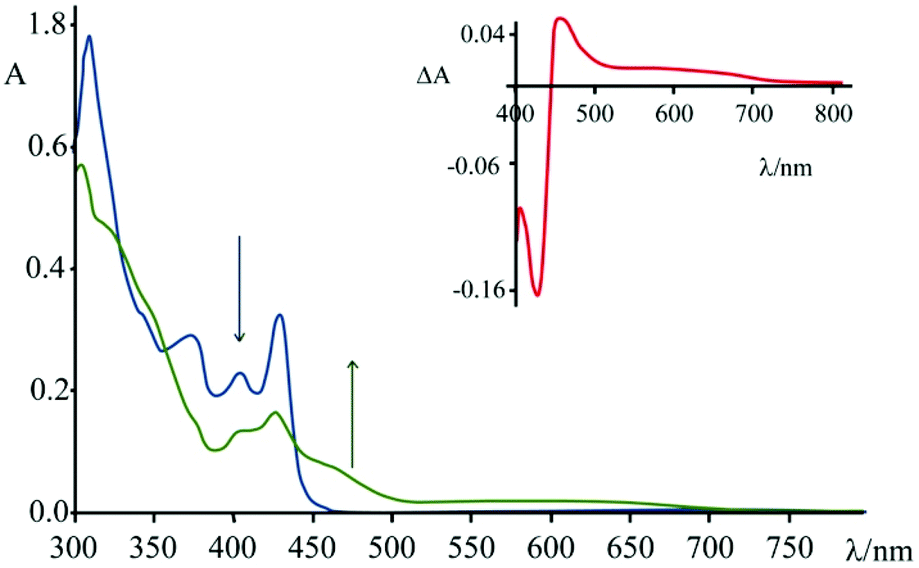
Spectroelectrochemical experiments have been conducted on 4, in acetonitrile solution and TBAPF6 as supporting electrolyte ([TBAPF6] = 5 × 10−2 M), upon applying −0.4 V vs. SCE (Fig. 8). At this potential, 4 undergoes a process of reduction which leads to the formation of the mono-cationic radical of 4.
 | ||
| Fig. 8 Absorbance spectrum of 4 (blue) in CH3CN ([TBAPF6] = 5 × 10−2 M) and absorbance spectrum of 4− (green) obtained by applying a potential of −0.4 V vs. SCE for 60 s. In the inset is reported the difference spectrum. | ||
As can be seen from Fig. 8, upon applying −0.4 V for about 60 s, the intensity of the absorption of the species 4 decreases in the region 310–430 nm and a new band (attributed to the reduced species) centred at 460 nm appears. In the inset of Fig. 8 it is possible to observe the calculated difference spectrum, which highlights the appearance of a new band in the region between 440 and 500 nm (with a minor contribution up to 700 nm). Unfortunately, this spectral region is also the one where characteristics of the guanine cation lie, so the proper contributions of the two species cannot be disentangled. The appearance of this absorption feature in the TAS, however, is a strong indication that a charge separated state is formed, involving the excited state of 4 as acceptor and the guanine as donor.
An apparently dissimilar behaviour is observed for 4 in the presence of [poly(dA-dT)2] (Fig. 7). In fact, differently from what was found in the previous case, the bleaching recorded right after excitation is slightly red-shifted, following the same trend as ground state absorption spectra of the corresponding mixtures (see Fig. 9). Unfortunately, as in the case of [poly(dG-dC)2], even with [poly(dA-dT)2] it is not possible to clearly isolate the transient absorption feature of 4 monoreduced, because its characteristic spectral area is covered by the bleaching. Nevertheless, it is possible to observe some spectral variations, albeit small, that indicate that in a very short time a new species is formed. In these conditions, in fact, 4 shows a first process (in less than 500 fs) which leads to a recovery of bleaching at 443 nm and an increase of the transient absorption at about 550 nm. An increased absorption is also apparent around 700 nm. From the comparison of the absorption spectrum of the monoreduced 4 with that of the adenine cation (which involves a low intensity absorption band at around 550 nm),17f,20 one can reasonably attribute this spectrum to that of the charge separated species obtained from an electron transfer process from the adenine nucleobase to the excited state of 4 (i.e.4*). This new species recombines to the ground state in about 25 ps.
