
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α,β-Unsaturated acyl ammonium species as reactive intermediates in organocatalysis: an update
Jacqueline
Bitai
,
Matthew T.
Westwood
and
Andrew D.
Smith
 *
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, Fife, Scotland, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 1st March 2021
Abstract
α,β-Unsaturated acyl ammonium species are versatile intermediates that have been applied in a variety of transformations including Michael additions, domino reactions and cycloadditions. Many of these transformations are promoted by chiral Lewis base catalysts, enabling the rapid generation of molecular complexity with high stereochemical control. This review highlights recent developments in the generation and application of α,β-unsaturated acyl ammonium intermediates reported since a previous review of this area in 2016. Particular emphasis will be placed on reports providing mechanistic insight into catalytic transformations and observed selectivities. A perspective on current challenges and potential future developments in the field of α,β-unsaturated acyl ammonium catalysis is also provided.
 Jacqueline Bitai | Jacqueline Bitai obtained her BSc in 2014 from the Vienna University of Technology followed by an MSc in 2017. She is currently a final year PhD student at the University of St. Andrews in the group of Prof. Andy Smith. Her research focuses on the development of enantioselective, dual catalytic processes using isothiourea organocatalysis in combination with palladium catalysis. |
 Matthew T. Westwood | Matthew Westwood obtained his MChem degree from Heriot-Watt University (2019) with his final year project under the supervision of Dr Ai-Lan Lee. During this time he undertook an exchange year at UNSW Sydney and research placements with Prof. Martina Stenzel, Dr. Ai-Lan Lee and Dr. Ruiraidh McIntosh. Matthew is currently a second year PhD student under the supervision of Prof. Andy Smith at the University of St Andrews, researching enantioselective organocatalysis. |
 Andrew D. Smith | Andy Smith was appointed at St Andrews in October 2005 and promoted to Professor in 2012. He was awarded the RSC Merck Award in 2014 and the RSC Charles Rees Award in 2018. His research programme is focused on catalytic enantioselective reaction processes using Lewis base catalysts and developing a comprehensive mechanistic understanding of these transformations. |
1. Introduction
The development and advancement of catalytic transformations to generate complex molecular scaffolds in a stereodefined manner is of great importance to the synthetic chemistry community. Organocatalysis has proven to be a versatile tool in this regard, as it can provide selective “activation” of simple substrates under mild reaction conditions and allow the preparation of stereodefined functional molecules.1 The transformation of substrates at the carboxylic acid oxidation level by tertiary amine Lewis base catalysis has seen increasing interest within the last decade and is the focus of this review.2 Amongst the family of species accessible through covalent activation using tertiary amine Lewis bases (Fig. 1), acyl ammonium3 and C(1)–ammonium enolate4 intermediates have found widespread application (e.g. in acyl transfer reactions, and in the α-functionalization of carboxylic acids and esters, respectively). However, the use of α,β-unsaturated acyl ammonium intermediates has received comparatively little attention. Initially accessed in the 1960s by Yamamura and co-workers,5 it took nearly 40 years before their utility in enantioselective catalysis was explored. In 2006, seminal work by the group of Fu demonstrated the synthetic potential of α,β-unsaturated acyl ammonium intermediates in a (3 + 2) annulation between acyl fluorides 1 and silylated indenes 2 catalysed by planar-chiral DMAP derivative 3 to generate the corresponding diquinane products 4 (Scheme 1).6 Subsequent to this, α,β-unsaturated acyl ammonium intermediates have been explored in a variety of enantioselective organocatalytic processes.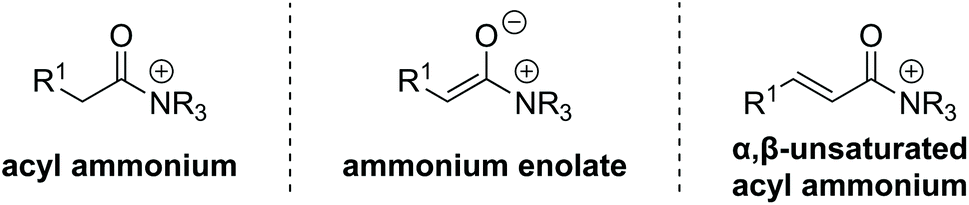 | ||
| Fig. 1 Intermediates accessible from the carboxylic acid oxidation level using tertiary amine Lewis bases (NR3). | ||
 | ||
| Scheme 1 First report of α,β-unsaturated acyl ammonium intermediates in enantioselective catalysis. | ||
In general, the formation of α,β-unsaturated acyl ammonium intermediates proceeds through acylation of the Lewis base catalyst by an appropriate precursor, forming an α,β-unsaturated acyl ammonium ion pair (Fig. 2a). These intermediates display increased electrophilicity at the C(3)-position relative to the parent substrate, increasing their reactivity. α,β-Unsaturated carboxylic acid derivatives used as precursors include acid chlorides, acid anhydrides, thioesters and electron deficient aryl esters. Among potential Lewis base catalysts suitable for the preparation of α,β-unsaturated acyl ammonium intermediates, scaffolds based on pyridine (e.g. DMAP 5 or PPY 6),7 cinchona alkaloids,8 amidines and isothioureas9 are commonly used (Fig. 2b). Chiral isothiourea derivatives have proven particularly viable and are the most common catalysts used to access α,β-unsaturated acyl ammonium intermediates for enantioselective transformations to date. The stereochemical outcome for each class of catalyst relies on facial differentiation of the catalyst bound substrate.
 | ||
| Fig. 2 (a) Common precursors to generate α,β-unsaturated acyl ammonium intermediates; (b) common tertiary amine Lewis base catalysts. | ||
This review provides an update on recent progress in the field of catalytic processes that proceed via α,β-unsaturated acyl ammonium intermediates. For a comprehensive discussion on processes developed prior to early 2016, readers are directed to a previous excellent review from Romo and co-workers.10 For ease of navigation, this review will be divided into two main parts based on the initial bond forming step: conjugate additions and cycloadditions (Scheme 2). Within conjugate additions, further distinctions are made based upon selective employment of the potential reactive sites within the α,β-unsaturated acyl ammonium intermediate during the catalytic process. These include Michael additions featuring aryloxide facilitated catalyst turnover (one reactive site used), Michael addition–annulation sequences (two reactive sites used) and domino reactions (three reactive sites used). In addition, a section highlighting miscellaneous processes that have suggested the involvement of α,β-unsaturated acyl ammonium intermediates is included. Where applicable, particular emphasis will be placed on mechanistic insights.
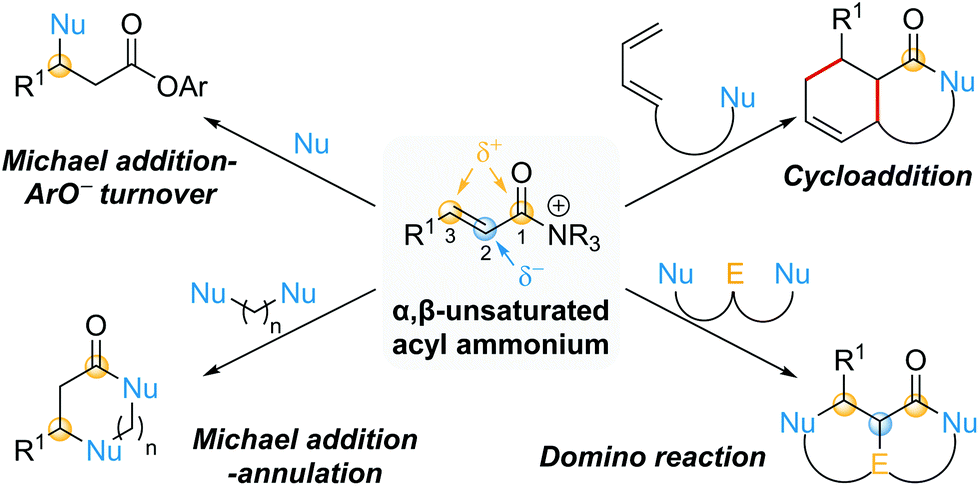 | ||
| Scheme 2 Review organisation based on exploited reactive sites (Nu = nucleophile, E = electrophile). | ||
2. Conjugate additions
2.1. Aryloxide facilitated catalyst turnover
The most conceptually simple method of functionalising α,β-unsaturated acyl ammonium intermediates involves addition of a nucleophile at the β-position to generate a monofunctionalised product. This transformation requires the counterion generated upon catalyst acylation to facilitate catalyst turnover without interfering in the conjugate addition step. Electron deficient aryloxide ions have proven particularly suitable for this task, making “activated” electron deficient aryl esters (such as 4-nitrophenyl or pentafluorophenyl esters) ideal precursors for simple conjugate addition reactions. This aryloxide facilitated catalyst turnover strategy is showcased in a generalised catalytic cycle (Scheme 3). Starting from an unsaturated aryl ester, acylation of the catalyst gives an α,β-unsaturated acyl ammonium ion pair, releasing the aryloxide counterion. Reaction of the aryloxide with the post-reaction acyl ammonium ion regenerates the catalyst and yields the corresponding aryl ester product. As aryl esters can prove rather unstable to isolation and purification, an in situ, post catalysis transformation into more stable and isolable ester or amide products is often necessary. This strategy has been successfully implemented in other tertiary amine Lewis base catalysed processes that proceed via C(1)–ammonium enolates4c and related ammonium ylide rearrangements11 and allows for the use of simple monofunctional reaction partners. Unification of this strategy with α,β-unsaturated acyl ammonium chemistry allows for the construction of acyclic β-functionalised products.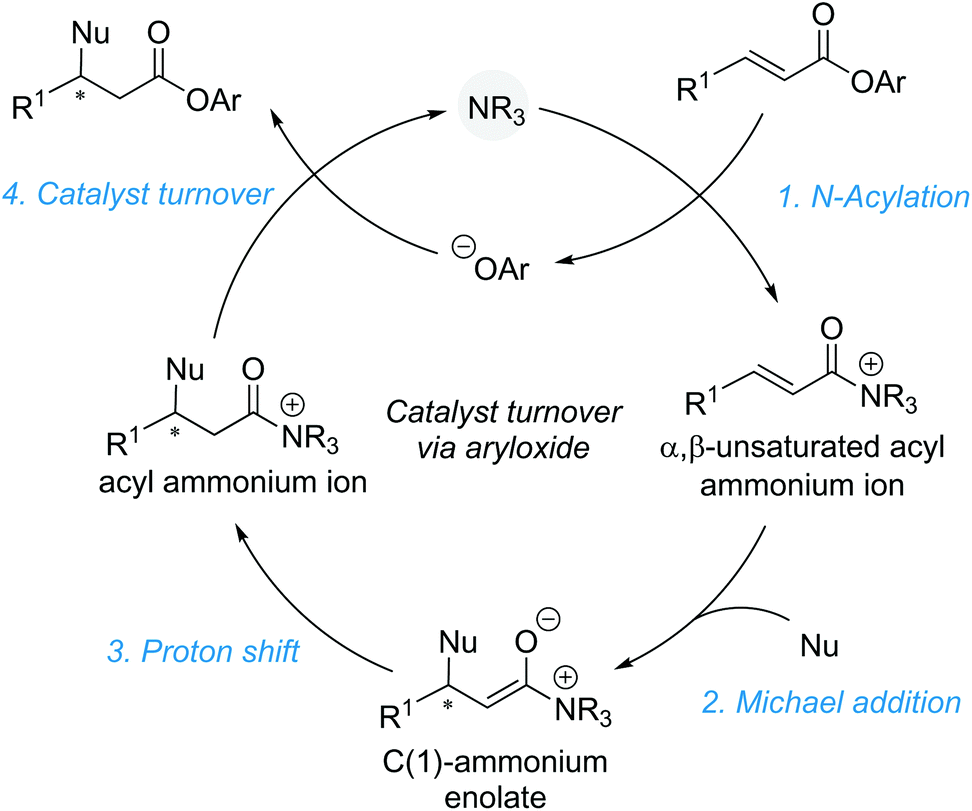 | ||
| Scheme 3 Aryloxide facilitated catalyst turnover strategy. | ||
In 2017, Smith and co-workers utilised this strategy in the enantioselective conjugate addition of simple nitroalkanes to α,β-unsaturated aryl ester Michael acceptors (Scheme 4a).12 A range of electron deficient aryl esters 7 underwent reaction with nitroalkane pronucleophile 8 as solvent to give β-functionalised products 10. High enantioselectivity was observed for simple symmetric nitroalkanes (R2 = R3). However, when unsymmetrical, disubstituted nitroalkanes (R2 ≠ R3) were employed, poor diastereoselectivity was observed, presumably due to post reaction equilibration. Mechanistic studies elucidated the reaction mechanism (Scheme 4b). Quantitative in situ reaction monitoring using 19F{1H} NMR spectroscopy, employing 19F labelled aryl ester 11 and (2R,3S)-8F-HyperBTM 12 identified acyl ammonium species 14 and its corresponding post conjugate addition species 15 as reaction intermediates as well as catalyst deactivation via protonation (Scheme 4b). Kinetic analysis using the variable time normalisation graphical analysis method developed by Burés13 revealed the reaction to be first order with respect to both aryl ester 11 and catalyst 12. Product inhibition via competitive acylation of catalyst 12 was also observed. Owing to the lability of the p-nitrophenyl ester moiety during isolation, a nucleophilic quench using an amine or alcohol was performed to give the stable amide or ester products, respectively.
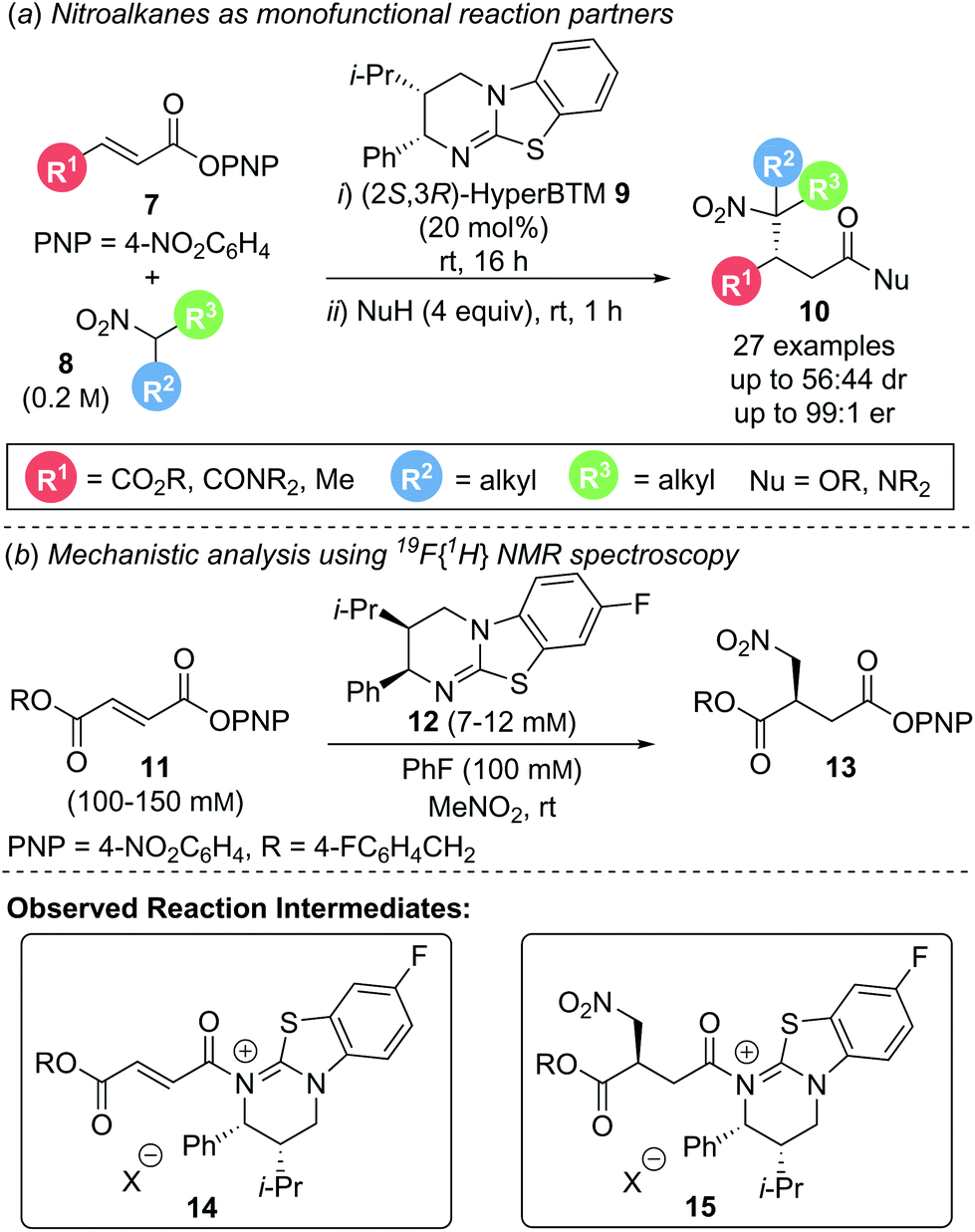 | ||
| Scheme 4 (a) Aryloxide turnover in the conjugate addition of nitroalkanes; (b) mechanistic investigation. | ||
The stereochemical outcome observed for these isothiourea catalysts is significantly influenced by a 1,5-O⋯S interaction between the acyl oxygen atom and sulfur atom of the catalyst (nO to σ*C–S), which restricts the conformational freedom of the catalyst bound intermediate (Scheme 5a).14 This not only increases the electrophilicity at the C(3)-position, but also induces facial differentiation of the substrate. N-acylation to generate the α,β-unsaturated acyl ammonium intermediate forces the stereodirecting phenyl substituent to adopt a pseudoaxial position to minimize 1,2 strain. This effectively blocks the Re face of the intermediate and thus, nucleophilic attack occurs preferentially from the Si face. As part of a study into the significance of this 1,5-Ch⋯Ch (Ch = chalcogen) interaction within isothiourea organocatalysis, two structural analogues of isothiourea catalyst (2R,3S)-HyperBTM 9 were reported (Scheme 5b).14g Replacement of the sulfur atom with either oxygen or selenium led to isourea 16 and isoselenourea 17, respectively. The strength of the intramolecular 1,5-Ch⋯O interaction within an acylated intermediate was predicted to be substantially weaker when Ch = O and significantly enhanced when Ch = Se.15 This interaction is postulated to be key to guiding reactivity and selectivity using isothiourea catalysts, with an enhanced 1,5-Ch⋯Ch interaction expected to result in increased reactivity. This postulate was borne out experimentally using the previously reported conjugate addition reaction between aryl ester 18 or 11 and nitromethane as a benchmark reaction. The isourea 16 showed only trace reactivity and essentially gave racemic conjugate addition product 19 or 13 (Scheme 5b, entry 1). Isothiourea 9 and isoselenourea 17 both performed well with similar enantioselectivity (∼95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 er) and yield (55%) (Scheme 5b, entries 2 and 3). Significantly, when monitored by 19F{1H} NMR spectroscopy the reaction catalysed by isoselenourea 17 showed a drastic increase in reaction rate (t1/2 = 8 min) relative to the same reaction catalysed by isothiourea 9 (t1/2 = 145 min).
