
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Borylation of conjugated carbonyl compounds with silylborane or bis(pinacolato)diboron catalyzed by Au nanoparticles†
Marios
Kidonakis
*,
Michael
Fragkiadakis
and
Manolis
Stratakis
 *
*
Department of Chemistry, University of Crete, Voutes 71003, Heraklion, Greece. E-mail: stratakis@uoc.gr
First published on 14th October 2020
Abstract
Conjugated aldehydes and ketones undergo reaction with Me2PhSiBpin (pin: pinacolato) catalyzed by Au nanoparticles supported on TiO2 forming exclusively the β-borylation products, via the intermediate formation of the labile silaboration adducts. This chemoselectivity pathway is complementary to the so far known analogous reaction catalyzed by other metals, where β-silylation occurs instead. β-Borylation also occurs with pinBBpin under identical reaction conditions in a variety of conjugated carbonyl compounds, including esters and amides which are unreactive in their attempted Au-catalyzed silaboration.
Introduction
Supported gold nanoparticles (Au NPs) and other nano Au(0) materials have emerged as powerful catalytic systems in a plethora of transformations of organic substrates, beyond aerobic oxidation or transfer hydrogenation.1 Among them, the activation and subsequent addition of interelement σ bond linkages (Si–H, Si–Si, B–B, B–Si) to alkynes, allenes and even strained σ systems represent a practical route for the synthesis of synthetically attractive organosilicon and organoboron compounds.2 Our group was the first to establish the ability of supported Au NPs to smoothly activate the Si–B bond of the fairly stable Me2PhSiBpin (pin: pinacolato) in its reaction with alkynes.3 Since then, a range of applications appeared using this silylborane and Au/TiO2 as the catalyst. Thus, we have additionally shown (Scheme 1) the regioselective silaboration of allenes,4 the regioselective ring opening of epoxides and oxetanes yielding β- or γ-silyloxy boronates respectively,5 the pinacol-type reductive dimerization of aryl substituted carbonyl compounds promoted by the silylborane,6 and more recently, the silaboration of 2-aryl-substituted cyclopropyl aldehydes, in which β-boronate silyl enol ethers are formed via ring-opening rearrangement of an intermediate α-silyloxy cyclopropyl radical.7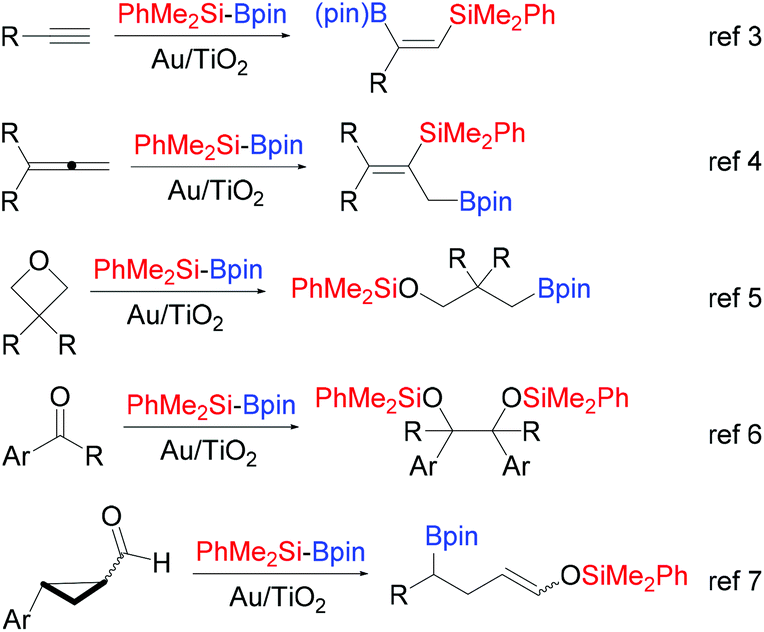 | ||
| Scheme 1 Reactivity motifs of silylborane Me2PhSi-Bpin with various organic compounds catalyzed by Au/TiO2, previously reported by our group. | ||
Results and discussion
Prompted by our intense interest in the Au NP-catalyzed reaction of silylboranes with organic compounds, we examined the reactivity of Me2PhSiBpin with conjugated carbonyl compounds. In our previous study regarding its reaction with simple carbonyl compounds, only aromatic benzaldehydes and acetophenones exhibited reactivity towards a radical-initiated dimerization pathway. With the exception of aryl-substituted cyclopropyl aldehydes which yield rearranged silaboration adducts,7 all simple aliphatic aldehydes or ketones that were examined did not exhibit any kind of reactivity.6 On the other hand, the reaction of conjugated carbonyl compounds with silylboranes in the presence of a proton source is