
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel pathway for the thermolysis of N-nitrosoanthranilates using flash vacuum pyrolysis leading to 7-aminophthalides†
Dragan
Zlatković
ab,
Doris
Dallinger
 *ac and
C. Oliver
Kappe
*ac
*ac and
C. Oliver
Kappe
*ac
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: do.dallinger@uni-graz.at; oliver.kappe@uni-graz.at
bDepartment of Chemistry, Faculty of Sciences and Mathematics, University of Niš, Višegradska 33, 18000 Niš, Serbia
cCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
First published on 13th October 2020
Abstract
Flash vacuum pyrolysis of methyl N-methyl-N-nitrosoanthranilate leads to elimination of nitric oxide and disproportionation of the formed N-radical to 7-(methylamino)phthalide and methyl N-methylanthranilate. This transformation was found to be a convenient, solvent-free method for the preparation of 7-(methylamino)phthalides. An alternative route through pyrolysis of N-benzyl-N-methyl anthranilates was also investigated.
Miltojević and Radulović have recently reported an interesting thermally induced degradation of methyl N-methyl-N-nitrosoanthranilate under gas chromatography (GC) and preparative (batch) thermolysis conditions (Scheme 1).1 The outcomes were quite different, although the homolytic cleavage of the N–NO bond was the predominant first step in both transformations. The fate of the obtained methyl N-methylanthranilate free radical during the GC-MS analysis was either a recombination (in the transfer line) to form 2a as major product or proton abstraction from the stationary phase (on the column) to generate methyl N-methyl anthtranilate (3a). On the other hand, large-scale thermolysis in a sealed vial at 220 °C yielded a highly complex mixture of mostly coupling products.1
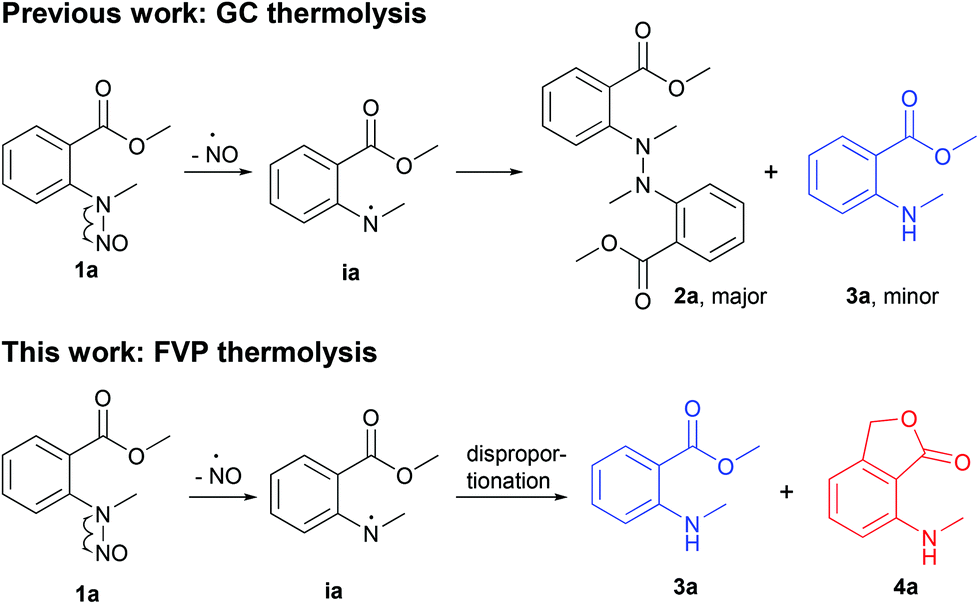 | ||
| Scheme 1 Thermolysis of N-methyl-N-nitrosoanthranilates. | ||
The extensive investigations by Miltojević and Radulović did not include high vacuum conditions, which can be achieved with the technique of flash vacuum pyrolysis (FVP). Here, intermolecular reactions are unlikely to occur and no external source of protons is present. FVP can be regarded as a gas-phase flow technique where substrate molecules are thermally activated by molecule-wall collisions at temperatures up to 1000 °C with extremely short contact times in the range of ms–s.2–5 In FVP, secondary reactions are minimized, because of the absence of a solvent and reagents (“chemistry without reagents“).3 Therefore, unimolecular transformations such as eliminations, rearrangements or cyclizations generally occur upon FVP thermolysis. In the past, FVP has been mostly employed for thermal reactions that were not possible under standard solution-phase conditions.5 Nowadays it is mostly used for reaction discovery and the synthesis of heterocyclic compounds, which cannot be easily obtained by other methods.6,7
Herein, we present the gas-phase thermolysis of N-nitroso derivatives of N-methylanthranilates applying FVP. To our knowledge, nitrosamines have not been employed so far in preparative FVP, even though the ease of N–NO bond cleavage is well known. This was surprising, because pyrolysis of alkyl nitrites is a standard procedure for the generation of alkoxy radicals (RO˙).2 We demonstrate that the FVP of nitrosamines is a convenient method for the generation of nitrogen radicals. In contrast to GC thermolysis,1 an unexpected disproportionation occurs, which leads to the corresponding anthranilate 3a and 7-(methylamino)phthalide 4a (Scheme 1).
