
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of the actinoallolides and a designed photoaffinity probe for target identification†
Matthew J.
Anketell
 ,
Theodore M.
Sharrock
and
Ian
Paterson
*
,
Theodore M.
Sharrock
and
Ian
Paterson
*
University Chemical Laboratory, University of Cambridge, Lensfield Road, CB2 1EW, UK
First published on 1st October 2020
Abstract
The actinoallolides are a family of polyketide natural products isolated from the bacterium Actinoallomurus fulvus. They show potent biological activity against trypanosomes, the causative agents of the neglected tropical diseases human African trypanosomiasis (sleeping sickness) and Chagas disease, while exhibiting no cytotoxicity against human cell lines. Herein, we give a full account of our strategy evolution towards the synthesis of this structurally unique class of 12-membered macrolides, which culminated in the first total synthesis of (+)-actinoallolide A in 20 steps and 8% overall yield. Subsequent late-stage diversification then provided ready access to the congeneric (+)-actinoallolides B–E. Enabled by this flexible and efficient endgame sequence, we also describe the design and synthesis of a photoaffinity probe based on actinoallolide A to investigate its biological mode of action. This will allow ongoing labelling studies to identify their protein binding target(s).
Introduction
Among communicable illnesses, parasitic diseases are the fourth largest cause of mortality worldwide, being responsible for over 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2015.1,2 Two such conditions are the neglected tropical diseases human African trypanosomiasis (sleeping sickness)3–5 and Chagas disease,6–10 both caused by protozoan parasites of the genus Trypanosoma. These illnesses are estimated to account for a loss of 455000 disability-adjusted life years annually.11 Trypanosomes are extremely adept at evading the immune system, and these neglected tropical diseases can only be cleared from the body with the help of chemotherapeutic agents.12 However, the ongoing development of effective pharmaceutical agents against the causative agents of these diseases has been challenging due to two major factors.13,14 First, the eukaryotic nature of protozoan parasites means that trypanosomes are biologically more closely related than bacteria to human cells. This renders the development of selective anti-trypanosomal drugs more challenging.15 Secondly, as these diseases mainly affect less economically developed countries, a lack of financial incentive to develop new drugs has led to an urgent need for better, more effective and safer treatments for these neglected diseases.16
000 deaths in 2015.1,2 Two such conditions are the neglected tropical diseases human African trypanosomiasis (sleeping sickness)3–5 and Chagas disease,6–10 both caused by protozoan parasites of the genus Trypanosoma. These illnesses are estimated to account for a loss of 455000 disability-adjusted life years annually.11 Trypanosomes are extremely adept at evading the immune system, and these neglected tropical diseases can only be cleared from the body with the help of chemotherapeutic agents.12 However, the ongoing development of effective pharmaceutical agents against the causative agents of these diseases has been challenging due to two major factors.13,14 First, the eukaryotic nature of protozoan parasites means that trypanosomes are biologically more closely related than bacteria to human cells. This renders the development of selective anti-trypanosomal drugs more challenging.15 Secondly, as these diseases mainly affect less economically developed countries, a lack of financial incentive to develop new drugs has led to an urgent need for better, more effective and safer treatments for these neglected diseases.16
Fortunately, the natural world possesses a rich and structurally diverse source of antiparasitic compounds.17 The actinoallolides (Fig. 1), a family of novel polyketides, are one such example, isolated by Iwatsuki and co-workers from a cultured strain (MK10-036) of the actinomycete bacterium Actinoallomurus fulvus, obtained from the roots of Capsicum fruitescens collected in Thailand.18 The 3D structure of actinoallolide A (1) was determined by spectroscopic analysis and X-ray crystallography, which revealed the presence of 10 stereocentres, two trisubstituted alkenes and a 12-membered macrolactone incorporating a five-membered hemiacetal. In addition, the structures of actinoallolides B–E (2–5) were confirmed by chemical correlation with actinoallolide A.
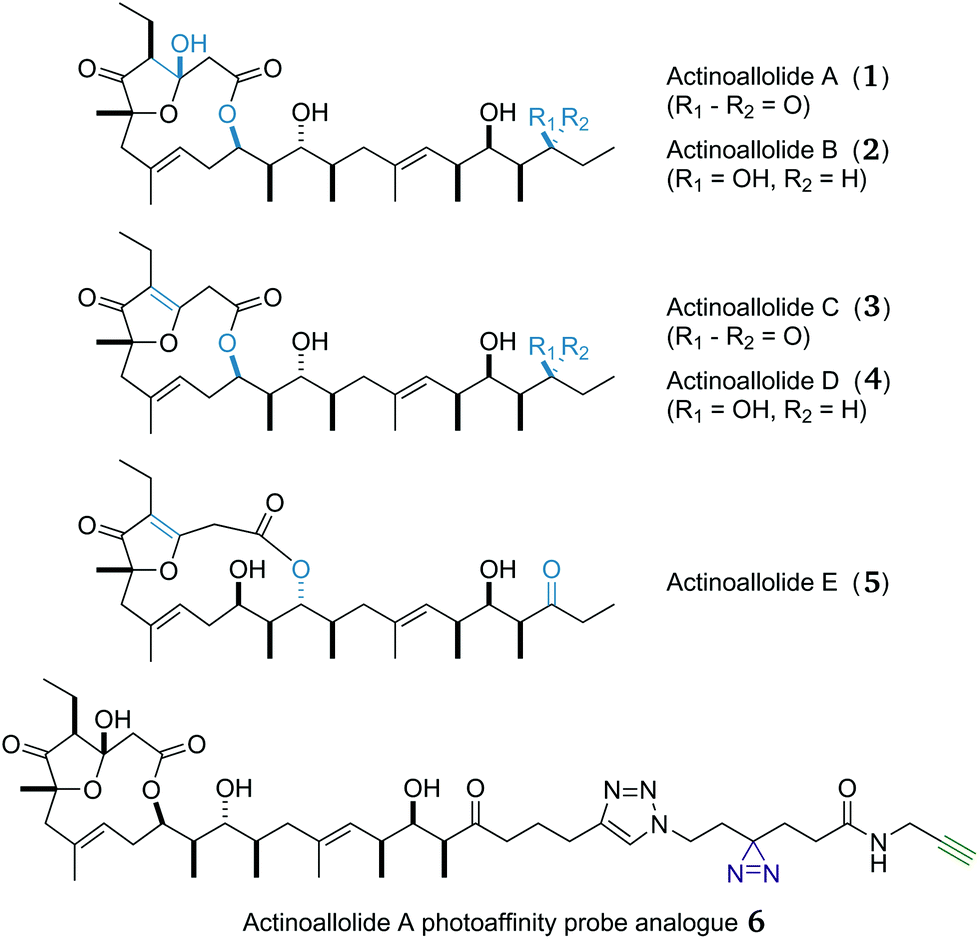 | ||
| Fig. 1 3D structures of the five actinoallolide congeners 1–5 and the designed photoaffinity probe analogue 6. | ||
The actinoallolides were tested against three strains of Trypanosoma and compared with several commonly used antitrypanosomal drugs. Actinoallolides A–E were also tested for biological activity against Trypanosoma brucei brucei, the strain responsible for animal trypanosomiasis (Nagana disease) and compared with three of the most commonly used drug treatments. They all compared favourably, with actinoallolide A determined as the most potent by two orders of magnitude, with an IC50 of 8.3 nM. Actinoallolide A (1) was also tested against Trypanosoma brucei rhodesiense, a strain responsible for human African trypanosomiasis and Trypanosoma cruzi, the causative agent of Chagas disease, showing double the potency of benznidazole. Actinoallolide A (1) showed no observable activity against MRC-5 human cells, giving it a high selectivity index (>20000).19 It was inactive against Gram-positive and negative bacteria and against yeast and fungi, indicating a highly specific mechanism of antitrypanosomal activity that is yet to be determined.