well known8 under various catalytic protocols, and leads to the exclusive conjugated addition of the silyl moiety as a nucleophile, forming β-silyl-substituted carbonyl compounds (Scheme 2). The group of Oestreich was the first to show that a cationic Rh(I) complex catalyzes silyl transfer from Me2PhSiBpin on cyclic conjugated ketones, obtaining additionally excellent enantioselectivity if an appropriate chiral ligand is added.9 This concept was later applied in the enantioselective conjugate silylation of α,β-unsaturated esters.10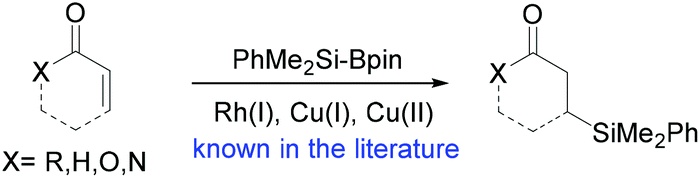 | ||
| Scheme 2 Known chemoselectivity in the metal-catalyzed reaction of Me2PhSi-Bpin with conjugated carbonyl compounds. | ||
Hoveyda's and Procter's groups reported the same highly enantioselective transformation in esters, amides, dienones and dienoates using chiral NHC Cu(I) complexes as catalysts,11 and immediately after a metal-free catalytic version using a chiral N-heterocyclic carbene.12 A few other catalytic protocols of β-silylation of α,β-unsaturated carbonyl compounds are also known, using a Cu(I) complex in the presence of a chiral amine,13 CuSO4 in the presence of a base,14 and more recently the Cu(I)-catalyzed reaction of silylborane with N-(2-iodoaryl)acrylamides forming silane-bearing oxindoles.15 In addition, trapping of the intermediates in the β-silylation of conjugated carbonyl compounds with aldehydes generates useful synthetic motifs.16 A common characteristic of the abovementioned metal-catalyzed procedures is the pre-activation of silylborane upon addition of a base on the B atom, which accelerates transmetallation of the silicon moiety onto the metal center, which finally delivers the silyl part as a nucleophile on the enone, in a 1,4-fashion. In general, in all metal-catalyzed reactions which make use of silylboranes, the silyl moiety behaves as a pseudo-nucleophile.17
We started the exploration of the possible Au NP-catalyzed silaboration using cyclic α,β-unsaturated ketones. To our delight, the treatment of 2-cyclohexen-1-one (1a) with 1.5 equiv. of Me2PhSiBpin (2) in the presence of Au/TiO2 (1 mol% in Au) in anhydrous benzene at 70 °C for 2 h resulted in the complete consumption of the enone and the formation of the β-boronate silyl enol ether 3a as the sole product, in 69% isolated yield (Scheme 3). The treatment of the enone with silylborane in the absence of the catalyst does not yield any products. Surprisingly in chlorinated solvents, such as 1,2-dichloroethane (DCE), the reaction does not occur. Similarly, conjugated cyclic ketones 1b and 1c were also subjected to silaboration forming the corresponding products 3b and 3c in 67% and 64% yields, respectively. These somehow moderate yields are rather associated with the instability of the silaboration adducts under chromatographic conditions. This chemoselectivity motif of silaboration is unknown in the literature, and is complementary to the cases of using Cu or Rh catalysts, for which β-silylation occurs instead.8
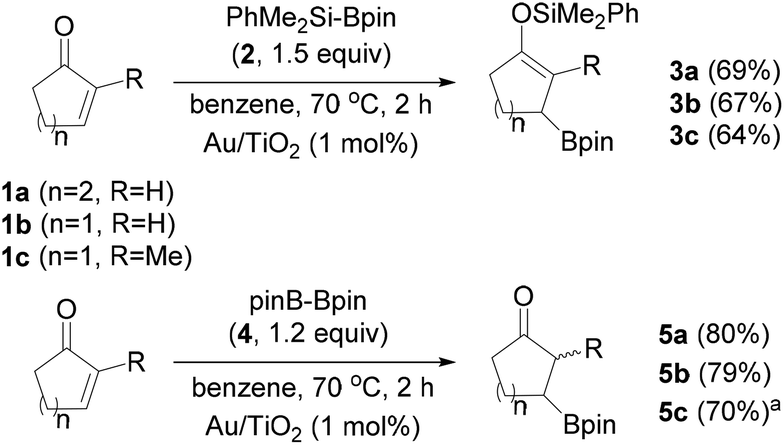 | ||
Scheme 3 Silaboration of cyclic enones 1a–1c with Me2PhSi-Bpin, and their β-borylation with pinB-Bpin catalyzed by Au/TiO2. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A mixture of diastereomers in a relative ratio of 1.1/1. A mixture of diastereomers in a relative ratio of 1.1/1. | ||