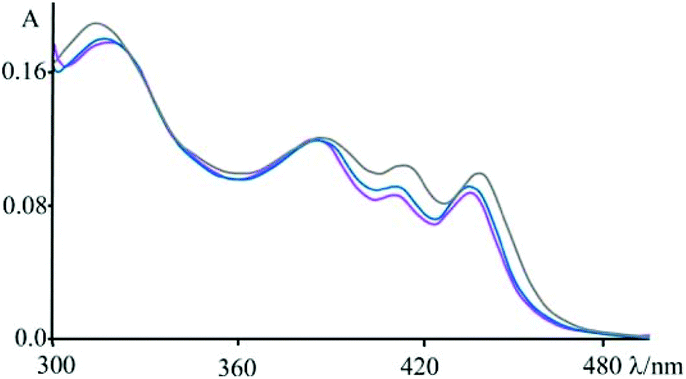 | ||
| Fig. 9 Absorbance spectra of 4 at pH 7 (phosphate buffer, 1 × 10−3 M; NaCl, 2.1 × 10−2 M) in the presence of an excess of DNA (blue), [poly(dG-dC)2] (magenta) and [poly(dA-dT)2] (grey); see also Fig. 1. | ||
Brought together, these findings allow to rationalize the behaviour observed for 4 in the presence of DNA (see Fig. 5). In particular, the spectrum formed in about 1 ps is the result of two contributions. In fact, it is possible to assign these spectral variations to the formation of two charge-separated states: one that involves 4* and the guanine fragments, and one that involves 4* and the adenine fragments. The species thus formed decays to the ground state bi-exponentially in 6.8 ps and 24 ps. In about 10 ps it is no longer possible to observe the contribution at 450 nm attributed to the guanine radical cation, while the species that involves the adenine radical cation recombines to the ground state in about 24 ps. Therefore, although it is not possible to clearly observe the two contributions that lead to the formation of the mixture of charge-separated states in less than 1 ps, the two different recombination processes leading to the formation of the ground state can be distinguished. The rate constants of the processes of charge recombination can be derived, which amount to 1.4 × 1012 s−1 in the case of the guanine-based contribution and to 5.9 × 1011 s−1 in the case of the adenine-based one.
Interestingly, such high rate constants suggest that these photoinduced processes do not imply damage of polynucleotides that would lead to irreversible accumulation of oxidized guanine and/or adenine bases.
Overall, a thorough analysis of the type of the present study can provide a guide for understanding the mechanisms that underpin the processes of charge separation involving DNA. On these bases, new photosensitizers could be designed that are capable either to achieve, if possible as a function of specific sequences of nucleotides, charge-separated states long-live enough to irreversibly damage the DNA, or – as useful for redox-optical devices (e.g. lab on chip devices) – to accelerate the charge recombination in order to obtain an output signal without damaging the DNA. In fact, for this purpose, it is of paramount importance to arrange the competition between the kinetics of charge recombination processes and that of inter-nucleotides hole transfers.20
Experimental
Compounds 3 and 4 were synthetized as previously reported.4oCalf thymus DNA, [poly(dA-dT)2], and [poly(dC-dG)2] were purchased from Sigma Chemical Co. DNA was purified as previously described,21 [poly(dA-dT)2] and [poly(dC-dG)2] were dissolved as received in the phosphate buffer containing the desired amount of NaCl to adjust the ionic strength. Their concentrations, expressed in base pairs, were determined spectrophotometrically using the molar absorptivities:22 1.32 × 104 M−1 cm−1 (258 nm), 1.32 × 104 M−1 cm−1 (262 nm), and 1.68 × 104 M−1 cm−1 (254 nm) for DNA, [poly(dA-dT)2], and [poly(dC-dG)2], respectively. All the experiments with DNA and synthetic polynucleotides were carried out at 298 K and pH 7, in a phosphate buffer 1 × 10−3 M and enough NaCl to give the desired ionic strength value.