We commenced our studies with the gas-phase thermolysis of 1a under GC-MS and GC-FID conditions (injector: 280 °C, column: 50–300 °C), which were comparable with those from the study of Miltojević and Radulović (injector: 250 °C, column: 70–315 °C).1 However, the heating rate in our experiment was higher (25 °C vs. 5 °C per minute). Surprisingly, the GC-MS chromatogram of 1a (Fig. S2 in the ESI†) obtained in our lab differed from that described by Miltojević and Radulović:1 Only minor amounts (ca. 25%, GC-FID peak area) of a dimeric compound (m/z 328 at 11.85 min) were detected, which was different from the dimer 2a, while the major thermolysis product was anthranilate 3a (m/z 165). A sharp peak at 7.05 min (10%) and an additional broad peak (7.50–7.80 min, 54%) were observed, indicating that 3a was formed in the injector and on the column, respectively.1 Another difference was the appearance of an additional signal at 8.27 min with m/z of 163, which is 2 units less than that of 3a. At that point we could not ascertain the exact structure of this compound.
To gain more information on the decomposition of 1a, a differential scanning calorimetry (DSC) measurement of methyl N-nitroso-N-methylanthranilate (1a) was performed before moving to the gas-phase thermolysis using FVP. The DSC analysis indicated a thermal decomposition at 219 °C (see Fig. S3 in the ESI†), which is in good agreement with the experimental decomposition data reported by Miltojević and Radulović.1 Therefore, with sublimation temperatures set to ≤110 °C in the FVP experiments, degradation of 1a should not occur in the sublimation flask.
As initial conditions for flash vacuum pyrolysis of 1a an oven temperature of 450 °C and 100–110 °C sublimation temperature were chosen (Table 1, entry 1). A mixture of 3a and the same compound with m/z of 163 that was also observed under GC thermolysis was obtained. This compound could be isolated by chromatography and its structure elucidated by a combination of 1D and 2D NMR analyses (see Fig. S4–7 in the ESI†). HSQC and HMBC experiments allowed us to unambiguously identify this compound as 7-(methylamino)phthalide (4a), which has never been reported before. The HMBC correlations of the H-3 signal (5.20 ppm) with C-1 (173.1 ppm) and C-3a (148.1 ppm) confirmed the presence of a lactone ring, and a 3-bond HMBC cross peak between the N–CH3 protons (2.93 ppm) and C-7 (149.1 ppm) confirmed the substitution pattern. In contrast to the GC thermolysis, compounds 3a and 4a were obtained in a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Table 1, entry 1), implying the pyrolysis to be a disproportionation reaction. To further investigate that observation, additional FVP experiments were performed varying the temperatures of sublimation (Tpre) and reaction (Toven, Tzone2), respectively, (Table 1). The ratio 3a/4a did not change significantly under the tested reaction conditions and was between 0.97 and 1.20. The total qNMR yield of products ranged from 66 to 76%, and isolated yields of chromatographically purified material correlated well with the qNMR yields (Table 1, entry 4).
