Synthesis and properties of chiral fluorescent helicene-BODIPY conjugates†‡
Received
1st September 2020
, Accepted 14th September 2020
First published on 24th September 2020
Abstract
A series of chiral fluorescent helicene-BODIPY conjugates was prepared by the regioselective formylation of aza[4]helicene precursors and then an efficient one-pot two-step BODIPY synthesis (13 examples, 28–82%). Fused conjugates exhibit absorption and fluorescence properties (ΦF 30–45%) in the red visible domain, and a CPL signature could be measured at 605 nm (glum ±5 × 10−4). Photophysical and electronic properties were investigated and rationalized through first principles.
Introduction
Azahelicenes are ortho-fused polyaromatics that contain at least one sp2 hybridized nitrogen atom embedded into the helical core. The presence of the heteroatom bestows these derivatives with added chemical reactivity and also electronic and (chir)optical properties that have been widely employed in asymmetric catalysis,1–3 biochemistry,4–7 solid-state self-assembly,8–13 material sciences,14–16 and chiral luminescence15,17–26 Azahelicenes usually absorb in the UV spectral domain with low fluorescence efficiencies which are attributed to an intersystem crossing from singlet to triplet excited states (leading to deexcitation by phosphorescence or by non-radiative processes)27 or to symmetry-forbidden S1 → S0 transitions.28 For the design of more effective azahelicene fluorophores, different synthetic strategies were considered,29–31 including their fusion with boron-dipyrromethene dyes (BODIPYs).31
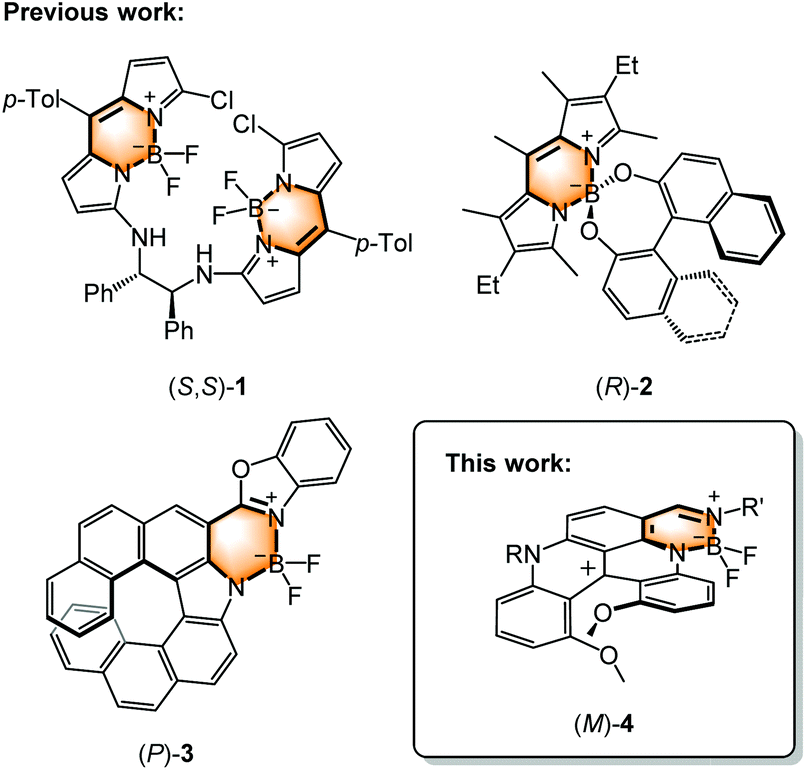
BODIPYs are generally characterized by their narrow absorption and emission bandwidths, covering the entire visible spectral range of light with high peak intensities, small Stokes shifts and high fluorescence quantum yields.32–34 These properties arise from the tetracoordinated B(III) atom that conformationally locks the two nitrogen ligands. The induced stiffness and planarity of the whole dipyrromethene core allows a maximal extension of the conjugation.35 Owing to their luminescence properties, BODIPYs have been actively explored over the last decades.32,33,35 In the context of this study, asymmetric versions of such fluorophores have been reported in the literature,29–31,36–43e.g.1–3 (Fig. 1), in which the chirality is brought by the presence of adjacent stereocenters, by the axial chirality of an allied BINOL ligand or even by the helical chirality of a fused helicene backbone. Herein, we report the synthesis and (chir)optical properties of a new family of chiral BODIPY derivatives 4 obtained by fusion with a cationic diaza [4]helicene scaffold; electronic, absorption and luminescence properties being rationalized using first principles.
 |
| | Fig. 1 Selected chiral BODIPYs. Configurations are shown arbitrarily. | |
Results and discussion
Late-stage functionalization of quinacridinium salts
Previously, our group developed the two-step synthesis of NH-quinacridiniums of type 5 (Scheme 1).44 These dyes are configurationally stable (ΔG‡ = 30.7 ± 4.0 kcal mol−1 at 140 °C) and their enantiomers can be separated via chiral stationary phase (CSP) HPLC. Protonated and neutral conjugates display a perfectly reversible acid–base equilibrium (pKa ∼ 9.0) and their (chir)optical properties are modulated by the pH. For the quinacridinium salts, absorbance at low energy around 625 nm and emission around 690 nm with a fluorescence quantum yield of 0.6% and a lifetime of 0.45 ns in water were noted. As it was deemed important to improve the emission properties, fusion with a BODIPY skeleton was considered using a derivatization strategy and a late-stage functionalization approach. In fact, on chemically-related cationic helical DMQA+ salts,45,46 it was shown that these compounds are surprisingly electron-rich and behave as effective nucleophiles in SEAr reactions. Consequently, NO2 and CHO groups can be introduced regioselectively at the periphery of the helical scaffold.23 With these results in mind, compounds 5 were subjected to Vilsmeier–Haack conditions and novel formylated derivatives 6 were obtained as single regioisomers (CHO introduced at position 6, Scheme 1). Alkyl-substituted 5a (R = Pr) reacts more readily than 5b (R = Ph); the formylation requiring only room temperature with 5a. This is favorable to avoid bis- and tris-CHO functionalization and it simplifies purification. In terms of optical properties, both compounds 6a and 6b exhibit a hypsochromic shift compared to their precursors with absorption and emission maxima still in the orange-red domain (e.g. λabs 563 nm, λem 613 nm for 6b, Fig. S1‡), as well as a notable increase in fluorescence quantum yields (ΦF 39% for 6b).