Absorption spectra were recorded with a JASCO V570 or a Cintra 3030 GBC spectrophotometers. For luminescence spectra, a Jobin Yvon-Spex Fluoromax P spectrofluorimeter was used, equipped with a Hamamatsu R3896 photomultiplier, and the spectra were corrected for photomultiplier response using a program purchased with the fluorimeter. Luminescence lifetimes were determined by time-correlated single-photoncounting (TCSPC) with an Edinburgh OB900 spectrometer (light pulse: Hamamatsu PL2 laser diode, pulse width 59 ps at 408 nm). Experimental uncertainties are as follows: absorption maxima, 2 nm; emission maxima, 5 nm; excited state lifetimes, 10%, unless otherwise stated.
The thermal denaturation temperature of compound–DNA mixtures (1:10) was determined in 1 × 10−3 M phosphate buffer (pH 7) solutions containing the compound (7.8 × 10−6 M) and 2 × 10−3 M NaCl. Melting curves were recorded at 260 nm. The temperature has been increased at a rate of 0.5 K min−1 by using a GBC Peltier system.
Spectrophotometric titrations were performed by adding to a complex solution [(3.1–4.7) × 10−5 M] successive aliquots of DNA, containing also the complex, in a 10 mm stoppered quartz cell and recording the spectrum after each addition. Data have been reported in the form of Scatchard plots of r/m as a function of m, where r is the binding ratio [compound]bound/[DNA]tot and m is the concentration of the unbound species. The data were analysed by a nonlinear least-squares fitting program, applied to the McGhee and von Hippel equation.16 The binding constant, KB, was determined by the program, using the extinction coefficient of the compounds, the free complex concentration, and the ratio of bound complex per mole of DNA. Extinction coefficient for bound compound was determined by Beer's law plots in the presence of a large excess of DNA.
Viscosity titrations were performed by means of a Cannon-Ubbelhode semimicrodilution viscometer (series no. 75, Cannon Instrument Co.), thermostatically maintained at 298 K in a water bath. The viscometer contained 2 mL of sonicated DNA solution, in 1 × 10−3 M phosphate buffer (pH = 7) and 1.0 × 10−2 M NaCl. The compound solution ((1.2–2.0) × 10−4 M), containing also DNA (6.0 × 10−4 M) at the same concentration as that in the viscometer, was delivered in increments of 90–570 μL from a micropipet. Solutions were freed of particulate material by passing them through nylon Acrodisc syringe filters before use. Flow times were measured by hand with a digital stopwatch. Reduced viscosities were calculated by established methods and plotted as ln η/η0 against ln (1 + r) for rodlike DNA (600 base pairs) (η = reduced viscosity of the biopolymer solution in the presence of compound; η0 = reduced viscosity of the biopolymer solution in the absence of compound; r = [compound]bound/[biopolymer]tot).
Time-resolved transient absorption experiments were performed using a pump–probe setup based on the Spectra-Physics MAI-TAI Ti:sapphire system as the laser source and the Ultrafast Systems Helios spectrometer as the detector. The pump pulse was generated using a Spectra-Physics 800 FP OPA instrument. The probe pulse was obtained by continuum generation on a sapphire plate (spectral range 450–800 nm). The effective time resolution was around 200 fs, and the temporal chirp over the white-light 450–750 nm range around 150 fs; the temporal window of the optical delay stage was 0–3200 ps. In order to cancel out orientation effects on the dynamics, the polarization direction of the linearly polarized probe pulse was set at a magic angle of 54.7° with respect to that of the pump pulse. Please note that all the transient spectra shown in the present paper are chirp corrected. This correction was done by using the pump induced absorption signals themselves in the same conditions (same cuvette, solvent, temperature, stirring frequency…) used for each single experiment. All the time-resolved data were analyzed with the Ultrafast Systems Surface Xplorer Pro software.