:1 ratio (Table 1, entry 1), implying the pyrolysis to be a disproportionation reaction. To further investigate that observation, additional FVP experiments were performed varying the temperatures of sublimation (Tpre) and reaction (Toven, Tzone2), respectively, (Table 1). The ratio 3a/4a did not change significantly under the tested reaction conditions and was between 0.97 and 1.20. The total qNMR yield of products ranged from 66 to 76%, and isolated yields of chromatographically purified material correlated well with the qNMR yields (Table 1, entry 4).
|
|
||||
|---|---|---|---|---|
| Entry |
T
preb (°C) |
T
ovenb (°C) |
ΔTzone2b (°C) |
3a/4ac (%) |
|
a Conditions: 0.5 mmol of 1a, vacuum: 4–5 × 10−3 mbar, quartz tube (QT): 3.5 cm inner diameter and 60 cm length.
b See Fig. S1 in the ESI† for further details.
c
1H NMR yield.
d Isolated yields.
|
||||
| 1 | 100–110 | 450 | +20 | 34/32d |
| 2 | 80–90 | 450 | +20 | 33/34 |
| 3 | 70–80 | 450 | +20 | 37/36 |
| 4 | 60–70 | 400 | 0 | 40/36 (39/35)d |
| 5 | 70–80 | 350 | +20 | 37/33 |
| 6 | 50–70 | 300 | +20 | 36/30 |

GC-MS and GC-FID analyses of the mixture that was collected in the cold trap revealed the additional presence of o-toluidine, N-methyl-o-toluidine and methyl anthranilate (in total up to 5%). Non-transformed 1a was also detected and quantified by NMR (1–3% with respect to 1a in the sublimation flask). We assume that the rest of 1a was lost either by decomposition to volatile compounds and/or complete degradation inside of the quartz tube.
An interesting observation was the increase in pressure during the FVP experiments, which can be attributed to the liberated NO. Once the starting nitroso compound was consumed, the pressure dropped to the initial 4–5 × 10−3 mbar (Fig. 1). This allowed us to monitor the progress of the thermolysis: depending on Tpre and Toven, complete sublimation of 1a through the tube was achieved within 25–50 min (Fig. 1).
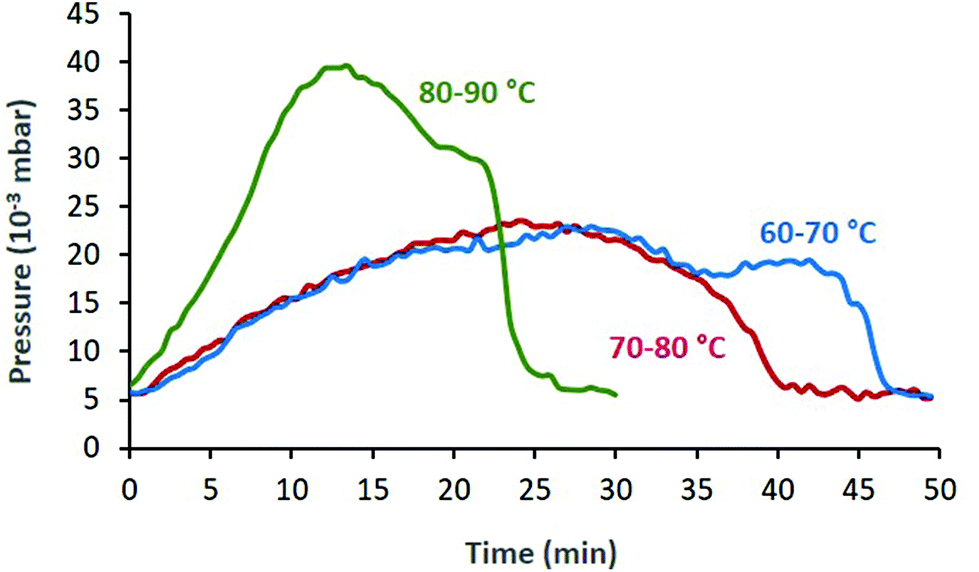 | ||
| Fig. 1 The pressure of the FVP system changes because of NO gas being released. Higher sublimation temperatures lead to shorter total sublimation times. The three curves correspond to entries 2 (green), 4 (blue) and 5 (red) from Table 1. | ||
For mechanistic insights toward the disproportionation pathway in the thermolysis of 1a, the deuterated methyl ester derivate 1b was subjected to FVP experiments employing the conditions from entry 2 in Table 1 (Scheme 2). In the initial step – the homolysis of the N–NO bond – a nitrogen radical (intermediate ib) is formed which abstracts the deuterium from the methyl ester (intermediate ii) and subsequently cyclizes to intermediate iii. The second equivalent of intermediate ib quenches iii by abstraction of the C-3a hydrogen and anthranilate 3b is generated. The deuteration degree at the CD3 group in 3b was 100%, which implies that the proton and deuterium abstraction step, respectively, occurred from the N-atom of intermediate ib. The alternative mechanism (radical transfer from N to C in intermediate ib and proton abstraction by the C-centered radical ib′, Scheme 2)8 could not be observed, because a deuteration degree of 0% was detected at the NMe group in the phthalide product.