 |
| | Scheme 1 Synthesis of fused helicene-BODIPYs 4 – Initial results. Reagents and conditions: (a) DMF (12 equiv.), POCl3 (24 equiv.) and then H2O 0–25 °C. (b) Ar–NH2 (1.1 equiv.), CH3CN, 60 °C, 1 h, MS (4 Å). (c) i-Pr2NEt (5.5 equiv.), BF3·OEt2 (7.0 equiv.), CH2Cl2, 25 °C, 8 h. (d) NaBF4 0.2 M. | |
Initial results
With compound 6a in hand, the formation of BODIPY analogues was started. A two-step one-pot protocol was considered via (i) a direct imine formation and then (ii) subsequent chelation with boron trifluoride. In a first set of experiments, six anilines were selected with various para-electron-donating and electron-withdrawing groups, EDGs and EWGs respectively. Standard conditions were selected involving a treatment of 6a with 1.1 equivalents of ArNH2 reagent in the presence of molecular sieves (4 Å). After 1 hour at 60 °C, the solvent was changed from acetonitrile to dichloromethane and the crude mixtures were immediately subjected to 5.5 equivalents of Hünig's base and 7.0 equivalents of BF3·OEt2. Mixed results were obtained as reactions with electron-rich anilines yielded the corresponding BODIPY derivatives (4aA 70%, 4aB 54%) while low yield or no reactivity was obtained with aniline itself and derivatives substituted with EWGs (Scheme 1). For the latter problematic reactions, we could readily demonstrate that the first step of the protocol was the issue as only partial formation of the necessary imine intermediates 7 was observed in NMR spectroscopic analyses of crude reaction mixtures (data not shown).
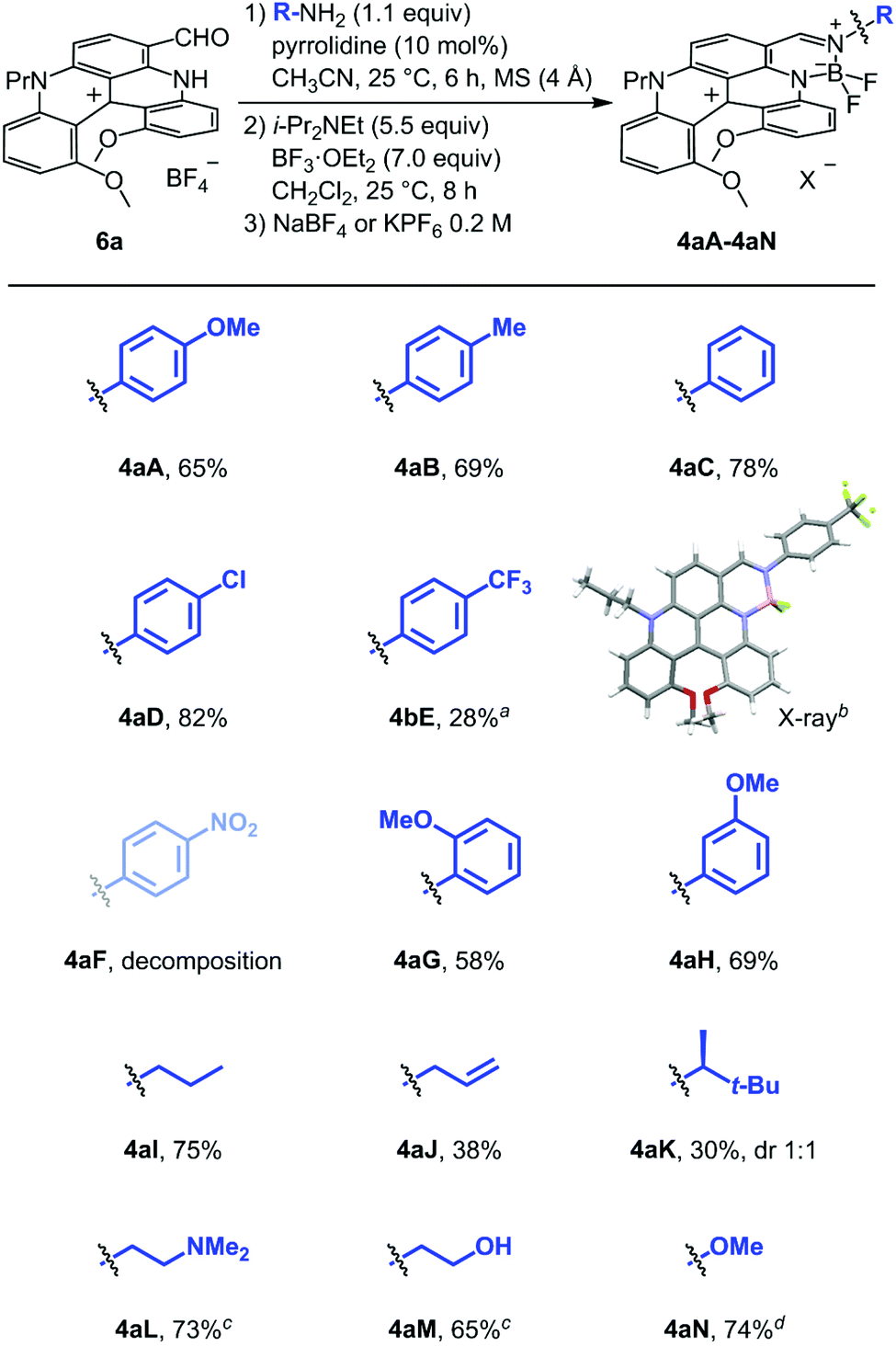
Optimized two-step one-pot procedure
Modified conditions were thus looked to promote the imine formation with EWG-anilines. p-Nitroaniline F was selected for the screening study and the results are detailed in the ESI (Scheme S1‡). An optimal procedure was identified using the protocol reported by García Ruano and Cid with pyrrolidine as catalyst (10 mol%);47 instantaneous formation of intermediate 7F occurring now at room temperature. With these conditions in hand for the first step, the scope of the one-pot procedure could be extended and a series of fused helicene-BODIPYs was synthesized (Scheme 2). To a single exception, the two-step procedure is general and applicable for all types of amines, whether aryl, alkyl, or heterosubstituted. Compounds made from para-anilines were isolated in good to modest yields (4aA to 4aE, 82–28%); the lowest being still obtained with EWGs due to the sensitivity of the products to chromatographic conditions which precluded the isolation of 4aF specifically. Monocrystals of 4aE were obtained and the