UV-vis-NIR spectroelectrochemical measurements were obtained with a SPECAC Omni Cell System: an optically transparent thin-layer electrode (OTTLE) cell with the working Pt-mesh, twinned Ag-wire reference and Pt-mesh auxiliary electrodes melt-sealed into a polyethylene spacer – CaF2 windows and 0.25 mm path length. The UV-vis-NIR spectra were registered with a JASCO V570 spectrophotometer concurrently applying a potential by using an Autolab multipurpose equipment interfaced to a PC. TBAPF6, (+99%) supporting electrolyte and acetonitrile solvent (anhydrous, 99.8%) were supplied by Aldrich.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Italian Ministero degli Affari Esteri e per la Cooperazione Internazionale (MAECI) is acknowledged by S. C.; M. L. Di P., F. N. and F. P. thanks the University of Messina for the FFABR program.Notes and references
- (a) A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, Molecular light switch for DNA: Ru(bpy)2(dppz)2+, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS; (b) E. Trovato, M. L. Di Pietro and F. Puntoriero, Shining a New Light on an Old Game – An OsII-Based Near-IR Light Switch, Eur. J. Inorg. Chem., 2012, 3984–3988 CrossRef CAS; (c) S. Campagna, M. Cavazzini, M. Cusumano, M. L. Di Pietro, A. Giannetto, F. Puntoriero and S. Quici, Luminescent Ir(III) Complex Exclusively Made of Polypyridine Ligands Capable of Intercalating into Calf-Thymus DNA, Inorg. Chem., 2011, 50, 10667–10672 CrossRef CAS PubMed; (d) F. Puntoriero, S. Campagna, M. L. Di Pietro, A. Giannetto and M. Cusumano, Luminescence of a Pt(II) complex in the presence of DNA. Dependence of luminescence changes on the interaction binding mode, Photochem. Photobiol. Sci., 2007, 6, 357–360 RSC.
- (a) S. Conoci, A. Mascali and F. Pappalardo, Synthesis, DNA binding properties and electrochemistry towards an electrode-bound DNA of a novel anthracene–viologen conjugate, RSC Adv., 2014, 4, 2845–2850 RSC; (b) A. Mancuso, A. Barattucci, P. Bonaccorsi, A. Giannetto, G. La Ganga, M. Musarra-Pizzo, T. M. G. Salerno, A. Santoro, M. T. Sciortino, F. Puntoriero and M. L. Di Pietro, Carbohydrates and Charges on Oligo(phenylenethynylenes): Towards the Design of Cancer Bullets, Chem. – Eur. J., 2018, 24, 16972–16976 CrossRef CAS PubMed; (c) S. Barrois and H.-A. Wagenknecht, Diarylethene-modified nucleotides for switching optical properties in DNA, Beilstein J. Org. Chem., 2012, 8, 905–914 CrossRef CAS PubMed.
- (a) T. Defever, M. Druet, D. Evrard, D. Marchal and B. Limoges, Real-Time Electrochemical PCR with a DNA Intercalating Redox Probe, Anal. Chem., 2011, 83, 1815–1821 CrossRef CAS PubMed; (b) S. Petralia, M. E. Castagna, E. Cappello, F. Puntoriero, E. Trovato, A. Gagliano and S. Conoci, A miniaturized silicon based device for nucleic acids electrochemical detection, Sens. Biosens. Res., 2015, 6, 90–94 Search PubMed; (c) S. Petralia, E. L. Sciuto, M. L. Di Pietro, M. Zimbone, M. G. Grimaldi and S. Conoci, An innovative chemical strategy for PCR-free genetic detection of pathogens by an integrated electrochemical biosensor, Analyst, 2017, 142, 2090–2093 RSC; (d) K. E. Augustyn, V. C. Pierre and J. K. Barton, Metallointercalators as Probes of DNA Recognition and Reactions, Wiley, New York, 2008 Search PubMed; (e) X. Qinfeng, D. Jing, M. Xiya, Z. Yanni, L. Chen-chen and Z. Chun-yang, Structurally Defined Ru(II) Metallointercalators for Real-Time Monitoring of DNA Amplification Reactions, Anal. Chem., 2019, 91, 8777–8782 CrossRef PubMed.