Notably, the reaction of cyclic ketones 1a–1c with bis(pinacolato)diboron (pinBBpin, 4, pin: pinacolato) also proceeds smoothly under the same catalytic conditions, and yields the β-boronate substituted ketones 5a–5c in very good yields (Scheme 3). Apparently, analogous to the enol silyl ethers 3a–3c, boron enolates are extremely labile and undergo hydrolysis under the reaction conditions to yield the single-borylation products 5a–5c. In the case of borylation of 1a with pinBBpin, the catalyst was separated after the end of the reaction from the slurry by filtration, and after washing with an organic solvent and drying in an oven it was reused without any loss in its activity. This is a typical characteristic of Au/TiO2 in a series of reactions involving silylboranes or bis(pinacolato)diboron.3–7 The activation of the diboron reagent pinBBpin with nano Au(0) materials has been well established in its reaction with alkynes,18 allenes4 and methylene cyclopropanes.19 The β-borylation of conjugated carbonyl compounds using pinBBpin as the boron source is also known in the literature applying several catalytic protocols.20 These include transition metals such as Rh(I),21 Ni(0),22,23a and Pd(0),23 primarily Cu(I) catalysts,24 Cu(II),25 Cu NPs or Cu(0),26 as well as organocatalysts.27
In Scheme 4, we propose that after the insertion of Me2PhSiBpin on Au NPs (denoted as Aun), primarily at its low coordinated Au atoms (corners and edges)2 to form Me2PhSi–(Aun)–Bpin species, and coordination of the substrate on them, the boron and silicon moieties are selectively delivered on the C and O atoms, respectively, forming the silaboration products. Similar arguments have been invoked in the Au NP-catalyzed silaboration of strained cyclic ethers5 and cyclopropyl aldehydes7 by our group, with the Bpin part of silylborane acting as a pseudo-nucleophile. The driving force of this specific mode of addition is the formation of the thermodynamically stable O–Si and C–B28 bonds, and the fact that boron is more electronegative compared to silicon. Contrary to this mode of addition, in all other metal catalyzed reactions as already mentioned,17 between silylborane and enones, the silicon moiety acts as a pseudo-nucleophile, being bound on the metal after an external basic reagent is bonded by the boron atom of silylborane, breaking therefore the Si–B bond. Analogous is the proposed mechanistic scenario of using pinBBpin (Scheme 4). The O–B bond of the diborylation products is extremely labile and via hydrolysis under the reaction conditions/work up, β-borylated ketones are formed. If in the borylation of 1a one equiv. of D2O is added along with B2pin2, >95% single deuteration is observed on the α-carbon (product 5a-d1), indicative of the reaction between the transient boron enolate and D2O. Note that no D-incorporation was detected when any of the single borylated products 5a–5c was treated with D2O under the same reaction conditions. Unfortunately, any other electrophile added, such as aldehydes or allyl halides, was inefficient in trapping this putative enol intermediate.