:5 er) and yield (55%) (Scheme 5b, entries 2 and 3). Significantly, when monitored by 19F{1H} NMR spectroscopy the reaction catalysed by isoselenourea 17 showed a drastic increase in reaction rate (t1/2 = 8 min) relative to the same reaction catalysed by isothiourea 9 (t1/2 = 145 min).
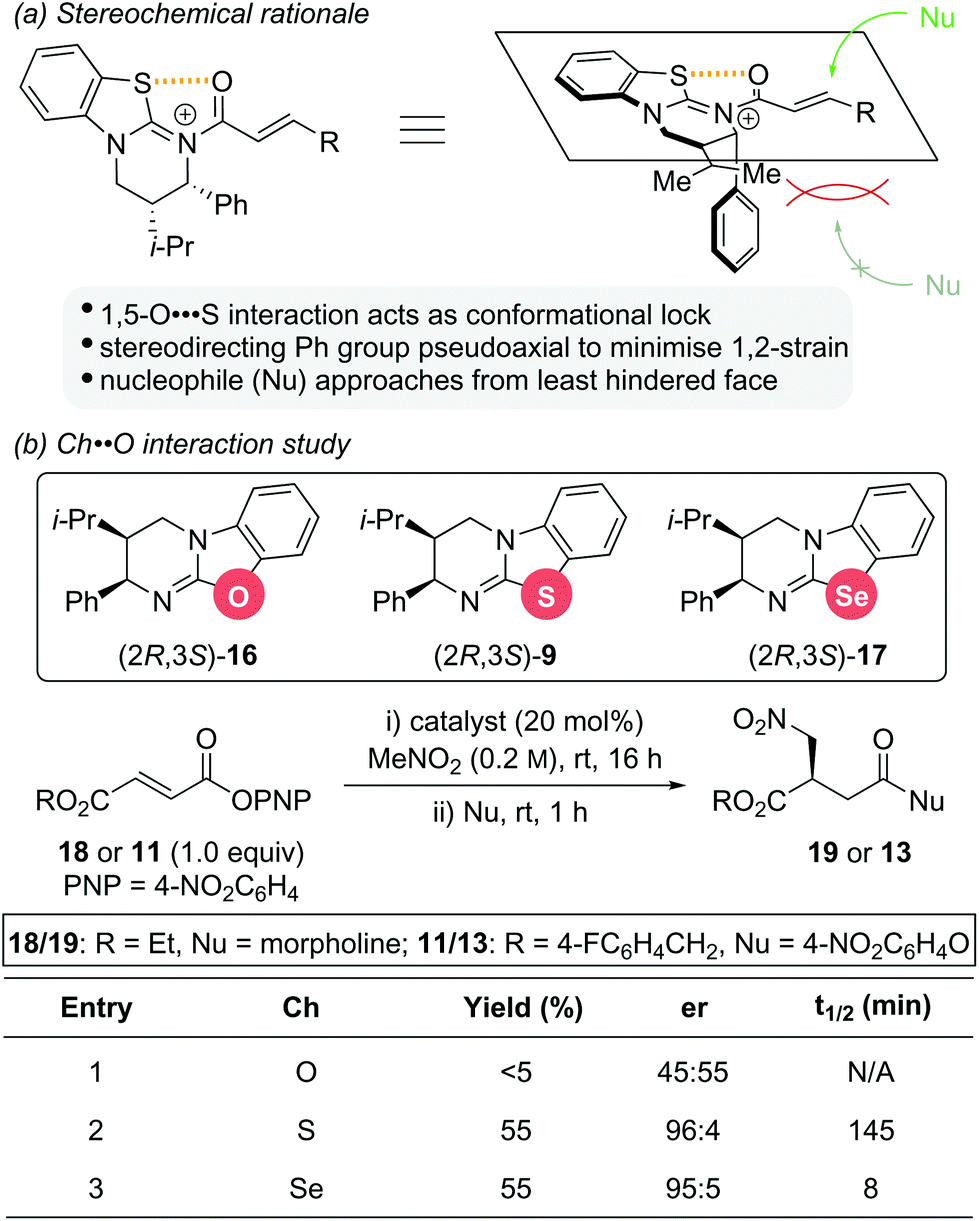 | ||
| Scheme 5 (a) Stereochemical rational for isothiourea catalysis; (b) structural analogues of HyperBTM 9 and their catalytic activity. | ||
In a related reaction, Smith and Lupton utilised silyl nitronates 24 as both nucleophile and to facilitate catalyst turnover via silyl ester formation (Scheme 6b).16 This unexpected silyl group migration was first observed in the reaction of aryl ester 18 with silyl nitronate 20, yielding a trace amount of silyl ester 22 (5%) alongside the expected conjugate addition product 21 (Scheme 6a). The use of symmetric anhydrides 23 as an α,β-unsaturated acyl ammonium precursor proved optimal, allowing access to a range of γ-nitro substituted silyl esters 25 with excellent enantioselectivity (>94:6 er) with reaction times of less than an hour. Intriguingly, the presence of a β-phenyl substituent did not give the expected silyl ester product but instead gave the nitroso ester 26. Silyl nitronates appear to show greater reactivity than the corresponding nitroalkanes previously described in Scheme 4. Use of a range of unsymmetrically substituted silyl nitronates (R2 ≠ R3) generated products with two contiguous stereogenic centres with excellent enantioselectivity and higher diastereoselectivity than observed with the corresponding nitroalkanes. Epimerisation studies were performed to gain insight into the origin of the diastereoselectivities observed. For disubstituted nitronates (where R2 ≠ R3 ≠ H), no epimerisation could be observed, indicating that the diastereomeric ratio is kinetically controlled. In contrast, monosubstituted nitronates (R3 = H) give products that readily epimerise α- to the nitro group, resulting in a thermodynamic ratio of diastereoisomers. Whilst several control experiments were performed to elucidate the formation of the silyl ester product, the mechanism of the silyl group transfer remains unclear.
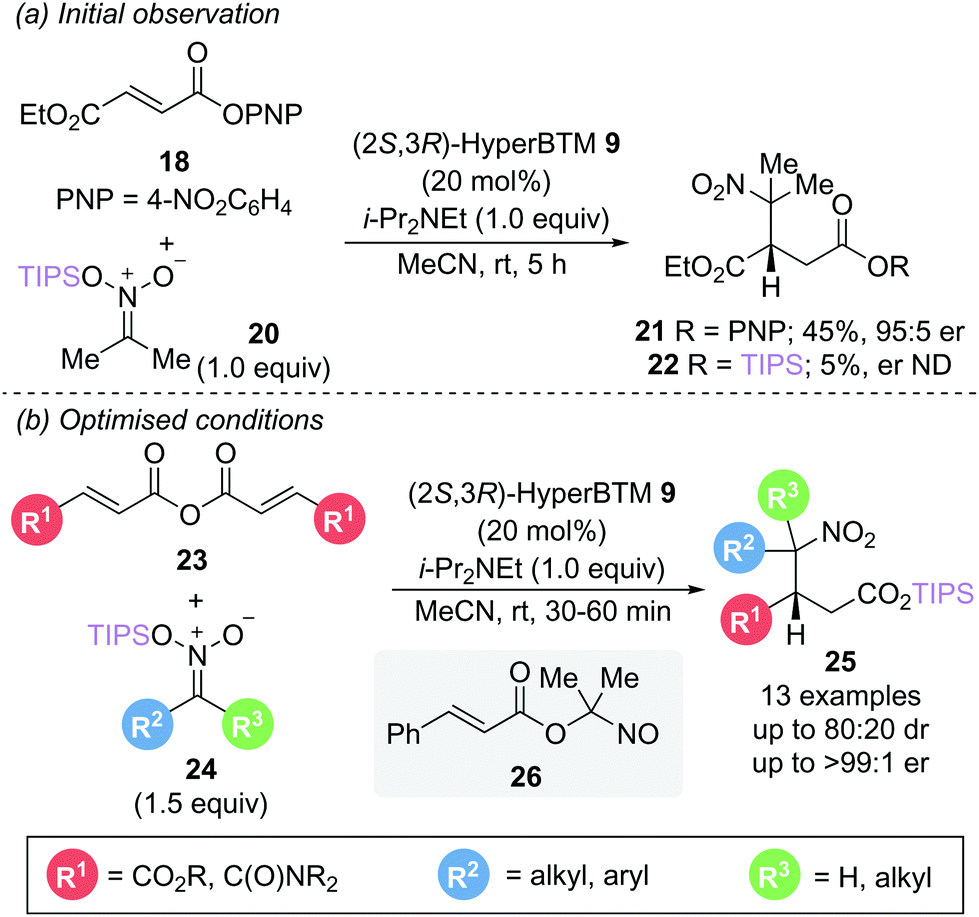 | ||
| Scheme 6 Michael addition using silyl nitronate nucleophiles: (a) initial observation and (b) optimised conditions. | ||
In 2020, Smith and co-workers reported the use of heterocyclic pro-nucleophiles as monofunctional reaction partners, expanding the applicability of the aryloxide turnover strategy (Scheme 7a).17 Dihydropyrazol-3-one 27 and 3-phenyloxindole 29 proved to be viable reaction partners for conjugate addition to a range of α,β-unsaturated aryl esters. Reaction of dihydropyrazol-3-ones 27 with aryl ester 7 in the presence of (2R,3S)-HyperBTM 9 gave the corresponding products 28 in uniformly excellent enantioselectivity (>92:8 er), but with modest diastereoselectivity. The reaction was high yielding and tolerant of various substituents within the nucleophile, including alkyl, allyl, and both aromatic and heteroaromatic substitution (Scheme 7a, top). Addition of 3-aryloxindoles 29 to aryl esters 7 also proceeded smoothly with excellent enantio- and uniformly high diastereocontrol, with the majority of examples showing >90:10 dr and >95:5 er. A range of functionalities were tolerated on each reaction partner with only minor variations in enantioselectivity and yield observed (Scheme 7a, bottom). Notably, medicinally interesting polyhalogenated substituents within the aryl ester (R1, X = F, Cl, Br) gave the corresponding products in high yields, and excellent enantio- and diastereoselectivity. Moreover, crotonic and cinnamic acid derived substrates (R1 = Me or Ph, respectively), typically unreactive in these processes, were also successfully employed. Other heterocyclic pro-nucleophiles showed almost no reactivity, attributed to their comparatively higher pKa values disfavouring enolate formation. The observed diastereoselectivity can be rationalised through a pre-transition state assembly with a potential stabilising C–H⋯O interaction between the enolate oxygen and the catalyst α-C–H (Scheme 7b).