- (a) V. Balzani, G. Bergamini and P. Ceroni, From the photochemistry of coordination compounds to light-powered nanoscale devices and machines, Coord. Chem. Rev., 2008, 252, 2456–2469 CrossRef CAS; (b) C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS; (c) P. Chen and T. Meyer, Medium Effects on Charge Transfer in Metal Complexes, Jpn. Chem. Rev., 1998, 98, 1439–1478 CrossRef CAS PubMed; (d) U. Narang, C. F. Zhao, J. D. Bhawalkar, F. V. Bright and P. N. Prasad, Characterization of a New Solvent-Sensitive Two-Photon-Induced Fluorescent (Aminostyryl)pyridinium Salt Dye, J. Phys. Chem., 1996, 100, 4521–4525 CrossRef CAS; (e) S. R. Marder, J. W. Perry and W. P. Schaefer, Synthesis of Organic Salts with Large Second-Order Optical Nonlinearities, Science, 1989, 245, 626–628 CrossRef CAS PubMed; (f) B. J. Coe, Switchable Nonlinear Optical Metallochromophores with Pyridinium Electron Acceptor Groups, Acc. Chem. Res., 2006, 39, 383–393 CrossRef CAS PubMed; (g) M. Konstantaki, E. Koudoumas, S. Couris, P. Lainé, E. Amouyal and S. Leach, Substantial Non-linear Optical Response of New Polyads Based on Ru and Os Complexes of Modified Terpyridines, J. Phys. Chem. B, 2001, 105, 10797–10804 CrossRef CAS; (h) S. R. Marder, J. W. Perry, B. G. Tiemann and W. P. Schaefer, Organometallic salts with large second-harmonic-generation powder efficiencies: (E)-1-ferrocenyl-2-(1-methyl-4-pyridiniumyl)ethylene salts, Organometallics, 1991, 10, 1896–1901 CrossRef CAS; (i) S. Hünig and H. Berneth, Two step reversible redox systems of the weitz type, Top. Curr. Chem., 1980, 92, 1–44 CrossRef; (j) S. R. Marder, J. W. Perry and C. P. Yakymyshyn, Organic Salts with Large Second-Order Optical Nonlinearities, Chem. Mater., 1994, 6, 1137–1147 CrossRef CAS; (k) F. Puntoriero, A. Arrigo, A. Santoro, G. la Ganga, F. Tuyèras, S. Campagna, G. Dupeyre and P. P. Lainé, Photoinduced Intercomponent Processes in Selectively Addressable Bichromophoric Dyads Made of Linearly Arranged Ru(II) Terpyridine and Expanded Pyridinium Components, Inorg. Chem., 2019, 9, 5807–5817 CrossRef PubMed; (l) C. L. Bird and A. T. Kuhn, Electrochemistry of the Viologens, Chem. Soc. Rev., 1981, 10, 49–82 RSC; (m) M.-P. Santoni, A. Santoro, T. M. G. Salerno, F. Puntoriero, F. Nastasi, M. L. Di Pietro, M. Galletta and S. Campagna, Photoinduced Charge Separation in a Donor–Spacer–Acceptor Dyad with N-Annulated Perylene Donor and Methylviologen Acceptor, ChemPhysChem, 2015, 15, 3147–3150 CrossRef PubMed; (n) J. Fortage, F. Tuyèras, P. Ochsenbein, F. Puntoriero, F. Nastasi, S. Campagna, S. Griveau, F. Bedioui, I. Ciofini and P. P. Lainé, Expanded Pyridiniums: Bis-cyclization of Branched Pyridiniums into Their Fused Polycyclic and Positively Charged Derivatives—Assessing the Impact of Pericondensation on Structural, Electrochemical, Electronic, and Photophysical Features, Chem. – Eur. J., 2010, 16, 11047–11063 CrossRef CAS PubMed; (o) J. Fortage, C. Peltier, F. Nastasi, F. Puntoriero, F. Tuyèras, S. Griveau, F. Bedioui, C. Adamo, I. Ciofini, S. Campagna and P. P. Lainé, Designing Multifunctional Expanded Pyridiniums: Properties of Branched and Fused Head-to-Tail Bipyridiniums, J. Am. Chem. Soc., 2010, 132, 16700–16713 CrossRef CAS PubMed; (p) C. Peltier, C. Adamo, P. P. Lainé, S. Campagna, F. Puntoriero and I. Ciofini, Theoretical Insights into Branched and Fused Expanded Pyridiniums by the Means of Density Functional Theory, J. Phys. Chem. A, 2010, 114, 8434–8443 CrossRef CAS PubMed; (q) J. Fortage, F. Tuyeras, C. Peltier, G. Dupeyre, A. Calborean, F. Bedioui, P. Ochsenbein, F. Puntoriero, S. Campagna, I. Ciofini and P. P. Lainé, Tictoid Expanded Pyridiniums: Assessing Structural, Electrochemical, Electronic, and Photophysical Features, J. Phys. Chem. A, 2012, 116, 7880–7891 CrossRef CAS PubMed.