The isolation group have also identified the actinoallolide biosynthetic gene cluster, proposed a biosynthesis,20 and reported preliminary synthetic studies.21 Herein, we give a full account of the first total synthesis of the actinoallolides,22 which was enabled by a challenging ring-closing metathesis reaction to form the macrocyclic trisubstituted alkene, the most complex example of its kind.23 As the biological target and mechanism of action of the actinoallolides are unknown, we sought to leverage our total synthesis to shed light on these unanswered questions. Based on a highly efficient endgame, we now report the design and synthesis of the actinoallolide A-based probe 6 for target identification using photoaffinity labelling.24,25
Results and discussion
Retrosynthetic analysis of actinoallolide A
Our proposed key bond disconnections (Scheme 1) were of the C1 ester and the C8–C9 alkene to form two fragments of roughly equal size and complexity: the macrocycle precursor 7 and the side chain 8. A major advantage of this approach is that it allows a flexible fragment coupling strategy whereby the two coupling steps could be undertaken in either order; cross metathesis could be followed by macrolactonisation, or esterification could be followed by ring-closing metathesis as desired.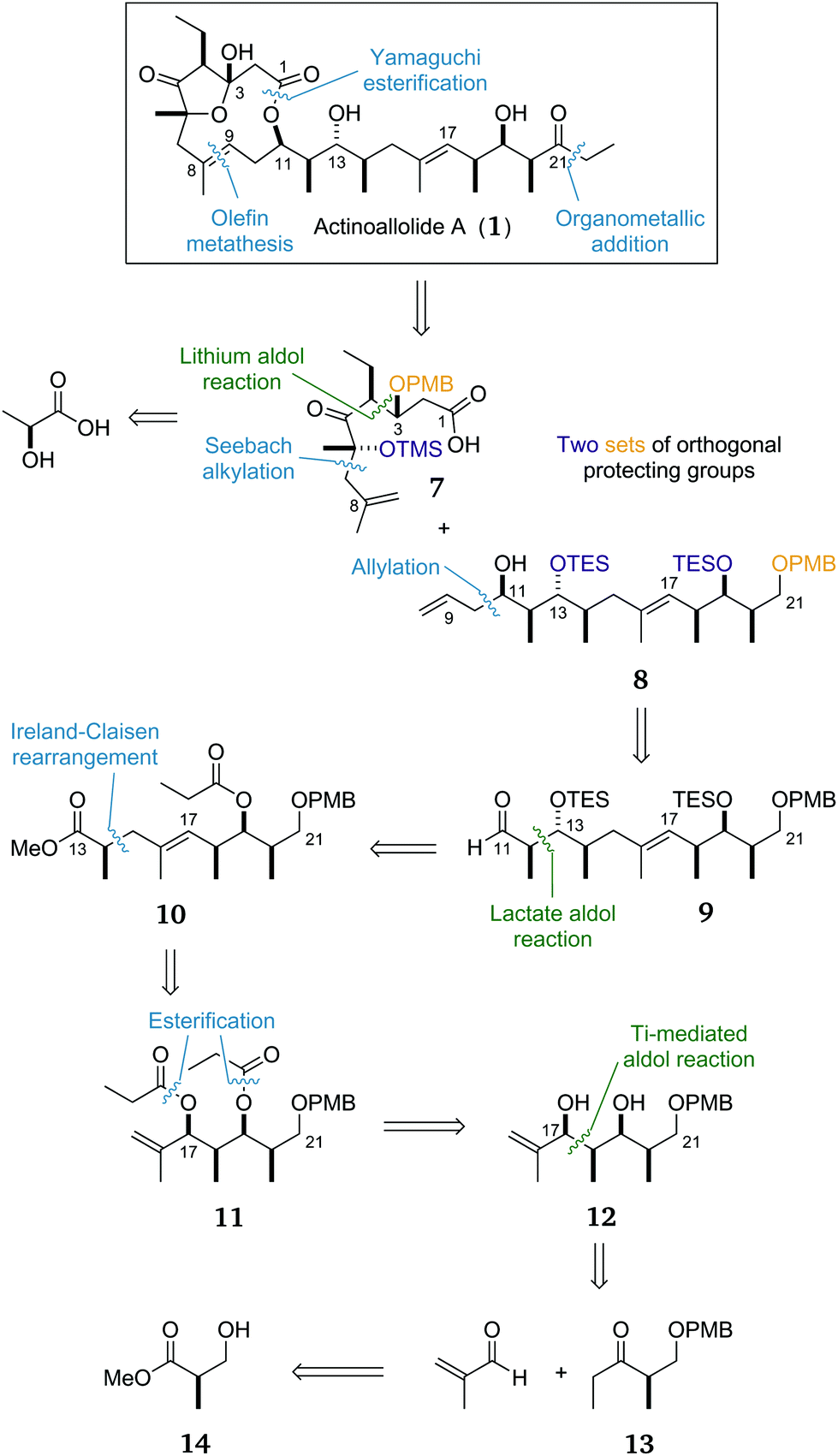 | ||
| Scheme 1 Initially proposed retrosynthesis of actinoallolide A with the three key aldol reactions highlighted in green. | ||
The C1–C8 fragment 7 would be forged from natural (S)-lactic acid with key bonds formed via a Seebach alkylation and a diastereoselective aldol reaction. Protection of the C3 and C6 alcohols would allow late-stage unmasking of the delicate five-membered hemiacetal after deprotection and oxidation at C3 to give actinoallolide A.
The side chain contains two distinct stereoclusters. The C11–C14 stereotetrad was envisaged to be constructed by a substrate-controlled allylation of 9 preceded by a lactate aldol reaction to reveal, after redox adjustment, ester 10. The isolated C14 stereocentre in 10 could then be installed by an Ireland–Claisen rearrangement from the diester 11. The ‘all syn’ stereotetrad in 12 can then be formed by a titanium-mediated aldol reaction/in situ reduction between methacrolein and ethyl ketone 13, itself synthesised in three steps from (R)-Roche ester (14). The decision to attach the terminal ethyl group at such a late stage was made in order to form the C21 ketone as late as possible to facilitate late-stage diversification. A more complex organometallic reagent could be used to add any modified tail in place of the ethyl group.
Finally, the decision to utilise two sets of orthogonal protecting groups was informed by our planned endgame. The hydroxyl groups at C21 and C3 require oxidation, whereas those at C6, C13 and C19 do not. Thus it was planned to use benzyl ether-based groups for the former and silyl ethers for the latter hydroxyl groups.
Synthesis of the C9–C21 side chain fragment (8)
The synthesis of side chain fragment 8 commenced with ethyl ketone 13.26 In previous work, the ‘all-syn’ aldol selectivity required for the construction of 12 had been achieved by use of tin(II) triflate as a Lewis acid.27,28 While effective, the use of tin(II) triflate was procedurally cumbersome, especially on scale owing to its capricious preparation. We sought to improve upon this process by combining two recent developments in aldol chemistry. Romea and Urpí have reported the use of titanium Lewis acids for the highly selective aldol reactions of protected β-hydroxy ketones29 and α-hydroxy ketones with an in situ reduction to afford the syn-1,3-diol.30 It was reasoned that combining these two processes would afford the desired ‘all-syn’ stereotetrad in diol 12. In the event, application of this in situ reduction with LiBH4 to the aldol addition of ketone 13 and methacrolein provided diol 12 in 90% yield as a single diastereomer (Scheme 2). Owing to the oxophilicity of the titanium and boron complexes formed in the reaction, an extended workup incorporating treatment with hydrogen peroxide and multiple washings with Rochelle salt solution was found to be necessary for release of 12.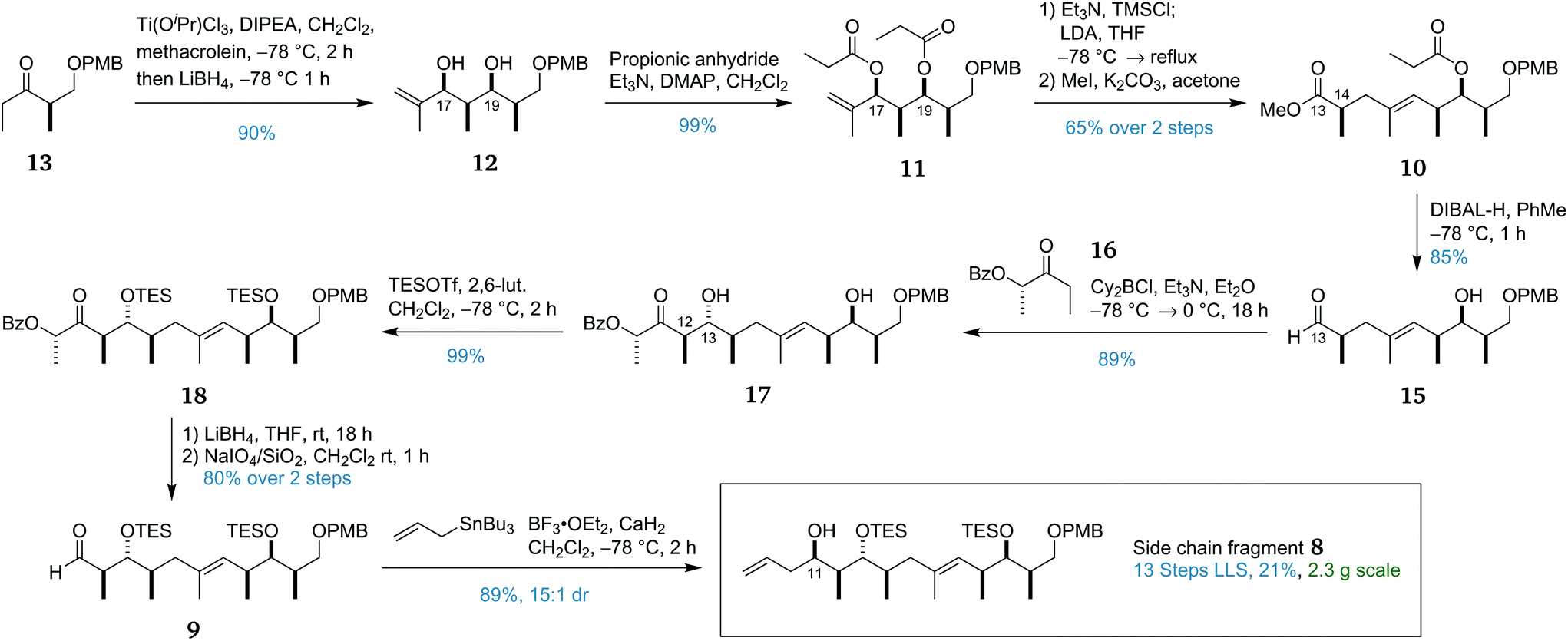 | ||
| Scheme 2 Synthesis of the side chain fragment 8. | ||