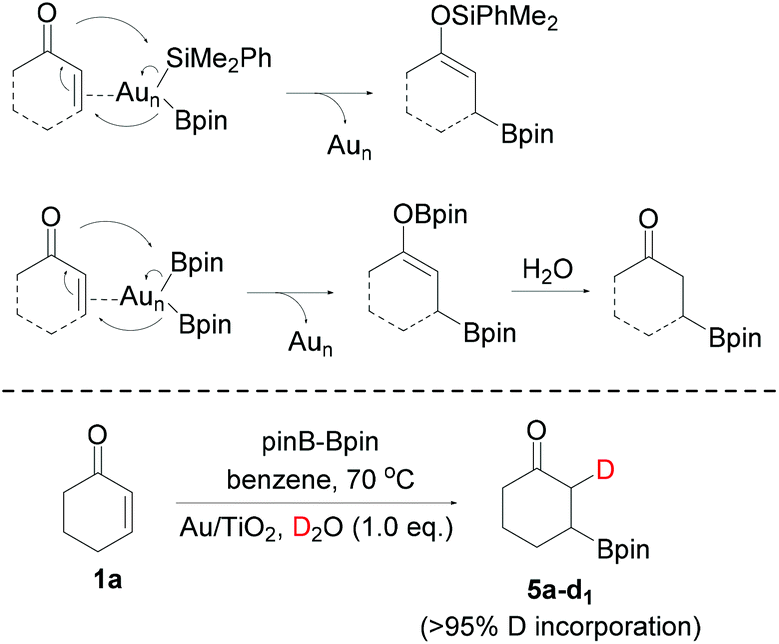 | ||
| Scheme 4 Proposed mechanisms of the Au NP-catalyzed silaboration/borylation. | ||
Following these very interesting results, we extended our silaboration studies with a series of acyclic α,β-unsaturated aldehydes and ketones (Table 1). With this kind of substrate the reaction proceeds smoothly, just as with the cyclic ketones shown in Scheme 3. Yet, no silaboration products could be seen. Instead, the corresponding β-borylated carbonyl compounds were isolated in good yields. The acyclic silaboration adducts can be detected by GC-MS in samples taken directly from the reaction flask. For example in the silaboration of crotonaldehyde (1l) in benzene-d6, the labile silaboration product 3l was detected having a cis-configuration because the 3JHH of the olefinic protons is 6.0 Hz (see page S22 of the ESI†). The cis-silaboration adducts appear to be more prone towards hydrolysis under the reaction conditions/workup more possibly because the Lewis-acidic boron moiety interacts intramolecularly with the silyl enol ether facilitating thus its hydrolysis. As a limitation, the more sterically hindered β,β-disubstituted conjugated aldehydes and ketones are completely unreactive against silylborane, as well as a series of the less electron deficient α,β-unsaturated esters and an amide.
| a Mixture of diastereomers in a relative ratio of 1.1/1. |
|---|

|
Contrary, however, to the unachieved reaction between Me2PhSiBpin and conjugated esters or amides (Table 1), their β-borylation occurs in good to excellent isolated yields using pinBBpin. In addition, all conjugated aldehydes and ketones whose reaction with silylborane was studied, as given in Table 1, afforded the borylated products 5d–5o in slightly higher yields relative to the use of Me2PhSiBpin. The additional results from the borylation of the rest of the conjugated carbonyl compounds are summarized in Table 2. Amide 1u reacts slowly and requires excess borylating reagent (2–3 equiv.) and a prolonged reaction time (overnight).
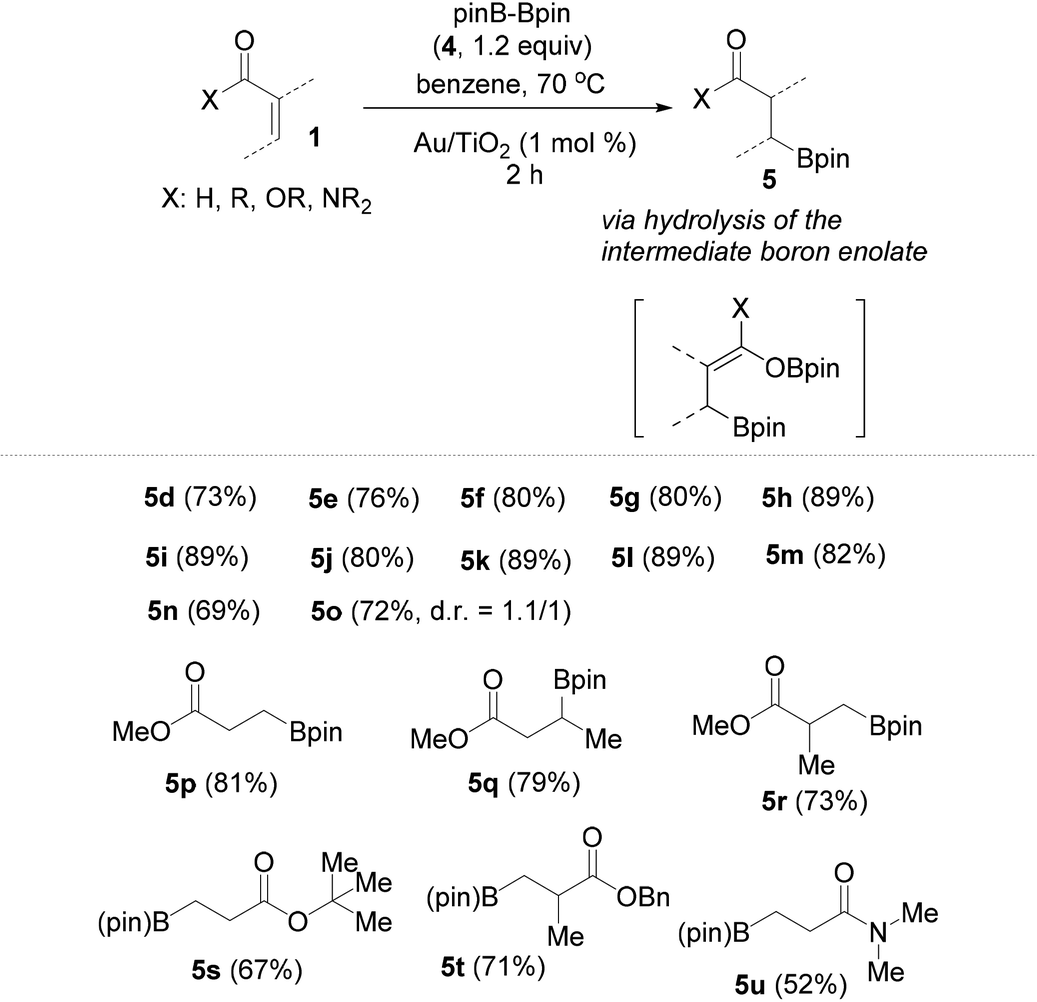
|
Conclusions
In conclusion, we report herein the so far unrecognized ability of supported Au(0) nanoparticles on TiO2 in catalyzing the silaboration of conjugated aldehydes and ketones with Me2PhSiBpin and the quite general β-borylation of α,β-unsaturated carbonyl compounds with pinBBpin. In contrast to all of the known metal-catalyzed analogous reactions with silylborane, in which the silyl moiety acts as a pseudo-nucleophile, under our Au NP-catalyzed conditions, the boryl moiety behaves as a nucleophile, leading to conjugate β-borylation. These results, exemplify once more the unique potency of supported Au nanoparticles in catalyzing organic transformations of high synthetic utility.Experimental section
The reactions were monitored by thin-layer chromatography (TLC) carried out on silica gel plates (60F-254). Benzene was passed though silica gel and kept over 4 Å molecular sieves. NMR spectra were recorded on a Bruker Avance-500 instrument. HRMS spectra were recorded on an Agilent Q-TOF mass spectrometer, G6540B model with the Dual AJS ESI-MS instrument. GC analyses were performed on a Shimadzu GC-17A model equipped with a 60 m HP-5 capillary column. Flash column chromatography for the purification of compounds was carried out on SiO2 (silica gel 60, particle size 0.040–0.063 mm) using mixtures of hexane or petroleum ether/ethyl acetate as the eluent. Au/TiO2 (1 wt% in Au) was provided by Strem Chemicals and has an average gold crystallite size of ∼2–3 nm.Conjugated carbonyl compounds 1a–1c, 1e–1f, 1h, and 1k–1u are commercially available. Enones 1d, 1g, 1i and 1j were synthesized as described in the ESI.†
General procedure of the Au/TiO2-catalyzed β-borylation
To a tube containing a solution of 0.2 mmol of the conjugate carbonyl compound in benzene (0.5 mL) were added 0.30 mmol of Me2PhSiBpin or 0.24 mmol of pinBBpin followed by the addition of 47.3 mg of Au/TiO2 (1.0 mol% in Au) and the mixture was heated to 70 °C for a certain period of time until the reaction was complete (TLC and GC-MS). The slurry was filtered with the aid of dichloromethane under a low pressure through a short pad of silica gel and the filtrate was evaporated. The desired product was isolated after column chromatography, using petroleum ether/ethyl acetate (typically ∼15/1). The borylation was also performed on a larger scale using 96.1 mg (1.0 mmol) of enone 1a, 305 mg of bis(pinacolato)diboron (4, 1.2 mmol), 3 mL of benzene and 236.4 mg of Au/TiO2 (2.4 mg of Au, as the catalyst contains 1 w/w% Au). After 2 h at 70 °C the reaction was complete and following the above workup, compound 5a was isolated in 76% yield (170 mg) after column chromatography, using petroleum ether/ethyl acetate = 15/1 as an eluent.Spectroscopic data of silaboration products
Spectroscopic data of β-borylated products
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under the HFRI PhD fellowship grant (GA No 31449). M. K. ProFI (FORTH, Heraklion, Greece) is acknowledged for obtaining the HRMS spectra of the unknown compounds.Notes and references
- Selected review articles: (a) M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469 CrossRef CAS; (b) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS; (c) T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636 RSC; (d) B. S. Takale, M. Bao and Y. Yamamoto, Org. Biomol. Chem., 2014, 12, 2005 RSC; (e) X. Liu, L. He, Y.-M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793 CrossRef CAS; (f) B. S. Takale, M. Bao, Y. Yamamoto, A. I. Almansour, N. Arumugam and R. S. Kumar, Synlett, 2015, 26, 2355 CrossRef CAS; (g) T. Jin, M. Terada, M. Bao and Y. Yamamoto, ChemSusChem, 2019, 12, 2936 CrossRef CAS.
- M. Stratakis and I. N. Lykakis, Synthesis, 2019, 51, 2435 CrossRef CAS.
- C. Gryparis and M. Stratakis, Org. Lett., 2014, 16, 1430 CrossRef CAS.
- M. Kidonakis and M. Stratakis, ACS Catal., 2018, 8, 1227 CrossRef CAS.
- E. Vasilikogiannaki, A. Louka and M. Stratakis, Organometallics, 2016, 35, 3895 CrossRef CAS.
- M. Kidonakis, A. Mullaj and M. Stratakis, J. Org. Chem., 2018, 83, 15553 CrossRef CAS.
- V. Kotzabasaki, M. Kidonakis, E. Vasilikogiannaki and M. Stratakis, Eur. J. Org. Chem., 2019, 7233 CrossRef CAS.
- (a) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS; (b) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29 CrossRef CAS.
- C. Walter, G. Auer and M. Oestreich, Angew. Chem., Int. Ed., 2006, 45, 5675 CrossRef CAS.
- C. Walter and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 3818 CrossRef CAS.
- (a) K.-S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef CAS; (b) V. Pace, J. P. Rae, H. Y. Harb and D. J. Procter, Chem. Commun., 2013, 49, 5150 RSC; (c) V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2014, 16, 476 CrossRef CAS; (d) K.-S. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823 CrossRef CAS.
- J. M. O′ Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712 CrossRef.
- I. Ibrahem, S. Santoro, F. Himo and A. Cordova, Adv. Synth. Catal., 2011, 353, 245 CrossRef CAS.
- J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 2090 CrossRef CAS.
- R.-X. Liang, R.-Y. Chen, C. Zhong, J.-W. Zhu, Z.-Y. Cao and Y.-X. Jia, Org. Lett., 2020, 22, 3215 CrossRef CAS.
- A. Welle, J. Petrignet, B. Tinant, J. Wouters and O. Riant, Chem. – Eur. J., 2010, 16, 10980 CrossRef CAS.
- Selected examples: (a) W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070 CrossRef CAS; (b) M. Cui and M. Oestreich, Org. Lett., 2020, 22, 3684 CrossRef CAS; (c) T. Seihara, S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Org. Lett., 2019, 21, 2477 CrossRef CAS; (d) Y. Takeda, K. Shibuta, S. Aoki, N. Tohnai and S. Minakata, Chem. Sci., 2019, 10, 8642 RSC; (e) A. Hensel, K. Nagura, L. B. Delvos and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 4964 CrossRef CAS; (f) C. Kleeberg, M. S. Cheung, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 19060 CrossRef CAS.
- (a) Q. Chen, J. Zhao, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2013, 15, 5766 CrossRef CAS; (b) M. Kidonakis and M. Stratakis, Eur. J. Org. Chem., 2017, 4265 CrossRef CAS.
- Q. Chen, X. Zhang, S. Su, Z. Xu, N. Li, Y. Li, H. Zhou, M. Bao, Y. Yamamoto and T. Jin, ACS Catal., 2018, 8, 5901 CrossRef CAS.
- (a) E. Hartmann, D. J. Vyas and M. Oestreich, Chem. Commun., 2011, 47, 7917 RSC; (b) J. A. Schiffner, K. Muther and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 1194 CrossRef CAS.
- T. Shiomi, T. Adachi, K. Toribakate, L. Zhou and H. Nishiyama, Chem. Commun., 2009, 5987 RSC.
- K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 5031 CrossRef CAS.
- (a) V. Lillo, M. J. Geier, S. A. Westcott and E. Fernandez, Org. Biomol. Chem., 2009, 7, 4674 RSC; (b) A. Bonet, H. Gulyas, I. O. Koshevoy, F. Estevan, M. Sanau, M. A. Ubeda and E. Fernandez, Chem. – Eur. J., 2010, 16, 6382 CrossRef CAS.
- Selected examples: (a) S. Mun, J.-E. Lee and J. Yun, Org. Lett., 2006, 8, 4887 CrossRef CAS; (b) X. Feng and J. Yun, Chem. Commun., 2009, 6577 RSC; (c) X. Feng and J. Yun, Chem. – Eur. J., 2010, 16, 13609 CrossRef CAS; (d) Q. Jiang, T. Guo, R. Gao, Q. Wang, J. Lou and Z. Yu, J. Org. Chem., 2018, 83, 7981 CrossRef CAS; (e) J. George, H. Y. Kim and K. Oh, Org. Chem. Front., 2020, 7, 709 RSC.
- A. B. Thorpe, J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 1918 CrossRef.
- (a) X.-F. Zhou, Y.-Y. Sun, Y.-D. Wu, J.-J. Dai, J. Xu, Y. Huang and H.-J. Xu, Tetrahedron, 2016, 72, 5691 CrossRef CAS; (b) T. Kitanosono and S. Kobayashi, Asian J. Org. Chem., 2013, 2, 961 CrossRef CAS.
- (a) K.-S. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253 CrossRef CAS; (b) A. Bonet, H. Gulyas and E. Fernandez, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS; (c) A. Bonet, C. Sole, H. Gulyas and E. Fernandez, Chem. – Asian J., 2011, 6, 1011 CrossRef CAS.
- S. Sakaki, B. Biswas, Y. Musashi and M. Sugimoto, J. Organomet. Chem., 2000, 611, 288 CrossRef CAS.
- S. W. Reilly, G. Akurathi, H. K. Box, H. U. Valle, T. K. Hollis and C. E. Webster, J. Organomet. Chem., 2016, 802, 32 CrossRef CAS.
- C. Medina, K. P. Carter, M. Miller, T. B. Clark and G. W. O’ Neil, J. Org. Chem., 2013, 78, 9093 CrossRef CAS.
- A. Pujol, A. D. Calow, A. S. Batsanov and A. Whiting, Org. Biomol. Chem., 2015, 13, 5122 RSC.
- H. Wu, S. Radomkit, J. M. O’ Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277 CrossRef CAS.
- M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H and 13C NMR spectra of all products (PDF). See DOI: 10.1039/d0ob01806f |
| This journal is © The Royal Society of Chemistry 2020 |