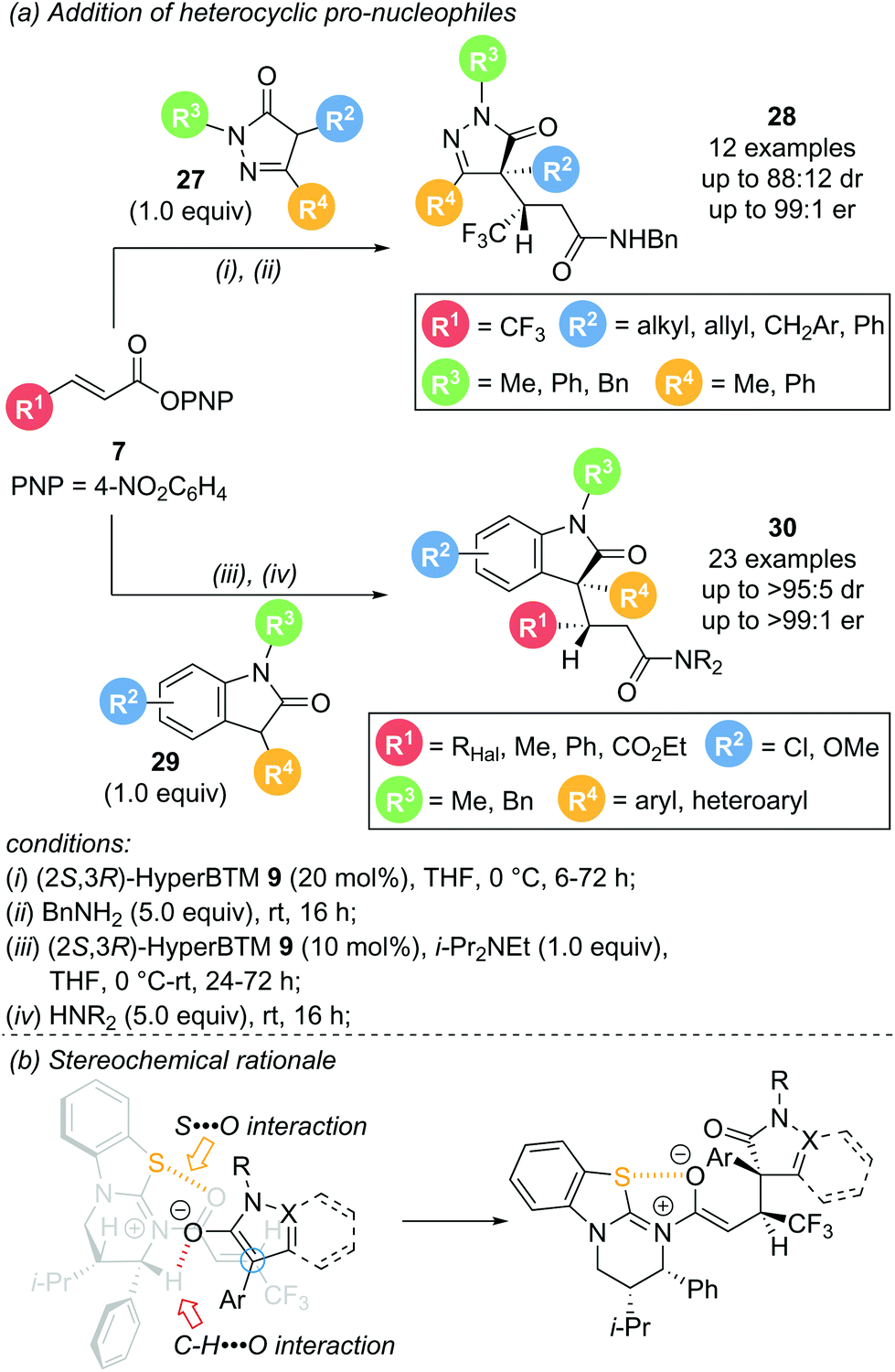 | ||
| Scheme 7 (a) Conjugate addition of heterocyclic pro-nucleophiles; (b) pre-transition state assembly as stereochemical rational. | ||
2.2. Michael addition–annulation reactions
A Michael addition–annulation approach harnesses the electrophilic reactivity of the α,β-unsaturated acyl ammonium intermediate at the C(1) and C(3) positions, employing bis-(pro)nucleophiles to generate cyclic products. A generalised catalytic cycle (Scheme 8) demonstrates the overall reaction, starting with acylation of the Lewis base by addition to an appropriate precursor to generate an α,β-unsaturated acyl ammonium ion pair. Michael addition of a nucleophile (Nu−) generates an intermediate C(1)–ammonium enolate. Proton transfer leads to an acyl ammonium ion, which undergoes intramolecular nucleophilic attack by the second nucleophile (Y), promoting catalyst release and generating the cyclic product. Importantly, the use of a bis-nucleophile leads to potential regioselectivity issues, which must be controlled generally by either employing nucleophiles with distinct reactivities or generating/unveiling the second nucleophile in situ. This Michael addition–annulation sequence is a convenient way of generating functionalised lactones and lactams in a stereodefined manner, as showcased by various research groups.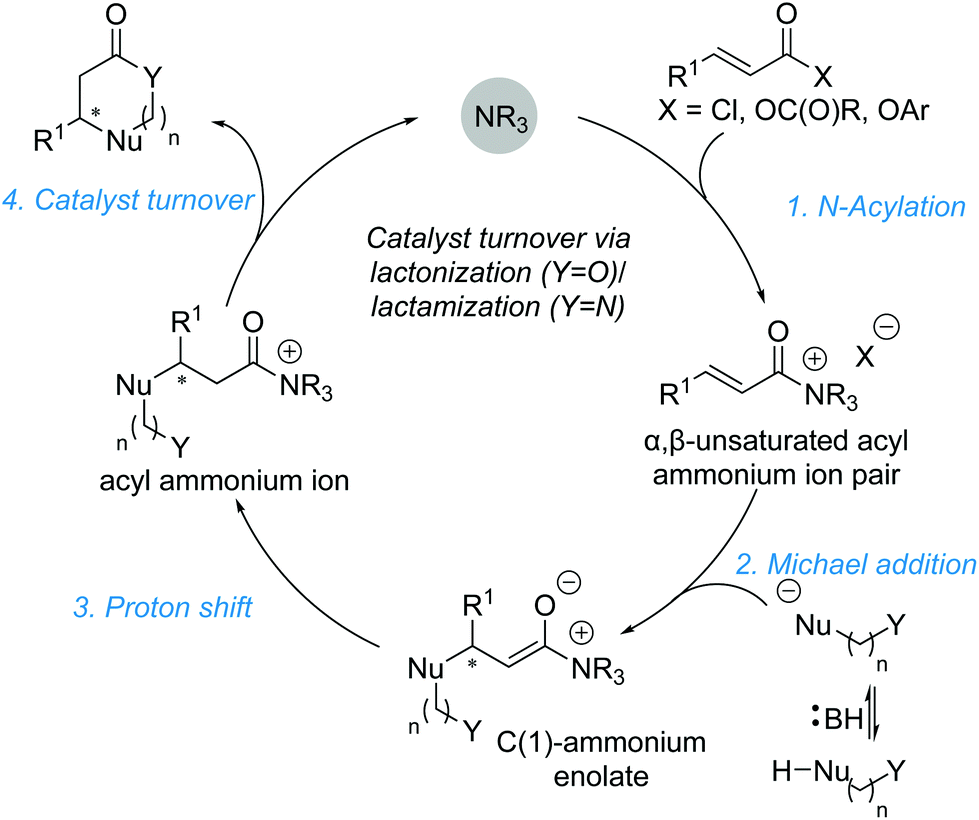 | ||
| Scheme 8 General catalytic cycle for a Michael addition–annulation reaction. | ||
In 2013, Smith and co-workers investigated the use of symmetric anhydrides as precursors in α,β-unsaturated acyl ammonium catalysis employing 1,3-dicarbonyl and 2-phenacylbenzothiazole nucleophiles (31 and 34, respectively) (Scheme 9a).18 The use of the latter led to the formation of dihydropyridinone products 35 through lactamization rather than the expected dihydropyranone products 36 through lactonization. Intrigued by this result, the groups of Smith and Cheong reported a combined experimental and computational study probing the chemo- and enantioselectivity observed in the Michael addition – annulation processes using benzazole nucleophiles.14d,18,19 Employing catalytic (2S,3R)-HyperBTM 9 (5 mol%), three types of benzazole nucleophiles (2-phenacylbenzoxazole 37, 2-phenacylbenzothiazole 34 and 2-N,N-dimethylacetamido-benzothiazole 38) were investigated in the reaction with a range of symmetric anhydrides 23 (Scheme 9b). Employing 2-phenacylbenzoxazoles 37 exclusively afforded the respective lactone products 39 (>95:5 lactone:lactam) with excellent enantiocontrol (>95:5 er). 2-Phenacylbenzothiazoles 34, on the other hand, led to preferential lactam formation (typically >80:20 lactam:lactone) with up to 95:5 er, consistent with the original observation by Smith and co-workers. Attempts to interconvert the minor lactone product 36 into the corresponding lactam 35 were unsuccessful, indicating that the product ratio results from kinetic control. Employing benzothiazole amides 38 led to the exclusive formation of the corresponding lactam product 40 (>95:5 lactam:lactone) with excellent enantioselectivity (>96:4 er). This increase in chemoselectivity within the benzothiazole series can be attributed to the preferred formation of the aza-enolate on the benzothiazole rather than the corresponding enolate on the amide functionality, favouring reaction through the benzothiazole N-atom. Computational analysis of the involved intermediates and transition structures using the M06-2X DFT method elucidated the importance of long-range 1,5-S⋯O interactions in determining the chemoselectivity (Scheme 9c). In the case of both benzoxazole and benzothiazole nucleophiles, a 1,5-S⋯O interaction can be found in the acylated catalyst between the isothiourea S-atom and the acyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. In addition, benzoxazoles exhibit a non-traditional hydrogen bonding C–H⋯O interaction between the catalyst α-C–H and the benzoxazole enolate, pre-organizing the cyclization transition state in favour of lactonization. However, for benzothiazoles a second 1,5-S⋯O interaction between the enol O-atom and the benzothiazole S-atom is energetically more favourable than a C–H⋯O interaction. With the enol O-atom unavailable to displace the catalyst, the cyclisation is forced to occur through the benzothiazole N-atom, leading to the corresponding lactam product.
O group. In addition, benzoxazoles exhibit a non-traditional hydrogen bonding C–H⋯O interaction between the catalyst α-C–H and the benzoxazole enolate, pre-organizing the cyclization transition state in favour of lactonization. However, for benzothiazoles a second 1,5-S⋯O interaction between the enol O-atom and the benzothiazole S-atom is energetically more favourable than a C–H⋯O interaction. With the enol O-atom unavailable to displace the catalyst, the cyclisation is forced to occur through the benzothiazole N-atom, leading to the corresponding lactam product.
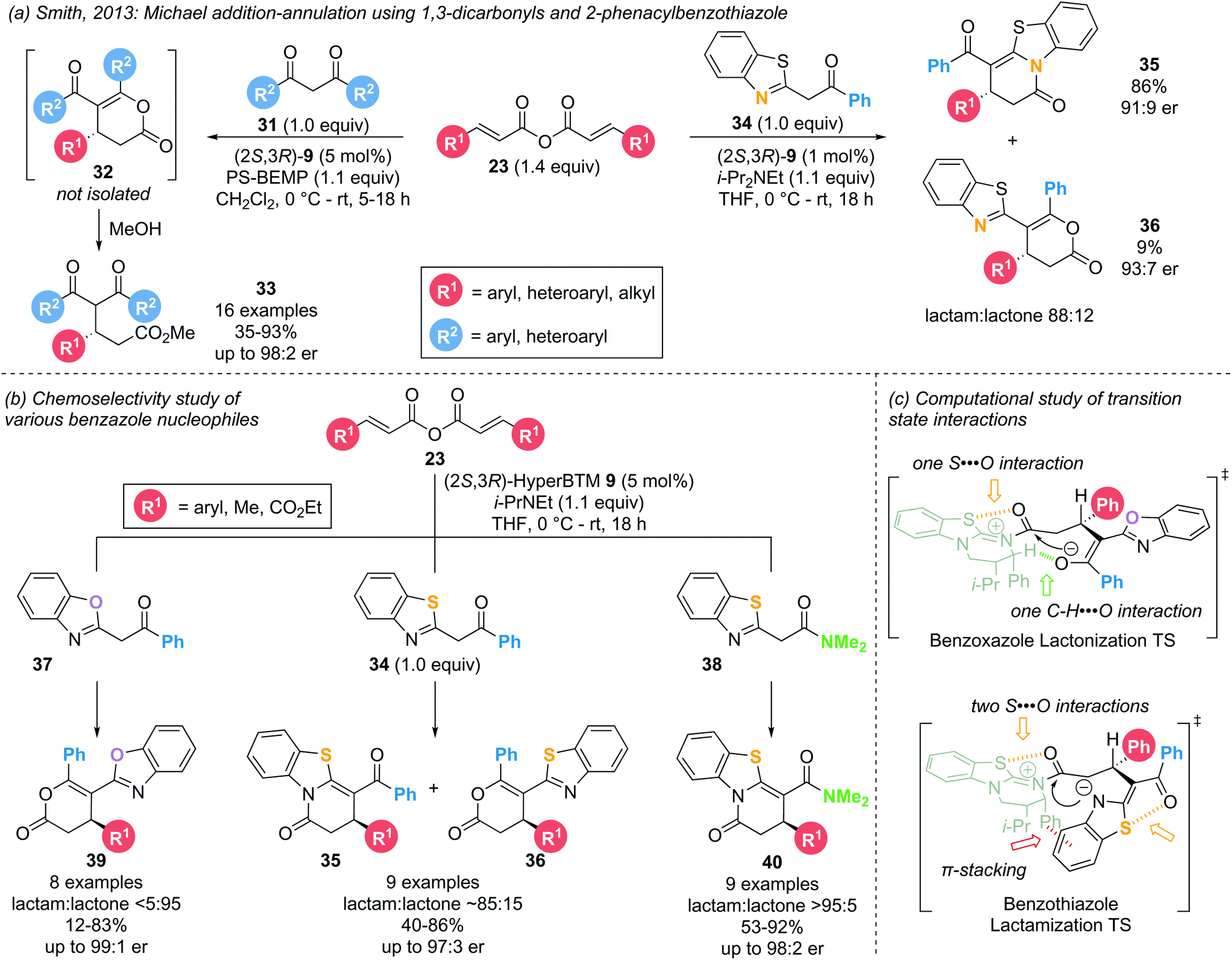 | ||
| Scheme 9 (a) Original Michael addition–annulation sequence; (b) Michael-lactamization vs. Michael-lactonization governed by non-bonding interactions; (c) computed interactions in cyclisation transition states governing chemoselectivity. | ||
This protocol was later extended to incorporate polyfluorinated substituents using polyfluoroalkyl-substituted α,β-unsaturated aryl esters 41 as precursors (Scheme 10a).19 Reaction with various benzothiazole (42) and benzoxazole (43) nucleophiles in the presence of (2S,3R)-HyperBTM 9 (5 mol%) furnished the corresponding lactam or lactone products 44–46 in high yield and excellent enantioselectivity following a Michael addition–annulation sequence. Notably, incorporation of an isomerisation step at the end of the reaction drastically increased the chemoselectivity in benzothiazole derived products 44 to exclusively yield the corresponding lactam. Application of this strategy to benzoxazole nucleophiles allowed for the selective formation of the lactonization product 45 (without isomerisation) or lactamization product 46 (with isomerisation) with excellent enantioselectivities. Symmetric anhydrides 23 used as α,β-unsaturated acyl ammonium precursors in the previous report were also compatible with this annulation–isomerisation protocol, yielding exclusively the corresponding lactam product 35 (Scheme 10b).
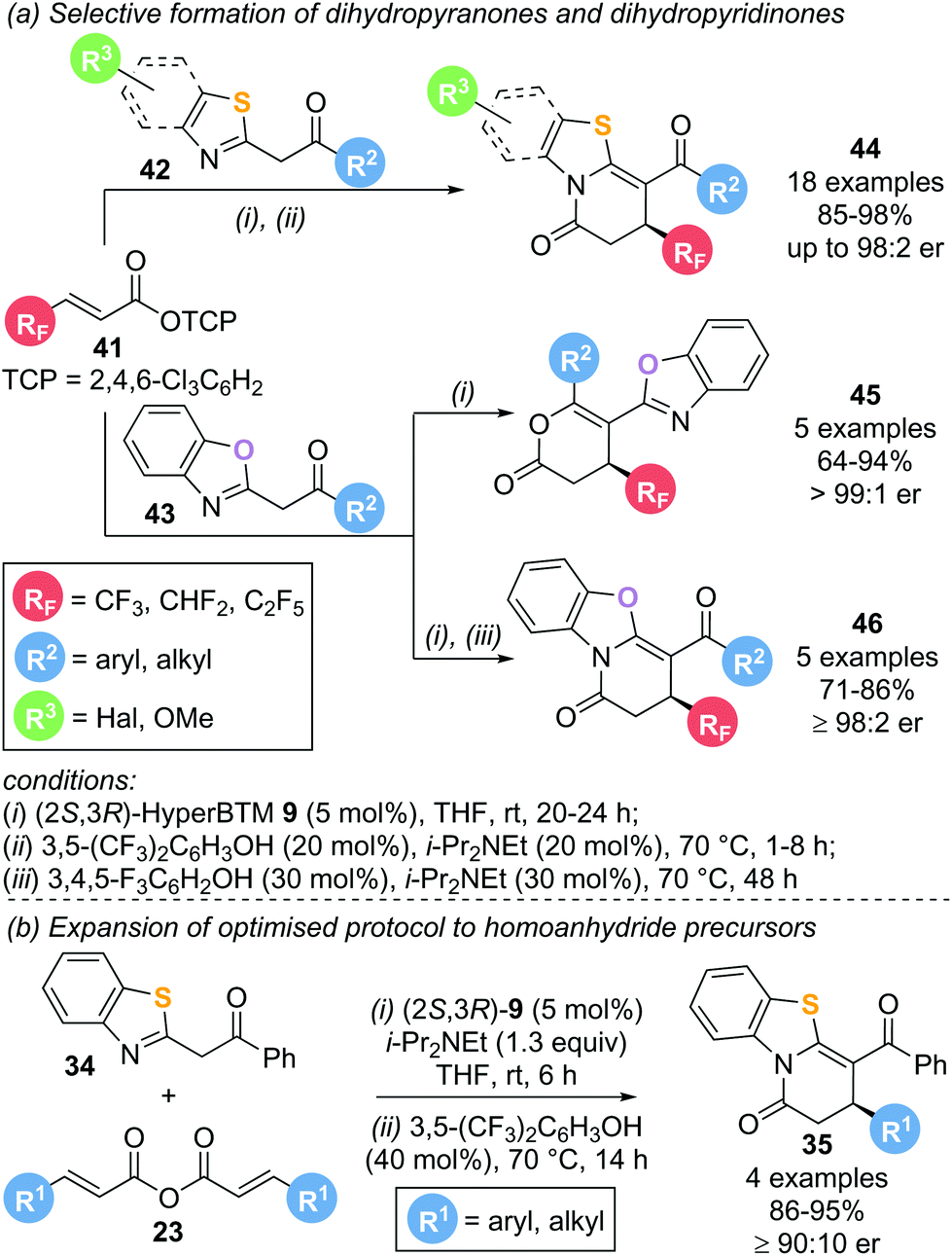 | ||
| Scheme 10 Selective Michael-lactamization or Michael-lactonization using aryl esters. | ||
Mechanistic experiments revealed the importance of the aryloxide counterion, generated in situ upon formation of α,β-unsaturated acyl ammonium intermediate 47, for the high chemoselectivity observed (Scheme 11). The aryloxide was found to play multiple roles in this annulation–isomerisation reaction:
(i) It can act as a Brønsted base to form the benzazole enolate 49 from benzazole 48, circumventing the need for an auxiliary base. Trichlorophenol (TCP) proved optimal in this regard, as it did not promote a base-mediated background reaction, giving the products in high enantioselectivity.
(ii) It can act as a Lewis base, facilitating isomerisation of lactone 52 into lactam 54via intermediate 53. Notably, 3,5-bis(trifuoromethyl)phenoxide or 3,4,5-trifluorophenoxide proved optimal in the isomerisation step, as ortho-substituted phenoxides were ineffective.
(iii) For benzoxazole derived lactones, the aryloxide can also act as a Lewis acid in an alternative isomerisation pathway. Activation of the lactone through a hydrogen bonding interaction (55) enables a (2S,3R)-HyperBTM 9 catalysed isomerisation into lactam 54via intermediate 56 in a kinetic resolution type process.