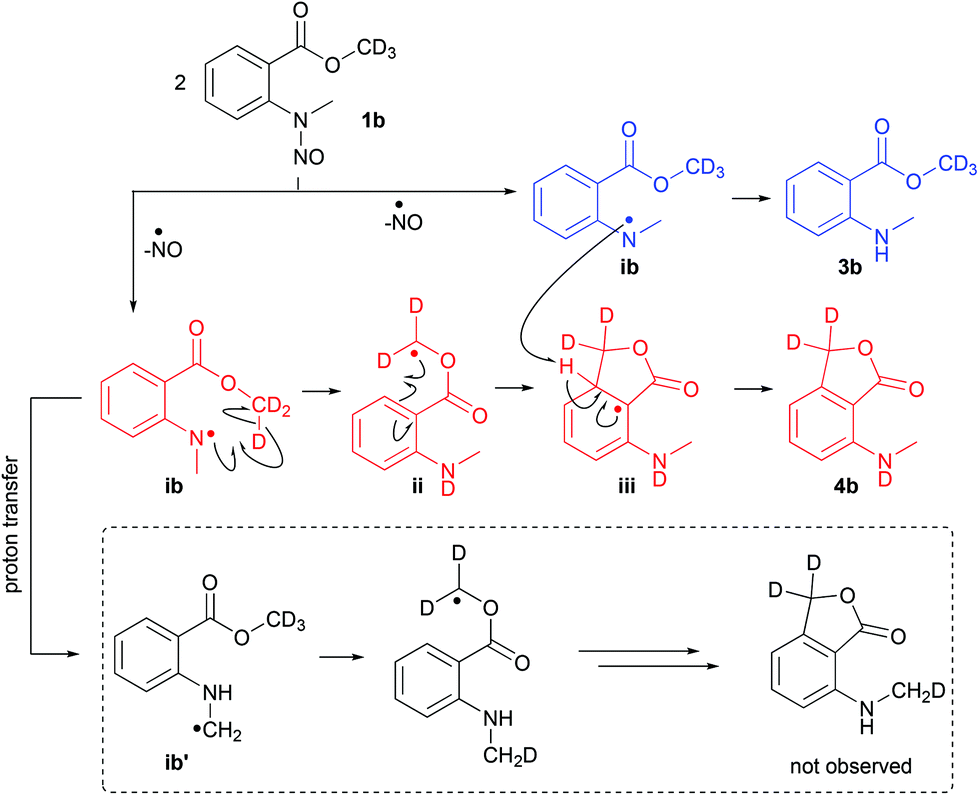 | ||
| Scheme 2 Proposed mechanistic pathway for the flash vacuum pyrolysis of methyl-d3N-methyl-N-nitrosoanthranilate (1b) leading to a disproportionation reaction. | ||
Additional FVP transformations were performed at 400 °C (Tpre = 60–90 °C, ΔTzone2 = +20 °C) using substituted N-methyl-N-nitrosoanthranilates 1c–1f (Scheme 3). Total isolated yields of disproportion products were between 49% and 60%, and the yields of the phthalide products 4c–4f ranged from 27% to 34%. FVP of benzyl N-methyl-N-nitrosoanthranilate was not successful, because of the low volatility of the substrate. Even at 170 °C sublimation temperature, the reaction did not occur, but decomposition inside the sublimation flask was observed. Hence, FVP works best for nitrosoanthranilate esters of lower alcohols (Scheme 3) and proved to be a convenient solvent- and catalyst-free preparation method of 3-substituted 7-(alkylamino)phthalides, even though the isolated yields of these compounds were only moderate. The other pyrolysis products (N-methylanthranilates 3c–3f) could be recycled and reused for the process after nitrosation. Other reported routes for the synthesis of the 7-aminophthalide scaffold are less sustainable: i.e. 7-amino-3-methylphthalide – an intermediate in the synthesis of the herbicide pyriftalid – has been prepared in a 4–6-step route starting from ortho-nitrophthalic acid, comprising a metal (RANEY®-Ni or Pd/C) catalyzed reduction as the last step.9 When starting directly from the rather expensive 2-acetyl-6-nitrobenzoic acid, the synthesis has been reduced to 1–2 steps.10 7-Aminophthalide has been generated in only 17% yield by applying the oxidation of 2-amino-6-methylbenzoic acid with NaBrO3/NaHSO3.11
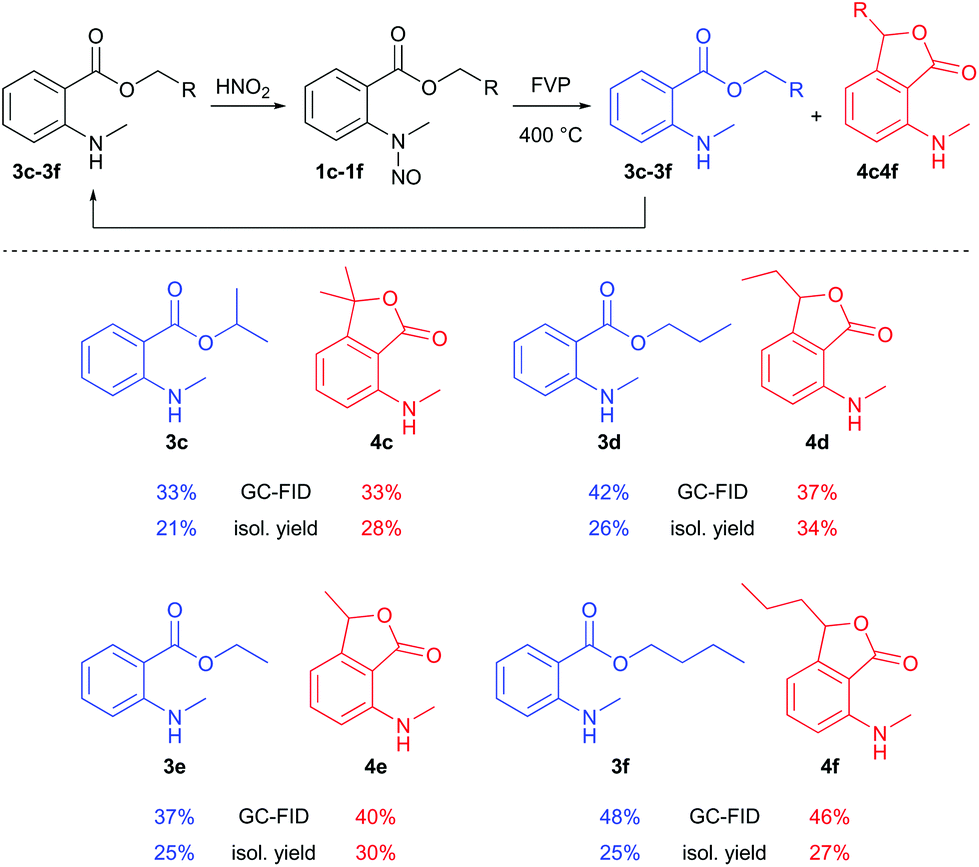 | ||
| Scheme 3 Reaction scope of the FVP of N-methyl-N-nitrosoanthranilates. GC-FID refers to GC-FID area% of the collected product after the FVP experiments. | ||