- (a) P. P. Lainé, S. Campagna and F. Loiseau, Conformationally gated photoinduced processes within photosensitizer–acceptor dyads based on ruthenium(II) and osmium(II) polypyridyl complexes with an appended pyridinium group, Coord. Chem. Rev., 2008, 252, 2552–2571 CrossRef; (b) J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS and references therein; (c) V. Balzani, L. Moggi and F. Scandola, in Supramolecular Photochemistry, ed. V. Balzani, D. Reidel Publishing Co., Dordrecht, The Netherlands, 1987, p. 1 Search PubMed; (d) V. Balzani and F. Scandola, in Supramolecular Photochemistry, Ellis Horwood, Chichester, 1991 Search PubMed.
- (a) J. Fortage, G. Dupeyre, F. Tuyèras, V. Marvaud, P. Ochsenbein, I. Ciofini, M. Hromadová, L. Pospísil, A. Arrigo, E. Trovato, F. Puntoriero, P. P. Lainé and S. Campagna, Molecular Dyads of Ruthenium(II)– or Osmium(II)–Bis(terpyridine) Chromophores and Expanded Pyridinium Acceptors: Equilibration between MLCT and Charge-Separated Excited States, Inorg. Chem., 2013, 52, 11944–11955 CrossRef CAS PubMed; (b) J. Fortage, F. Puntoriero, F. Tuyèras, G. Dupeyre, A. Arrigo, I. Ciofini, P. P. Lainé and S. Campagna, Photoinduced Electron Transfer in Os(terpyridine)-biphenylene-(bi)pyridinium Assemblies, Inorg. Chem., 2012, 51, 5342–5352 CrossRef CAS PubMed; (c) F. Puntoriero, A. Arrigo, A. Santoro, G. La Ganga, F. Tuyèras, S. Campagna, G. Dupeyre and P. P. Lainé, Photoinduced Intercomponent Processes in Selectively Addressable Bichromophoric Dyads Made of Linearly Arranged Ru(II) Terpyridine and Expanded Pyridinium Components, Inorg. Chem., 2019, 58, 5807–5817 CrossRef CAS PubMed.
- (a) P. Ceroni and M. Venturi, Photoactive and Electroactive Dendrimers: Future Trends and Applications, Aust. J. Chem., 2011, 64, 131–146 CrossRef CAS; (b) M. Marchini, M. Baroncini, G. Bergamini, P. Ceroni, M. D'Angelantonio, P. Franchi, M. Lucarini, F. Negri, T. Szreder and M. Venturi, Hierarchical Growth of Supramolecular Structures Driven by Pimerization of Tetrahedrally Arranged Bipyridinium Units, Chem. – Eur. J., 2017, 23, 6380–6390 CrossRef CAS PubMed; (c) C. M. Ronconi, J. F. Stoddart, V. Balzani, M. Baroncini, P. Ceroni, C. Giansante and M. Venturi, Polyviologen Dendrimers as Hosts and Charge-Storing Devices, Chem. – Eur. J., 2008, 14, 8365–8373 CrossRef CAS PubMed; (d) A. Juris, Recent developments in photo- and redox-active dendrimers, Annu. Rep. Prog. Chem., Sect. C, 2003, 99, 177–241 RSC; (e) R. Toba, J. M. Quintela, C. Peinador, E. Roman and A. E. Kaifer, Dendritic cavitands: preparation and electrochemical properties, Chem. Commun., 2002, 1768–1769 RSC; (f) W. S. Baker, B. I. Lemon and R. M. Crooks, Electrochemical and Spectroscopic Characterization of Viologen-Functionalized Poly(Amidoamine) Dendrimers, J. Phys. Chem. B, 2001, 105, 8885–8894 CrossRef CAS; (g) P. Ceroni and M. Venturi, in Electrochemistry of Functional Supramolecular Systems, ed. P. Ceroni, A. Credi and M. Venturi, Wiley, Hoboken, NJ, 2010, p. 145 Search PubMed.