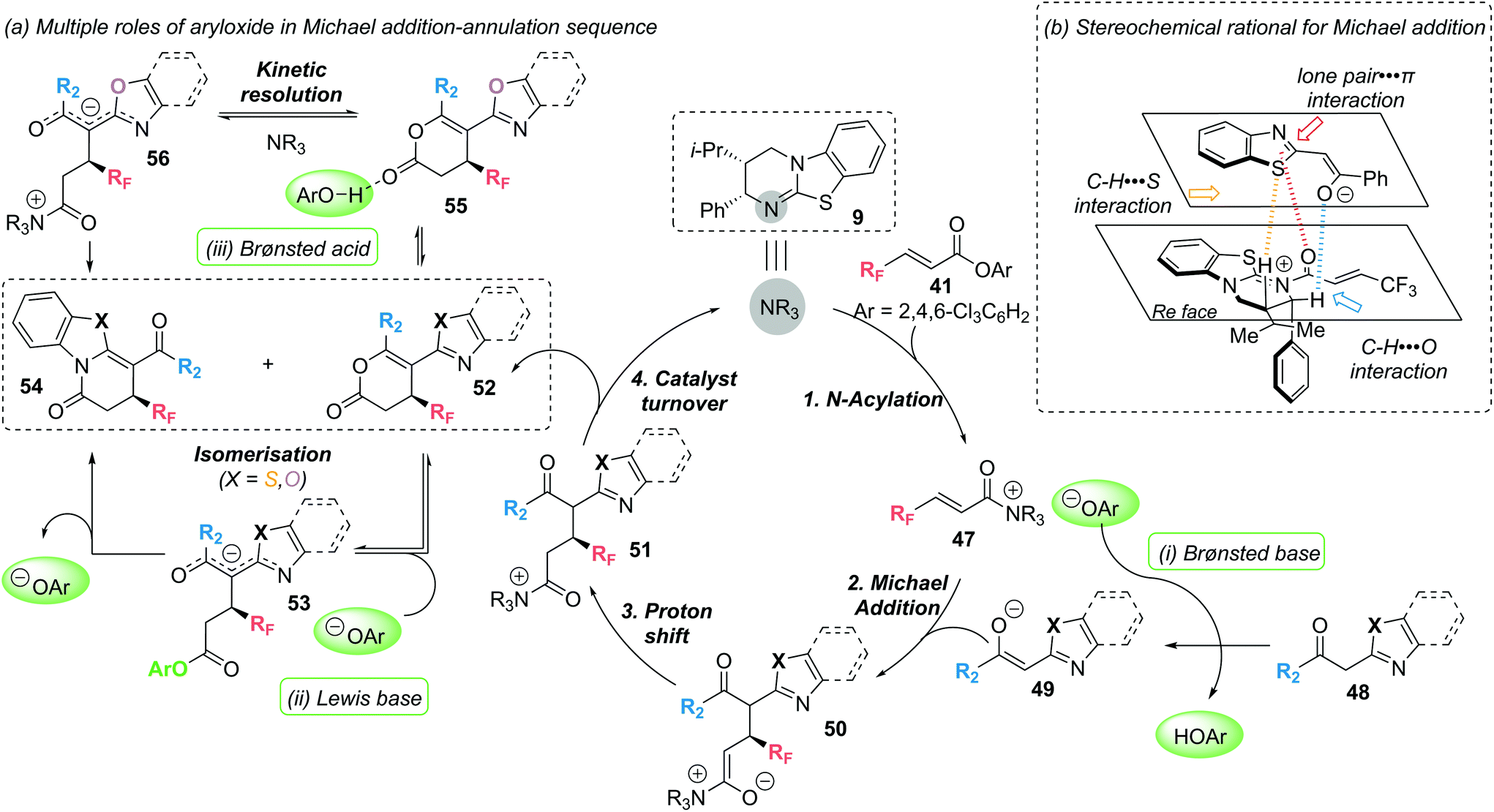 | ||
| Scheme 11 Michael addition-annulation using 2-acylbenzazoles and fluoroalkyl substituted unsaturated aryl esters: (a) proposed catalytic cycle highlighting multiple roles of aryloxide; (b) stereochemical rational for enantiodetermining Michael addition. | ||
In 2020, these mechanistic experiments were complemented by a computational study by the groups of Wei and Ding to gain further mechanistic insight.20 DFT calculations confirmed the proposed role of the aryloxide as Brønsted base/acid facilitating proton transfer. Additional calculations highlighted the nucleophilicity of N vs. O in the corresponding intermediates as being important in determining the observed lactam vs. lactone product formation. In addition, Michael addition was identified as the enantiodetermining step, with non-covalent interaction (NCI) analysis highlighting three additional non-bonding interactions (classified as lone pair-π; C–H⋯S and C–H⋯O), as well as the previously identified 1,5-S⋯O and CH⋯O interactions, in governing the observed chemo- and stereoselectivity (Scheme 11b).
In 2017, Matsubara and co-workers reported a protocol for the selective formation of substituted 1,5-benzothiazepines.21 Based on earlier work from the same group,22 aminothiophenol 58 and mixed anhydride 57 could be converted into the corresponding 1,5-benzothiazepines 62–64 using isothiourea catalysts (Scheme 12). By choosing the appropriate anhydride precursor, 1,5-benzothiazepines bearing substituents in either the 2-position (62), the 3-position (63) or both 2- and 3-positions (64) were accessible in generally high yields (up to 99%) and enantioselectivities (up to 99:1 er). Notably, mechanistic investigations into the formation of 3-substituted 1,5-benzothiazepines revealed that thioester 59 is formed reversibly in situ and is likely to be the functioning α,β-unsaturated acyl ammonium precursor, as no formation of α,β-unsaturated acyl ammonium intermediate was observed when only mixed anhydride and catalyst were present. Moreover, thia-Michael addition is postulated to be reversible, making this process a dynamic kinetic asymmetric transformation with the enantioselectivity dictated by the different cyclization rates of diastereomeric intermediates.
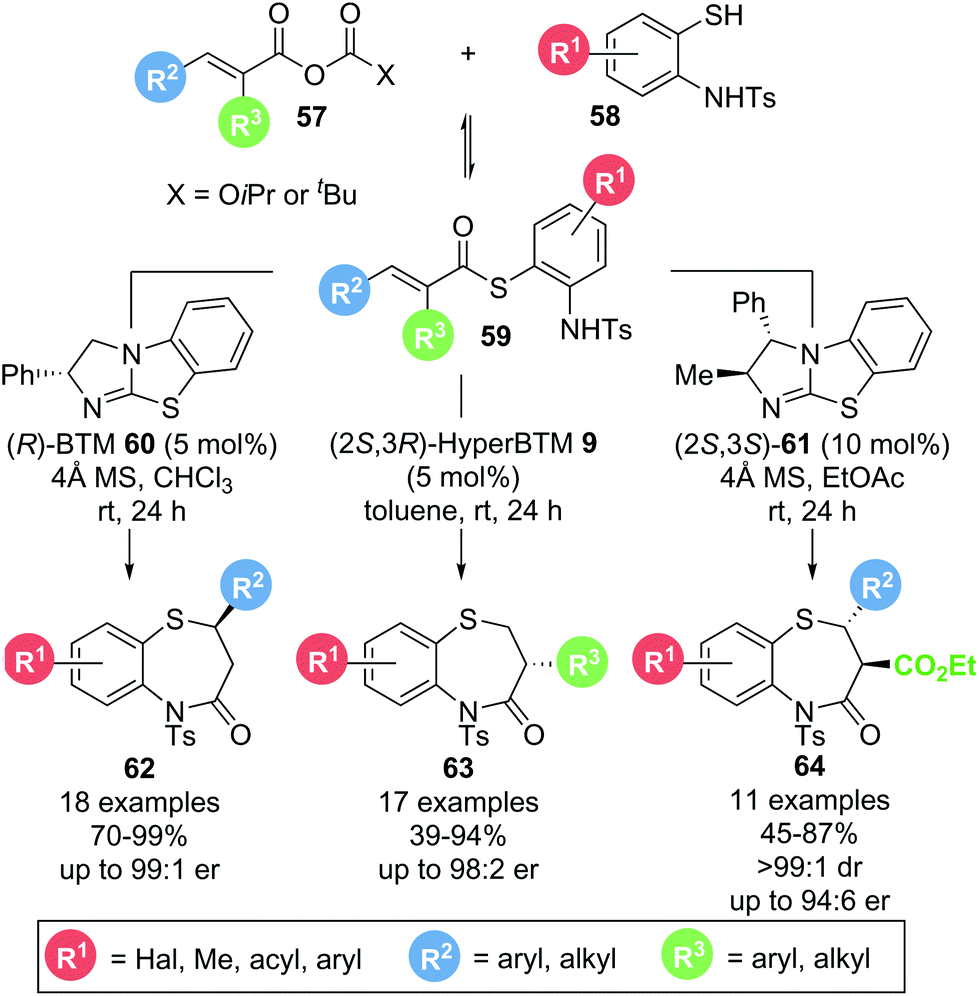 | ||
| Scheme 12 Thia-Michael addition–lactamization to generate 1,5-benzothiazepines. | ||
In 2018, Romo and co-workers reported a related transformation for the formation of medium sized lactams using amino malonate 65 as bis-nucleophile in combination with acid chlorides 66 as α,β-unsaturated acyl ammonium precursors (Scheme 13).23 The use of cinchona alkaloid TMSQD 69 as Lewis base catalyst and thermodynamic enolization conditions (i-Pr2NEt and LiCl) were essential for obtaining good yields (up to 87%) and high enantioselectivities (>92:8 er). Although only a limited scope of this Michael addition–lactamization process was reported, derivatization of the obtained medium-sized lactam products 67 and 68 to ring opened or ring contracted derivatives demonstrated the synthetic utility of the process.
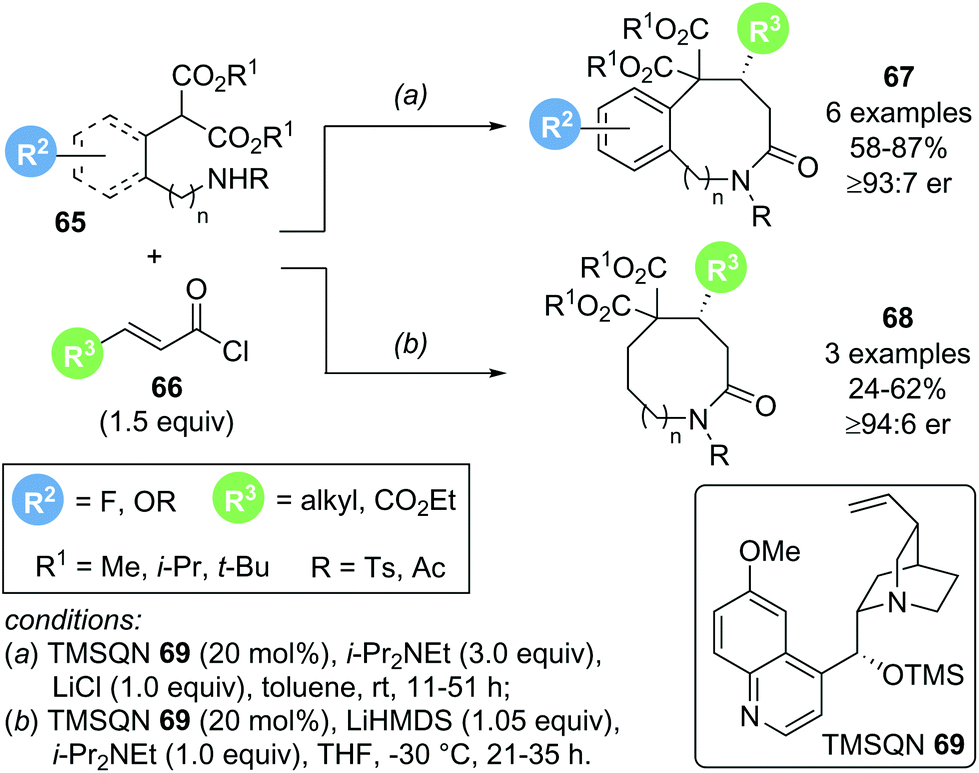 | ||
| Scheme 13 Michael-lactamization sequence to generate medium sized lactams. | ||
In 2019, Birman used achiral, electron-rich amidine organocatalyst DHIP 71 in the Michael addition–annulation of aliphatic S-phenacyl thiocinnamate 70.24 Based on their earlier work on thiochromenes25 (see Scheme 24) and thiochromanes26 (see Scheme 25) they predicted the formation of β-lactam 77 or dihydrothiophene 78via an analogous domino pathway. However, dihydrothiophene 72 was the only product observed. Its formation can be rationalised proceeding through a thia-Michael addition followed by proton transfer and subsequent ring closure via a Dieckmann-like cyclisation (Scheme 14a). Substrates with electron donating, electron withdrawing and heteroaromatic aryl substituents (R1, R3) all reacted smoothly. A β-dimethyl substituted Michael acceptor also gave the desired product, whereas α-substitution was not tolerated. As the synthesis of certain α,β-unsaturated thioester substrates proved troublesome, an alternative two component strategy starting with α,β-unsaturated TCP ester 79 and thiol 80, was devised, giving the corresponding dihydrothiophenes 81 in comparable yields (Scheme 14b). Unfortunately, studies towards an enantioselective variant of this protocol by using chiral analogues of the amidine catalyst were unsuccessful. This was speculated to be due to a racemic background reaction operating via an alternative reaction pathway without the involvement of an organocatalyst.
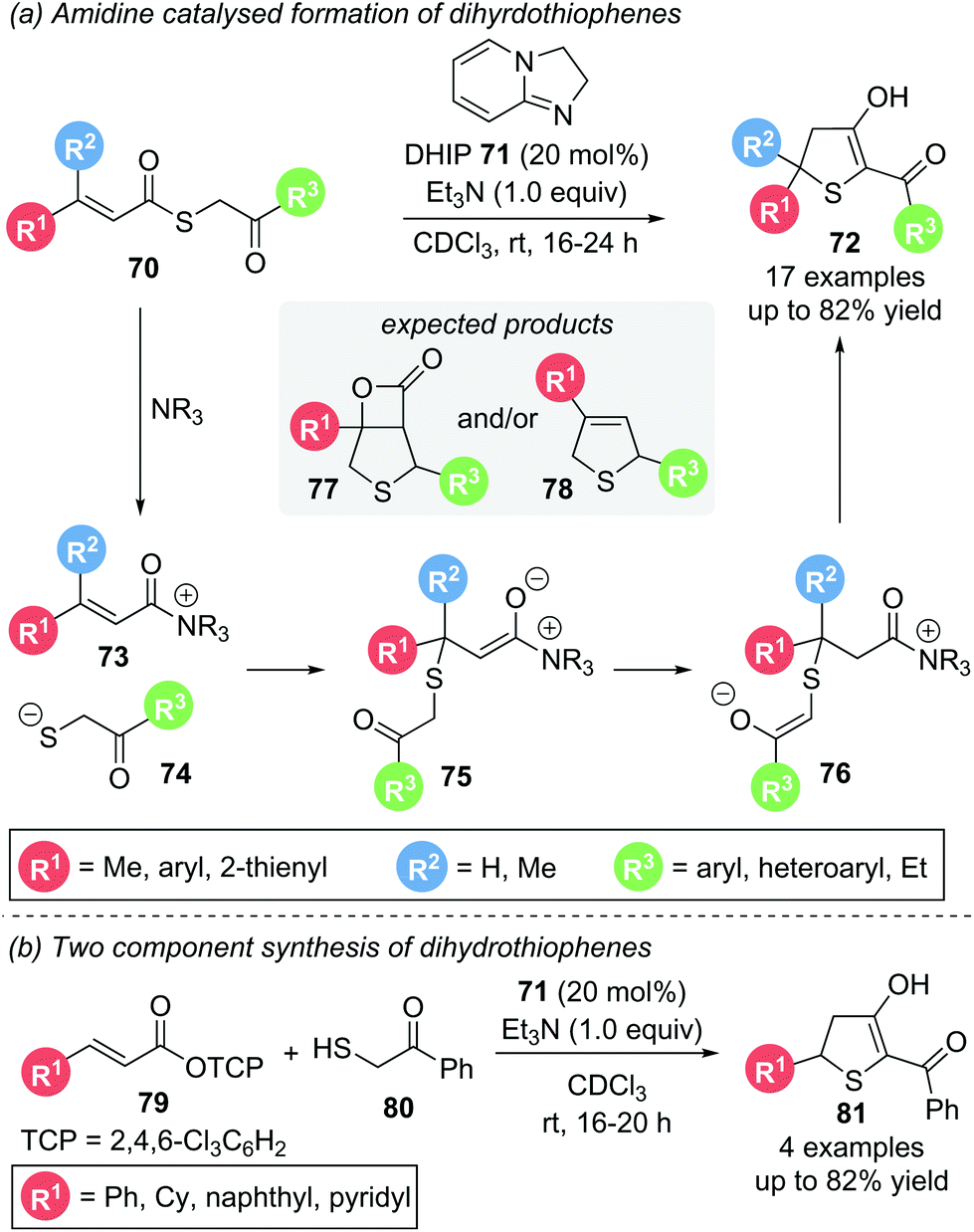 | ||
| Scheme 14 Thia-Michael addition – Dieckmann cyclisation for the synthesis of dihydrothiophenes. | ||
The applicability of such Michael addition–annulation sequences to the construction of heterocyclic molecules was further demonstrated by Luo, Deng and co-workers.27 Using symmetric anhydrides 23 as α,β-unsaturated acyl ammonium precursors and indoline-2-thiones 82 as pronucleophiles with isothiourea catalyst (2S,3R)-HyperBTM 9 furnished indolo[2,3-b]-dihydrothiopyranones 83 in high yields (61–97%) and generally excellent enantioselectivities (up to 99:1 er) (Scheme 15). Notably, the reaction did not require any auxiliary base as the in situ generated carboxylate anion was assumed to deprotonate the indoline-2-thione to generate the thioenolate nucleophile required for initial Michael addition.