There were, however, some drawbacks of our method related to the use of N-nitroso compounds. Nitrosamines are known to be potent carcinogens, and extreme care must be taken during the preparation and handling of these compounds.12,13 Consequently, we decided to generate the nitrogen radicals i (Scheme 1) via the pyrolysis of N-benzyl- (5) or N-allyl-N-methyl-anthranilates (6), respectively. A similar approach for the generation of N-radicals via the FVP of N-benzyl-N-methylamino ylides has been reported by Aitken and co-workers.8 McNab and co-workers generated phenoxyl (PhO˙) and thiophenoxyl (PhS˙) radicals by FVP of allyloxy- and allylthiobenzoates at 650 °C.14,15 In theory, yields higher than 50% are possible with this method since benzyl or allyl radicals (formed by the homolysis of the N-allyl or N-benzyl bond, respectively) could rearomatize intermediate iii (see Scheme 2).16 The synthesis of N-benzyl- or N-allyl-N-methyl-anthranilates was not that straightforward as expected, because of the electron-withdrawing effect of the carboxy group and the low nucleophilicity of the N-atom. The reductive amination of benzaldehyde with methyl N-methylanthranilate (3a), or vice versa of formaldehyde with N-benzylanthranilate, using sodium triacetoxyborohydride (NaBH(OAc)3) failed. Benzylation of 3a with benzyl chloride/K2CO3 in acetone did not yield any product, even after 48 h of reflux, but subjecting the reaction mixture to higher temperatures (220 °C) by employing sealed vessel microwave heating, the benzylated anthranilates 5 were successfully synthesized (Scheme 4). The reactions were fairly clean (no compound other than the starting material and the product was detected) and the products could be isolated via column chromatography in 52–62% yield. This method for the synthesis of N-benzyl-N-methylanthranilates proved convenient compared to the only other published procedure that involves heating of 2-fluorobenzoate and benzylmethylamine in toluene/K2CO3 under reflux for 5 days.17 Microwave-assisted methylation of N-benzylanthranilate using CD3I, surprisingly yielded the N,N-dimethylated anthranilate as the major product.18 Our method also worked for the N-allylation, but at a lower temperature of 200 °C (Scheme 4).
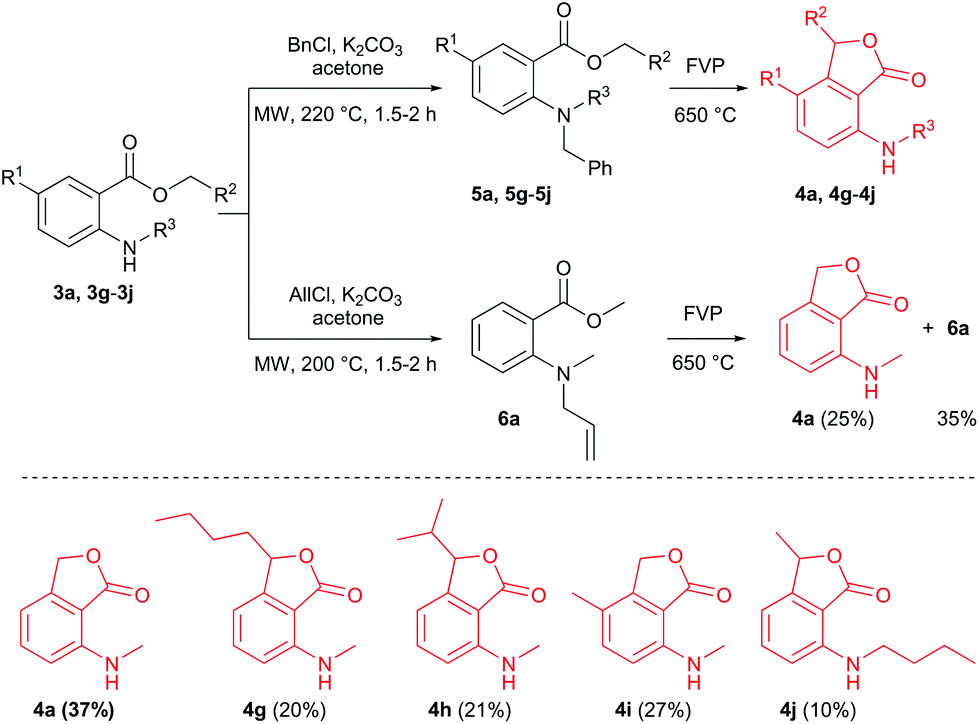 | ||
| Scheme 4 Synthesis and FVP of N-benzyl and N-allylanthranilates. | ||
FVP of N-allyl derivative 6a at 650 °C with a sublimation temperature of 70–80 °C did not produce satisfactory results as the 1H NMR yield of product 4a was only 25%. The unreacted substrate 6a was recovered from the cold trap in 35%, indicating that the temperature of the preheater was too high. However, performing the pyrolysis with a lower sublimation temperature of 50–60 °C did not resolve the problem as the yields of 4a and 6a were similar. FVP of the N-benzyl derivative 5a (Toven = 650 °C, Tpre = 100–110 °C) was more successful: no starting material was recovered from the cold trap and the 37% yield of 4a was comparable to the yield obtained via FVP of the N-nitroso counterpart 1a. The same reaction conditions were applied for the synthesis of (methylamino)phthalides 4g–i (Scheme 4), which were obtained in yields ≤30%. Pyrolysis of the N-butyl compound 5j was also attempted, but product 4j was isolated in low yield (10% NMR yield) as an inseparable mixture together with ethyl anthranilate, indicating that the scope was limited to N-methyl derivatives of 5.