crystallographic structure was determined by X-ray diffraction analysis (Fig. S9‡). ortho- and meta-OMe substituents did not lead to any reactivity difference and compounds 4aG and 4aH were isolated in good yields, 58% and 69% respectively. The protocol was then extended to BODIPY derivatives derived from alkylamines, compounds 4aI to 4aM. Their synthesis was achieved in good to modest yields (30–75%). In the case of 4aK made from (S)-3,3-dimethylbutyl amine (ee 99.7%), 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers was obtained which could not be separated by crystallization or chromatography. With aminoalkyl L and hydroxyalkyl M side chains, the corresponding products 4aL and 4aM displayed a strong increase in polarity and a counter-ion exchange from BF4 to PF6 anion was then necessary to improve their elution on silica gel. The ion metathesis was readily achieved after a wash with KPF6 (0.2 M aqueous solution). Finally, synthesis of 4aN made from NH2OMe could also be achieved and the product was isolated in 74% yield.
:1 mixture of diastereoisomers was obtained which could not be separated by crystallization or chromatography. With aminoalkyl L and hydroxyalkyl M side chains, the corresponding products 4aL and 4aM displayed a strong increase in polarity and a counter-ion exchange from BF4 to PF6 anion was then necessary to improve their elution on silica gel. The ion metathesis was readily achieved after a wash with KPF6 (0.2 M aqueous solution). Finally, synthesis of 4aN made from NH2OMe could also be achieved and the product was isolated in 74% yield.
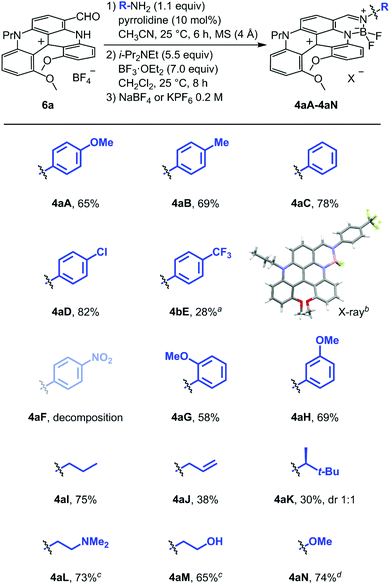 |
| | Scheme 2 Synthesis of 4aA to 4aN salts – optimized conditions. Unless otherwise noted, compounds 4 are isolated as tetrafluoroborate salts. a2.2 equiv. of p-(trifluoromethyl)aniline were used. bStick view of the crystal structure of 4aE (BF4− omitted for clarity). cPF6 salt. d1.1 equivalents of MeONH2·HCl + 1.1 equivalents of Et3N. | |
(Chir)optical properties
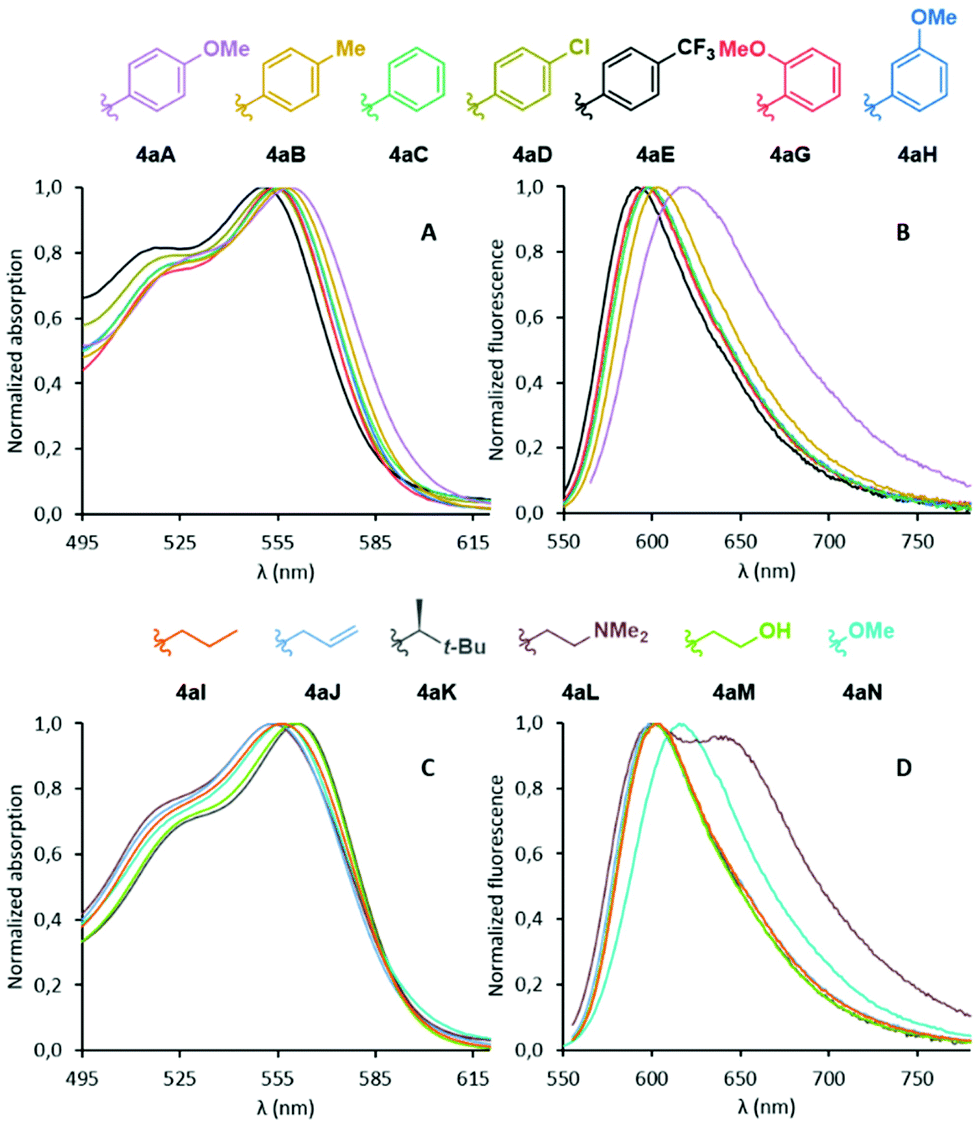
With compounds 4aA to 4aN in hand, we turned our attention to their properties. Absorption and emission spectra were measured and the corresponding data is compiled in Fig. 2 and Table 1. In short, the optical properties are quite uniformed across the various derivatives. Differences are observed but the amplitude is modest (Δλabs 11 nm and Δλem 29 nm). In the case of aryl-substituted imines 4aA to 4aH (Fig. 2A and B), both absorption and fluorescence spectra showed a slight bathochromic shift in the presence of EDGs and, to the contrary, a small hypsochromic shift for EWGs. This behavior