After selective monoesterification of the allylic alcohol in diol 12 was found to be problematic, it was decided to proceed through the required Ireland–Claisen rearrangement31 with diester 11. Only the O17 ester was correctly positioned to undergo the [3,3]-sigmatropic rearrangement while the O19 ester would be removed during the later reduction of the C13 methyl ester to the aldehyde. Guided by a similar Ireland–Claisen rearrangement used in the total synthesis of ebelactone,32 we began investigating this key step. Initial attempts at performing this reaction led to appreciable formation of the C14 α-silylated by-product. This TMS group could be removed with K2CO3 in MeOH but, as would be expected, led to significant epimerisation. The epimer was visible by NMR and provided evidence that the Ireland–Claisen product had been obtained as a single diastereomer. This side reaction is proposed to occur via enolisation of the formed silyl ester by excess LDA and trapping of this enolate with TMSCl. As such, it was important to precisely control the equivalents of LDA used. In order to avoid quenching any LDA, it is necessary to remove all traces of HCl from the large excess of TMSCl used. In the ebelactone work, this was accomplished by premixing TMSCl with Et3N and centrifuging the resulting suspension to remove the precipitate. In this work, removal of the precipitate was more conveniently accomplished via filtration through a syringe tip filter (PVDF membrane, 0.45 μm pores). Optimising the quantity of LDA used increased the yield to 73% with no observable formation of the α-silylated by-product. For ease of isolation, the carboxylic acid product was methylated without purification to afford methyl ester 10.
Pleasingly, the DIBAL-H reaction of this ester 10 proceeded well, reducing the methyl ester at C13 to the corresponding aldehyde and liberating the free hydroxyl group at C19, setting the stage for the second strategic aldol reaction. While substrate control was used to construct all other stereocentres in side chain fragment 8, the anticipated mismatch between the Felkin–Anh selectivity of aldehyde 15 and the desired configuration at the C13 hydroxyl stereocentre made it impossible in this case. Using boron aldol methodology developed in the group,33,34 we were able to couple aldehyde 15 with lactate-derived ketone 16 to form anti adduct 17 as a single diastereomer, overriding the inherent facial selectivity of aldehyde 15 to install the C12 and C13 stereocentres. Protection of both hydroxyl groups as TES ethers then afforded 18 which underwent a standard two-step auxiliary removal procedure to provide aldehyde 9.33 Addition of the allyl moiety to 9 to complete the side chain fragment 8 required some optimisation with the use of allylmagnesium chloride or allyltrimethylsilane leading to low diastereoselectivity. Fortunately, it was found that the utilisation of allyltributylstannane in a Hosomi–Sakurai reaction35,36 with the non-chelating Lewis acid BF3·OEt2 afforded 8 in high yield with 15:1 dr.
This completed the scalable and high-yielding synthesis of the side chain fragment 8 in 13 linear steps from (R)-Roche ester (14) with an overall yield of 21%. This excellent yield (an average of 89% per step) and the scalability of the route allowed 2.3 g to be prepared, providing ample material for development of the endgame.
Synthesis of the C1–C8 macrocycle precursor fragment
The synthesis of the macrocycle precursor fragment began with the construction of dioxolanone 19via a two-step procedure (Scheme 3),37 allowing an enolate alkylation38 with methallyl bromide to install the C6 stereocentre. This reaction proved difficult to optimise, with a 53% yield of 20, albeit as a single diastereomer. This was due to formation of a major by-product due to self-reaction of the dioxolanone.39 However, the modest yield was deemed sufficient and allowed the preparation of 20 on a multi-gram scale.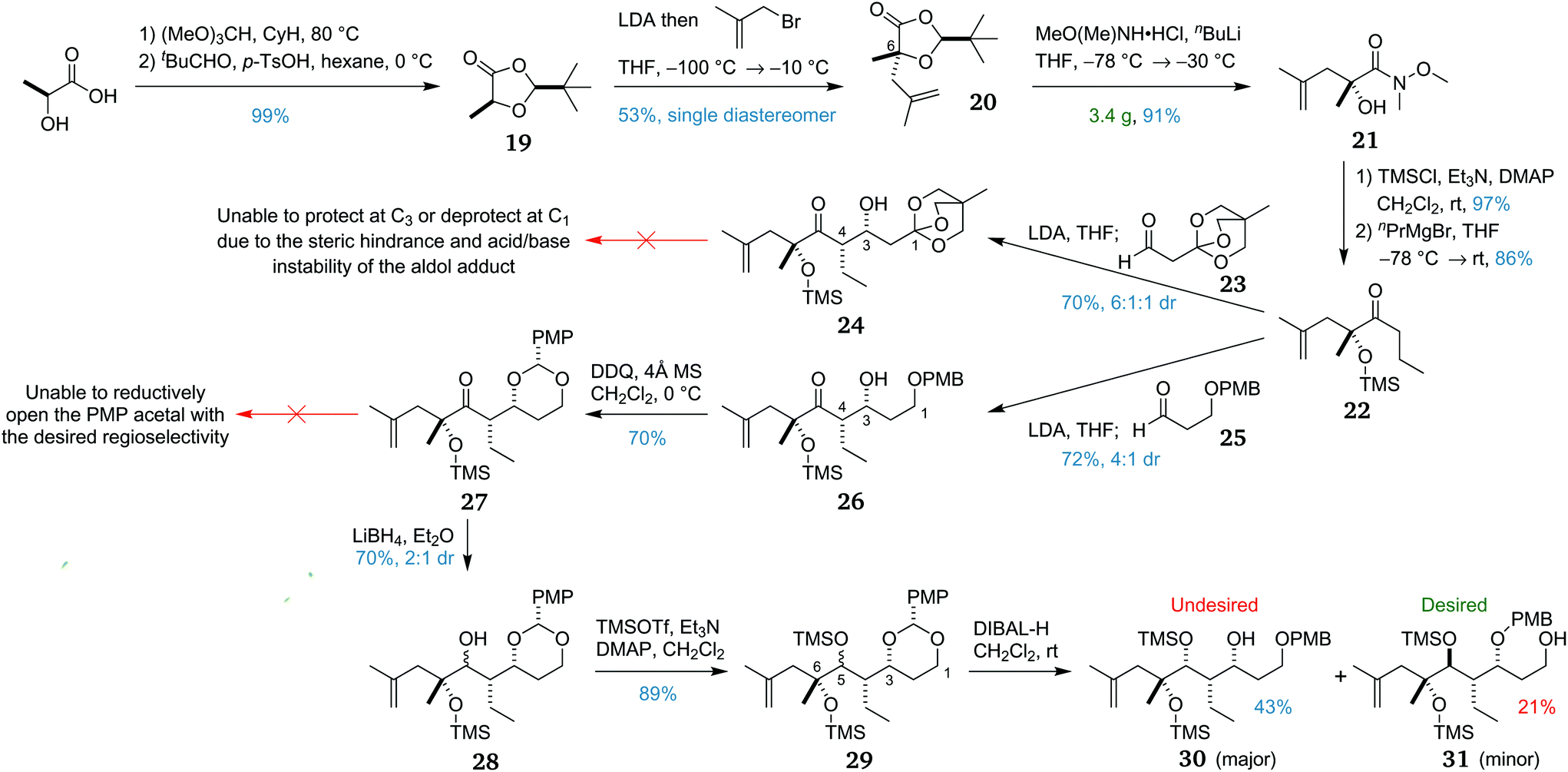 | ||
| Scheme 3 Synthesis of the key propyl ketone 22 and attempts at elaboration. | ||
The removal of the pivaldehyde-derived auxiliary via ring opening of dioxolanone 20 was the next task to be accomplished. In preliminary studies, the desired conversion to propyl ketone 22 was achieved using a four-step sequence commencing with ring opening to the methyl ester. We envisaged abridging this sequence by using a different ring-opening procedure. The first attempt to convert the dioxolanone to the propyl ketone directly by addition of propylmagnesium bromide, however, was unsuccessful, yielding either unreacted starting material or the double addition product. Ring opening to the Weinreb amide 21 was trialled next and proceeded in excellent yield using the in situ generated anion of N,O-dimethylhydroxylamine. With this result, a one-pot conversion to the desired propyl ketone was examined. However, the conversion of 21 to the corresponding propyl ketone in situ proved unsuccessful even with warming to room temperature. Weinreb amide 21 was next silyl protected and converted to propyl ketone 22, required for the planned C3–C4 bond-forming lithium-mediated aldol reaction. As the protecting group strategy utilised a silyl group at C6 and a PMB group at C3, the C1 protecting group was required to be orthogonal to both.