As part of this study, we also wanted to develop a method for the preparation of 3-alkyl-7-mercaptophthalides. A compound that was of special interest was the pyriftalid precursor 7-mercapto-3-methylphthalide 11 (Scheme 5).9 In theory, this compound could be formed from the thiyl radical of 2-mercaptomethylbenzoate in a mechanism analogous to the one shown in Scheme 2. A simple method for the generation of such radicals would be the FVP of disulfides,2 as it has been shown that vacuum pyrolysis of diaryl disulfides at 400–700 °C resulted in the homolytic cleavage of the S–S bond.19 This approach looked especially attractive, because the starting material, diethyl 2,2′-dithiobisbenzoate (7a), was easily prepared from low-cost 2,2′-dithiosalicylic acid. Only starting material was recovered from the cold trap when pyrolysis of 7a was performed at 475 °C, with the preheater set at 200 °C. At 625 °C, 2,3-dihydrobenzothiophene 8 (Scheme 5) was obtained as the only isolated product in 25% yield, but unfortunately, no mercaptophthalide 11 was observed. On the other hand, FVP of the corresponding dimethyl diester 7b at 650 °C (Tpre = 140 °C) did produce 7-mercaptophthalide 10, albeit in a very low yield of 9%. The same results were obtained for the FVP of methyl 2-(allylthio)benzoate (9a) and methyl 2-(benzylthio)benzoate (9b), respectively (Scheme 5).
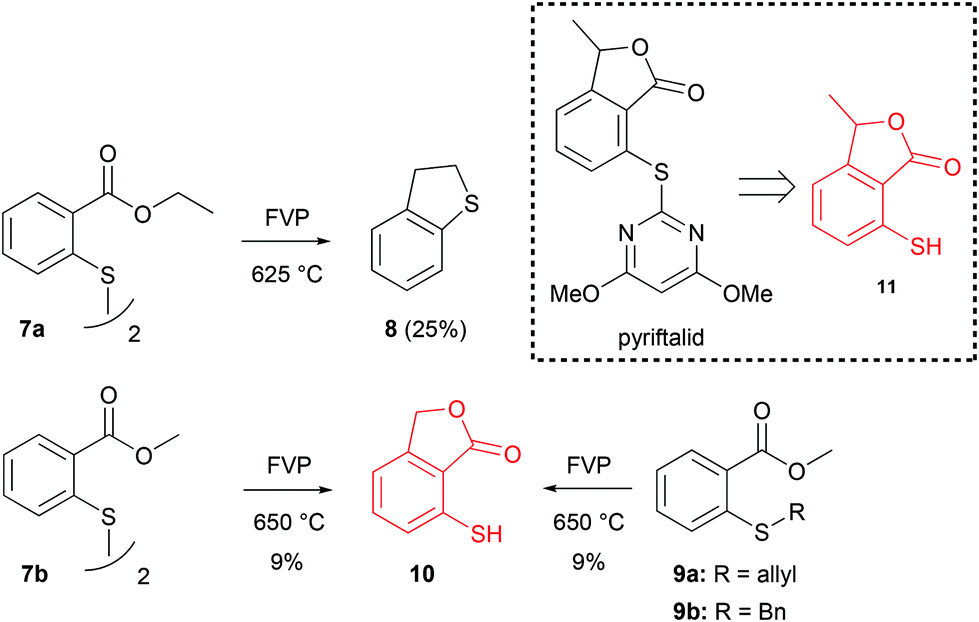 | ||
| Scheme 5 Attempted FVP synthesis of 7-mercaptophthalides. | ||
Conclusions
Flash vacuum pyrolysis of easily accessible N-methyl-N-nitrosoanthranilate esters offers a convenient and solvent-free route to the synthesis of otherwise not readily attainable 3-substituted 7-(methylamino)phthalides 4. The reaction mechanism was discovered to proceed via a disproportionation pathway, yielding N-methylanthranilate esters 3 as other products, which could be recycled to generate the corresponding N-nitroso precursor 1. 7-(Alkylamino)phthalides 4 could also be generated via an alternative route in comparable yields by FVP of less toxic N-benzyl derivatives 5, although this route comes at a cost of a reduced atom economy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CC Flow Project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation, and Technology (BMVIT), the Austrian Federal Ministry of Science, Research and Economy (BMWFW), and by the State of Styria (Styrian Funding Agency SFG). This work was supported by a postdoctoral grant to D. Z. (contract number 451-03-877/2019-14) provided by the Ministry of Education, Science and Technological Development of the Republic of Serbia. We thank ThalesNano for the provision of the Flash Pyrolysis Platform, Wolfgang Sindler (RCPE) for performing DSC measurements and Philipp Neu and Sigrid Lagarde (University of Graz) for the HRMS measurements.Notes and references
- A. B. Miltojević and N. S. Radulović, RSC Adv., 2015, 5, 53569–53585 RSC.