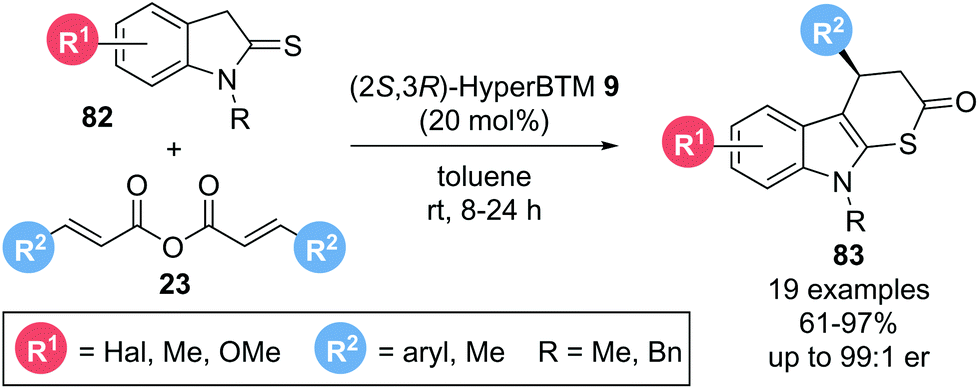 | ||
| Scheme 15 Michael addition-thioester formation to generate dihydrothiopyranones. | ||
In a related study, Smith and co-workers reported the enantioselective synthesis of tetrahydro-α-carbolinones.28 Employing α,β-unsaturated p-nitrophenyl esters 7 and indoline-2-imines 84 as starting materials in the presence of isothiourea catalyst (2S,3R)-HyperBTM 9, the corresponding products 85 could be obtained in good yields (32–99%) and excellent enantioselectivities (up to 99:1 er) (Scheme 16). In this case the use of NaHCO3 as auxiliary base was necessary to generate an enamine nucleophile for the desired Michael addition. Notably, variation of the β-substituent within the ester (R2) allowed successful incorporation of a variety of polyhalogenated substituents. In addition, generally challenging aliphatic substituents (R2) were also tolerated, furnishing the desired products with excellent enantiocontrol, albeit in reduced yield (38–59%).
 | ||
| Scheme 16 Michael-lactamization for the synthesis of tetrahydro-α-carbolinones. | ||
In 2016, Du showcased a dual Lewis acid/base strategy in the regioselective synthesis of 4H-pyran-4-ones 91 from in situ activated alkynyl acids 86 and 1,3-dicarbonyl compounds 87 (Scheme 17).29 The reaction proceeds through Michael addition to acyl ammonium intermediate 88via an O-centred enolate nucleophile, giving the corresponding 4H-pyran-4-ones 91 in good yields (18–83%). Interestingly, the formation of isomeric lactone 2H-pyran-2-one 90via C-centred enolate nucleophilic addition was only observed in two cases (R1 = H or Me). Aromatic and heteroaromatic substituents (R1) exclusively gave 4H-pyran-4-one 91. With R1 being the only variable in this series of substrates, these results suggest that the substitution present on the alkynyl acid (R1) is important in determining product selectivity. Notably, though theoretically catalytic in Lewis base, stoichiometric DMAP 5 was required to obtain acceptable yields. The role of the Lewis acid (Sc(OTf)3) remains unclear, as the reaction also proceeds in its absence, albeit with lower yield (47% without Sc(OTf)3vs. 65% with Sc(OTf)3).
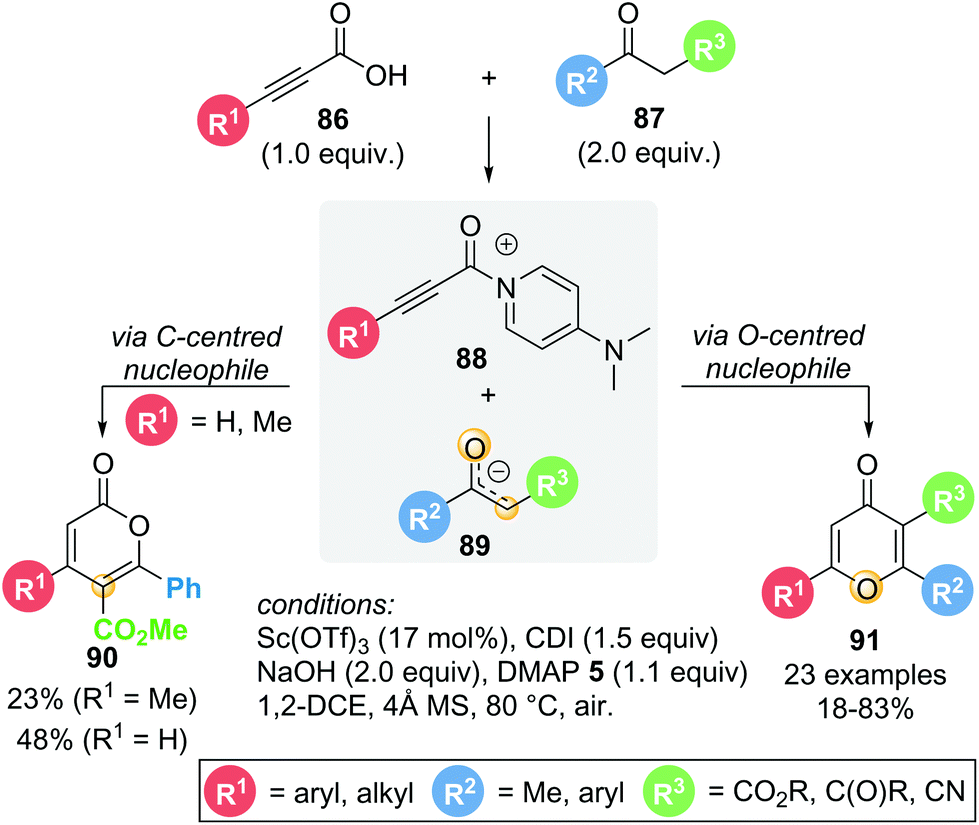 | ||
| Scheme 17 Dual Lewis acid/base strategy for the synthesis of 4H-pyran-4-ones. | ||
In 2020, Gong and co-workers presented a dual isothiourea/Brønsted acid catalysed Michael addition–lactamization sequence using α,β-unsaturated trichlorophenyl (TCP) esters 79 and isolated, acyclic enamines 92 to yield enantioenriched functionalised 3,4-dihydropyridin-2-ones 94 (Scheme 18).30 The presence of a Brønsted acid co-catalyst (Ph2PO2H) led to a significant rate enhancement in product formation as demonstrated by 1H NMR experiments. This was attributed to a hydrogen bonding interaction between the phosphate counterion and the enamine, enhancing the nucleophilicity of the latter. A high degree of substitution on both α,β-unsaturated ester 79 and enamine 92 were tolerated, including aryl and alkyl substituents (R2, R3), and electron withdrawing groups (R1, R3), with excellent yields and enantioselectivities maintained throughout. Notably, a selection of polyfluorinated substituents (R1) could be incorporated with high yields (71–87%) and excellent stereocontrol (>96:4 er). In addition, usually challenging crotonic (R1 = Me) and cinnamic (R1 = Ph) ester derivatives also gave the desired products in moderate yields (77% and 57%) but high enantioselectivities (91:9 er and 95:5 er, respectively).
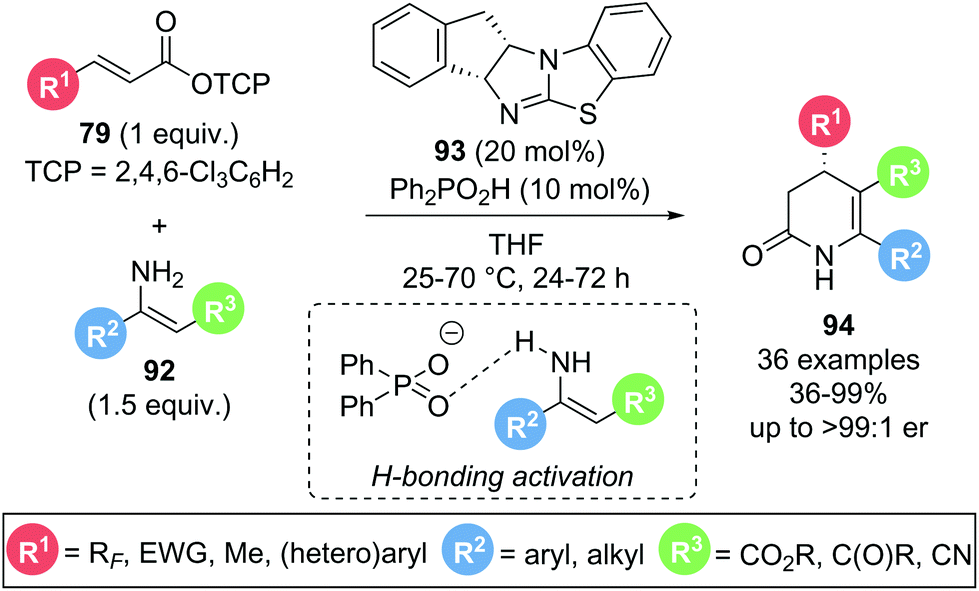 | ||
| Scheme 18 Cooperative Lewis base/Brønsted acid catalysis for the synthesis of dihydropyridinones. | ||
2.3. Domino (cascade) reactions
Domino reactions are becoming increasingly popular as they allow for the rapid generation of molecular complexity, often in a chemo- and stereoselective fashion. In the context of α,β-unsaturated acyl ammonium catalysis, a general domino sequence depicted in Scheme 19 can be envisioned. Nucleophilic addition of the Lewis base catalyst (NR3) into an appropriate precursor generates an α,β-unsaturated acyl ammonium ion pair. Michael addition of a nucleophile (Nu−) initiates the domino sequence, as the generated C(1)–ammonium enolate can react with a pendant electrophile (typically a carbonyl derivative or Michael acceptor). This generates another nucleophile (Y−), which can displace the catalyst (often via lactonization or lactamization). This strategy can rapidly generate polycyclic structures in a stereoselective fashion and has been utilised recently by the groups of Romo, Smith and Birman.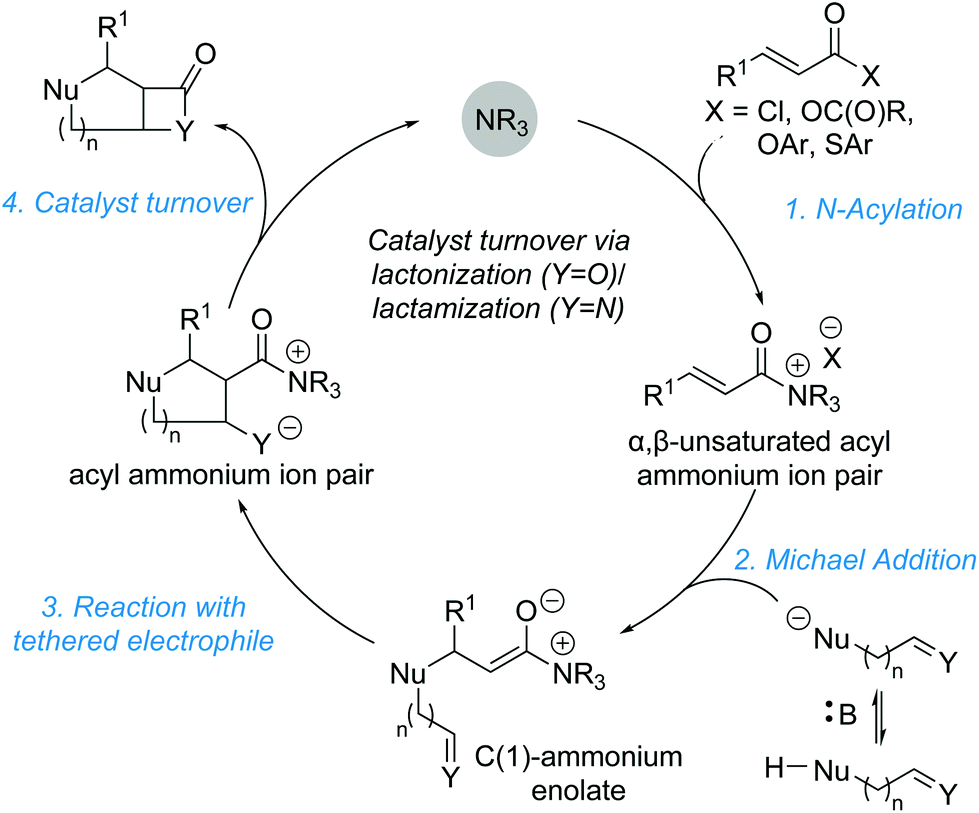 | ||
| Scheme 19 General catalytic cycle for domino reactions. | ||
In 2013, Romo and co-workers reported a domino Michael-aldol-lactonization sequence forming bicyclic β-lactones using readily available acid chlorides 96 and keto malonates 95 (Scheme 20).31 This protocol allowed the incorporation of various substituents, generating products with up to 4 contiguous stereocentres, either in racemic or enantioenriched form, in good yields (up to 95%) and high selectivities (up to >95:5 dr and 99:1 er). Importantly, the use of a lithium base was necessary to obtain good reactivity, as more weakly coordinating counter ions (Na+ or K+) led to drastically lower yields.
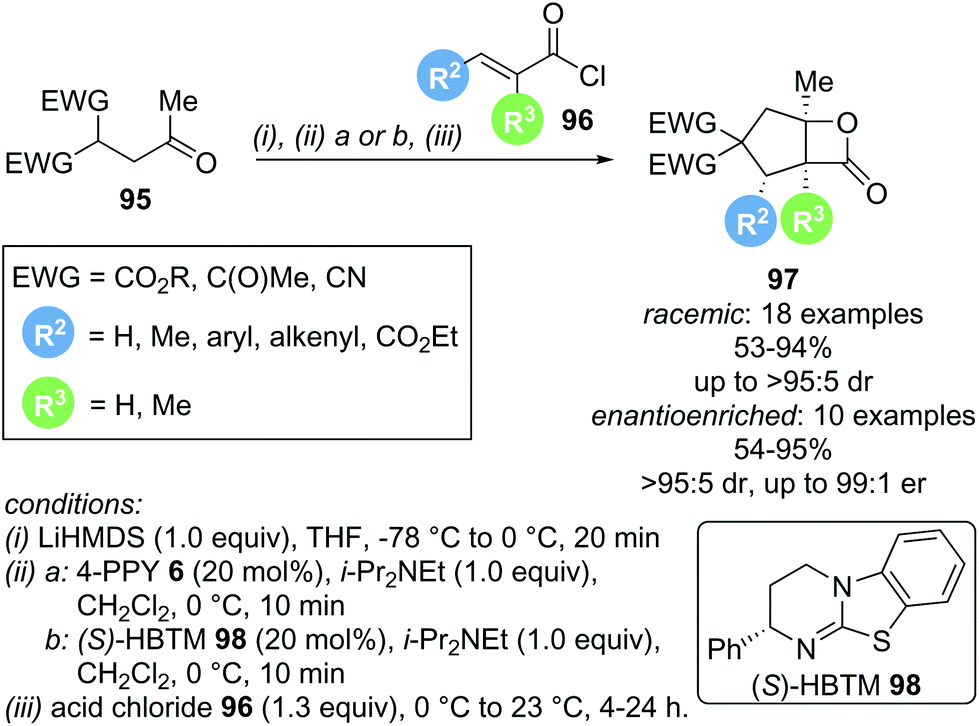 | ||
| Scheme 20 Michael-aldol-lactonization cascade. | ||
As an expansion to Romo's work, Smith and co-workers reported the enantioselective synthesis of cyclopentane annulated δ-lactones 100via a Michael–Michael-lactonization cascade (Scheme 21a).32 This protocol uses Michael donor–acceptor substrates 99 containing a malonate and a tethered enone, in combination with acid chlorides 66 as α,β-unsaturated acyl ammonium precursors. A range of malonate ester nucleophiles (variation of R1) and electron rich aryl substituents on the enone (R2) and the acid chloride (R3) were well tolerated, giving the desired lactones in good yield (up to 64%) and with good stereocontrol (>81:19 dr, up to 95:5 er). However, incorporation of aryl substituents with electron withdrawing groups (R3) required the isolation of the corresponding products as the ring opened cyclopentanes 101, as the δ-lactone reaction products proved unstable to chromatographic purification. The use of (2S,3R)-HyperBTM 9 as catalyst and LiHMDS as stoichiometric base was crucial for selective promotion of initial 1,4-addition over 1,2-addition. Notably, an increase in enantioselectivity with increasing reaction temperature was observed, prompting kinetic investigations using Eyring analysis. The rate of formation of the two product enantiomers (K) is related to the differential activation enthalpy (ΔΔH‡) and differential activation entropy (ΔΔS‡) according to the differential Eyring equation (Scheme 21b). Plotting the natural logarithm of the enantiomeric ratio as a function of reciprocal temperature gave the activation parameters ΔΔH‡ (−0.865 kJ mol−1) and ΔΔS‡ (+4.42 J mol−1 K−1). A dominant entropic term suggests that enantioselectivity is entropically controlled, indicating conformational flexibility in the transition state. Hence, conformational control through chelation may play a key role in determining enantioselectivity and would explain the importance of a lithium base for obtaining high reactivity and selectivity (Scheme 21c).
 | ||
| Scheme 21 (a) Michael–Michael-lactonization cascade for the synthesis of cyclopentanes; (b) kinetic analysis using an Eyring plot; (c) substrate organisation in transition state. | ||
In 2018, Romo reported a related cascade reaction, generating the reactive substrate 104in situ from a 1,3-dicarbonyl pronucleophile 102 and an alkylidene malonate 103 that undergoes a domino reaction with a catalytically generated α,β-unsaturated acyl ammonium species to form tricyclic β-lactone 105 (Scheme 22).33 Preliminary investigations with achiral Lewis base catalyst 4-pyrrolidinopyridine 6 (4-PPY) showed that the scope for this process is rather limited. While acyclic ketoesters gave the desired β-lactone in promising yield (54–75%), the reaction was not diastereoselective. The use of cyclic β-ketoesters greatly improved the observed diastereoselectivity (>95:5 dr), albeit with drastically reduced yield (22%). The use of highly reactive acid chlorides 66 (R3 = H or CO2Et) led to improved yield (up to 58%) while maintaining high diastereoselectivity. Further optimisation with chiral Lewis base catalysts revealed that the product yields generally did not exceed 50%. It was postulated that this may indicate the occurrence of a kinetic resolution, as the initially formed Michael adduct 104 is obtained as a racemate. Attempts to further increase the yields through a dynamic kinetic asymmetric transformation (DYKAT) of 104 by increasing the reversibility of the Michael adduct formation were unsuccessful. Although limited in scope, this Michael–Michael-aldol-lactonization sequence furnished valuable tricyclic β-lactones 105 in excellent diastereoselectivity and, when using chiral Lewis base (2S,3R)-HyperBTM 9, generally high enantioselectivity (>95:5 dr, up to 94:6 er).