mimics the outcome of the functionalization of DMQA+ at position 6 with EDGs and EWGs.23 Compounds 4aB to 4aH present relatively strong quantum yields of fluorescence (24–36%, CH3CN, 10−5 M) around λem 591–603 nm; only derivative 4aA displaying a slightly red-shifted spectrum (λem 620 nm) and drastically lowered fluorescence efficiency (ΦF 4%). This fluorescence quenching will be rationalized later based on first principles. With N-alkyl derivatives 4aI to 4aN (Fig. 2C and D), almost identical optical properties were also obtained, with slightly higher quantum yields than the aryl series, to the exception of 4aL that displays a second transition at 639 nm in its fluorescence spectrum. This specific behavior is attributed to the formation of aggregates as a lowering of the concentration of 4aL results in the fading of this band (Fig. S3‡). As a whole, absorption and emission values for compounds 4 exhibit a slight bathochromic shift compared to that reported by Maeda and Ema for their azahelicene-fused BODIPY analogues.31
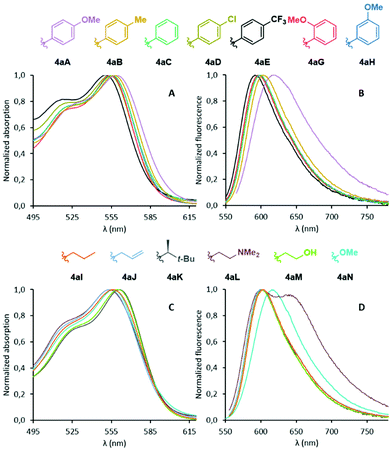 |
| | Fig. 2 Normalized absorption (A) and emission (B) spectra of compounds 4aA to 4aH. Normalized absorption (C) and emission (D) spectra of compounds 4aI to 4aN. Solutions in CH3CN (10−5 M), λexc 560 nm. | |
Table 1 Photophysical properties of BODIPY derivatives 4
| Compound |
λ
abs (nm) |
ε
max (L mol−1 cm−1) |
λ
em (nm) |
Stokes shift (cm−1) |
Φ
Fa (%) |
| Solutions in CH3CN (10−5 M). Relative to cresyl violet (ΦF = 54% in MeOH). Estimated error ±10%. See ref. 35. See Fig. S2.‡ |
|
5
|
625 |
12700 |
690 |
1507 |
0.6 |
|
4aA
|
559 |
12700 |
620 |
1760 |
4 |
|
4aB
|
557 |
16300 |
603 |
1370 |
31 |
|
4aC
|
555 |
10300 |
597 |
1268 |
32 |
|
4bC
|
559 |
5600 |
598 |
1167 |
26 |
|
4aD
|
553 |
11200 |
598 |
1361 |
31 |
|
4aE
|
550 |
10900 |
591 |
1261 |
30 |
|
4aG
|
554 |
10700 |
598 |
1328 |
24 |
|
4aH
|
555 |
9200 |
599 |
1324 |
36 |
|
4aI
|
556 |
13900 |
604 |
1429 |
36 |
|
4aJ
|
554 |
13200 |
601 |
1412 |
29 |
|
4aK
|
561 |
13500 |
601 |
1186 |
41 |
|
4aL
|
553 |
11600 |
602 |
1472 |
44 |
|
4aM
|
560 |
17200 |
602 |
1246 |
46 |
|
4aN
|
556 |
10600 |
616 |
1752 |
40 |
For the study of the chiroptical properties, compound 4bC was selected. Direct CSP-HPLC resolution of this adduct was not easily achievable and, hence, an enantiospecific synthesis starting from the single enantiomers (+)-(P)-6b and (−)-(M)-6b precursors was considered; the resolution of racemic 6b being easier to achieve by CSP-HPLC than that of derivative 6a.44 Treatment of each enantiopure fraction with regular aniline and then BF3·OEt2 afforded (+)-(P)-4bC and (−)-(M)-4bC in 80–88% yields. Electronic circular dichroism (ECD) spectra of the two samples displayed perfect mirror image from each other with three Cotton effects in the UV region (230, 275 and 345 nm) and only one transition in the visible domain (395 nm) (Fig. 3A). Like for DMQA+ and other related [4]helicene derivatives, the S0 → S1 transition centered around 560 nm does not have a strong chiroptical signature in ECD and we were not able to measure a reliable gabs value for this transition.23,48
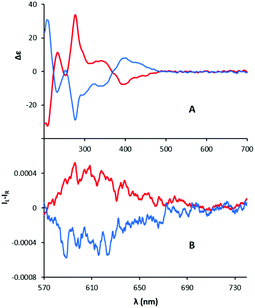 |
| | Fig. 3 ECD (A, ca. 10−5 M) and CPL (B, ca. 6.7 × 10−6 M) spectra of (+)-(P)-4bC (red line) and (−)-(M)-4bC (blue line). Solutions in CH3CN. λexc 517 nm. | |
In circularly polarized luminescence (CPL), (+)-(P)-4bC and (−)-(M)-4bC displayed weak monosignate transitions with the maximum around 605 nm (Fig. 3B). The CPL spectra closely retrace the fluorescence spectrum (Fig. S5‡). The glum factor is about ± 5 × 10−4 (see Fig. S4‡), in agreement with previously reported cationic diaza [4]helicenes.17,23,24 However, these data highlighted a particularly intriguing result. Usually, for a single enantiomer, the sign of the CPL signal is identical to that of the lowest energy Cotton effect observed in ECD.49 In the present case, the first measurable Cotton effect from the red-end of the spectrum falls at 395 nm and it is not associated with the S0 → S1 transition.50 Therefore, it cannot be compared to the S1 → S0 transition observed in CPL.
Electrochemical properties
In addition, cyclic voltammetry (CV) analyses were performed vs. Fc/Fc+ in anhydrous and degassed CH3CN solutions containing [Bu4N+][PF6−] (0.1 M) as supporting electrolyte. Some examples of cyclic voltammograms are presented in Fig. 4 and all redox potential data are summarized in Table 2. At first glance, we can observe that all derivatives of type 4 show analogous CV profiles, with two irreversible one-electron reductions around −1.15 and −1.45 V, and one irreversible one-electron oxidation around 1.25 V. Redox values are similar and little affected by the nature of the N-substituent, contrarily to DMQA+ and its functionalized analogues.23 In the aryl series, small variations are noticed nevertheless. Eox1 values gently increase from 4aA to 4aE along with the electron-withdrawing ability of the substituents. Finally, two compounds behave differently with (i) p-MeO-phenyl-substituted derivative 4aA exhibiting an extra irreversible one-electron oxidation at 1.34 V and (ii) Pr-substituted compound 4aI presenting a different reduction profile (pseudo-reversible first and second negligible waves).
 |
| | Fig. 4 Voltammetric curves of 4aA, 4aC and 4aE (10−3 M) in acetonitrile ([TBA][PF6] 10−1 M) solutions recorded vs. Fc/Fc+ at a Pt working electrode (∅ = 3 mm, ν = 0.1 V s−1). | |
Table 2 Cathodic and anodic potential values of derivatives 4a
| Compound |
Reduction |
Oxidation |
|
E
red1 (V) |
E
red2 (V) |
E
ox1 (V) |
E
ox2 (V) |
| Values measured in CH3CN (10−3 M), recorded experimentally vs. Fc/Fc+ at a Pt working electrode (∅ = 3 mm, ν = 0.1 V s−1) and containing [TBA][PF6] as a supporting electrolyte (10−1 M). Eredn and Eoxn represent the n successive reduction and oxidation processes, respectively. Unless otherwise noted, all processes are irreversible. Pseudo-reversible process. |
|
4aA
|
−1.14 |
−1.48 |
1.19 |
1.34 |
|
4aB
|
−1.14 |
−1.48 |
1.23 |
— |
|
4aC
|
−1.11 |
−1.41 |
1.24 |
— |
|
4aD
|
−1.09 |
−1.46 |
1.26 |
— |
|
4aE
|
−1.06 |
−1.36 |
1.28 |
— |
|
4aG
|
−1.13 |
−1.41 |
1.24 |
— |
|
4aH
|
−1.14 |
−1.49 |
1.24 |
— |
|
4aI
|
−1.14a |
— |
1.20 |
— |
|
4aJ
|
−1.16 |
−1.42 |
1.21 |
— |
|
4aK
|
−1.20 |
−1.48 |
1.19 |
— |
|
4aL
|
−1.20 |
−1.43 |
1.29 |
— |
|
4aM
|
−1.18 |
−1.47 |
1.20 |
— |
|
4aN
|
−1.11 |
−1.40 |
1.20 |
— |
(TD)DFT calculations
To gain a better understanding of the spectroscopic and electrochemical properties of BODIPY derivatives 4, DFT calculations were carried out using the Gaussian 09 software package.51 The structure optimization of all compounds was carried out at the B3LYP52/6-311G(d,p) level of theory in gas phase. The analysis of the computed geometries highlights that all para-substituted aromatic rings in derivatives 4aA to 4aE show a 49° tilt angle with respect to the attached BODIPY framework, in agreement with the crystallographic data of 4aE. ortho- and meta-Substituted aromatic rings in derivatives 4aG and 4aH display a slightly less pronounced inclination (ca. 45°).
To investigate the nature of the excited states involved in the absorption and emission bands, ab initio calculations were carried out (see ESI‡ for additional information). TD-DFT (Time Dependent Density Functional Theory) calculations were performed at the same level of theory and the first ten electronic vertical excitations were computed and compared to the experimental data (Fig. 5 and Table 3). It is known that hybrid functionals tend to provide accurate estimations with error ranges of 0.20–0.25 eV.53 Consequently, B3LYP was chosen as the operating functional for this project. In order to make experimental λabs values concur with their S0 → S1 transitions, a particularly low offset of 0.15 eV was needed. Range-separated hybrid functionals are also known to be particularly effective for charge-transfer electronically excited-states (EESs),53 and CAM-B3LYP was then tested with derivative 4aC as an example. Unfortunately, this functional proved a poorer precision (error = 0.70 eV) and it was hence discarded. Finally, calculations in acetonitrile as an implicit solvent were also carried out for derivatives 4aA to 4aC, but a slightly worse accuracy was obtained (error = 0.2 eV). Vertical excitation analysis indicates that the lowest energy band corresponds to HOMO → LUMO transition for most compounds, with the exception of 4aH and 4aL that present prevalent HOMO−1 → LUMO transition (see Fig. S7 and Tables S1–S17‡ for details).
 |
| | Fig. 5 Examples of computed excited states and experimental absorption spectra of BODIPY derivatives 4a. 0.15 eV offset applied to all λcalc axes. Computed at the B3LYP/6-311G(d,p) level of theory in gas phase. | |
Table 3 Experimental and computed transitions for compounds 4a
| Compound |
Exp. |
Calculateda |
|
λ
abs (eV) |
λ
abs (eV) |
f (cgs) |
HOMO–LUMO gap (eV) |
|
Computed at the B3LYP/6-311G(d,p) level of theory in gas phase.
HOMO−1 → LUMO transition.
|
|
4aA
|
2.218 |
2.199 |
0.0695 |
2.60 |
|
4aB
|
2.226 |
2.364 |
0.0797 |
2.86 |
|
4aC
|
2.234 |
2.388 |
0.0711 |
2.91 |
|
4aD
|
2.242 |
2.381 |
0.0758 |
2.90 |
|
4aE
|
2.254 |
2.402 |
0.0666 |
2.94 |
|
4aG
|
2.238 |
2.344 |
0.0752 |
2.81 |
|
4aH
|
2.234 |
2.323 |
0.0427 |
2.71 |
| 2.433 |
0.0283 |
2.98b |
|
4aI
|
2.230 |
2.408 |
0.0589 |
2.95 |
|
4aJ
|
2.242 |
2.409 |
0.0591 |
2.95 |
|
4aK
|
2.210 |
2.408 |
0.0647 |
2.94 |
|
4aL
|
2.242 |
1.946 |
0.0059 |
2.29 |
| 2.415 |
0.0649 |
2.85b |
|
4aM
|
2.214 |
2.412 |
0.0559 |
2.96 |
|
4aN
|
2.230 |
2.388 |
0.0567 |
2.92 |
Not surprisingly, a discrepancy was observed for compound 4aA, for which S0 → S1 transition is not fitting the experimental UV-Vis spectrum; the corresponding HOMO–LUMO gap being particularly low compared to the rest of the series (2.60 eV vs. 2.81–2.98 eV). As a consequence, frontier molecular orbitals (FMOs) responsible for the transition at the λabs were calculated of each compound of type 4 (Fig. S8‡). Their examination indicates that the nature of the N-substituent has an impact on the HOMO distribution, while the LUMO always remains unchanged. Aromatic-substituted derivatives increase the HOMO delocalization beyond the helical framework, the effect being more pronounced with EDGs than with EWGs (cf.Fig. 6). Also, the partial localization of the HOMO in the p-MeO-phenyl substituent of derivative 4aA suggests a possible photoinduced electron transfer (PET) process from this electron-rich aromatic group to the helical framework after UV light irradiation. This process would account for the observed fluorescence quenching of 4aA. In support of this hypothesis, this effect can be seen, to a lesser extent, with derivative 4aG. Conversely, non-aromatic substituents do not alter this distribution and the HOMO resides mainly in the helicene framework.
 |
| | Fig. 6 Examples of HOMO (red and blue lobes) and LUMO (orange and green lobes) representations of compounds 4a computed at the B3LYP/6-311G(d,p) level of theory in gas phase. Isovalues ±0.02. | |
Rationale of the electronic properties
Computational studies were also performed to provide some insight into the electrochemical properties. As mentioned earlier, most derivatives 4 display two one-electron reductions and one one-electron oxidation processes; only compound 4aA displaying an extra one-electron oxidation (Fig. S6‡ and Table 2). For this reason, with a wish to cover a maximum of redox processes, 4aA was selected for the theoretical study. To locate the single unpaired electron after reduction or after oxidation, a calculation of the doublet spin state of 4aA was thus carried out. The second set of calculations was performed for the doubly reduced and oxidized species respectively, and both singlet and triplet spin state energies were compared (Table 4). These analyses indicate that the first reduction takes place mostly at the central carbon 12 of the helicene (Fig. 7, left, blue dot). Regarding the second one-electron reduction, the energetic comparison within singlet and triplet spin states shows that the latter is slightly more stable (ΔE = 3.9 kcal mol−1) and that a structure with two unpaired electrons is hence favored. In this case, the second unpaired electron is found at carbon 41 and, to a lesser extent, delocalized between carbons 5–11–26 (green dots). In the same manner, the oxidation processes also occur in two different regions, the first unpaired electron being delocalized between carbons 18–24–44 (Fig. 7, right, red dots) and the second one mostly at carbon 15 (orange dot).
 |
| | Fig. 7 Positions subjected to the 1st reduction (blue dot), 2nd reduction (green dots), 1st oxidation (red dots) and 2nd oxidation (orange dot) of BODIPY derivative 4aA. Computed at the B3LYP/6-311G(d,p) level of theory in gas phase. Hydrogen atoms have been omitted for clarity. | |
Table 4 Redox processes: energetic values for compound 4aA
| Process |
Charge |
Multiplicity |
Spin state |
Energy (kcal mol−1) |
| Computed at the B3LYP/6-311G(d,p) level of theory in gas phase. |
| 1st red |
0 |
2 |
Doublet |
— |
| 2nd red |
−1 |
1 |
Singlet |
0.0 |
| −1 |
3 |
Triplet |
−3.9 |
| 1st ox |
+2 |
2 |
Doublet |
— |
| 2nd ox |
+3 |
1 |
Singlet |
0.0 |
| +3 |
3 |
Triplet |
−1.1 |
Conclusions
Thanks to a late-stage Vilsmeier–Haack reaction, the regioselective formylation of diaza [4]helicenes 5 was achieved. Then, a one-pot two-step derivatization of 6 into fused helicene-BODIPY conjugates 4 was rendered possible by the use of pyrrolidine as a catalyst in the first imine formation step. The series of fluorophores were obtained in moderate to good yields (13 examples, 28–82%). These derivatives benefit from a strongly enhanced fluorescence compared to unfunctionalized precursors 5 and they display optical properties in the red visible domain. While an ECD signature could not be obtained at low energy, a CPL signal was nevertheless measured at 605 nm (glum ± 5 × 10−4). The nature of the various substituents introduced on the BODIPY N-atom has, unfortunately, minor effects on their optical and electrochemical properties, as confirmed through first principles analyses.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (SNF 200020-172497 and 200020-184843). We acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. We thank Dr Amalia I. Poblador-Bahamonde, Dr Johann Bosson and Dr Adiran de Aguirre for their advice concerning the computational experiments.
Notes and references
- J. Chen and N. Takenaka, Chem. – Eur. J., 2009, 15, 7268–7276 Search PubMed.
- N. Takenaka, J. Chen, B. Captain, R. S. Sarangthem and A. Chandrakumar, J. Am. Chem. Soc., 2010, 132, 4536–4537 Search PubMed.
- P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263–15278 Search PubMed.
- R. Passeri, G. G. Aloisi, F. Elisei, L. Latterini, T. Caronna, F. Fontana and I. N. Sora, Photochem. Photobiol. Sci., 2009, 8, 1574–1582 Search PubMed.
- O. Kel, A. Fürstenberg, N. Mehanna, C. Nicolas, B. Laleu, M. Hammarson, B. Albinsson, J. Lacour and E. Vauthey, Chem. – Eur. J., 2013, 19, 7173–7180 Search PubMed.
- A. Babič, S. Pascal, R. Duwald, D. Moreau, J. Lacour and E. Allémann, Adv. Funct. Mater., 2017, 27, 1701839 Search PubMed.
- C. Bauer, R. Duwald, G. M. Labrador, S. Pascal, P. Moneva Lorente, J. Bosson, J. Lacour and J.-D. Rochaix, Org. Biomol. Chem., 2018, 16, 919–923 Search PubMed.
- E. Murguly, R. McDonald and N. R. Branda, Org. Lett., 2000, 2, 3169–3172 Search PubMed.
- C. Bazzini, T. Caronna, F. Fontana, P. Macchi, A. Mele, I. Natali Sora, W. Panzeri and A. Sironi, New J. Chem., 2008, 32, 1710–1717 Search PubMed.
- W. Shen, S. Graule, J. Crassous, C. Lescop, H. Gornitzka and R. Réau, Chem. Commun., 2008, 7, 850–852 Search PubMed.
- S. Graule, M. Rudolph, N. Vanthuyne, J. Autschbach, C. Roussel, J. Crassous and R. Réau, J. Am. Chem. Soc., 2009, 131, 3183–3185 Search PubMed.
- I. Alkorta, F. Blanco, J. Elguero and D. Schröder, Tetrahedron: Asymmetry, 2010, 21, 962–968 Search PubMed.
- S. Graule, M. Rudolph, W. Shen, J. A. G. Williams, C. Lescop, J. Autschbach, J. Crassous and R. Réau, Chem. – Eur. J., 2010, 16, 5976–6005 Search PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 Search PubMed.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 Search PubMed.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 Search PubMed.
- J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808–11809 Search PubMed.
- R. Hassey, E. J. Swain, N. I. Hammer, D. Venkataraman and M. D. Barnes, Science, 2006, 314, 1437 Search PubMed.
- C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 Search PubMed.
- N. Saleh, B. Moore II, M. Srebro, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, K. K. Deol, G. Muller, J. Autschbach and J. Crassous, Chem. – Eur. J., 2015, 21, 1673–1681 Search PubMed.
- N. Saleh, M. Srebro, T. Reynaldo, N. Vanthuyne, L. Toupet, V. Y. Chang, G. Muller, J. A. G. Williams, C. Roussel, J. Autschbach and J. Crassous, Chem. Commun., 2015, 51, 3754–3757 Search PubMed.
- J. Bosson, G. M. Labrador, S. Pascal, F.-A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem. – Eur. J., 2016, 22, 18394–18403 Search PubMed.
- I. Hernández Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 Search PubMed.
- S. Pascal, C. Besnard, F. Zinna, L. Di Bari, B. Le Guennic, D. Jacquemin and J. Lacour, Org. Biomol. Chem., 2016, 14, 4590–4594 Search PubMed.
- G. M. Upadhyay, H. R. Talele and A. V. Bedekar, J. Org. Chem., 2016, 81, 7751–7759 Search PubMed.
- J. R. Brandt, L. Pospíšil, L. Bednárová, R. C. da Costa, A. J. P. White, T. Mori, F. Teplý and M. J. Fuchter, Chem. Commun., 2017, 53, 9059–9062 Search PubMed.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. Vander Donckt, Chem. Phys. Lett., 1976, 43, 33–36 Search PubMed.
- H. Kubo, T. Hirose and K. Matsuda, Org. Lett., 2017, 19, 1776–1779 Search PubMed.
- E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, J. Bañuelos, T. Arbeloa, I. López-Arbeloa, M. J. Ortiz and S. d. l. Moya, Chem. Commun., 2013, 49, 11641–11643 Search PubMed.
- E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. d. l. Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 Search PubMed.
- C. Maeda, K. Nagahata, T. Shirakawa and T. Ema, Angew. Chem., 2020, 59, 7813–7817 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 Search PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 Search PubMed.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 Search PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 Search PubMed.
- Y. Wu, S. Wang, Z. Li, Z. Shen and H. Lu, J. Mater. Chem. C, 2016, 4, 4668–4674 Search PubMed.
- H. Lu, J. Mack, T. Nyokong, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2016, 318, 1–15 Search PubMed.
- F. Zinna, T. Bruhn, C. A. Guido, J. Ahrens, M. Bröring, L. Di Bari and G. Pescitelli, Chem. – Eur. J., 2016, 22, 16089–16098 Search PubMed.
- M. Saikawa, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 10727–10730 Search PubMed.
- C. Ray, E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, Í. López-Arbeloa, J. Bañuelos, K. D. Cohovi, J. L. Lunkley, G. Muller and S. de la Moya, Chem. – Eur. J., 2016, 22, 8805–8808 Search PubMed.
- R. B. Alnoman, S. Rihn, D. C. O'Connor, F. A. Black, B. Costello, P. G. Waddell, W. Clegg, R. D. Peacock, W. Herrebout, J. G. Knight and M. J. Hall, Chem. – Eur. J., 2016, 22, 93–96 Search PubMed.
- R. Clarke, K. L. Ho, A. A. Alsimaree, O. J. Woodford, P. G. Waddell, J. Bogaerts, W. Herrebout, J. G. Knight, R. Pal, T. J. Penfold and M. J. Hall, ChemPhotoChem, 2017, 1, 513–517 Search PubMed.
- J. Jiménez, L. Cerdán, F. Moreno, B. L. Maroto, I. García-Moreno, J. L. Lunkley, G. Muller and S. de la Moya, J. Phys. Chem. C, 2017, 121, 5287–5292 Search PubMed.
- A. Wallabregue, P. Sherin, J. Guin, C. Besnard, E. Vauthey and J. Lacour, Eur. J. Org. Chem., 2014, 6431–6438 Search PubMed.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 Search PubMed.
- B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773–1783 Search PubMed.
- S. Morales, F. G. Guijarro, J. L. García Ruano and M. B. Cid, J. Am. Chem. Soc., 2014, 136, 1082–1089 Search PubMed.
- R. Duwald, J. Bosson, S. Pascal, S. Grass, F. Zinna, C. Besnard, L. Di Bari, D. Jacquemin and J. Lacour, Chem. Sci., 2020, 11, 1165–1169 Search PubMed.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 Search PubMed.
- In most instances, the band around 400 nm is associated to the transition from HOMO−1 to LUMO+1.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 Search PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 Search PubMed.
Footnotes |
| † The dataset for this article can be found at the following DOI: 10.26037/yareta:qs3vif5ggbc6jpanuricxbkrbm. It will be preserved for 10 years. |
| ‡ Electronic supplementary information (ESI) available: Experimental conditions, full characterizations, 1H NMR, 13C NMR, 19F NMR and IR spectra of all new compounds; Rf and HRMS. CCDC 2024041. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01809k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Francesco
Zinna
a,
Francesco
Zinna