- R. F. C. Brown, Pyrolytic Methods in Organic Chemistry: Application of Flow and Flash Vacuum Pyrolytic Techniques, Academic Press, New York, USA, 1980 Search PubMed.
- H. McNab, Aldrichimica Acta, 2004, 37, 19–26 CAS.
- C. Wentrup, Angew. Chem., Int. Ed., 2017, 56, 14808–14835 CrossRef CAS.
- L. T. Scott, J. Org. Chem., 2016, 81, 11535–11547 CrossRef CAS.
- R. A. Aitken and Y. Boubalouta, Adv. Heterocycl. Chem., 2015, 115, 93–150 CrossRef CAS.
- L. C. Lengyel, G. Sipos, T. Sipőcz, T. Vágó, G. Dormán, J. Gerencsér, G. Makara and F. Darvas, Org. Process Res. Dev., 2015, 19, 399–409 CrossRef CAS.
- Such hydrogen atom transfer and the subsequent attack of NHCH2˙ on a triple bond was observed during the pyrolysis of N-benzyl-N-methylamino ylides: R. A. Aitken, L. Murray and A. M. Z. Slawin, Molecules, 2018, 23, 2153 CrossRef.
- C. Lüthy, H. Zondler, T. Rapold, G. Seifert, B. Urwyler, T. Heinis, H. C. Steinrücken and J. Allen, Pest Manage. Sci., 2001, 57, 205–224 CrossRef.
- Z. Li, B. Li, A.-J. Yang and F.-L. Zhang, Org. Process Res. Dev., 2017, 21, 1528–1532 CrossRef CAS.
- E. Bayer, S. Hayat, Atta-ur-Rahman, M. I. Choudhary, K. M. Khan, S. T. A. Shah, M. Imran-ul-Haq, M. U. Anwar and W. Voelter, Arzneim.-Forsch./Drug Res., 2005, 55, 588–597 CAS.
- Most nitrosamines are carcinogens, mutagens, and teratogens. Nitrosoanthranilate 1a was reported to be non-mutagenic: A. Sakai, K. Yoshikawa, H. Kurata and A. Tanimura, J. Food Hyg. Soc. Jpn., 1978, 19, 85 CrossRef CAS . This, however, does not imply that this compound is non-cancerogenic.
- M. A. Armour, Hazardous Laboratory Chemicals Disposal Guide, CRC Press, Boca Raton, FL, 3rd edn, 2003 Search PubMed.
- M. Black, J. I. G. Cadogan and H. McNab, J. Chem. Soc., Perkin Trans. 1, 1994, 155–159 RSC.
- M. Black, J. I. G. Cadogan and H. McNab, Org. Biomol. Chem., 2010, 8, 2961–2967 RSC.
- The attempted pyrolysis of ethyl N-benzylanthranilate failed to give the desired 7-amino-3-methylphthalide. N-phenylformamide and benzophenone imine were identified as main products by GC-MS. It appears that the reaction is possible only with secondary nitrogen radicals.
- R. A. Aitken and L. Murray, J. Org. Chem., 2008, 73, 9781–9783 CrossRef CAS.
-
. - I. V. Koval’, Russ. Chem. Rev., 1994, 63, 735–750 CrossRef.

Footnote |
| † Electronic supplementary information (ESI) available: Procedures, product characterization and NMR spectra. See DOI: 10.1039/d0ob01946a |
| This journal is © The Royal Society of Chemistry 2020 |