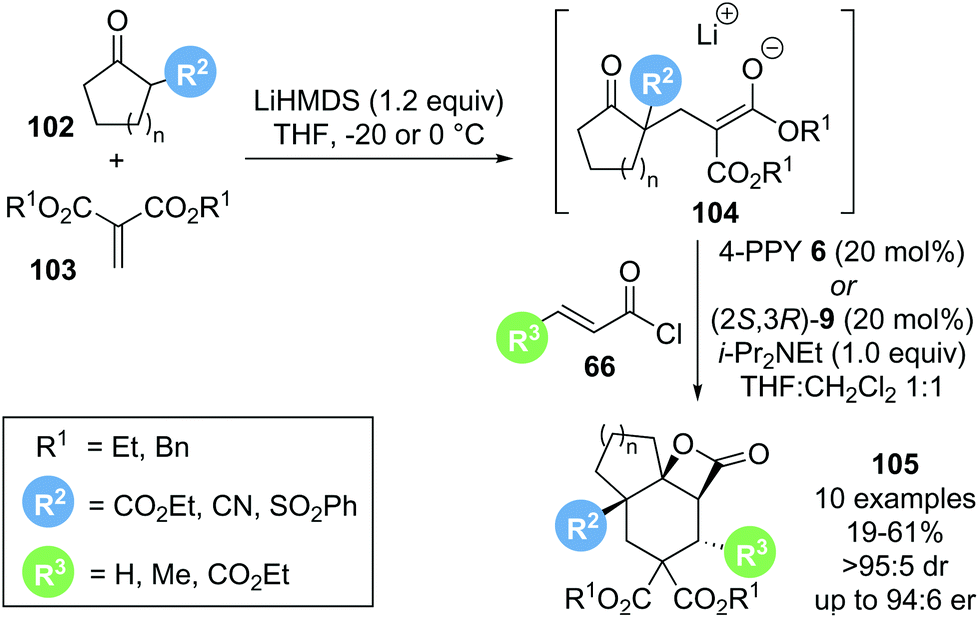 | ||
| Scheme 22 Michael–Michael-aldol-lactonization cascade to generate tricyclic β-lactones. | ||
In 2016, Smith and co-workers demonstrated that bench-stable activated aryl esters are viable precursors for isothiourea catalysed domino processes.34 α,β-Unsaturated acyl ammonium precursor with a tethered Michael acceptor 106 were employed for the formation of complex, polycyclic products in high selectivity (Scheme 23). Three classes of nucleophile were used, with each resulting in a unique class of polycyclic structures. Employing 1,3-dicarbonyls 31 as nucleophiles resulted in the formation of tricyclic δ-lactones 108 in good yields (46–79%) with excellent diastereo- and enantiocontrol (>75:25 dr, up to 99:1 er) via a Michael–Michael-lactonization pathway. Using acyl benzothiazole nucleophiles 42 under the same conditions resulted in pentacyclic products 109 being formed in good yields (20–83%) with excellent stereocontrol (>95:5 dr, up to 97:3 er). Importantly, the major product observed results from lactamization through the benzothiazole N-atom rather than lactonization through the pendant enone, as has been observed in other methodologies using benzothiazole nucleophiles. This reaction outcome is rationalised through a Michael-lactamization-Michael pathway being operative, with the pentacyclic lactam products 109 being formed with >90% selectivity for most examples. As a third class of nucleophiles, acyl benzimidazoles 107 were also investigated. A significant base promoted background reaction was observed, leading to the corresponding racemic pentacyclic lactam products 110 in good yields (49–94%) and diastereoselectivity (>70:30 dr) in the absence of the Lewis base catalyst. Further optimization allowed a small set of products to be obtained in enantiomerically enriched form (>78:22 er). By analogy to the domino pathway proposed for acyl benzothiazole nucleophiles 42, a similar Michael-lactamization-Michael reaction pathway is likely to be operative for acyl benzimidazole nucleophiles 107. However, due to the different heterocycle used, the second Michael addition proceeds via a different enolate, resulting in another distinct product topology.
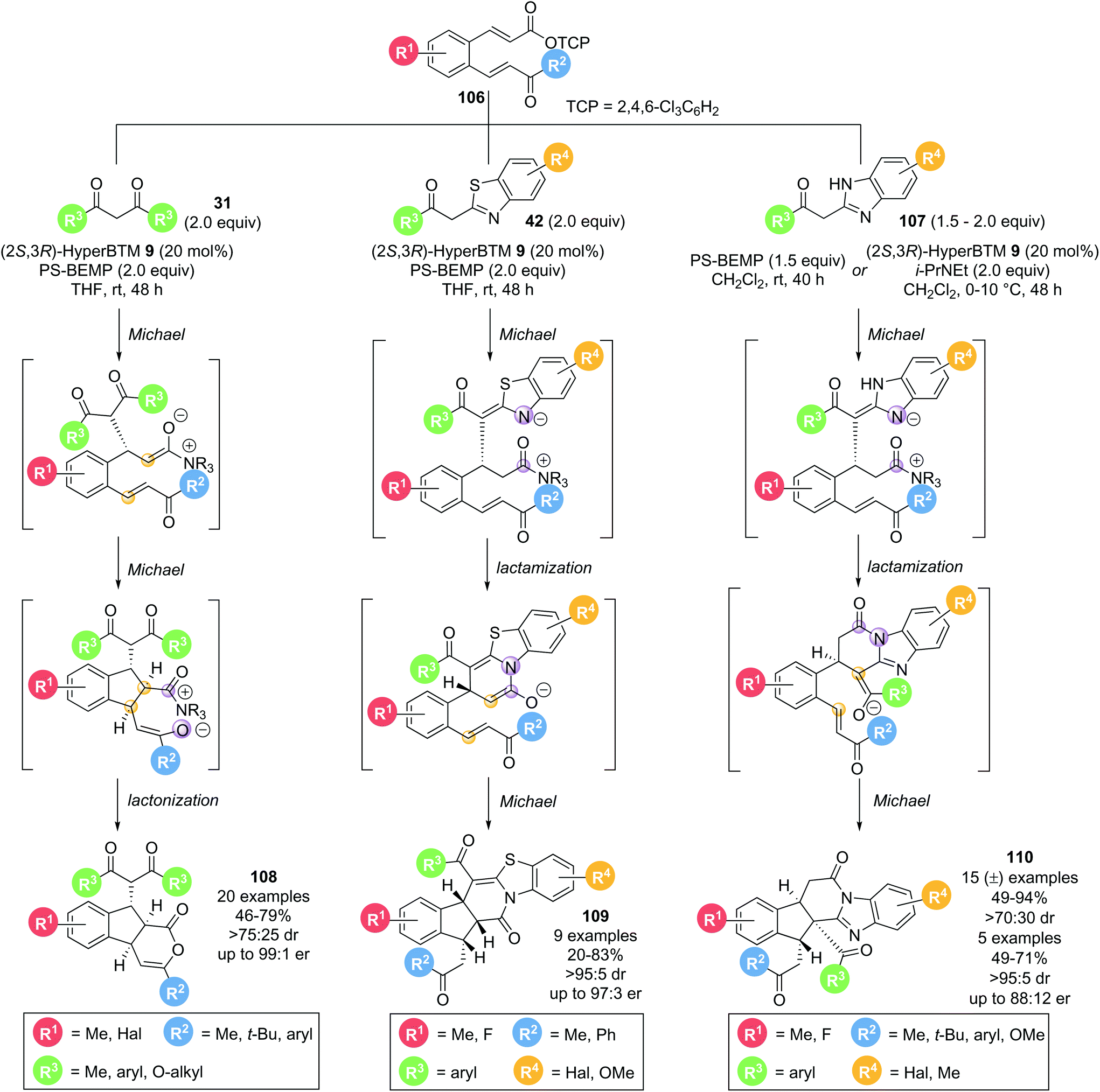 | ||
| Scheme 23 Stereodivergent nucleophile dependent Michael addition–cyclisation cascade to generate polycyclic compounds. | ||
In 2016, Birman and co-workers reported the enantioselective synthesis of 2-substituted thiochromenes 114 from α,β-unsaturated thioesters 111via a thia-Michael addition-aldol-lactonization cascade, followed by decarboxylation of the formed β-lactone 113 (Scheme 24).25 Notably, this report marked the first use of thioesters as substrates with isothiourea catalysts. Employing isothiourea catalyst (2R,3S)-HBTM-2 112 (10 mol%), various substituents on the aromatic core (R1) were well tolerated. Several different aryl and heteroaryl substituents on the Michael acceptor (R2) could also be incorporated, giving the corresponding thiochromenes 114 in excellent yield (up to 99%) and enantioselectivity (>97:3 er). As the required thiolate nucleophile and α,β-unsaturated acyl ammonium species are both generated in situ from the thioester substrate during catalyst acylation, this protocol remarkably allows for the formation of enantioenriched thiochromenes without any additives and with CO2 as the only by-product.
 | ||
| Scheme 24 Thia-Michael-aldol-lactonization cascade to generate thiochromenes. | ||
This protocol was extended to the synthesis of tricyclic thiochromanes 119 by employing α,β-unsaturated thioesters 118 bearing a pendant Michael acceptor (Scheme 25b).26 Initial investigations showed that isothiourea catalyst DHPB 115 could facilitate the desired transformation with good diastereoselectivity, but exhibited poor reactivity. A DFT study showed that electron rich amidine based catalysts exhibit greater Lewis basicity than DHPB, suggesting they might exhibit greater reactivity in the desired transformation. An extensive screen revealed electron-rich isothiourea 116 and amidine H-PIP 117 to be the most reactive and selective catalysts (Scheme 25a). Employing H-PIP 117 (10 mol%), a variety of aryl substituents (R1, R2) could be incorporated, yielding the corresponding thiochromane annulated δ-lactone products 119 in good yields (60–93%) with excellent diastereo- and enantioselectivities (up to 95:5 dr, >90:10 er).
 | ||
| Scheme 25 Michael–Michael-lactonization cascade to generate trisubstituted thiochromanes. | ||
3. Cycloadditions
Cycloadditions using α,β-unsaturated acyl ammonium species are highly effective processes for the rapid assembly of molecules with a high degree of structural complexity, often containing multiple contiguous stereogenic centres. A representative catalytic cycle is shown in Scheme 26. Reaction of the Lewis base catalyst with the corresponding acyl chloride or fluoride generates an α,β-unsaturated acyl ammonium species. Cycloaddition, followed by the reaction with a tethered nucleophile (Y) allows catalyst release and yields the corresponding cycloaddition product.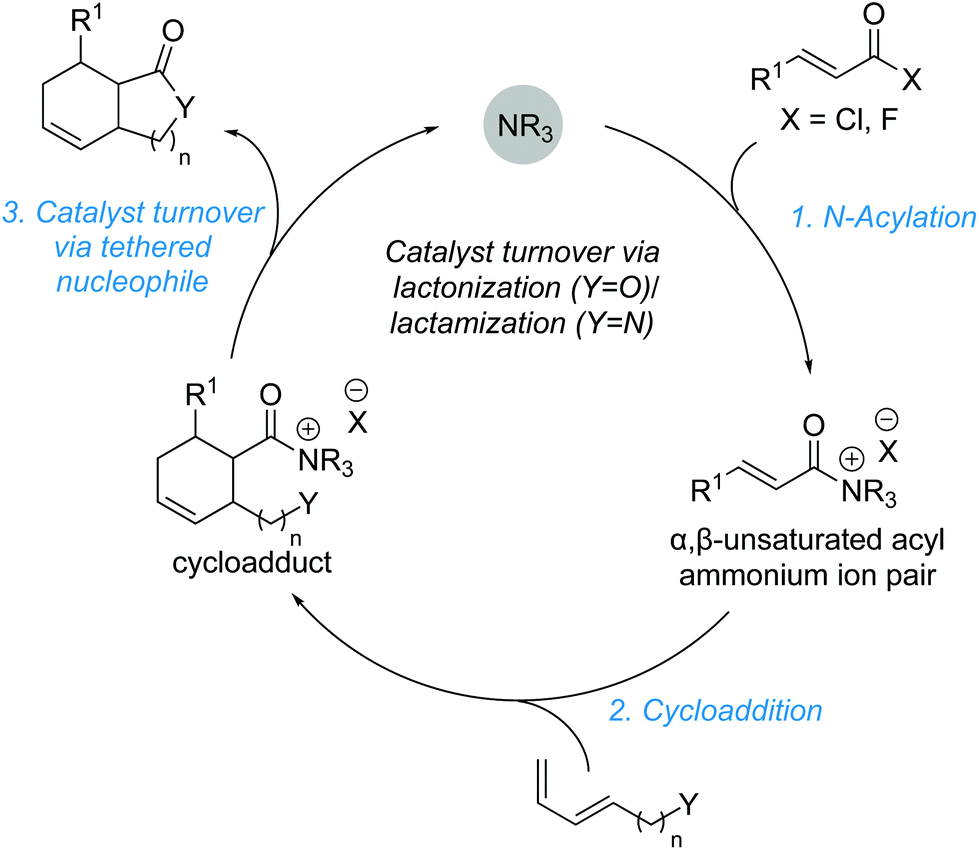 | ||
| Scheme 26 Catalyst turnover enabled by sequential cycloaddition-annulation. | ||
In 2017, as an expansion of their earlier Diels–Alder (DA)-lactonization strategy,14c Romo and Tantillo described an elegant stereodivergent DA-lactonization approach that gave access to all stereoisomeric members of bi- and tricyclic γ-lactones 121 and 122 (Scheme 27).35 Acid chloride 66 and O-silylated diene 120 undergo DA cycloaddition in the presence of (S)-BTM 60, followed by lactonization with the racemic pendant alcohol, giving bi- and tricyclic γ-lactones 121 and 122 with good yields and exceptional endo- and enantioselectivity (all >95:5 endo:exo and ≥99:1 er). Acid chlorides bearing an electron withdrawing group (R1 = CO2Et) or a methyl substituent (R1 = Me) reacted smoothly whereas acryloyl (R1 = H) and cinnamoyl chloride (R1 = Ph) were unsuccessful, as were non-siloxy substituted dienes. Brønsted base selection played a key role in determining the stereoselectivity, with improved enantioselectivity observed using 2,6-lutidine and 2,6-di-tert-butylpyridine (>99:1 er) compared to pyridine, Et3N and i-Pr2NEt (80:20 to 93:7 er). Interestingly, this was not proposed to be a result of a competitive racemic cycloaddition. Instead, computational studies indicated that the ability of the Brønsted base to simultaneously engage in C–H⋯π, π⋯π stacking and hydrogen bonding interactions may govern the observed stereoselectivity. Notably reaction with judiciously selected Lewis and Brønsted bases, and the resulting tuning of intermolecular interaction, allowed for full access to all stereoisomers. 13C NMR studies indicated that isothioureas may not lead to substrate activation via an appreciable lowering of the LUMO, but reduce the rate of intermolecular esterification, allowing for the DA cycloaddition to initiate the cascade. This is complemented by an earlier NMR spectroscopy study from the same group, which concluded that isothioureas increase the steric barrier for 1,2-addition to the carbonyl allowing for a chiral domain in which the latent 1,4-reactivity of the starting acid chloride can predominate.36 The products from this methodology could be used to greatly expedite the synthesis of natural products. For example, (+)-dihydrocompactin precursor (+)-124 could be accessed enantioselectively in just 2 steps from (−)-123 (6 steps overall) compared to the previous 14 step racemic synthesis (Scheme 27b).