From propyl ketone 22, two routes to macrocycle precursor fragment 7 were initially considered. The first plan was to utilise an orthoester protecting group on aldehyde 23, as this would allow access to C1 at the desired carboxylic acid oxidation level. This lithium-mediated aldol reaction afforded syn adduct 24, providing the desired diastereomer in good yield and with useful diastereoselectivity. We had planned to protect the C3 alcohol with a PMB group, however all attempted conditions led to no reaction or to degradation, presumably owing to steric congestion. Furthermore, removal of the orthoester protecting group could not be achieved without degradation due to the acid and base sensitivity of the aldol adduct.
With this approach proving to be unviable, C1 was next introduced at the protected alcohol oxidation state. Noting that PMB groups can be oxidatively transposed,40 this second plan was to utilise a PMB group on aldehyde 25 to produce aldol adduct 26. As before, the lithium-mediated aldol reaction afforded the syn adduct in good yield, in a 4:1 ratio of diastereomers. The major diastereomer was then converted to PMP acetal 27 by treatment with DDQ under anhydrous conditions. Disappointingly, subsequent attempts at reductive PMP opening to the primary alcohol were all unsuccessful, with the use of DIBAL-H40 or NaCNBH3/TMSCl41 causing side-reactions or reacting with the undesired regioselectivity to revert to secondary alcohol 26. This unexpected selectivity may be due to preferential chelation of the Lewis acid between the C3 oxygen and the ketone rather than the less hindered C1 oxygen as would normally be expected.
Given that this selectivity problem was likely due to the propensity of the C5 ketone to coordinate with the adjacent C3 oxygen, it was proposed that reducing the C5 ketone and transiently protecting the resulting alcohol as a TMS ether might circumvent this impasse. Ketone 27 was thus reduced using LiBH4 to form secondary alcohol 28 in 70% yield and 2:1 dr. The diastereomers were inseparable by chromatography so the alcohols were then silylated to form 29. This mixture of diastereomeric PMP acetals then underwent reductive opening with DIBAL-H. Surprisingly, the two epimeric acetals reacted with opposing regioselectivities, with the major diastereomer forming the undesired secondary alcohol 30 and the minor diastereomer forming the desired primary alcohol 31.
In light of this mixed result, it was reasoned that if we could override the inherent substrate selectivity of ketone 27 to reduction, and obtain the 1,3-anti diastereomer selectively, this might provide a viable route to the macrocycle fragment. Testing a range of simple reducing agents was fruitless, with each one either giving the undesired selectivity or no reaction. It was next decided to attempt the reduction of the C5 ketone prior to PMP acetal formation, using the C3 alcohol in 26 to direct a 1,3-anti Evans–Tishchenko42 or Evans–Saksena reduction.43 With prolonged reaction time and an excess of the samarium catalyst,42 it was possible to achieve a low yield of 10% with 2:1 dr for the former reaction, but no product was obtained under the latter conditions. This was attributed to the steric hindrance of the C5 ketone with its quaternary α stereocentre incorporating a bulky OTMS group.
These frustrations en route to the macrocycle precursor fragment led us to consider more substantial modifications to the route. The efforts thus far had focused on installing a PMB protecting group at C3, either by a ‘PMB transposition'of the PMB group at C1, or by direct protection of the alcohol at C3. The former method had failed due to the selectivity problems with opening the intermediate PMP acetal and the latter had failed due to the steric hindrance of aldol adduct 26 and its instability under forcing conditions. To circumvent these problems within the constraints of the overall protecting group strategy – that is to allow for the chemoselective oxidative removal of the C3 and C21 protecting groups in one operation, it was proposed to switch the C3 protecting group from PMB to PMBM (para-methoxybenzyloxymethyl).44 Like the PMB group, it can be oxidatively removed orthogonally to silyl groups but can be installed under far milder conditions. However, as the PMBM group was liable to cleavage under oxidative conditions, this alteration necessitated a revision of the C1 protecting group such that it could be removed orthogonally to the PMBM group. Given that the aromatic ring in the DMB (3,4-dimethoxybenzyl)45 group is more electron rich than in the PMBM group, it was anticipated that selective oxidative cleavage of the DMB group would be possible.46
The corresponding DMB-protected aldehyde 32 required for the aldol coupling was prepared via a three-step procedure.47 This aldehyde then underwent an analogous lithium-mediated aldol reaction with propyl ketone 22, providing syn adduct 33 (Scheme 4) in similar yield and dr to that obtained from the PMB-protected variant. This was subsequently PMBM protected using the mild base DIPEA in refluxing acetonitrile overnight to afford PMBM ether 34 in excellent yield. With triply-protected aldol adduct 34 in hand, selective DMB cleavage was next attempted. Fortunately, one equivalent of DDQ with careful monitoring afforded primary alcohol 35 in excellent yield, with complete retention of the PMBM group. This then underwent a double oxidation sequence; first was a Swern oxidation to give aldehyde 36, then a Pinnick oxidation provided the complete macrocycle precursor fragment 37.
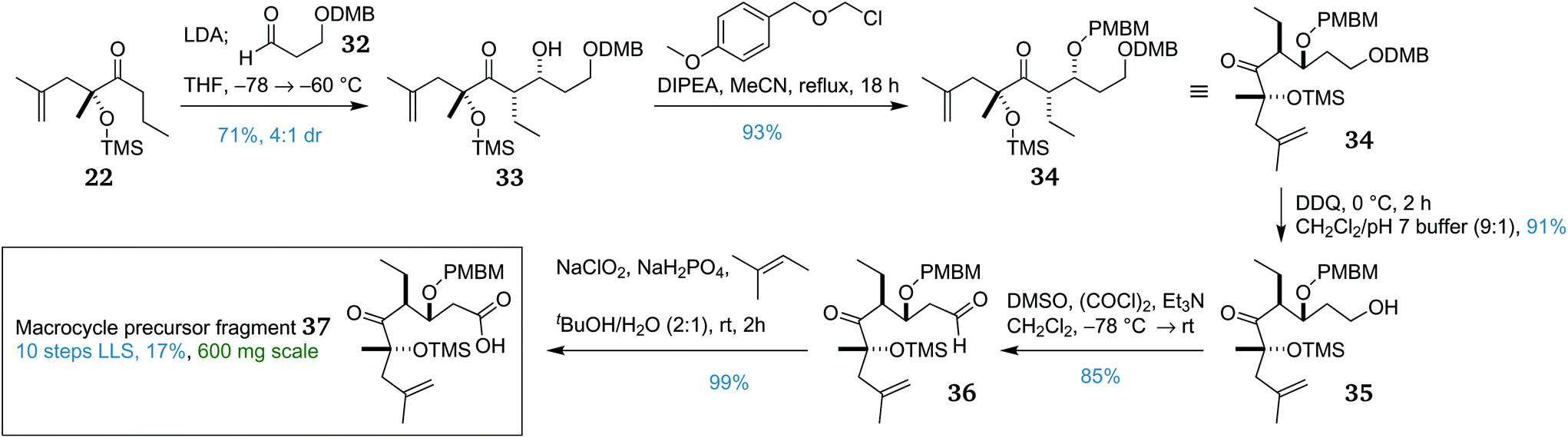 | ||
| Scheme 4 Completion of the synthesis of macrocycle precursor fragment 37. | ||
Overall, macrocycle precursor fragment 37 was synthesised in 10 linear steps from (S)-lactic acid with an overall yield of 17%. This represents an average of 84% per step, and the scalability of the route allowed 600 mg to be prepared, providing ample material for development of the endgame.
Fragment union and endgame
With key fragments 8 and 37 in hand, the stage was set to explore the pivotal fragment union sequence. The retrosynthetic plan (Scheme 1) had left the ordering of these steps flexible. However, preliminary investigations had indicated that an efficient and stereoselective cross metathesis to give the trisubstituted 8E alkene was unlikely to succeed, so we proceeded with exploring the esterification/RCM sequence. The fragments 37 and 8 were coupled together under standard Yamaguchi conditions48 to afford ester 38 in an excellent yield of 99%, enabling investigation of the “do-or-die” macrocycle-forming RCM step (Scheme 5).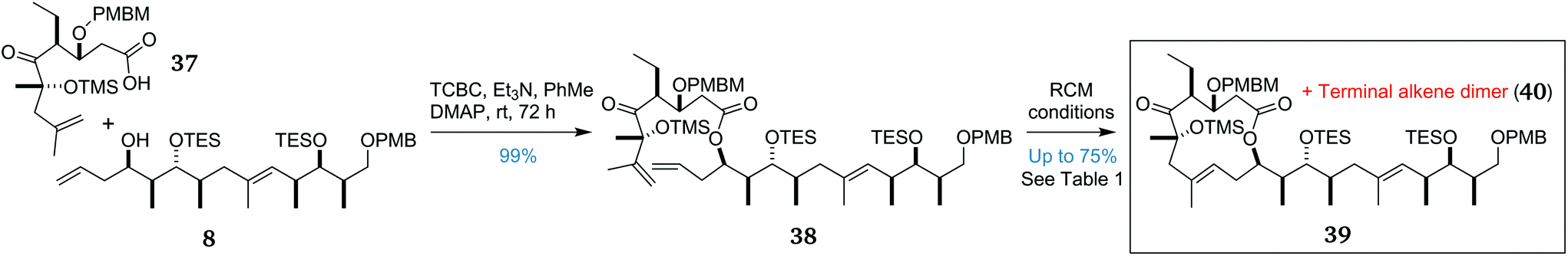 | ||
| Scheme 5 Fragment union and RCM-mediated formation of the 12-membered macrocycle 39. | ||
The first attempt at cyclisation of 38 using Grubbs second-generation catalyst (G-II)49,50 in refluxing degassed CH2Cl2 provided only the undesired dimer 40 in quantitative yield (Table 1, entry 1). Increasing the catalyst loading to 50 mol% (entry 2) or the use of higher-boiling solvents (entries 3 and 4) failed to provide any macrocyclic product, in each case returning only dimer 40. As an additional measure, a solution of catalyst was added portionwise over the initial few hours of the reaction to reduce catalyst decomposition, although the colour change from purple to brown was observed within 30 min of each addition, indicating rapid catalyst degradation. In light of these failures, we switched to the more reactive Hoveyda–Grubbs second-generation catalyst (HG-II).51,52 Gratifyingly, an initial RCM reaction of 38, overnight in refluxing toluene, afforded an inseparable 1:1 mixture of dimer 40 and desired macrocycle 39 (entry 5). Increasing the catalyst loading to 40 mol% and the reaction time to 72 h increased the product:dimer ratio to 1.5:1 (entry 6). In an attempt to increase the reaction rate and yield, the solvent was changed to o-xylene to allow reflux at a higher temperature. Surprisingly, this led to a reduction in the product:dimer ratio to 1.2:1 (entry 9), indicating that there was a ‘sweet spot’ temperature at around 110–140 °C, below which the reaction is prohibitively slow and above which catalyst decomposition occurs.
| Entry | Cat. (mol%) | Solvent (mM) | Temp. | Time | 39:40 |
|---|---|---|---|---|---|
| 1 | G-II (20) | CH2Cl2 (5) | 40 °C | 18 h | 0:1 |
| 2 | G-II (50) | CH2Cl2 (10) | 40 °C | 18 h | 0:1 |
| 3 | G-II (20) | Benzene (1) | 80 °C | 18 h | 0:1 |
| 4 | G-II (20) | Toluene (1) | 110 °C | 18 h | 0:1 |
| 5 | HG-II (20) | Toluene (1) | 110 °C | 18 h | 1:1 |
| 6 | HG-II (40) | Toluene (1) | 110 °C | 72 h | 1.5:1 |
| 7 | HG-II (40) | Toluene (10) | 110 °C | 72 h | 1.3:1 |
| 8 | HG-II (40) | Toluene (0.25) | 110 °C | 72 h | 2:1 |
| 9 | HG-II (40) | Xylene (0.25) | 145 °C | 72 h | 1.2:1 |
| 10 | HG-II (40) | Toluene (1) | 110 °C | 7 d | 3:1 |
By this point, a mechanistic hypothesis was proposed whereby the terminal alkene present in 38 rapidly dimerises. The ruthenium catalyst can then reinsert into the new disubstituted alkene in this dimer and can then undergo one of two processes. In most cases, this undergoes an intermolecular reaction with another molecule of dimer 40 to reversibly return to the dimer. Much more slowly, but irreversibly, this ruthenium carbene can undergo an intramolecular reaction with the 1,1-disubstituted alkene to afford desired macrocycle 39. To help encourage the slow, irreversible RCM step, the duration of the reaction was increased to seven days, increasing the product:dimer ratio to 3:1 and providing the desired product with an improved yield of 75% (entry 10). To our knowledge, this challenging and remarkable transformation is the most complex example of an RCM reaction to form a trisubstituted alkene in a medium-sized ring.23
Having prepared an abundant supply of advanced intermediate 39, the removal of both PMB-containing protecting groups was accomplished by treatment with DDQ, affording diol 41 in 94% yield (Scheme 6). Chemoselective oxidation of the primary alcohol at C21 in the presence of the C3 secondary alcohol was achieved using a TEMPO/BAIB oxidation53,54 which afforded aldehyde 42 in 90% yield. Subsequently, chemoselective addition of ethylmagnesium bromide to the newly formed aldehyde in the presence of the C5 ketone proceeded smoothly, affording an inseparable epimeric mixture of 43 in 91% yield, with no observable attack at the ketone.
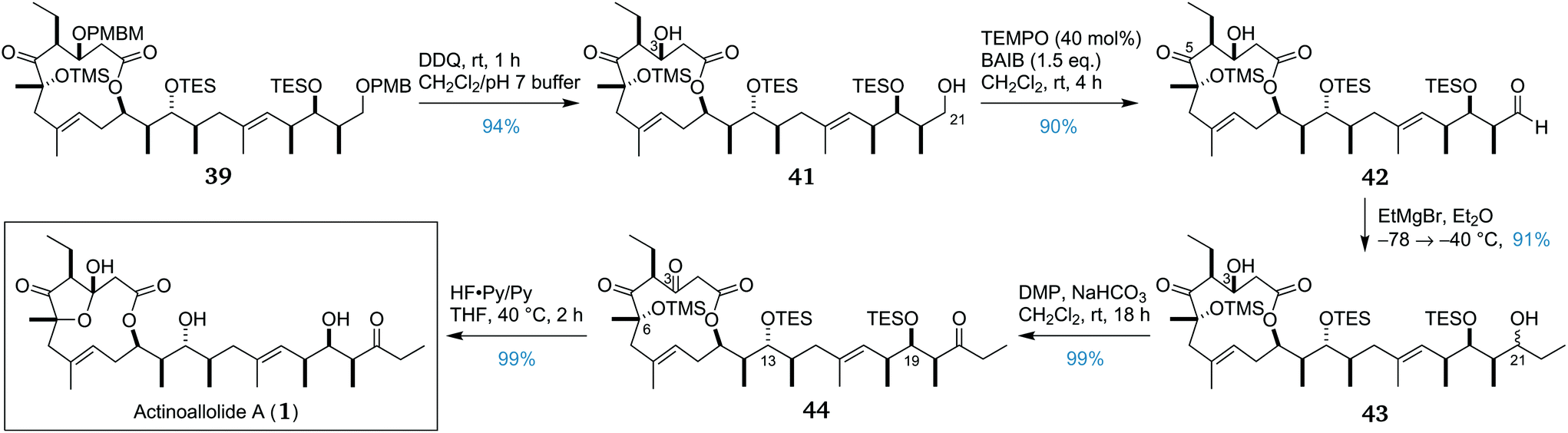 | ||
| Scheme 6 Endgame and completion of the total synthesis of actinoallolide A (1). | ||
Having completed the full carbon backbone of the actinoallolides, our attention now turned to the final two steps of oxidation and global deprotection. Oxidation of both secondary alcohols at C3 and C21 was initially attempted under Swern conditions, however, this was found to be unreliable. Alternatively, the oxidation was performed using the milder Dess–Martin periodinane.55 Double oxidation under these conditions was found to be clean and reliable, providing triketone 44 in 99% yield.
The only obstacle now remaining was the final deprotection. The conditions should ensure complete cleavage of the C6 TMS ether and the C13 and C19 TES ethers, while also forming the transannular hemiacetal. The conditions were also required to be sufficiently mild to avoid any possible deleterious side reactions. Preliminary experience with the lability of the hemiacetal motif led to the hypothesis that actinoallolides C–E may be artefacts of the elaborate chromatographic isolation process. Consequently, we were particularly cautious to avoid elimination across the C3–C4 bond and transesterification of the macrolactone in 44. Initial attempts using TBAF were unsuccessful, leading either to incomplete deprotection or degradation. Pleasingly, acidic fluorous deprotection conditions proved successful, with treatment of 44 with a 1:3 mixture of HF·py/py with warming to 40 °C effecting quantitative conversion to (+)-actinoallolide A (1). The resulting synthetic actinoallolide A possessed comparable specific rotation and identical NMR spectra to those of the natural product (see the ESI†).
The total synthesis of (+)-actinoallolide A (1) was thus achieved in 20 linear steps from (R)-Roche ester (14) with an overall yield of 8%, representing an average yield of 88% per step. Importantly, this first total synthesis gave an orthogonal validation of the full 3D structure and absolute configuration of actinoallolide A and will enable further biological investigations of this highly potent family of natural products.
Conversion of actinoallolide A to actinoallolides B–E
We next sought to access the other four congeners, actinoallolides B–E (2–5), by replication of the conversion protocols in the isolation paper (Scheme 7).18 Actinoallolide A (1) underwent a 1,3-syn hydroxyl-directed reduction using triethyl–borane followed by sodium borohydride to afford actinoallolide B (2) in 93% yield and as a single diastereomer. Subsequent treatment of actinoallolide B (2) with trifluoroacetic acid caused dehydration of the cyclic hemiacetal to provide actinoallolide D (4) in 99% yield.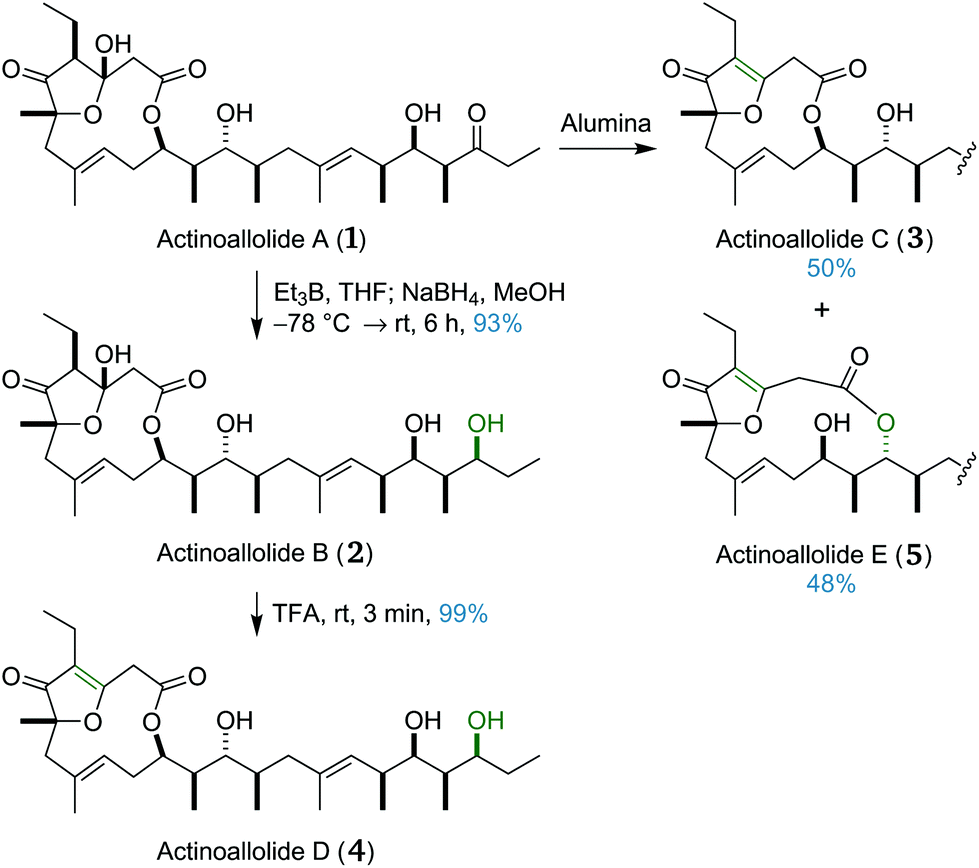 | ||
| Scheme 7 Conversion of actinoallolide A (1) to actinoallolides B–E (2–5). | ||
During the initial purification of actinoallolide A using silica chromatography, some minor by-product formation was observed. Using alumina instead, attempted purification of actinoallolide A led to conversion to a 1:1 mixture of actinoallolide C (3) and actinoallolide E (5). This unexpected result indicated the instability of the cyclic hemiacetal moiety to basic conditions, perhaps explaining why global deprotection was unsuccessful under basic fluorous conditions. This spontaneous conversion of actinoallolide A (1) to actinoallolides C (3) and E (5) on alumina also provides evidence that these compounds are isolation artefacts rather than genuine natural products. This serendipitous result completed our synthesis of all five members of the actinoallolides.
Based on the assumption that actinoallolides C and E are isolation artefacts, it is likely that actinoallolide D is also an artefact, arising from dehydration of actinoallolide B. This would leave actinoallolides A and B as the only true natural products, differing only in the oxidation level at C21. In examining the proposed biosynthesis of actinoallolide A,20 the authors had identified an inactive ketoreductase domain (KR1) in the polyketide synthase responsible for the biosynthesis of actinoallolide A. They commented that “although KR1 has catalytic amino acids and the NADPH binding motif, it seems to be inactive, as predicted from the structure of actinoallolide A that has a ketone at C21”. If, rather than being inactive, the KR1 domain were instead partially active, this identified polyketide synthase would be able to generate both actinoallolides A and B. When the KR1 domain performs the C21 reduction, actinoallolide B, containing the C21 alcohol, is produced, and when transfer to module 2 occurs before the reduction can take place, the product is instead actinoallolide A, with the unreduced C21 ketone. On inspection of the amino acid sequence of the KR1 domain, it possesses both sets of characteristic amino acids which determine whether reduction affords the L- or D-configuration, potentially explaining its partial inactivity.