- (a) B. Colasson, A. Credi and G. Ragazzon, Light-driven molecular machines based on ruthenium(II) polypyridine complexes: Strategies and recent advances, Coord. Chem. Rev., 2016, 325, 125–134 CrossRef CAS; (b) W. Sliwa, B. Bachowska and T. Girek, Viologens as Components of Supramolecular Structures, Curr. Org. Chem., 2007, 11, 497–513 CrossRef CAS.
- (a) C. Peltier, P. P. Lainé, G. G. Scalmani, M. J. Frisch, C. Adamo and I. Ciofini, Environmental effects on electronic absorption spectra using DFT: An organic and positively charged fused polycyclic chromophore as a case study, J. Mol. Struct.: THEOCHEM, 2009, 914, 94–99 CrossRef CAS; (b) V. Balzani, A. Credi, S. J. Langford, A. Prodi, J. F. Stoddart and M. Venturi, Spectroscopic and Electrochemical Properties of Catenanes Containing the 2,7-Diazapyrenium Unit, Supramol. Chem., 2001, 13, 303–311 CrossRef CAS; (c) R. Ballardini, A. Credi, M. T. Gandolfi, C. Giansante, G. Marconi, S. Silvi and M. Venturi, Photophysical, photochemical and electrochemical properties of a series of aromatic electron acceptors based on N-heterocycles, Inorg. Chim. Acta, 2007, 360, 1072–1082 CrossRef CAS.
- M. L. Di Pietro, F. Puntoriero, F. Tuyèras, P. Ochsenbein, P. P. Lainé and S. Campagna, Photochemically driven intercalation of small molecules into DNA by in situ irradiation, Chem. Commun., 2010, 46, 5169–5171 RSC.
- E. C. Long and J. K. Barton, On demonstrating DNA intercalation, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS.
- (a) R. F. Pasternack, P. Garrity, B. Ehrlich, C. B. Davis, E. J. Gibbs, G. Orloff, A. Giartosio and C. Turano, The influence of ionic strength on the binding of a water soluble porphyrin to nucleic acids, Nucleic Acids Res., 1986, 14, 5919–5931, DOI:10.1093/nar/14.14.5919; (b) C. V. Kumar, R. S. Turner and E. H. Asuncion, Groove binding of a styrylcyanine dye to the DNA double helix: the salt effect, Photochem. Photobiol., 1993, 74, 231–238 CrossRef CAS; (c) V. S. Chirvony, V. A. Galievsky, S. N. Terekhov, B. M. Dzhagarov, V. V. Ermolenkov and P.-Y. Turpin, Binding of the Cationic 5-Coordinate Zn(II)-5,10,15,20-tetrakis(4-N-methylpyridyl)porphyrin to DNA and ModelPolynucleotides: Ionic-Strength Dependent Intercalationin [Poly(dG-dC)]2, Biospectroscopy, 1999, 5, 302–312 CrossRef CAS.
- (a) M. T. Record, C. F. Anderson and T. M. Lohman, Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity, Q. Rev. Biophys., 1978, 11, 103–178 CrossRef CAS PubMed; (b) G. S. Manning, The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides, Q. Rev. Biophys., 1978, 11, 179–246 CrossRef CAS PubMed.
- (a) M. J. Waring, Complex formation between ethidium bromide and nucleic acids, J. Mol. Biol., 1965, 13, 269–282 CrossRef CAS PubMed; (b) J. B. LePecq and C. Paoletti, A fluorescent complex between ethidium bromide and nucleic acids: Physical-Chemical characterization, J. Mol. Biol., 1967, 27, 87–106 CrossRef CAS PubMed.
- M. Cusumano, M. L. Di Pietro and A. Giannetto, DNA Interaction of Platinum(II) Complexes with 1,10-Phenanthroline and Extended Phenanthrolines, Inorg. Chem., 2006, 45, 230–235 CrossRef CAS PubMed.
- J. D. McGhee and P. H. Von Hippel, Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS PubMed.
- (a) K. E. Erkkila, D. T. Odom and J. K. Barton, Recognition and Reaction of Metallointercalators with DNA, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS PubMed; (b) D. García-Fresnadillo, N. Boutonnet, S. Schumm, C. Moucheron, A. Kirsch-De Mesmaeker, E. Defrancq, J. F. Constant and J. Lhomme, Luminescence Quenching of Ru-Labeled Oligonucleotides by Targeted Complementary Strands, Biophys. J., 2002, 82, 978–987 CrossRef; (c) J. M. Kelly, A. Tossi, D. McConnell and C. OhUigin, A study of the interactions of some polypyridylruthenium(II) complexes with DNA using fluorescence spectroscopy, topoisomerisation and thermal denaturation, Nucleic Acids Res., 1985, 13, 6017–6034 CrossRef CAS PubMed; (d) S. Campagna, M. Cavazzini, M. Cusumano, M. L. Di Pietro, A. Giannetto, F. Puntoriero and S. Quici, Luminescent Ir(III) Complex Exclusively Made of Polypyridine Ligands Capable of Intercalating into Calf-Thymus DNA, Inorg. Chem., 2011, 50, 10667–10672 CrossRef CAS PubMed; (e) Charge Transfer in DNA: From Mechanism to Applications, ed. H. A. Wagenknecht, Wiley, New York, 2005 Search PubMed; (f) F. D. Lewis, R. M. Young and M. R. Wasielewski, Tracking Photoinduced Charge Separation in DNA: from Start to Finish, Acc. Chem. Res., 2018, 51, 1746–1754 CrossRef CAS PubMed.
- S. Steenkßen and S. V. Jovanovic, How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution, J. Am. Chem. Soc., 1997, 119, 617–618 CrossRef.
- K. Ohkubo, K. Yukimoto and S. Fukuzumi, Direct detection of nucleotide radical cations produced by electron-transfer oxidation of DNA bases with electron-transfer state of 9-mesityl-10-methylacridinium ion and resulting efficient DNA cleavage without oxygen, Chem. Commun., 2006, 2504–2506 RSC.
- (a) C. Dohno, E. D. A. Stemp and J. K. Barton, Fast Back Electron Transfer Prevents Guanine Damage by Photoexcited Thionine Bound to DNA, J. Am. Chem. Soc., 2003, 125, 9586–9587 CrossRef CAS PubMed; (b) J. Yoo, S. Delaney, E. D. A. Stemp and J. K. Barton, Rapid Radical Formation by DNA Charge Transport through Sequences Lacking Intervening Guanines, J. Am. Chem. Soc., 2003, 125, 6640–6641 CrossRef CAS PubMed; (c) T. Takada, K. Kawai, S. Tojo and T. Majima, Kinetics of Multistep Hole Transfer in DNA by Monitoring the Transient Absorption of the Pyrene Radical Cation, J. Phys. Chem. B, 2003, 107, 14052–14057 CrossRef CAS.
- M. Cusumano and A. Giannetto, The interaction of mixed-ligand square-planar complexes with calf thymus DNA, J. Inorg. Biochem., 1997, 65, 137–144 CrossRef CAS PubMed.
- C. Hiort, P. Lincoln and B. Norden, DNA binding of Δ- and Δ-Ru(phen)2DPPZ]2+, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9pp00418a |
| This journal is © The Royal Society of Chemistry and Owner Societies 2020 |