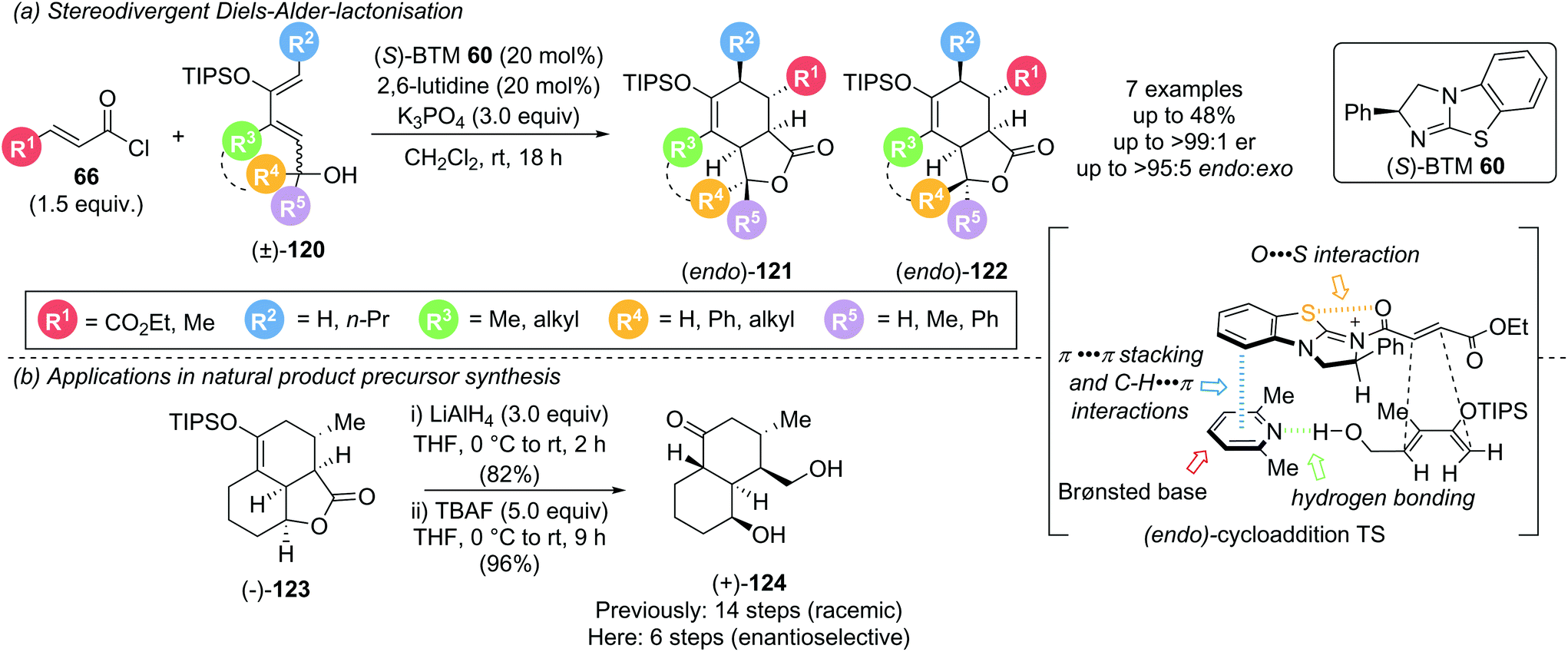 | ||
| Scheme 27 Stereodivergent Diels–Alder-lactonization and its use in natural product precursor synthesis. | ||
Romo later incorporated the furan motif into a DA-lactamization cascade reaction (Scheme 28a).37 The aromaticity of furans often renders them unproductive in DA cycloadditions as they readily undergo retro-DA processes. To counteract this reversibility, a pendant sulfonamide on the furan 125 was necessary for an irreversible exo-selective lactamization step following the DA cycloaddition. The choice of this pendant nucleophile was key, as replacement of the sulfonamide with an alcohol gave effectively racemic product, rationalised by either slow lactonization leading to a retro-DA after the initial cycloaddition, or by initial intermolecular ester formation followed by racemic DA cycloaddition. Only highly electron deficient and reactive dienophiles were able to overcome the barrier imposed by the inherently unreactive furan. Computational studies indicated that due to the presence of both reversible DA and lactamization steps, the manifold is operating under thermodynamic control (Scheme 28b). The retro-DA step was calculated to have an energy barrier of ∼19–23 kcal mol−1, indicating facile reversibility at room temperature. A potentially reversible lactamization step was identified, particularly for the endo-diastereoisomer meaning potential funnelling of the endo-diastereoisomer to the more thermodynamically favoured exo-diastereoisomer. The reversibility of the endo-diastereoisomer 128 was not experimentally tested as it could not be isolated. However, the isolable exo-diastereoisomer 127 did not appear to undergo retro-DA as no erosion of enantiopurity was observed after re-subjection to the reaction conditions.
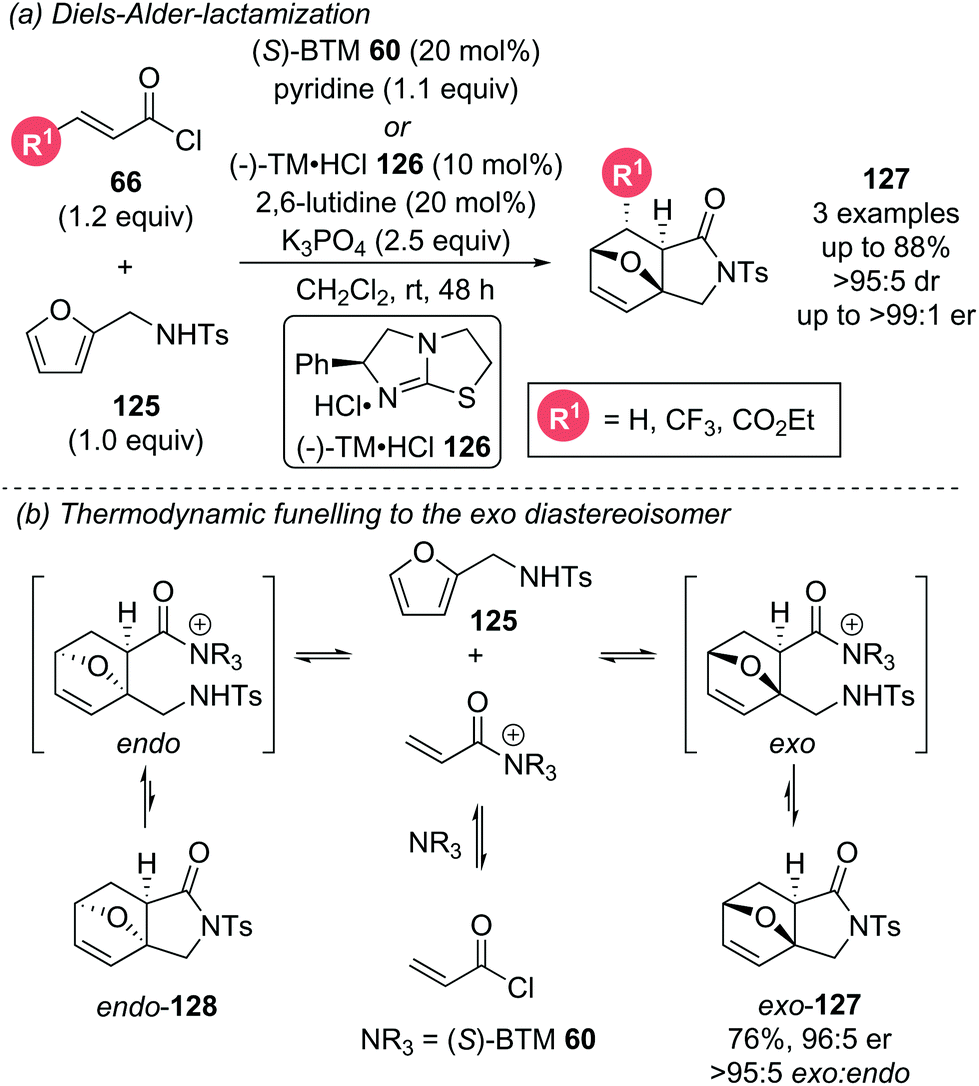 | ||
| Scheme 28 Organocatalytic Diels–Alder-lactamization. | ||
In 2018, Ye and co-workers demonstrated the use of DBU as a dual Lewis and Brønsted base in the [4 + 2] annulation of in situ activated carboxylic acids 129 and α-cyano-β-methylenones 130 to give the corresponding 1,3,5-trisubstituted benzenes 131 with generally good to excellent yields (Scheme 29).38 Generation of a reactive acyl imidazole 132in situ by treatment of the carboxylic acid 129 with 1,1′-carbonyldiimidazole (CDI), followed by N-acylation of DBU, yields α,β-unsaturated acyl ammonium intermediate 133. [4 + 2] DA cycloaddition between this intermediate and the enolate 134 of α-cyano-β-methylenone 130 affords cyclohexene 135. Subsequent β-lactonization, followed by decarboxylation and aromatisation afforded benzene derivative 131. Deuterium labelling studies supported a deprotonation/protonation aromatisation sequence from 137. A large degree of functionality on both the α,β-unsaturated carboxylic acid 129 and enone 130 were tolerated, including alkyl, aromatic and heteroaromatic substituents. However, carboxylic acids bearing aliphatic substituents in the β-position (R3) were unreactive. Intriguingly, during a control experiment, reaction of a preformed acyl imidazole with a methylenone could be carried out with a catalytic quantity of DBU (20 mol%) and a stoichiometric amount of Cs2CO3 (2 equiv.) to give 131 in 77% yield, indicating that Lewis base catalytic turnover is possible within this system.
 | ||
| Scheme 29 [4 + 2] cycloaddition between α,β-unsaturated carboxylic acids and methylenones. | ||
4. Miscellaneous
This section covers reports that invoke α,β-unsaturated acyl ammonium intermediates in functionalisation strategies but are not easily characterised into the sections discussed above.In 2014, Bonne and Rodriguez investigated the use of α,β-unsaturated acyl cyanides 139 as bis-electrophiles for a Michael addition–annulation reaction with β-ketoamides 138 as bis-nucleophiles (Scheme 30a).39 The transformation was conducted using Takemoto's thiourea catalyst 140 primarily as a hydrogen-bonding catalyst. However, involvement of the tertiary amine motif in the formation of an acyl ammonium intermediate was also tentatively proposed. Notably, Michael acceptors other than acyl cyanides, such as cinnamaldehyde, cinnamoyl chloride or 4-nitrophenol cinnamate, were unreactive under these conditions. To rationalise the stereochemical outcome, the substrate-catalyst interactions were considered (Scheme 30b). The enolate nucleophile is coordinated to the thiourea moiety via hydrogen bonding while activation of the electrophile was proposed to proceed through one of two possible modes. A second hydrogen-bonding interaction (Scheme 30b, pre-TS A) and formation of a covalently bound acyl ammonium species (Scheme 30b, pre-TS B) were both proposed and would lead to the same stereochemical outcome. To gain insight into the activation mode operative under catalysis conditions, 1H NMR experiments were performed. In deuterated acetonitrile, the formation of an acyl ammonium species was clearly observed and further verified by HRMS analysis. However, when the same NMR experiment was performed in deuterated benzene, no acyl ammonium was observed, indicative of substrate activation via hydrogen bonding in this case. As the catalysis was conducted in m-xylene, activation of the electrophile via hydrogen bonding will likely be operative in the reported reaction. However, these findings indicate that the activation mode for the electrophile with this type of catalyst is highly solvent dependent, so both possibilities ought to be considered.
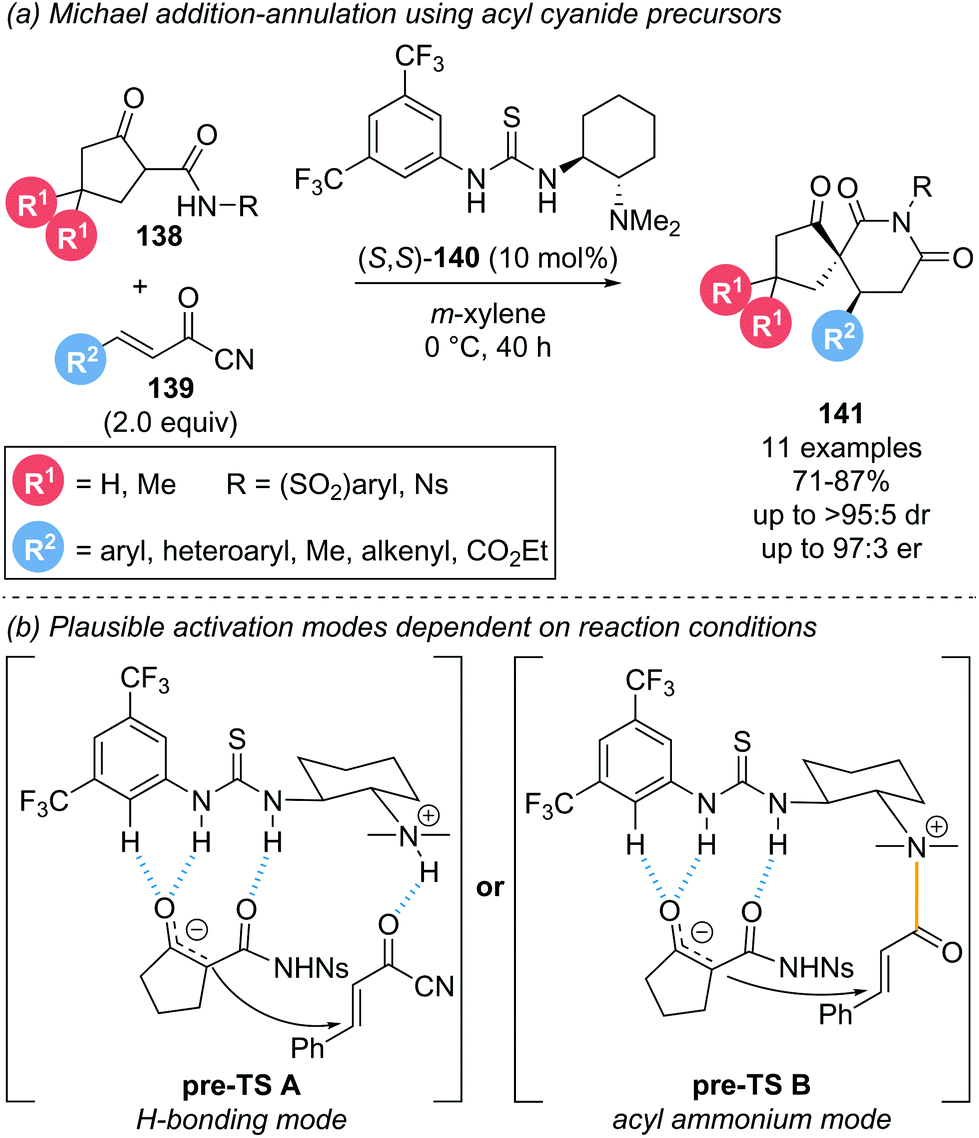 | ||
| Scheme 30 (a) Use of acyl cyanides in hydrogen bonding catalysis; (b) proposed transition states depending on catalyst mode of activation. | ||
In 2017, the groups of Lu and Du investigated the organocatalytic activation of cyclopropenones 143 in the presence of isatins 142 to obtain spirooxindoles 144 and 145 (Scheme 31a).40 Interestingly, the nature of the catalyst was not only important for reactivity, but also determined the regioselectivity of the spirooxindole products obtained. Using DMAP 5 as catalyst, spirooxindole 144 was formed, with only trace amounts of isomeric 145 detectable. By employing a chiral DMAP derivative, promising enantioselectivities of up to 84:16 er were observed. In contrast, isothiourea catalyst DHPB 115 lead to the preferred formation of regioisomeric 145. However, the reaction was generally lower yielding and less selective than DMAP catalysed reactions. Unfortunately, no enantioinduction could be achieved for this series by using chiral isothiourea catalysts. With Lewis base catalysts, activation of the cyclopropenone can be achieved via either 1,2-addition or 1,4-addition (Scheme 31b). Employing DMAP, 1,2-addition is postulated to give an α,β-unsaturated acyl ammonium intermediate 146. The obtained intermediate is nucleophilic at the β-carbon, initiating attack on the isatin, followed by lactonization to give spirooxindole 144. With DHPB as catalyst, 1,4-addition is postulated to lead to acyl anion intermediate 147. Nucleophilic attack of C(1) of the intermediate on the isatin and subsequent oxa-Michael addition results in spirooxindole 145 as the major product. However, both pathways are highly speculative.