Synthesis of an actinoallolide A photoaffinity probe analogue
Having achieved a practical, convergent and high-yielding total synthesis of the actinoallolides, we moved onto the design and synthesis of a photoaffinity probe analogue (Scheme 8) to identify their biological target. In order to minimally perturb the structure of the native natural product, the photoaffinity probe 6 was chosen to extend from the C23 terminus.56 The photoreactive moiety is a diazirine, decomposing to a highly-reactive carbene upon irradiation at 365 nm, facilitating cross-linking to the bound target protein. The functional handle is a terminal alkyne, allowing the facile conjugation of any desired reporter tag using a copper-catalysed azide–alkyne cycloaddition.25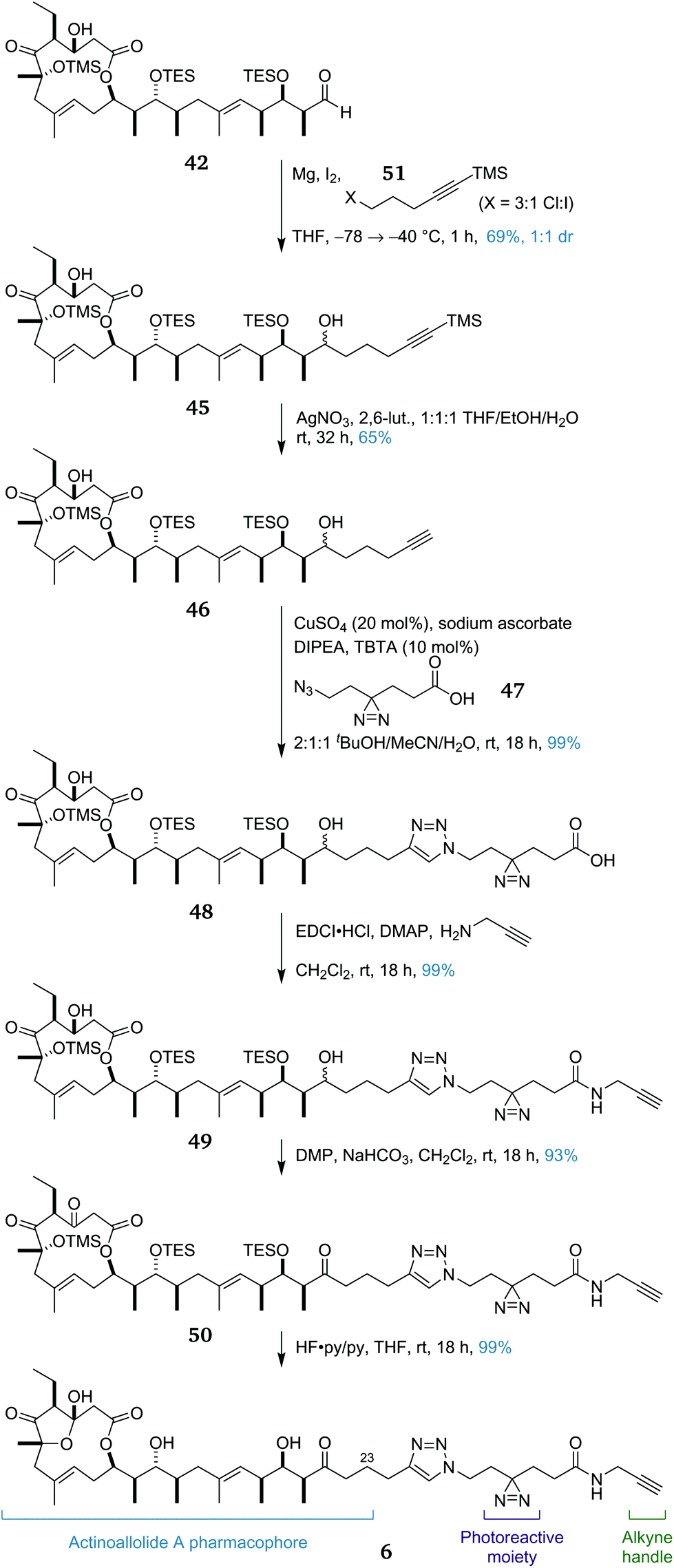 | ||
| Scheme 8 Diversion of advanced intermediate 42 towards the synthesis of photoaffinity probe analogue 6 of actinoallolide A. | ||
The synthesis of probe 6 began by intercepting the advanced intermediate 42. In the total synthesis, addition of an ethyl group was required to complete the actinoallolide carbon skeleton. Instead, the addition of the Grignard reagent derived from halide 45 afforded diol 46 in 69% yield as an inconsequential 1:1 mixture of diastereomers. Chemoselective removal of the alkynyl TMS group was achieved by treatment with AgNO3 and gave 47 in 65% yield. Next, a copper-catalysed azide–alkyne cycloaddition between alkyne 47 and linker fragment 48 (prepared from 1,4-cylohexanedione in eight steps)57 proceeded to afford triazole 49 (99%).
The EDCI/DMAP-mediated amidation of 49 with propargylamine proceeded smoothly, affording amide 50 in 99% yield. This amide then underwent a double DMP-mediated oxidation to afford triketone 51 (93%). Finally, global deprotection under acidic fluorous conditions proceeded with an excellent yield of 99%, cleaving all three silyl ethers and inducing formation of the transannular five-membered hemiacetal to form photoaffinity probe 6. To date, this route has provided 26 mg of 6, sufficient material for the required photoaffinity experiments. In preliminary studies, the biological activities of synthetic actinoallolide A (1) and photoaffinity probe 6 have been confirmed and photoaffinity studies are currently ongoing.
Conclusions
This account details the first total synthesis of the actinoallolides, a family of polyketide natural products prized for their promising anti-trypanosomal properties.58 This feat has been accomplished in a scalable, high-yielding and stereoselective fashion, giving actinoallolide A (1) in 20 linear steps from (R)-Roche ester (14) with an overall yield of 8%. This was achieved in a highly convergent manner, utilising two fragments 8 and 37 of similar complexity, which were obtained in 13 and 10 steps respectively. The stubbornness of C3 manipulation to standard ‘PMB transposition’ conditions was frustrating and necessitated modifications to the original strategy. Fortunately, the judicious choice of alternative protecting groups allowed us to overcome this obstacle.Fragment union was accomplished by esterification followed by a highly challenging ring-closing metathesis, gratifyingly forming the macrocyclic trisubstituted alkene as a single geometrical isomer. This “do-or-die” transformation required significant optimisation and is the most complex example of its kind. A high-yielding endgame finally afforded actinoallolide A. Actinoallolide A was then converted to actinoallolides B–E, completing the total synthesis of the entire natural product family. The alumina-mediated conversion of actinoallolide A to actinoallolides C and E provides evidence that actinoallolides C–E are isolation artefacts, with actinoallolides A and B as the only true natural products. On review of the biosynthetic data for the actinoallolides, a partially active ketoreductase domain in the actinoallolide A polyketide synthase has been postulated to be responsible for generating these two congeneric natural products.
With the secondary aim of probing how actinoallolide A exerts its highly specific biological effects, this route was diverted to give 6 as a designed photoaffinity probe. This was achieved in six steps from advanced intermediate 42. Ongoing work is focused on using 6 in various assays with the aim of identifying the biological target of actinoallolide A. Identification of this target and knowledge of the mechanism of action may allow the generation of simplified, more synthetically tractable or more potent actinoallolide analogues in the ongoing effort to develop new, effective medicinal agents to treat neglected tropical diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Herchel Smith Fund (studentship to M. J. A.), Dr Rachel J. Porter and Dr Simon Williams for assistance, and Thomas Hayhow (AstraZeneca) for his interest. We are grateful to Professor Terry Smith and Dr Gordon Florence at the University of St Andrews for performing the preliminary bioactivity assays. We also acknowledge the National Mass Spectrometry Facility at Swansea University.References
- C. Mathers, G. Stevens, W. R. Mahanani, J. Ho, D. M. Fat and D. Hogan, Global Health Estimates 2015: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2015, World health organization technical report, 2016.
- P. J. Hotez and A. Kamath, PLoS Neglected Trop. Dis., 2009, 3, e412 CrossRef.
- P. Büscher, G. Cecchi, V. Jamonneau and G. Priotto, Lancet, 2017, 390, 2397–2409 CrossRef.
- R. Brun, J. Blum, F. Chappuis and C. Burri, Lancet, 2010, 375, 148–159 CrossRef.
- Trypanosomes and Trypanosomiasis, ed. S. Magez and M. Radwanska, Springer-Verlag, Wien, 2014 Search PubMed.
- J. Bermudez, C. Davies, A. Simonazzi, J. P. Real and S. Palma, Acta Trop., 2016, 156, 1–16 CrossRef CAS.
- J. A. Pérez-Molina and I. Molina, Lancet, 2018, 391, 82–94 CrossRef.
- C. Bern, N. Engl. J. Med., 2015, 373, 456–466 CrossRef CAS.
- J. D. Stanaway and G. Roth, Global Heart, 2015, 10, 139–144 CrossRef.
- P. J. Hotez, E. Dumonteil, L. Woc-Colburn, J. A. Serpa, S. Bezek, M. S. Edwards, C. J. Hallmark, L. W. Musselwhite, B. J. Flink and M. E. Bottazzi, PLoS Neglected Trop. Dis., 2012, 6, e1498 CrossRef.
- WHO Disease burden and mortality estimates, http://www.who.int/healthinfo/global_burden_disease/estimates/en/, (accessed September 2020).
- D. J. Barry and R. McCulloch, Adv. Parasitol., 2001, 49, 1–70 Search PubMed.
- M. C. Field, D. Horn, A. H. Fairlamb, M. A. J. Ferguson, D. W. Gray, K. D. Read, M. De Rycker, L. S. Torrie, P. G. Wyatt, S. Wyllie and I. H. Gilbert, Nat. Rev. Microbiol., 2017, 15, 217–231 CrossRef CAS.
- P. E. Cockram and T. K. Smith, J. Nat. Prod., 2018, 81, 2138–2154 CrossRef CAS.
- A. Strelkauskas, A. Edwards, B. Fahnert, G. Pryor and J. Strelkauskas, Microbiology: A Clinical Approach, Garland Science, 2015 Search PubMed.
- A. R. Renslo and J. H. McKerrow, Nat. Chem. Biol., 2006, 2, 701–710 CrossRef CAS.
- L. Paloque, A. Triastuti, G. Bourdy and M. Haddad, Natural Antimicrobial Agents, Springer International Publishing, Cham, 2018, pp. 215–245 Search PubMed.
- Y. Inahashi, M. Iwatsuki, A. Ishiyama, A. Matsumoto, T. Hirose, J. Oshita, T. Sunazuka, W. Panbangred, Y. Takahashi, M. Kaiser, K. Otoguro and S. Omura, Org. Lett., 2015, 17, 864–867 CrossRef CAS.
- B. L. Roth, The Serotonin Receptors: From Molecular Pharmacology to Human Therapeutics, Springer Science and Business Media, 2008 Search PubMed.
- Y. Inahashi, T. Shiraishi, A. Také, A. Matsumoto, Y. Takahashi, S. Omura, T. Kuzuyama and T. Nakashima, J. Antibiot., 2018, 71, 749–752 CrossRef CAS.
- J. Oshita, Y. Noguchi, A. Watanabe, G. Sennari, S. Sato, T. Hirose, D. Oikawa, Y. Inahashi, M. Iwatsuki, A. Ishiyama, S. Omura and T. Sunazuka, Tetrahedron Lett., 2016, 57, 357–360 CrossRef CAS.
- For a preliminary communication, see: M. J. Anketell, T. M. Sharrock and I. Paterson, Angew. Chem., 2020, 59, 1572–1576 CrossRef CAS.
- C. Lecourt, S. Dhambri, L. Allievi, Y. Sanogo, N. Zeghbib, R. B. Othman, M.-I. Lannou, G. Sorin and J. Ardisson, Nat. Prod. Rep., 2018, 35, 105–124 RSC.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS.
- L. B. Tulloch, S. K. Menzies, A. L. Fraser, E. R. Gould, E. F. King, M. K. Zacharova, G. J. Florence and T. K. Smith, PLoS Neglected Trop. Dis., 2017, 11, e0005886 CrossRef.
- I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott and N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535–9544 CrossRef CAS.
- I. Paterson, S. J. Fink, L. Y. W. Lee, S. J. Atkinson and S. B. Blakey, Org. Lett., 2013, 15, 3118–3121 CrossRef CAS.
- I. Paterson and R. D. Tillyer, Tetrahedron Lett., 1992, 33, 4233–4236 CrossRef CAS.
- J. G. Solsona, J. Nebot, P. Romea and F. Urpí, J. Org. Chem., 2005, 70, 6533–6536 CrossRef CAS.
- J. Esteve, S. Matas, M. Pellicena, J. Velasco, P. Romea, F. Urpí and M. Font-Bardia, Eur. J. Org. Chem., 2010, 3146–3151 CrossRef CAS.
- R. E. Ireland and R. H. Mueller, J. Am. Chem. Soc., 1972, 94, 5897–5898 CrossRef CAS.
- I. Paterson and A. N. Hulme, J. Org. Chem., 1995, 60, 3288–3300 CrossRef CAS.
- I. Paterson and D. J. Wallace, Tetrahedron Lett., 1994, 35, 9087–9090 CrossRef CAS.
- I. Paterson, D. J. Wallace and C. J. Cowden, Synthesis, 1998, 639–652 CrossRef CAS.
- J. H. Lee, Tetrahedron, 2020, 76, 131351 CrossRef CAS.
- A. B. Smith, T. J. Beauchamp, M. J. LaMarche, M. D. Kaufman, Y. Qiu, H. Arimoto, D. R. Jones and K. Kobayashi, J. Am. Chem. Soc., 2000, 122, 8654–8664 CrossRef CAS.
- R. Nagase, Y. Oguni, T. Misaki and Y. Tanabe, Synthesis, 2006, 3915–3917 CAS.
- D. Seebach, R. Naef and G. Calderari, Tetrahedron, 1984, 40, 1313–1324 CrossRef.
- J. S. Kingsbury and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 13813–13815 CrossRef CAS.
- G. Ehrlich, J. Hassfeld, U. Eggert and M. Kalesse, J. Am. Chem. Soc., 2006, 128, 14038–14039 CrossRef CAS.
- R. Johansson and B. Samuelsson, J. Chem. Soc., Perkin Trans. 1, 1984, 2371–2374 RSC.
- D. A. Evans and A. H. Hoveyda, J. Am. Chem. Soc., 1990, 112, 6447–6449 CrossRef CAS.
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS.
- A. P. Kozikowski and J.-P. Wu, Tetrahedron Lett., 1987, 28, 5125–5128 CrossRef CAS.
- Y. Oikawa, T. Tanaka, K. Horita, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1984, 25, 5393–5396 CrossRef CAS.
- D. A. Evans, P. H. Carter, E. M. Carreira, A. B. Charette, J. A. Prunet and M. Lautens, J. Am. Chem. Soc., 1999, 121, 7540–7552 CrossRef CAS.
- O. P. Anderson, A. G. Barrett, J. J. Edmunds, S.-I. Hachiya, J. A. Hendrix, K. Horita, J. W. Malecha, C. J. Parkinson and A. VanSickle, Can. J. Chem., 2001, 79, 1562–1592 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- A. K. Chatterjee and R. H. Grubbs, Org. Lett., 1999, 1, 1751–1753 CrossRef CAS.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- S. Gessler, S. Randl and S. Blechert, Tetrahedron Lett., 2000, 41, 9973–9976 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- A. E. J. d. Nooy, A. C. Besemer and H. v. Bekkum, Synthesis, 1996, 1153–1176 CrossRef.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- S. Ho, D. L. Sackett and J. L. Leighton, J. Am. Chem. Soc., 2015, 137, 14047–14050 CrossRef CAS.
- J. D. Mortison, M. Schenone, J. A. Myers, Z. Zhang, L. Chen, C. Ciarlo, E. Comer, S. K. Natchiar, S. A. Carr, B. P. Klaholz and A. G. Myers, Cell Chem. Biol., 2018, 25, 1506–1518 CrossRef CAS.
- For recent work by our group on other complex bioactive macrolides, see: (a) A. W. Phillips, M. J. Anketell, T. Balan, N. Y. S. Lam, S. Williams and I. Paterson, Org. Biomol. Chem., 2018, 16, 8286–8291 RSC; (b) S. Williams, J. Jin, S. B. J. Kan, M. Li, L. J. Gibson and I. Paterson, Angew. Chem., Int. Ed., 2017, 56, 645–649 CrossRef CAS; (c) N. Anžiček, S. Williams, M. P. Housden and I. Paterson, Org. Biomol. Chem., 2018, 16, 1343–1350 RSC; (d) N. Y. S. Lam, G. Muir, V. R. Challa, R. Britton and I. Paterson, Chem. Commun., 2019, 55, 9717–9720 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation of new compounds, together with NMR comparisons of the synthetic and natural actinoallolides. See DOI: 10.1039/D0OB01831G |
| This journal is © The Royal Society of Chemistry 2020 |