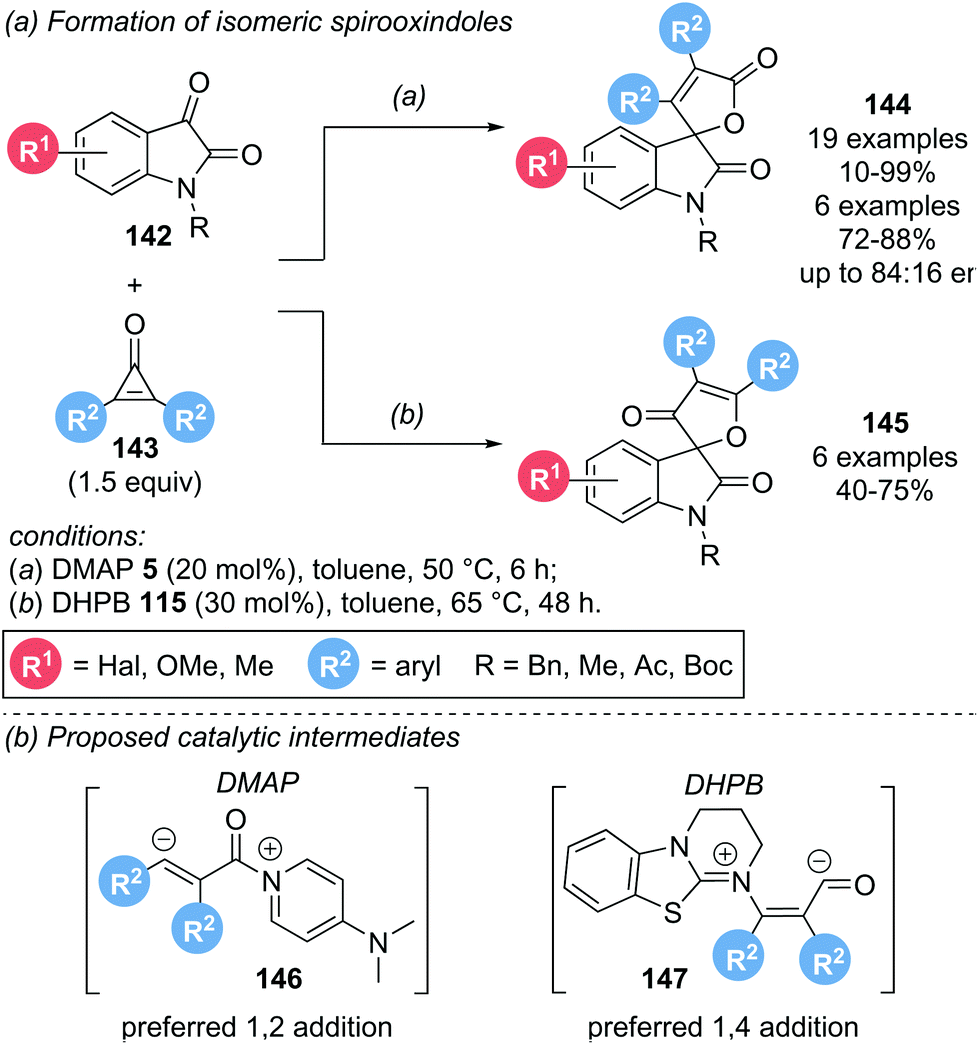 | ||
| Scheme 31 (a) Use of cyclopropenones for the synthesis of spirooxindoles; (b) proposed catalytic intermediates dependent on Lewis base used. | ||
In 2020, Zhang and co-workers reported the γ-carboxylation of butenoate 148 using CO2 which after cyclisation from α,β-unsaturated acyl ammonium intermediate 150 led to glutaconic anhydride 149 (Scheme 32).41 DBU is proposed to play multiple roles within the reaction, acting as both a Lewis and Brønsted base as well as activating CO2via formation of a DBU-CO2 adduct. A variety of leaving groups (X) could be tolerated on butenoate 148, namely acid chlorides, acyl imidazole and several aryl esters. Notably the reaction proceeded smoothly with an atmosphere of CO2 provided by a balloon. 13C isotopic labelling studies using 13CO2 were used to identify which carbonyl group in product 149 originated from CO2.
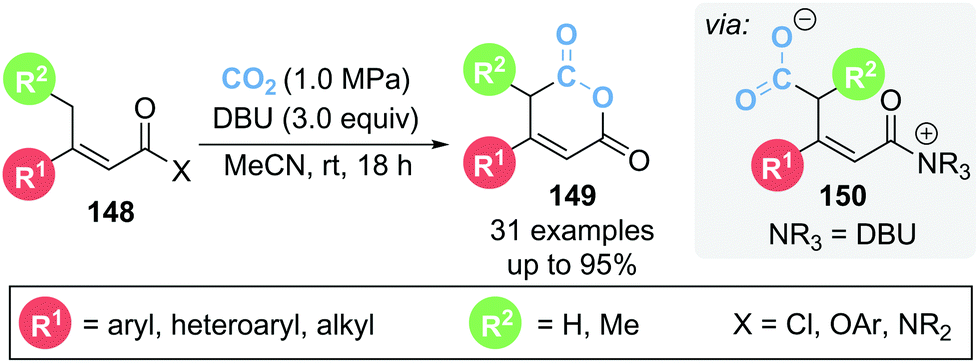 | ||
| Scheme 32 The use of CO2 as C(1) building block in the synthesis of glutaconic anhydride. | ||
An alternative use of α,β-unsaturated acyl ammonium species exploits the electrophilic character at the C(1)-position without involving reactive sites at the C(2) or C(3)-position utilised in Michael addition reactions. In 2020, Dong and co-workers demonstrated the use of α,β-unsaturated acyl ammonium intermediates in the formation of axially chiral anilides via atroposelective N-acylation (Scheme 33).42 Employing α,β-unsaturated mixed anhydrides 57 and sulfonamides 151 in the presence of (R)-HBTM 98, the desired anilides 152 were obtained in high yields (61–95%) and generally high enantioselectivities (>86:14 er). A variety of aryl substituents on the acyl donor (R1) were well tolerated. Notably, alkyl substituents also gave the corresponding anilides in excellent enantioselectivity. Different sulfonyl groups could also be incorporated. Importantly, when the bulky tert-butyl group (R3) on the sulfonamide nucleophile was replaced with a smaller methyl or halogen substituent (R3, R4), a drop in enantioselectivity was observed. This was attributed to the tert-butyl group being required for effective facial differentiation of the sulfonamide through steric interactions.
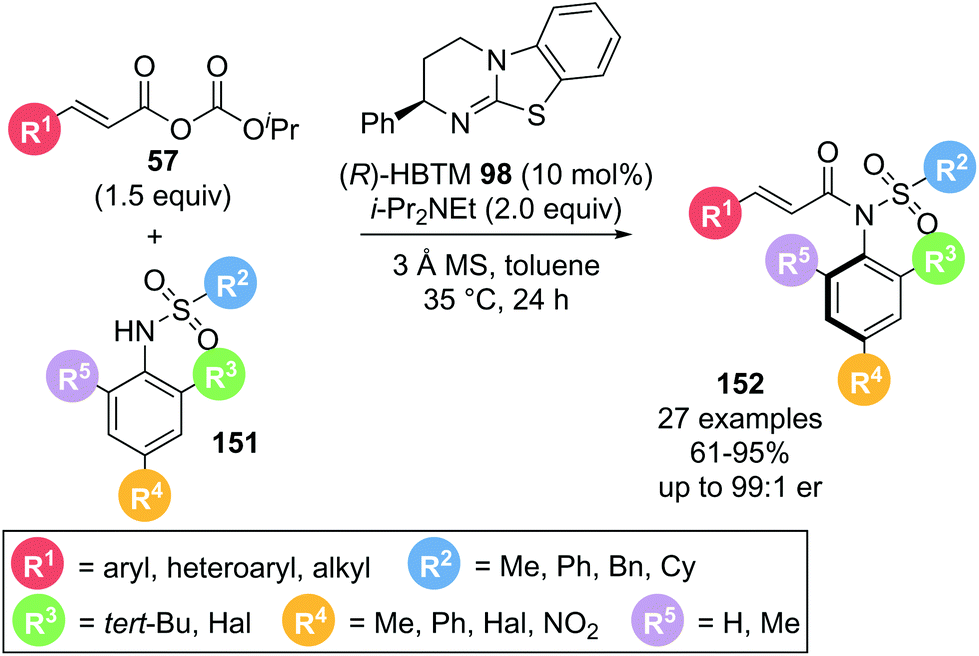 | ||
| Scheme 33 α,β-Unsaturated acyl ammonium intermediates used in acyl transfer reaction using mixed anhydrides. | ||
In a complementary study, Zhao and co-workers also reported the atroposelective synthesis of anilides 152 (Scheme 34).43 Reaction of symmetric anhydride 23 with a sulfonamide 151 possessing an ortho-halogen substituent in the presence of (S)-HBTM 98 yielded axially chiral sulfonamide 152 with excellent yields and good enantioselectivities. A wide range of both anhydrides and sulfonamides bearing a variety of aryl, heteroaryl and alkyl substituents were tolerated with minor variation in yield and enantioselectivity. The reduced size of the halogen substituents widely employed in this report likely explain the lower enantioselectivities observed as similarly substituted sulfonamides under Dong and co-workers conditions gave comparable enantioselectivities. In a notable application, the use of the products as catalysts was also explored.
 | ||
| Scheme 34 α,β-Unsaturated acyl ammonium intermediates used in acyl transfer reaction using symmetric anhydrides. | ||
5. Conclusions
α,β-Unsaturated acyl ammonium species are powerful synthetic intermediates in asymmetric reactions. Whilst many highly efficient methodologies using these intermediates have been established, several areas for future development are still to be explored. Application of these species in target molecule orientated synthesis remain rare and could further highlight their synthetic utility. The advent of aryloxide catalytic turnover and the subsequent use of monofunctional reaction partners, offers the opportunity for ‘simple’ Michael addition reactions that would otherwise be inaccessible at the carboxylic acid oxidation level. Whilst some mechanistic and computational studies have been reported, further contributions would allow for greater rational design of novel enantioselective reaction processes. Strategies to overcome the limitations on the commonly unreactive alkyl and aryl β-substituted Michael acceptors have yet to be developed and may require the design of alternative catalysts. Further exciting opportunities may arise from the merging of α,β-unsaturated acyl ammonium intermediates with other intermediates arising from either transition metal or photoredox catalysis. Whilst previous work has shown the compatibility of these dual catalytic reaction modes with ammonium enolate chemistry, their combination with α,β-unsaturated intermediates has yet to be explored.Note added in proof (December 07, 2020):
While this manuscript was at the proof stage, Romo and co-workers reported the application of enantioselective α,β-unsaturated acyl ammonium catalysis in the formal synthesis of (+)-neooxazolomycin44 and Smith and co-workers demonstrated its applicability to the transfer hydrogenation of α,β-unsaturated para-nitrophenyl esters.45 Birman also demonstrated further cascade cyclization reactions of α,β-unsaturated thioesters.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge The University of St Andrews (JB) and the EaSICAT Centre for Doctoral Training (MTW) for funding.Notes and references
- (a) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC; (b) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS; (c) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638–4660 CrossRef CAS; (d) P. Vogel, Y.-H. Lam, A. Simon and K. N. Houk, Catalysts, 2016, 6, 128 CrossRef; (e) D. L. Hughes, Org. Process Res. Dev., 2018, 22, 574–584 CrossRef CAS; (f) S.-H. Xiang and B. Tan, Nat. Commun., 2020, 11, 3786 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS.
- C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012–6042 CrossRef.
- (a) M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS; (b) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (c) C. McLaughlin and A. D. Smith, Chem. – Eur. J., 2020 DOI:10.1002/chem.202002059.
- K. Kato, Y. Shizuri, Y. Hirata and S. Yamamura, Chem. Commun., 1968, 324–325 RSC.
- E. Bappert, P. Müller and G. C. Fu, Chem. Commun., 2006, 2604–2606 RSC.
- R. P. Wurz, Chem. Rev., 2007, 107, 5570–5595 CrossRef CAS.
- (a) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS; (b) T. Marcelli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 142–152 CAS.
- (a) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; (b) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589–5610 CrossRef CAS.
- S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef CAS.
- (a) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef CAS; (b) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef CAS.
- A. Matviitsuk, M. D. Greenhalgh, D.-J. B. Antúnez, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef CAS.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef.
- (a) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS; (b) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS; (c) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS; (d) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC; (e) M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS; (f) D.-J. B. Antúnez, M. D. Greenhalgh, A. C. Brueckner, D. M. Walden, P. Elías-Rodríguez, P. Roberts, B. G. Young, T. H. West, A. M. Z. Slawin, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2019, 10, 6162–6173 RSC; (g) C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. D. L. Houpliere, K. B. Ling, T. K. Smith, A. M. Z. Slawin, P. H. Willoughby, S. L. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705–3710 CrossRef CAS; (h) Y. Nagao, S. Miyamoto, M. Miyamoto, H. Takeshige, K. Hayashi, S. Sano, M. Shiro, K. Yamaguchi and Y. Sei, J. Am. Chem. Soc., 2006, 128, 9722–9729 CrossRef CAS; (i) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS; (j) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS; (k) M. Breugst and J. J. Koenig, Eur. J. Org. Chem., 2020, 5473–5487, DOI:10.1002/ejoc.202000660.
- (a) C. Bleiholder, R. Gleiter, D. B. Werz and H. Köppel, Inorg. Chem., 2007, 46, 2249–2260 CrossRef CAS; (b) A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357–4416 CrossRef CAS; (c) R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS; (d) S. Kolb, G. A. Oliver and D. B. Werz, Angew. Chem., Int. Ed., 2020, 59, 22306–22310 CrossRef CAS.
- A. Matviitsuk, M. D. Greenhalgh, J. E. Taylor, X. B. Nguyen, D. B. Cordes, A. M. Z. Slawin, D. W. Lupton and A. D. Smith, Org. Lett., 2020, 22, 335–339 CrossRef CAS.
- C. Shu, H. Liu, A. M. Z. Slawin, C. Carpenter-Warren and A. D. Smith, Chem. Sci., 2019, 11, 241–247 RSC.
- E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC.
- M. D. Greenhalgh, S. Qu, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909–4918 RSC.
- C. Wang, S.-J. Li, Q.-C. Zhang, D. Wei and L. Ding, Catal. Sci. Technol., 2020, 10, 3664–3669 RSC.
- Y. Fukata, K. Yao, R. Miyaji, K. Asano and S. Matsubara, J. Org. Chem., 2017, 82, 12655–12668 CrossRef CAS.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS.
- G. Kang, M. Yamagami, S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2018, 57, 6527–6531 CrossRef CAS.
- D. M. Leace, M. R. Straub, B. A. Matz and V. B. Birman, J. Org. Chem., 2019, 84, 7523–7531 CrossRef CAS.
- N. A. Ahlemeyer and V. B. Birman, Org. Lett., 2016, 18, 3454–3457 CrossRef CAS.
- N. A. Ahlemeyer, E. V. Streff, P. Muthupandi and V. B. Birman, Org. Lett., 2017, 19, 6486–6489 CrossRef CAS.
- J.-H. Jin, X.-Y. Li, X. Luo and W.-P. Deng, Tetrahedron, 2018, 74, 6804–6808 CrossRef CAS.
- H. Liu, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2020, 22, 1301–1305 CrossRef CAS.
- S. Dong, C. Fang, W. Tang, T. Lu and D. Du, Org. Lett., 2016, 18, 3882–3885 CrossRef CAS.
- Y.-C. Zhang, R.-L. Geng, J. Song and L.-Z. Gong, Org. Lett., 2020, 22, 2261–2265 CrossRef CAS.
- G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef CAS.
- E. R. T. Robinson, A. B. Frost, P. Elías-Rodríguez and A. D. Smith, Synthesis, 2017, 409–423 CAS.
- K. N. Van and D. Romo, J. Org. Chem., 2018, 83, 632–643 CrossRef CAS.
- A. Matviitsuk, J. E. Taylor, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Chem. – Eur. J., 2016, 22, 17748–17757 CrossRef CAS.
- M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, Chem. Sci., 2017, 8, 1511–1524 RSC.
- S. Vellalath, K. N. Van and D. Romo, Tetrahedron Lett., 2015, 56, 3647–3652 CrossRef CAS.
- M. E. Abbasov, B. M. Hudson, W. Kong, D. J. Tantillo and D. Romo, Org. Biomol. Chem., 2017, 15, 3179–3183 RSC.
- C.-L. Zhang, Z.-F. Zhang, Z.-H. Xia, Y.-F. Han and S. Ye, J. Org. Chem., 2018, 83, 12507–12513 CrossRef CAS.
- S. Goudedranche, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Chem. – Eur. J., 2014, 20, 410–415 CrossRef CAS.
- J. Xu, J. Cao, C. Fang, T. Lu and D. Du, Org. Chem. Front., 2017, 4, 560–564 RSC.
- K. Zhang, W.-Z. Zhang, X.-Y. Tao, M. Zhang, W.-M. Ren and X.-B. Lu, J. Org. Chem., 2020, 85, 11579–11588 CrossRef CAS.
- D. Li, S. Wang, S. Ge, S. Dong and X. Feng, Org. Lett., 2020, 22, 5331–5336 CrossRef CAS.
- J.-Y. Ong, X. Q. Ng, S. Lu and Y. Zhao, Org. Lett., 2020, 22, 6447–6451 CrossRef CAS.
- C. M. Chaheine, P. T. Gladen, M. E. Abbasov and D. Romo, Org. Lett., 2020, 22, 9282–9286 CrossRef CAS.
- W. Jiufeng, C. M. Young and A. D. Smith, Tetrahedron, 2020, 131758, DOI:10.1016/j.tet.2020.131758.
- N. A. Ahlemeyer, M. R. Straub, D. M. Leace, B. A. Matz and V. B. Birman, J. Org. Chem., 2021, 86, 1191–1197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |